

ABSTRACT
DNA methylation is an epigenetic modification critical to normal genome regulation and development. The vitamin folate is a key source of the one carbon group used to methylate DNA. Because normal mammalian development is dependent on DNA methylation, there is enormous interest in assessing the potential for changes in folate intake to modulate DNA methylation both as a biomarker for folate status and as a mechanistic link to developmental disorders and chronic diseases including cancer. This review highlights the role of DNA methylation in normal genome function, how it can be altered, and the evidence of the role of folate/folic acid in these processes.
Introduction
Folate is an essential water-soluble vitamin occurring naturally in select foods as well as in the synthetic form (folic acid) used in supplements and in food fortification programs (1–3). There are many critical cellular pathways dependent on folate as a 1-carbon source including DNA, RNA, and protein methylation as well as DNA synthesis and maintenance. Folate can be a limiting factor in all these reactions (Fig. 1).
Figure 1.

Folic acid metabolism. This schematic shows the process by which folate/folic acid is used for DNA methylation. The MTHFR 677C→T variant reduces enzyme activity (175) and may help to divert the available methyl groups from the DNA methylation pathway toward the DNA synthesis pathway (176–178). The pathway is complex and highly regulated. with feedback loops and interactions not shown in the schematic. Gene names for enzymes are in italics and cofactors are in parentheses. DHF, dihydrofolate; DHFR, dihydrofolate reductase; DNMT, DNA methyltransferase; dTMP, thymidylate; dUMP, deoxyuridine monophosphate; MS, methionine synthase; MTHFR, methylenetetrahydrofolate reductase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate; TS, thymidylate synthase.
Epigenetics is the study of heritable changes in phenotype or gene expression that do not result from changes in the primary DNA sequence (4, 5). Recognized mechanisms of epigenetic regulation in mammals include DNA methylation, post-translational modification of histones, chromatin remodeling, microRNAs, and long noncoding RNAs (6, 7). These epigenetic regulatory mechanisms modulate chromatin structure and contribute to regulation of the major molecular processes in the nucleus including transcription, replication, repair, and RNA processing. DNA methylation is a covalent modification of genomic DNA that modifies gene expression and provides a mechanism for transmitting and perpetuating epigenetic information through DNA replication and cell division. The role of DNA methylation in cellular regulation has also provided the potential for a new paradigm of disease intervention and treatment. The development of various inhibitors of DNA methylation that alter methylation patterns within intact mammalian cells has led to the clinical use of some inhibitors in experimental therapies for human diseases such as hematological malignancies (8) and myelodysplastic disorders (9).
DNA methylation patterns are stable and are retained in purified genomic DNA; therefore, studies of DNA methylation are amenable to a wide variety of cell-free assays and technologies including DNA methylation analysis at single-nucleotide resolution, next-generation sequencing, and genome-wide methylation profiling (10) (Table 1). Currently, new DNA sequencing technologies are beginning to provide novel insight into genome-wide patterns of DNA methylation (10–13). Although high-resolution truly genome-wide studies have been limited to a very small sample size, a recent study described the DNA methylation level at 1505 individual sites (loci) in 808 genes in 1628 human genomic DNA samples (14). Even this tremendous data set only covers a tiny portion of the genome in a limited number of samples. Despite these recent advances, a basic understanding of normal variation in genomic patterns of DNA methylation in humans across tissues, age, populations, disease, or environmental conditions (including dietary intakes) have not been well described. The methods needed to undertake these types of studies and the infrastructure to do these analyses are rapidly emerging (10, 15, 16).
Table 1.
Representative list of commonly used methodologies for analyzing DNA methylation1
| Description | Pros | Cons | References | |
|---|---|---|---|---|
| Global | ||||
| Liquid chromatography tandem mass spectrometry | Digestion/hydrolysis of genomic DNA to nucleosides and fractionation by LC-MS | Highly sensitive, quantitative, accurate, and reproducible | Requires costly instrumentation; requires ~ 1 μg amounts of DNA | Shellnut et al. (137) |
| HPLC | Digestion/hydrolysis of genomic DNA to nucleotides or bases and fractionation on HPLC | Highly quantitative, accurate, and reproducible | Requires costly instrumentation; requires μg amounts of DNA | Armstrong et al. (186) |
| Methyl acceptance | Global methylation determined by level of in vitro methylation of genomic DNA by SssI methylase | Technical simplicity | Lacks sensitivity; imprecise; weak reproducibility | Balaghi and Wagner (187) |
| Gene specific | ||||
| Methyl-sensitive restriction enzyme digestion | Assesses methylation at specific restriction sites using Southern blotting | Standard molecular biology techniques | Limited to analysis of individual restriction sites; not strictly quantitative | Wolf and Migeon (188) |
| Methylation-specific polymerase chain reaction | Specifically amplifies methylated genomic DNA sequences after sodium bisulfite treatment of genomic DNA | Sensitivity; technical simplicity | Not quantitative; limited sequence coverage | Herman et al. (189) |
| Pyrosequencing | Determines level of methylation at specific CpG sites | Highly quantitative; single-nucleotide resolution; technically straightforward | Requires costly instrumentation; restricted sequence coverage | Tost and Gut (190) |
| Microarray | Large numbers of individual CpG sites from specific genes are assayed using standard microarray equipment after bisulfite treatment; 1500 > 450K sites per sample | Quantitative; specific; technically straightforward; large number of CpGs at a time | Requires costly instrumentation; requires ~0.5 μg DNA | Bibikova et al. (191), Christensen et al. (131), Fernandez et al. (14) |
| Sodium bisulfite genomic sequencing | Determines methylation status of each cytosine in a region of interest within a single DNA molecule | Single-nucleotide resolution; reveals DNA methylation patterns of individual DNA molecules | Labor intensive; time-consuming; restricted sequence coverage | Clark et al. (192) |
| Genome-wide profiling | ||||
| MethylC-seq | Determines methylation status of each cytosine in the genome after chemical conversion of unmethylated cytosines to uracils in genomic DNA | Single-nucleotide resolution; full genome coverage; the most comprehensive method for genome-wide profiling of DNA methylation | Requires sequencing of full genome at ~15-fold coverage; cost | Lister et al. (45) |
| meDIP/meDNA pull-down/MIRA | Uses antimethylated cytosine antibody or methylated DNA-binding protein to enrich for methylated DNA | Successfully used in a wide variety of published studies; technically accessible | Low resolution, not full genome coverage; efficiency dependent on CpG density | Weber et al. (193), Nair et al. (194), Rauch et al. (195) |
| RRBS | A size-selected portion of the genome is isolated after restriction enzyme digestion and sequenced after sodium bisulfite treatment | Single-nucleotide resolution | Biased toward analyzing CpG islands; lacks full genome coverage | Gu et al. (196) |
| CHARM | Uses restriction enzyme that specifically cleaves methylated DNA | Quantitative | Lacks full genome coverage | Irizarry et al. (197) |
| HELP | Compares the relative representation of MspI and HpaII fragments at individual loci | Assays both CpG islands and non-CpG island sites; quantitative | Lacks full genome coverage | Khulan et al. (198) |
| MSCC | Assesses methylation status at all recognition sites of a methylation-sensitive restriction enzyme | High resolution; not restricted to CpG islands | Not strictly quantitative; indirect assessment of methylation by detecting unmethylated CpG sites; lacks full genome coverage | Ball et al. (199) |
For sodium bisulfite conversion used in methylation-specific polymerase chain reaction, pyrosequencing, sodium bisulfite genomic sequencing, full genome sequencing, RRBS, etc., all cytosines except methylated cytosines in genomic DNA are chemically converted uracil by sodium bisulfite; methylated cytosines are resistant to conversion and can be detected by various methods. (This table is not a comprehensive list of methods used to assay DNA methylation.) CHARM, comprehensive high-throughput arrays for relative methylation; HELP, HpaII tiny fragment enrichment by ligation-mediated polymerase chain reaction; HPLC, high-performance liquid chromatography; LC-MS, liquid chromatography-mass spectrometry; meDIP, methylated DNA immunoprecipitation; meDNA, methylated DNA; MIRA, methylated CpG island recovery assay; MSCC, methylation-sensitive cut counting; RRBS, reduced representation bisulfite sequencing.
This review was written to provide the nutritional scientist with a synopsis of the mechanistic aspects of DNA methylation as a background for understanding the potential for the nutrient folate to affect these same molecular processes. Specifically, highlights of the following are covered: the roles and mechanisms of DNA methylation and demethylation; the mechanisms of gene silencing by DNA methylation; the function of DNA methlyation; reprogramming of DNA methylation patterns during development and differentiation; and the importance of changes in DNA methylation (hyper- and hypomethylation). Following this, the potential role of folate in the DNA methylation process and its assessment as a folate status biomarker and link to disease outcomes are covered: folate's role in 1-carbon metabolism related to DNA methylation; low folate status and DNA methylation; background of DNA methylation, cancer, and folate; studies of cancer patients and folate status and global DNA methylation; studies of healthy adults and folate status; changes in DNA methylation in response to the environment and diet—the importance of the developmental timing of exposure; studies of folate in the fetus, infants, cord blood; and high folate and folic acid intake and DNA methylation. The review concludes with a focus on research issues including methodological considerations that are key in planning and interpreting research investigations related to assessing differences in DNA methylation in response to changes in folate status. Clearly there are many challenging research issues that need to be addressed to fill in the gaps in our knowledge related to the potential for folate to modulate DNA methylation and potentially have an impact on development and health maintenance.
Current status of knowledge
Part I: overview of DNA methylation
Introduction to DNA methylation.
Methylation of cytosine is common throughout the human genome. This covalent modification most commonly occurs at cytosines within a 5′-CpG-3′ dinucleotide when a methyl group from S-adenosylmethionine (SAM)6 is enzymatically transferred to the 5 position of cytosine to generate 5-methylcytosine (5-MC) in genomic DNA. DNA methylation patterns are a product of the frequency of cytosine DNA methylation at specific sites along a strand of DNA. Recent genome-wide high-resolution DNA methylation analysis of a primary human fibroblast cell line demonstrated that 4.25% of total cytosines in genomic DNA are methylated, 67.7% of CpGs are methylated, and 99.98% of DNA methylation occurs in CpG dinucleotides (10). Similar analysis of a human embryonic stem cell line showed that 5.83% of cytosines are methylated, 82.7% of CpG dinucleotides are methylated, and 25% of all cytosine methylation occurs at non-CpG sites (10); an earlier study also reported high levels of non-CpG methylation in mouse embryonic stem cells in contrast to methylation patterns in mouse somatic cells (17). Although the importance of CpG methylation is established, the role of non-CpG methylation is a new active area of research.
Interestingly, the frequency of CpG dinucleotides is lower than would be expected throughout most mammalian genomes (18). This is believed to be due to the spontaneous deamination of 5-MC to yield thymine and a cytosine-to-thymidine transition in DNA (19). However, certain regions of the genome have approximately a 10-fold higher frequency of the CpG dinucleotide than the rest of the genome. These regions are referred to as CpG islands and are often correlated with the location of genes, particularly promoter regions and other regulatory regions (20, 21). These islands compose <1% of the genome and are typically unmethylated (Fig. 2). Mapping CpG islands has been used as a tool for gene discovery because >50% of all mammalian genes are associated with CpG islands (19, 22–24). CpG islands were originally defined as genomic regions ∼200 base pairs in size with a C+G content of 50% and an observed CpG/expected CpG >0.6 (20), although this definition is somewhat arbitrary and has been refined more recently to correlate more closely with promoter regions and regulatory regions (21) (Fig. 2). Another functionally important region for DNA methylation are CpG island “shores,” which are CpG-containing regions that are within 2 kb of a CpG island, contain low densities of CpG sites, and have been shown in recent work to be a location of tissue-specific differential methylation in normal tissues (13). Methylation of CpG island shores has been correlated with gene expression, and certain shores have shown altered methylation patterns in colon cancer (13).
Figure 2.

DNA methylation patterns at different types of genetic loci and how the patterns change in cancer and in response to maternal diet. This is a cartoon of a hypothetical single section of double-stranded DNA. Each bar represents a 5′-CpG -3′ site. Methylated CpG are indicated by bars with balls at the end. The green arrow indicates transcription start and active transcription. The red X equals gene silencing. The numbered blue rectangles are exons. (A) DNA methylation patterns conducive to transcription:– patterns set during development. CpGs throughout the genome are targeted for methylation by DNA methyltransferases (DNMT1, DNMT3a, DNMT3b). CpG islands are regions with a concentration of CpGs often (but not always) associated with the promoter regions of genes. CpG islands generally are thought to be actively protected from DNA methylation to allow for appropriate regulation of transcription (179). (B) Dysregulation of DNA methylation seen in cancer post-transformation. Cancer cells have aberrant DNA methylation patterns. Overall DNA methylation levels are reduced (58, 180–182). A subset of genetic sites is hypermethylated, including some tumor suppressor genes; the specific loci hypermethylated vary among types of tumors (114, 131). (C) Maternal diet changes methylation levels at the metastable epiallele, which can change transcription of a neighboring region. Metastable epialleles are regions of the genome that have a variable methylation patterns that are semiheritable across generations, illustrated by the agouti mouse model (147). In mice, a maternal diet that includes additional methyl donors can lead to increased methylation at the metastable epiallele (the intracisternal A particle retrotransposon), a change in expression at nearby genetic loci, and permanent changes in the phenotype distribution of the offspring (147). The sequence of the retrotransposon is a candidate for methylation in the early embryo; however, the extent to which it becomes methylated varies in genetically identical individuals based on maternal diet (methyl donors as well as other dietary factors) (146, 183). A recent study found candidate loci in the human genome that show differential methylation dependent on season of birth (148).
DNA methylation and demethylation: roles and mechanisms.
Methylation at CpG dinucleotides provides a mechanism for transmitting DNA methylation patterns after DNA replication and perpetuating patterns of epigenetic regulation through subsequent cell generations. Methylation of CpG dinucleotides results in symmetrically methylated CpG sites (i.e., methylation of the CpG sequence on both strands of DNA) (as seen in Fig. 2). However, after DNA replication, the 2 resulting daughter DNA molecules become hemimethylated with the parental template strand maintaining its methylation and the newly synthesized strand lacking methylation. Continued DNA replication of these hemimethylated daughter molecules would eventually result in the loss of DNA methylation patterns in subsequent generations. In 1975, Riggs (25) and Holliday and Pugh (26) proposed a scheme by which DNA methylation patterns at CpG dinucleotides could be restored in newly synthesized DNA after DNA replication. They postulated that hemimethylated CpG sites generated by DNA replication could be specifically recognized by a maintenance DNA methyltransferase that would methylate CpG sites on the newly synthesized DNA strand immediately after DNA replication. This would regenerate the patterns of symmetrically methylated CpG dinucleotides in daughter cells and maintain the epigenetic states and patterns of gene regulation that existed in parental cells. In 1983, a DNA methyltransferase (DNMT) (now referred to as DNMT1) was identified that demonstrated the predicted preference for hemimethylated DNA (27, 28). Thus, the general mechanism is now widely accepted as a mechanism for maintaining DNA methylation patterns after DNA replication and cell division, although more recent evidence suggests that maintenance methylation alone may not be sufficient to fully perpetuate DNA methylation patterns genome-wide (29). Nonetheless, maintenance DNA methylation of hemimethylated substrates provides a clear example of a mechanism that allows the transmission and retention of epigenetic information through multiple cell generations. It has been postulated that the availability of folate as a source of methyl groups may affect the ability to maintain DNA methylation patterns in replicating cells (30, 31).
Mammals have 3 types of DNMT: DNMT1, DNMT3a, and DNMT3b (32). DNMT1 is the most abundant DNMT in cells and, as previously described, is believed to act as the primary maintenance methyltransferase to methylate hemimethylated DNA after DNA replication and preserve parental DNA methylation patterns in daughter cells. In contrast, DNMT3a and DNMT3b function as de novo methyltransferases to methylate fully unmethylated CpG sites. They function primarily during mammalian development to establish DNA methylation patterns as epigenetic remodeling and reprogramming proceed during differentiation. DNMT3a and DNMT3b methylate different subsets of DNA sequences in the genome as evidenced by the different phenotypes of mice carrying a knockout of either methyltransferase (32). These de novo methyltransferases also appear to play a role in maintenance methylation to fully preserve parental DNA methylation patterns in daughter cells (29).
Although DNA methylation has usually been regarded as a relatively stable epigenetic mark, there are clear examples where DNA demethylation in mammalian cells occurs in the absence of DNA replication and cell division, suggesting the existence of mechanisms of active DNA demethylation (33). However, studies of DNA demethylation have been controversial and mechanisms of demethylation in mammals still are not well understood (33). One mechanism of passive DNA demethylation involves inhibition or loss of maintenance methylation at CpG sites so that multiple rounds of DNA replication and cell division eventually result in the loss of DNA methylation at these sites in subsequent cell generations. Several mechanisms of active DNA demethylation also have been postulated and reported, but none have been conclusively demonstrated. One potential mechanism of active demethylation involves direct removal of the methyl group via cleavage of a highly stable C-C bond; however, reports demonstrating this mechanism have not been subsequently confirmed or reproduced (34–36). Other potential mechanisms of active demethylation involve base excision repair (BER) of DNA after events such as removal of 5-MC bases in DNA by glycosylase activity or enzymatic deamination of 5-MC to thymidine and formation of a T:G mismatch; in either case, the excised base would be replaced with an unmethylated cytosine by BER (37). Nucleotide excision repair has also been reported to play a role in active DNA demethylation, although other studies seem to contradict this mechanism (38–40). Most recently, the appreciation that notable levels of 5-hydroxymethylcytosine (5-HMC) are present in mammalian genomic DNA and that conversion of 5-MC to 5-HMC in DNA can be catalyzed by mammalian TET (ten-eleven translocation) proteins has generated interest in the possible role of 5-HMC in mechanisms of DNA demethylation (41–43). One potential mechanism of 5-HMC–mediated demethylation would involve recognition and removal of 5-HMC and replacement with an unmethylated cytosine by BER. Thus, what was once regarded as a highly stable and terminal epigenetic state, DNA methylation is becoming increasingly viewed as a more dynamic process. As more genome-wide methylation studies are reported, there is evidence of both increases and decreases in DNA methylation on differentiation (44–46). Furthermore, the finding of variation in DNA methylation between the sexes and changes in methylation during aging suggests that there is more interindividual variation and variation in DNA methylation patterns than was previously expected (47–50).
Mechanisms of gene silencing by DNA methylation.
Methylation of CpG islands, especially those islands colocalized with promoters or other regulatory regions, is generally associated with gene repression. The mechanisms by which DNA methylation silences transcription are not fully understood, although several mechanisms have been proposed. First, methylation of a specific regulatory DNA sequence and the attendant insertion of the methyl group into the major groove of DNA may prevent stable binding of a regulatory protein (e.g., transcription factor) to that sequence, thereby directly preventing gene activation by a transcription factor. In vitro binding studies have identified sequence-specific DNA-binding transcription factors whose interaction with their cognate DNA recognition sequence is negatively affected by methylation of a CpG dinucleotide within the binding sequence (51). However, many transcription factors are not sensitive to DNA methylation and not all DNA sequences bound by a sequence-specific transcription factor will contain a CpG dinucleotide. Therefore, other more indirect mechanisms must also be involved in repressing transcription by DNA methylation. One such indirect mechanism is via DNA-binding proteins containing methylated DNA-binding domains (52). These methylated DNA-binding proteins, such as methyl CpG–binding protein 2 and methylated DNA–binding domain 2, recognize and bind to 1 or more methylated CpG dinucleotides. These proteins in turn interact with or recruit transcriptional silencing complexes containing chromatin remodeling complexes and/or histone modification enzymes (e.g., histone deacetylases) that act to form a transcriptionally repressive condensed chromatin structure at the associated gene (53–55). DNMTs have also been found as components of multisubunit chromatin modifying and remodeling complexes involved in transcriptional repression, suggesting that the DNA methylation machinery also may be recruited to specific genes by other mechanisms (similar to those that recruit histone modification and remodeling activities) and that chromatin modification/remodeling and DNA methylation act cooperatively to silence transcription. In contrast, intragenic DNA methylation of active genes is often higher than in promoter regions, suggesting that methylation in the body of genes is a positive regulator of gene expression; a recent study suggests that this may occur by suppressing the repressive effects of the polycomb complex, which is associated with chromatin remodeling (56).
Function of DNA methylation.
Cytosine methylation has been hypothesized to be an ancient component of the immune system designed to recognize and inactivate parasitic viral DNA sequences that infiltrate the genome (57). Mammalian genomes contain a large proportion of repetitive DNA sequences (e.g., nearly 50% of human genomic DNA) that include retroviral sequences, transposons, retrotransposons long interspersed elements (LINEs), short interspersed elements (SINEs), extended blocks of tandemly repeated DNA sequences (e.g., satellite DNA), etc. Most of the DNA methylation present in mammalian genomes is associated with these repetitive DNA sequences and serves to suppress the potential activity and deleterious effects of repetitive DNA (58–60). This includes suppressing mobility and propagation of repetitive elements capable of transposition and suppressing illegitimate recombination between related repetitive elements (58–60). These functions serve to prevent widespread genome instability and rearrangement mediated by repetitive DNA sequences (61).
It is hypothesized that over time the function of DNA methylation has evolved and now plays additional roles, including transcriptional regulation, X-chromosome inactivation, imprinting, and tissue-specific gene expression (19, 62, 63). Hypermethylation of CpG islands is highly correlated with transcriptional silencing of the associated gene, particularly for CpG islands associated with a gene promoter region (55). One clear example of this is the aberrant stable transcriptional silencing of certain tumor suppressor genes in cancer cells by the hypermethylation of their promoter CpG islands (64). However, methylation of a CpG island does not necessarily lead to gene silencing. For example, the gene for telomerase has been shown to be activated by methylation (65). DNA methylation also occurs within the body of genes and between genes; the function of methylation at CpG sites in these regions still is poorly understood. Also, it is not clear whether methylation of critical CpG sites within CpG islands triggers hypermethylation of the island and gene silencing (66), whether overall levels of hypermethylation within a CpG island govern silencing, or whether both mechanisms contribute to silencing. Recently, in contrast to CpG island methylation, reports indicate that methylation in the body of genes has a role as a positive regulator of transcription (56) and may also have a role in transcription elongation (67), alternative promoter use (68), and nucleosome positioning (69). Generally, DNA methylation is thought to maintain gene silencing versus initiating gene silencing (70) such that a gene is silenced initially by another mechanism and that silencing is stabilized by methylation of associated regulatory regions (e.g., CpG island, promoter region).
X-chromosome inactivation is the process by which female mammals compensate for the unequal dose of X-linked genes between male (XY) and female (XX) cells (71). Females randomly inactivate 1 of the 2 X chromosomes present in each and every somatic cell in a process that occurs in early female embryogenesis, resulting in monoallelic expression of genes subject to X-chromosome inactivation. Once a female cell in early embryogenesis randomly chooses which X chromosome to inactivate (i.e., the maternally or paternally inherited X chromosome), all mitotic progeny of that cell maintain inactivation of the same X chromosome throughout the remainder of development into adult tissues. One of the hallmarks of the inactive X chromosome is the hypermethylation of promoter CpG islands along the length of the inactive X, which contributes to the stable maintenance of transcriptional silencing of the associated genes on the inactive X chromosome (72); these same CpG islands are typically unmethylated on the active X chromosome.
Genomic imprinting is another mammalian epigenetic system of monoallelic gene expression in which the 2 alleles of a particular gene within the same cell are differentially regulated, in this case, in a parent-of-origin fashion (73). Thus, for some imprinted genes, only the paternally inherited allele is expressed, whereas the maternally inherited allele is stably silenced. Conversely, for other imprinted genes, the paternally inherited allele is stably silenced and only the maternally inherited allele is transcriptionally active. This differential monoallelic expression of the maternal or paternal allele is usually associated with differential DNA methylation of 1 or more regulatory regions of these genes. These so-called differentially methylated regions (DMRs) are generally (but not always) hypermethylated on the transcriptionally silenced allele and hypomethylated on the expressed allele. Imprinting control regions that regulate imprinting across an entire domain of imprinted genes are typically DMRs (74, 75). The differential parent-of-origin DNA methylation patterns at imprinted genes are established during gametogenesis where DNA of the male and female gametes acquire either maternal- or paternal-specific methylation patterns before fertilization (although some parent-of-origin methylation patterns may also continue to develop post-zygotically) (Supplemental Fig. 1A) (76). These differential gametic methylation imprints are then maintained and propagated during subsequent somatic development to facilitate the parent-of-origin patterns of monoallelic transcription associated with imprinted genes. Imprinted genes are thought to comprise a small proportion of mammalian genomes with <100 currently confirmed in humans and mice (77). In humans there is a limited number of known imprinted loci (<80), and only a subset of these are shared with the mouse (78, 79). However, a recent study of imprinting in embryonic and adult mouse brains found parent-of-origin bias in the expression of 1300 genes and transcript isoforms and, if confirmed, would suggest that imprinting (or parent-of-origin effects) may be more widespread and complex than originally believed (80).
Reprogramming of DNA methylation patterns during development and differentiation.
DNA methylation patterns undergo remodeling during the course of mammalian differentiation and development to eventually generate the cell type–specific methylation patterns found in adult somatic cells. Dynamic changes in methylation are particularly pronounced during gametogenesis and early embryogenesis (Supplemental Fig. 1). During the course of gametogenesis, DNA methylation patterns are erased (i.e., demethylated) within each developing germ cell (primordial germ cells) and then remethylated and reset to the genomic DNA methylation patterns specific to either the sperm or the egg as gametogenesis proceeds (81, 82). This process is particularly important for establishing the gamete-specific methylation patterns of imprinted genes in sperm and egg before fertilization. Current data suggest that other nonimprinted loci may also be subject to at least partial erasure, and resetting of DNA methylation patterns during gametogenesis (83–85). In early embryogenesis, DNA methylation patterns also undergo a genome-wide process of erasure, that in mouse embryogenesis begins shortly after fertilization and continues to the blastocyst stage (86). This wave of demethylation erases the methylation patterns of the gametes and is followed by DNA remethylation during the remaining course of embryogenesis to establish the genome-wide and cell-type specific DNA methylation patterns found in fully differentiated adult somatic cells (44, 87, 88). In small studies of human embryos, it has been shown that there is a wave of demethylation at the 4-cell stage followed by remethylation beginning as early as the late morula (89). An important exception to this dynamic reprogramming of DNA methylation patterns during embryogenesis occurs with imprinted genes that escape this process of erasure and resetting, thereby maintaining throughout embryogenesis the differential DNA methylation of the paternally and maternally inherited alleles in somatic cells characteristic of most imprinted loci (90).
These periods of dynamic reprogramming of DNA methylation patterns during gametogenesis and embryogenesis may also present windows of opportunity for environmental influences (e.g., diet and nutrition) on the developing embryo to alter the normal process of establishing DNA methylation (and other epigenetic) patterns (91–93). Such changes in DNA methylation patterns could affect normal patterns of gene expression or alter genome stability, thereby leading to an increased risk of diseases later in life, even if these changes in methylation occur in only a subset of cells of a given tissue.
It is also possible to alter DNA methylation levels and patterns within intact mammalian cells by treatment with various chemical inhibitors, most commonly cytidine analogs such 5-azadeoxycytidine. 5-Azadeoxycytidine (and its ribonucleotide analog 5-azacytidine) functions as a demethylating agent (9), resulting in hypomethylation of the genome in treated cells. These cytidine analogs and other DNA-demethylating drugs have recently been introduced as potential therapeutic agents for the treatment of human diseases, particularly myelodysplastic syndromes (9).
What does a change in DNA methylation reflect? Hypermethylation versus hypomethylation.
Any CpG site in a single DNA molecule can either be methylated or unmethylated, so the level of methylation at that site will either be 0% or 100%. However, within a single diploid cell, the methylation level of a given CpG site on 1 strand of DNA can be 0% (symmetrically unmethylated on both chromosomes), 50% (symmetrically methylated on 1 chromosome and unmethylated on the other chromosome, as often seen in DMRs of imprinted genes), or 100% (symmetrically methylated on both chromosomes). DNA methylation assays are usually performed on a population of cells; therefore, the level of methylation at a given CpG site in these assays reflects the collective methylation at this site in multiple cells (and multiple DNA molecules) and the variation in methylation seen at individual CpG sites. Thus, a finding that a particular CpG site is 50% methylated in a population of cells could mean that this site is 50% methylated in every cell (i.e., 1 allele methylated and the other allele unmethylated), that this site is unmethylated in 50% of the cells in the population and fully methylated in the other 50% of cells, or some other combination of methylation levels among cells in the population that yields a composite methylation level of 50%. The different functional consequences of these 3 different scenarios are important.
When considering the implications of changes in the percentage of methylation at a specific CpG site (e.g., in response to dietary conditions), it is important to consider that a 10% change in DNA methylation level at a site could be distributed across an entire population of cells (and not appreciably affect transcription), reflect a 100% change in a subpopulation of cells (and represent gene silencing or activation in these cells), or something in between. Each of these potential scenarios has different implications for the impact of the same 10% change in methylation level.
In the case of CpG islands, gene silencing is generally associated with multiple methylated CpG sites across a strand of DNA (Fig. 2). However, site-specific methylation can result in gene silencing if the methylated CpG blocks the binding of a DNA methylation-sensitive transcription factor. At most genetic loci, it is unknown which CpG sites or how many along the strand of DNA must be methylated to silence any given gene. When evaluating the impact of changes in DNA methylation, it is important to consider the exact location assayed and the potential of methylation at that site or region to change gene expression or other biological processes of interest.
Part II: Folate and DNA methylation
Folate's role in 1-carbon metabolism related to DNA methylation.
Under normal dietary conditions, absorbed folate is metabolized to 5-methyltetrahydrofolate (5-methylTHF, monoglutamyl form) in the intestine and/or liver. 5-MethylTHF is the primary folate constituent taken up by nonhepatic tissues, which then must be polyglutamated for cellular retention and 1 carbon cycle coenzyme function. Tetrahydrofolate (THF) is the most effective substrate for polyglutamate synthetase; therefore, 5-methylTHF must be converted to THF via the methionine synthase reaction (Fig. 1). When folic acid is consumed in fortified foods or supplements, it is metabolized primarily to 5-methylTHF during intestinal absorption and/or first pass in the liver, after which it behaves identically to natural dietary folate. Folic acid is normally first reduced to dihydrofolate by dihydrofolate reductase and subsequently to THF to enter the folate pool (Fig. 1). In some cases, the capacity of dihydrofolate reductase is exceeded and folic acid may appear in the circulation in the oxidized form (94). Once the THF coenzyme is formed from either folic acid or dietary folate, it is first converted to 5,10-methyleneTHF by the vitamin B-6–dependent enzyme serine hydroxymethyltransferase and subsequently irreversibly reduced to 5-methylTHF by methylenetetrahydrofolate reductase (MTHFR). This reaction is key to maintaining the flux of methyl groups for the remethylation of homocysteine to methionine via the vitamin B-12–dependent methionine synthase reaction. Methionine is the substrate for SAM or AdoMet, a cofactor and methyl group donor for numerous methylation reactions including the methylation of DNA, RNA, neurotransmitters, and other small molecules, phospholipids, and proteins, including histones (95). A number of SAM-dependent reactions have regulatory roles by affecting both genome stability and gene transcription (55), localization of protein (96), and small molecule degradation (97).
In addition to folate, a number of other dietary nutrients are required to maintain 1 carbon flux, ensuring normal homocysteine remethylation, SAM formation, and DNA methylation. These nutrients include vitamin B-6 (serine hydroxymethyltransferase activity), riboflavin (MTHFR stability), vitamin B-12 (methionine synthase function), and choline (betaine precursor as a hepatic methyl source via betaine:homocysteine methyltransferase) (98).
The 1-carbon pathway, and thus DNA methylation, functions under tight regulatory controls. SAM is the major regulator of folate-dependent homocysteine remethylation because it is a potent inhibitor of MTHFR. Under the condition of high SAM concentration, MTHFR is inhibited, which reduces the synthesis of 5-methylTHF and hence remethylation of homocysteine. Conversely, when SAM concentrations are low, remethylation of homocysteine is favored. MTHFR activity and thus 5-methylTHF formation may also be modified by the common genetic variant, 677C→T, which reduces enzyme activity (99). It is also recognized that S-adenosylhomocysteine (SAH) functions as a potent product inhibitor of SAM-dependent methyltransferases (100). For this reason, continual hydrolysis of SAH to homocysteine is essential to maintain normal DNA methylation (101). Moderate elevations in plasma homocysteine concentration have been reported to be associated with increased concentration of SAH, but not SAM, and increased SAH concentration has been associated with global DNA hypomethylation (102).
Low folate status and DNA methylation.
Low folate status (as defined by various measures including blood folate concentrations, folate intake, and/or folic acid intake) has been associated with an increased risk of cardiovascular disease, multiple cancers, and neural tube defects (103–105). The mechanisms by which low folate status contributes to these disorders remain unclear. During DNA replication, folate depletion can have destabilizing consequences. Inadequate folate availability during cell division can result in the compromised production of thymidine, such that uracil may be substituted in the DNA sequence (Fig. 1). This mutagenic event may trigger attempts to repair the defect and increases the frequency of chromosomal breaks (104). In tissue culture, it has been shown that low folic acid results in the formation of micronuclei (indicative of chromosome breakage) and that the MTHFR TT genotype leads to increased micronuclei formation under low folate conditions (106).
DNA methylation, cancer, and folate.
Global hypomethylation and targeted hypermethylation are considered defining characteristics of human tumors (14, 23, 58, 107, 108) (Fig. 2B). DNA methylation patterns are widely dysregulated in human cancer (Fig. 2B). Early in the study of DNA methylation, genome-wide hypomethylation was found in tumor tissues compared with matched healthy tissues (58). Additionally, ∼5% of genes were found to be hypermethylated in nonrandom patterns specific to the type of tumor (109). It was determined that these changes in DNA methylation in tumors resulted in silencing of tumor suppressor genes and chromosome instability (Fig. 2B) (110, 111). Hypomethylation of repetitive elements may be predictive of increased risk of cancer and increased mortality from cancer (112). In recent large-scale DNA methylation studies of multiple types of primary tumors (thousands of loci in hundreds of tumors), DNA methylation changes in tumors were associated with concomitant changes in gene expression and tumor characteristics (113, 114). The DNA methylation changes in cancer do not appear to be random because tumors of specific cell lineages show similar DNA methylation changes that are distinct from other tumor types and normal tissues. As tumors increase in severity, a progressive number of loci show increased DNA methylation primarily at CpG islands and decreased DNA methylation in non-CpG islands sites (14). The magnitude of DNA methylation changes observed in cancer tissues is substantially greater than any differences in DNA methylation between types of normal tissue (14).
Folic acid intake has been reported to prevent loss of heterozygosity of a tumor suppressor gene in a small supplementation trial in humans (115). Folate depletion of human tissue culture cells can result not only in the expected global hypomethylation (116, 117) but also targeted hypermethylation of the H-cadherin locus (117). It is unclear whether increases in dietary folate and/or folic acid result in changes in healthy tissues that can predispose to carcinogenesis. Many studies of the effects of folic acid supplementation have focused on either preventing or promoting cancer, especially colon cancer (Supplemental Table 1).
Studies of cancer patients and folate status and global DNA methylation.
There has been great interest in reversing epigenetic changes seen in early tumorigenesis (e.g., global hypomethylation) as rapidly dividing tissue tumors may be susceptible to low folate availability, resulting in global hypomethylation. Three clinical trials examined the impact of folic acid supplementation (0.4–10 mg/d) on global DNA methylation in colon cancer patients (30, 118, 119) (Supplemental Table 1). These studies have shown that DNA methylation might be a biomarker for colorectal cancer and that methylation of DNA in the colon and leukocytes can be increased by supplementation with folic acid in human patients (118, 120). Among the observational studies (e.g., cohort and case-control) of the association of folate status (measured as folate/folic acid intake and or blood folate concentration) and cancer, 5 studies examined the global DNA methylation level (121–125) (Supplemental Table 1). In these studies, an association was found between cancer and global DNA hypomethylation in circulating blood and/or tumor tissue; however, there was not a consistent association between global DNA methylation and folate status (intake or blood folate) across studies (121–125). This lack of a definitive association between low folate status and global DNA hypomethylation could be due to a limited sample size or imprecision of the assays. Across studies outlined in Supplemental Table 1, global hypomethylation was found in tumors and even normal colon cells and blood of cancer patients (121, 122, 124, 126).
The association of global hypomethylation with folate status (e.g., intake, blood folate concentration) has been inconsistent among studies of cancer patients (Supplemental Table 1). This is not unexpected given the variety of study designs, different global DNA methylation assays, and types of tissues examined. There are many techniques available to assess “global” DNA methylation level including those that look at repetitive element methylation level LINE, SINE, and Alu and those that examine various restriction sites throughout the genome. However, the correlation between these measures is actually quite low (127). This is not necessarily due to the accuracy of the measure but due to the fact that although they can be thought of as global, they are actually measuring different types and subsets of DNA sequences in disparate regions of the genome that may be regulated differently. Indeed, recent work suggests that there are different changes in DNA methylation between types of tumors and types of repetitive DNA elements often used as proxies for global methylation (128). Additionally, each of the early trials of global DNA methylation in colon cancer patients used the methyl acceptance assay (30, 118, 119). This assay provides an estimate of global DNA methylation by using an enzyme to methylate purified genomic DNA with radiolabeled tritium. Methyl acceptance is limited in precision and necessitates intact DNA (129) so that the use of damaged DNA (which may occur as a result of the outcome of interest or during preparation or handling) will result in inaccurate results. Careful consideration of assay methodology is critical for study design to ensure appropriate interpretation and reproducibility of the data.
Studies of cancer patients and folate status and site-specific DNA methylation.
For patients with a previous adenoma of the colon, to date, only 1 trial examined the effect of folic acid supplementation (1 mg/d for 3 y) on site-specific DNA methylation, and it was limited to 2 loci (49). In tumors, methylation of tumor suppressor CpG islands is generally either fully methylated and silenced (>80% methylated), or unmethylated (<20% methylated) depending on the exact location of the CpG sites (130). The changes in DNA methylation observed in association with variation in red blood cell (RBC) folate concentration in humans are much smaller than those present in tumors. Wallace et al. (49) evaluated DNA methylation levels at 2 CpG island–containing genes in colon tissue of persons with a previous adenoma after a placebo-controlled folic acid supplementation trial (1 mg/d for 3 y). They found a trend of greater methylation at each genetic locus as the quintile of RBC folate increased [ERα: lowest RBC folate quintile (Q1 43.3–520.9 μg/L) vs. highest RBC folate quintile (Q5 1081.2–2620.8); 10.3% vs. 11.3% methylated; P-trend = 0.03 and SFRP1: Q1 vs. Q5; 21.2% methylated vs. 23.0% methylated P-trend = 0.01]. Although statistically significant, the functional impact of these small changes in DNA methylation (<2%) on gene transcription or longer term health outcomes is unclear at this time. There was no association of DNA methylation at either of these loci with treatment with 1 mg/d folic acid for 3 y, plasma folate concentration, or dietary folate intake. Although age, protein intake, and race were strongly associated with DNA methylation at these loci. In this study, there were no statistically significant associations of methylation at either of these loci with hyperplasic polyps or adenomas after 3 y of treatment.
Two additional observational studies of cancer examined site-specific DNA methylation and folate status (131, 132). Kim et al. (132) examined DNA methylation at 3 genetic loci and found that high plasma folate concentration among cases (≥6.7 μg/L vs. < 4.1 μg/L) was associated with an increased methylation at p73 but not p16in4A or hMLH1. Christensen et al. (131) recently examined a cohort of women with breast cancer and the DNA methylation level at 1413 sites in 733 genes. They found that dietary folate intake was associated with DNA methylation class membership in primary breast tumors (131). This type of study examining thousands of loci simultaneously will likely produce a better understanding of the interaction between DNA methylation and folate, although substantial analytic and informatics challenges exist.
It is important to note that low folate status can lead to both hypo- and hypermethylation, resulting in misregulation of this complex system. The changes observed in cancer do not appear to be random such that not all CpG islands are susceptible to hypermethylation (133, 134). Some of these changes appear to precede transformation (initiation of cancer), and others may be a consequence of transformation. At this time, it is unclear whether subsets of genetic loci are susceptible to DNA methylation changes in response to folate/folic acid intake in cancer patients; the studies highlighted in Supplemental Table 1 show a number of loci that warrant examination in future studies. Large-scale genome-wide analysis may provide additional candidates.
Studies of healthy adults and folate status.
In healthy adults, dividing tissues such as the blood and the gut mucosa are likely to be most susceptible to low folate in the diet. In humans, 3 studies examined the effects of controlled folate depletion and repletion on global DNA methylation in leukocytes (135–137) (Supplemental Table 1). Folate depletion resulted in reductions in global DNA methylation in older women in controlled feeding studies (135, 136), a finding that was not observed by Shelnutt et al. (137) in younger female adults. Both Jacob et al. (136), and Shelnutt et al. (137) observed considerable increases in methylation with folate intake during the repletion period. In the Shelnutt et al. (137) study, the increases were limited to those with the MTHFR TT genotype (see Fig. 1 for location in the folic acid cycle). Axume et al. (138) did not find DNA methylation changes in response to depletion or repletion; however, the study was restricted to those with the MTHFR CC genotype. Although the results from the controlled feeding trials of healthy adults are somewhat inconsistent, these studies are limited by small sample size (n = 8–33 individuals per group), differences in age groups, different initial folate status, and varying lengths of intervention.
Two additional observational studies examined global DNA methylation and folate status in patients without cancer (126, 139) (Supplemental Table 1). Friso et al. (139) found an MTHFR genotype-dependent association between lower global DNA methylation and lower plasma folate concentration. Pufulete et al. (126) observed a trend toward lower DNA methylation with lower serum folate concentration. Taken together, these findings suggest that there may be a differential global DNA methylation response to folate depletion and repletion dependent on age, genotypes, duration, and magnitude of exposure. None of these studies of healthy adults examined site-specific DNA methylation changes; however, in a study of men with hyperhomocystinemia, global hypomethylation was reversed and monoallelic expression of imprinted genes restored in patients given folic acid therapy (140) (Supplemental Table 1).
Changes in DNA methylation in response to the environment and diet: the importance of the developmental timing of exposure.
There is increasing evidence of more interindividual variation and variation in DNA methylation patterns across the life span than was previously expected, and these changes may be modulated by environmental exposures. DNA methylation varies between the sexes and changes during aging (47–50). In addition, an increasing number of environmental exposures in humans such as air pollution (141), benzene exposure (142), and particulate pollution (141) have been linked to changes in DNA methylation (e.g., repetitive element hypomethylation) in adults. Furthermore, Friso et al. (143) found that estrogen replacement resulted in lowering of plasma homocysteine concentration and increases in global methylation in a small (n = 13 postmenopausal women) double-blind, placebo-controlled, double-crossover study, whereas no changes in methylation were found at the ERα, ERβ, and p16 genes. Thus, certain environmental exposures have been associated with changes in DNA methylation and changes in 1-carbon metabolites.
There has been increasing interest in in utero effects of environmental exposures (from toxins to nutrition) on long-term health outcomes, and it has been hypothesized that disease risk may in part be determined by maternal and paternal diet (87, 144, 145). Studies of cancers have found that there are changes in DNA methylation in the blood that reflect the cancer risk in the tissue of interest (119, 124). This may be because dividing tissues are susceptible to the same insults or that the change that resulted in the predisposition to cancer happened in the embryonic period and is reflected in many tissues (Supplemental Fig. 1C).
In mice it is clear that maternal dietary changes in 1-carbon availability can affect the DNA methylation patterns and phenotype of offspring (87). The best-studied example is the agouti mouse. The agouti mouse strain has an insertion of an intracisternal A particle (IAP) sequence in an upstream region of the agouti gene that encodes a paracrine signaling molecule. The agouti gene in this strain is now regulated by the promoter activity of the IAP. Increasing the availability of methyl donors in the mother's diet during pregnancy resulted in increased DNA methylation of the IAP and phenotypic changes associated with altered regulation of the agouti gene in offspring (146, 148). Recently, Waterland et al. (148) found loci in the human genome that also may act as metastable epialleles (similar to the agouti locus in the agouti mouse) that show differential methylation in response to season of birth in a small human study. In both of these studies, the identity of the causative agent for changes in DNA methylation is unclear because there were a pool of methyl donors supplied in the agouti mouse model, and multiple dietary factors were altered in the small human study.
Epigenetic effects of other dietary components are of interest as well. The offspring of male mice fed a low-protein diet (from weaning to sexual maturity) showed numerous although modest (typically 10–20%) changes in DNA methylation in the liver, with altered expression of genes involved in lipid and cholesterol biosynthesis, compared with control offspring of fathers fed a normal protein diet (149). Paternal high-fat diets were reported to reduce DNA methylation at the Il13ra2 gene in mouse pancreatic islet cells of female offspring (150). Interestingly, epigenetic effects of certain environmental conditions in utero appear to be abrogated by dietary folic acid supplementation. In rats, a maternal low-protein diet has been reported to increase DNA methylation of the imprinting control region of the imprinted Igf2/H19 domain in the livers of offspring compared with control offspring whose mothers were fed a normal protein diet. This increase in methylation in response to the low-protein diet was prevented by dietary supplementation of the low-protein diet with folic acid (151). A similar outcome was reported for prenatal exposure to the widespread environmental contaminant bisphenol A (BPA). In the agouti mouse strain, changes in DNA methylation at the agouti locus in response to BPA exposure in utero (via maternal dietary consumption of BPA during pregnancy) were negated by supplementation of the BPA-containing diet with folic acid (152). Thus, certain environmental conditions and diets of parents may have epigenetic effects on their offspring and dietary folic acid can, in some cases, counteract the epigenetic effects of these environmental conditions on offspring. This would suggest the potential for a complex interplay between the effects of maternal and paternal environments and diets on the epigenome of offspring.
Extreme exposures can be used as proof-of-principle to show plausibility of a cause-and-effect relationship. Studies of prenatal starvation have shown an association with many adverse health outcomes including both short-term and long-term consequences: neural tube defects (NTDs), metabolic syndromes, and increased risk of schizophrenia, depending on the precise timing of the exposure and the sex of the fetus (153–158). The period around conception may be one of the more sensitive periods and corresponds to the time when the epigenetic patterns are reset (Supplemental Fig. 1B). It has been hypothesized that epigenetic changes as a result of starvation exposure may be responsible for these effects later in life. However, it is not clear which macronutrient or micronutrient deficiency or combination thereof may be responsible for these health outcomes.
Studies in fetus, infants, cord blood, and folate status.
As a result of the erasure and resetting of DNA methylation patterns in early development and differentiation as well as the rapid growth rate in early development, early development could be particularly susceptible to folate intake. Two studies showed epigenetic changes in individuals prenatally exposed to famine during the Dutch Hunger Winter at the end of World War II (which might be indicative of extreme folate deficiency) (153, 159) (Supplemental Table 1). Heijmans et al. (159) found methylation changes in IGF2 associated with prenatal exposure to prenatal famine. Tobi et al. (153) examined 15 loci important for growth and metabolic disease and found that methylation was lower at INSIGF and higher at IL10, LEP, ABCA1, GNASAS, and MEG3 in persons exposed to famine periconceptionally compared with their sex-matched siblings (all P < 0.001). An interaction with sex was found for INSIGF, LEP, and GNASAS. Exposure to famine at later gestational ages resulted only in changes to GNASAS (P < 0.001). In men only LEP was significantly associated with later exposures.
The association between maternal use of folic acid and DNA methylation in offspring was examined in 1 study. Steegers-Theunissen et al. (160) found that maternal use of folic acid was associated with a 4.5% increase in IGF2 DMR methylation (49.5% vs. 47.4%, P = 0.014) in children. IGF2 DMR methylation in the child was associated with maternal SAM level and inversely with birth weight (−1.7% methylation per SD birth weight, P = 0.034). Although highly significant, the absolute changes observed in these studies were a <3% change in the DNA methylation level of loci that are normally unmethylated. It is unknown whether these DNA methylation changes would result in functionally relevant transcriptional changes.
A recent study by Chang et al. (161) compared the DNA methylation level in various tissues from terminated fetuses (18–28 wk) affected by NTDs with matched normal controls. There were differences in the level and patterns of global DNA methylation between the NTD-affected and control fetuses. There was hypomethylation in the brain of fetuses with NTDs compared with controls (P < 0.01). In addition to the lower mean serum folate concentration in mothers with NTD-affected pregnancies, a correlation between a woman's serum folate concentration and DNA methylation in the brain tissue of NTD-affected fetuses was found (161) (Supplemental Table 1).
Three recent studies examined the relationship between maternal folic acid supplementation in cord blood on methylation of the LINE-1 repetitive element (162), site-specific methylation (27,578 genetic loci) (163), IGF2 promoter methylation (164), and both IGF2 and H19 (165) (Supplemental Table 1). These studies showed no association of folate status (as defined as serum folate concentration or folic acid intake) with global or site- specific DNA methylation (162–164). However, a fourth study by Hoyo et al. (165) found an association between increased folic acid intake and decreased DNA methylation at H19. Additionally, there was an association of increasing maternal plasma homocysteine concentration and decreasing percentage of methylation of LINE-1 (r = −0.688, P = 0.001) (162), and significant correlation between methylation patterns (cluster in the modeling of the site-specific methylation) and plasma homocysteine concentration (P = 0.038), LINE-1 methylation (P = 0.028), and birth weight percentiles (P = 0.019) (163). Additionally, Ba et al. (164) found that maternal and cord blood vitamin B-12 concentrations were associated with P3 (third promoter CpG site) methylation (both P < 0.01). P2 was also associated with maternal weight gain and exposure to smoking (P = 0.03 and P = 0.02).
These studies show the possibility that folate/folic acid in the maternal diet could change DNA methylation levels in the offspring. However, these relationships are complex and can involve other components of the diet and/or substrates from 1-carbon metabolism, suggesting no definitive linear associations of folate/folic acid intake in the mother with increases in DNA methylation in the child.
High folate and folic acid intake and DNA methylation.
The purpose of many of the controlled feeding trials was to attempt to reverse the aberrant DNA methylation observed in cancer. The effect of folate insufficiency is clearly detrimental both to the embryo and its short-term risk of NTDs and the possible longer term risks of diabetes or other health outcomes (156, 157, 166, 167). Additionally, high blood folate concentrations (121) and high global DNA methylation have been shown in a number of studies to reduce the risk of cancer (123, 125). However, recent focus has shifted to concern about folic acid supplementation resulting in progression of existing tumors and altering normal DNA methylation patterns (31, 168, 169). Currently, there is no direct evidence of aberrant DNA methylation and change in gene expression in response to “high” levels of folate/folic acid intake. In addition, there is no consensus on a dose of folic acid or a blood folate concentration that would be associated with potential adverse health outcomes.
Measurement of DNA methylation is not indicative of a single process or phenomenon. Different regions of the genome are differentially regulated and would not be expected to respond similarly to any given exposure. DNA methylation is a highly regulated process dependent on time, tissue, DNA sequence, region of the genome, and a multitude of other regulated enzymes and proteins. Simple correlations, such as high folate status leading to increases in DNA methylation, are unlikely to apply broadly; indeed the conflicting results presented in Supplemental Table 1 illustrate the complexity of the process.
In the very limited number of studies to date, folic acid supplement use was not associated with the complete methylation and silencing of CpG island–associated tumor suppressor genes observed in cancer among adults (49) or children (160). Promoter regions of genes are highly regulated and CpG islands in promoters generally remain protected from DNA methylation under normal circumstances. Even in tumors, loci susceptible to hypermethylation vary among tumor types (108, 113, 114, 131, 170, 171). As previously discussed, a recent study by Hoyo et al. (165) examined the DNA methylation level in the cord blood of newly delivered infants at the H19 DMR and found significantly decreased DNA methylation level (toward the expected 50% level) with increasing maternal folic acid intake in pregnancy, suggesting to Hoyo et al. that folic acid intake in a fortified population may provide additional benefit. At this time, there are insufficient data to determine whether there is an effect of higher doses of folic acid at any particular locus, genomic region, specific tissue type, or developmental state and whether the change would result in increased risk or benefit.
Part III: Future research
Although several studies have begun to examine the relationship between DNA methylation and folate (Supplemental Table 1), many studies are underpowered in terms of the number of participants, tissues types, and number of epigenetic loci examined. None of the findings associating folate status (indicated by folate/folic acid intake or blood folate concentration) with changes in global or site-specific DNA methylation have been consistently replicated. It remains an open question whether folate/folic acid intake is associated with changes in DNA methylation and whether there is an interaction with other micronutrients or genetic variation. Even in experimental animal models that show changes in DNA methylation after increased intake of 1-carbon sources, it is unknown whether the effects are due to folic acid or one of the other 1-carbon sources (e.g., betaine, choline) or a combination thereof (147). Because folate is not the only vitamin in the 1-carbon cycle and DNA methylation is not the only methylation reaction that may affect health, studies designed to evaluate interactions are needed.
As technology enables the greater use of genome-wide analyses of DNA methylation at single nucleotide resolution, the previously held dogma surrounding aspects of DNA methylation and folate status may require modification. No population-based, randomized, controlled trial has examined the effects of folic acid on DNA methylation levels and patterns in healthy humans. In planning new studies, it is important to consider the appropriate assay, tissue to test, type of DNA methylation change anticipated, the folate/folic acid exposure of interest, DNA methylation patterning, and the timing of the folate/folic acid exposure (Table 2).
Table 2.
Questions to consider when planning or evaluating a study on the effect of folic acid or folate on DNA methylation
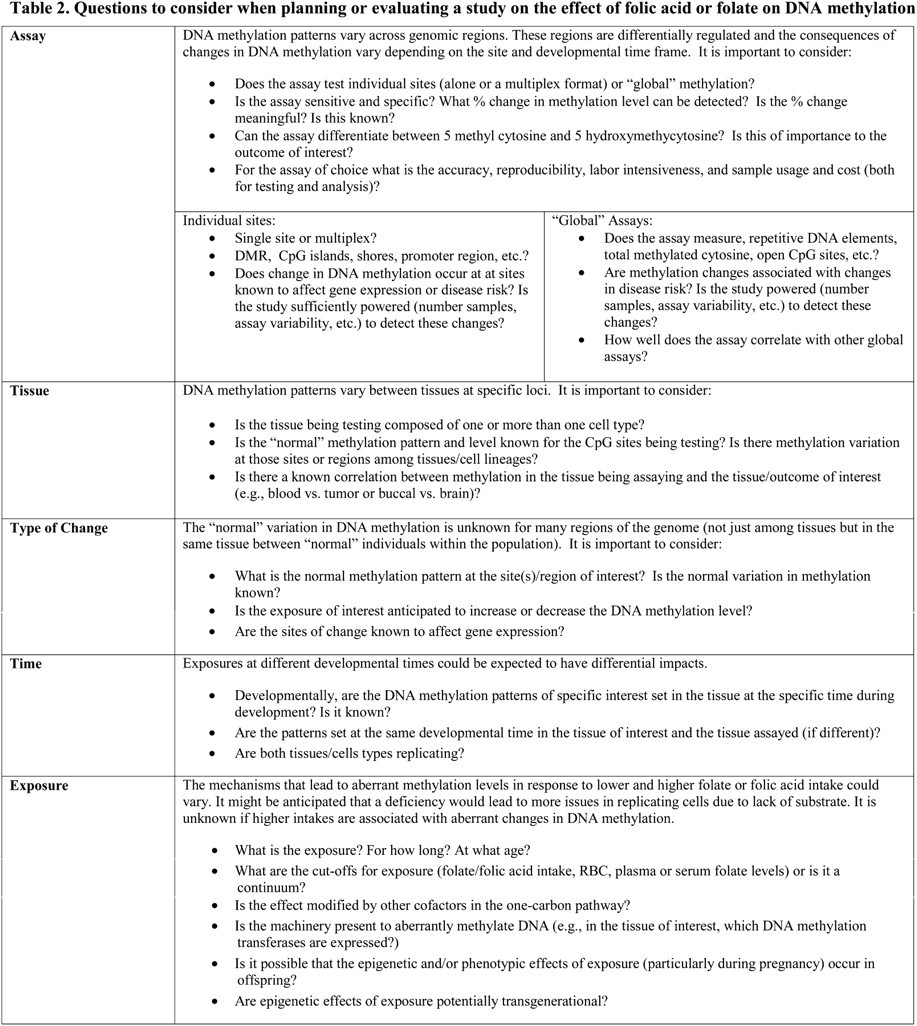
Enumerable questions about the relationship between folate status and DNA methylation need further exploration and include the following: What is the normal variation in DNA methylation level and pattern across the genome and between tissues and developmental stages? Are there systematic differences in DNA methylation between different populations that vary with micronutrient intake? Are there health consequences due to the small changes in DNA methylation levels/patterns observed in many of the existing studies? How does folate status interact with other nutrients to affect DNA methylation level/patterning and does this vary depending on the developmental timing or tissue assayed? How does DNA methylation vary in relation to timing and dose of folate or specifically folic acid intake?
Conclusions
Appropriate DNA methylation patterns are critical to normal genome function. Aberrant DNA methylation patterns are present in many human diseases, including cancer, imprinting disorders, and developmental disabilities (172–174). The human studies of DNA methylation and folate/folic intake acid vary widely in their study design, timing of exposure, and amount of folate/folic acid intake, tissue tested, assays, and, not surprisingly, the findings. At this time, the evidence suggests that low folate status is associated with decreases in global DNA methylation, which has in some studies has been associated with an increased risk of cancer. However, it is unclear how specific regions of the genome respond to higher or lower folate intakes. To date, the majority of studies have only examined a limited number of genetic loci and/or a small number of samples. There is no direct evidence that high dietary folate or folic acid intake leads to aberrant DNA methylation, changes in gene expression, or disease state. Given the conflicting results to date, it is clear that the current research does not support a linear relationship or dose response between folic acid supplementation and global or site-specific DNA methylation level. This is not unexpected given that DNA methylation is part of a complex, highly regulated system. Additional research is needed to elucidate the relationship between folate and DNA methylation.
Supplementary Material
Timing of DNA Methylation Patterning. The DNA methylation patterns in an individual are determined during critical windows during development. Many of the details of DNA methylation patterning during human development remain unknown. The purpose of this figure is to provide a very broad overview and illustrate (1) the times during development that DNA methylation patterns are set, and thus times that may be particularly sensitive and (2) the possible tissues impacted at specific developmental windows and the distance in time between exposures of interest and the impact on an individual. A. Imprinting patterns are set in the gamete of the parent, starting during the parents embryonic period; the precise timing for each loci is still under investigation (184). The DNA methylation patterns at imprinted loci may reflect exposures experienced by the grandmother when the parent was in utero through the gametogenesis period until fertilization, if the changes resulting from an exposure are inherited like a somatic mutation, this is under active investigation. B. The DNA methylation patterns of the majority of the genome are re-patterned in the early embryo (89). Exposures during this small window of time in the days after conception may be reflected in many tissues depending on the exact timing and affected cell lineages. C. Throughout development, a limited subset of loci change methylation patterns as cells differentiate (88, 185). Exposures later in development would only affect those limited loci that are differentiating in the affected tissues.
Studies in humans that have examined the association between folate/folic acid intake or blood folate levels and DNA methylation
Abbreviations
- BER
base excision repair
- BPA
bisphenol A
- DMR
differentially methylated region
- DNMT
DNA methyltransferase
- 5-HMC
5-hydroxymethylcytosine
- IAP
intracisternal A particle
- 5-MC
5-methylcytosine
- 5-methylTHF
5-methyltetrahydrofolate
- LINE
long interspersed element
- MTHFR
methylenetetrahydrofolate reductase
- NTD
neural tube defect
- RBC
red blood cell
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SINE
short interspersed element
- THF
tetrahydrofolate
Footnotes
Supplemental Table 1 and Supplemental Figure 1 are available from the “Online Supporting Material” link in the online posting of the article and from the same link in the online table of contents at http://advances.nutrition.org.
Literature Cited
- 1. Eitenmiller R, Landen W. Folate : REaW L.editor Vitamin Analysis for the Health and Food Science. Boca Raton: CRC Press; 1999. p. 411–65. [Google Scholar]
- 2. O'Broin JD, Temperley IJ, Brown JP, Scott JM. Nutritional stability of various naturally occurring monoglutamate derivatives of folic acid. Am J Clin Nutr. 1975;28:438–44. [DOI] [PubMed] [Google Scholar]
- 3. Berry RJ, Bailey L, Mulinare J, Bower C. Fortification of flour with folic acid. Food Nutr Bull. 2010;31:S22–35. [DOI] [PubMed] [Google Scholar]
- 4. Fazzari MJ, Greally JM. Introduction to epigenomics and epigenome-wide analysis. Methods Mol Biol. 2010;620:243–65. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. [DOI] [PubMed] [Google Scholar]
- 6. Kundu S, Peterson CL. Role of chromatin states in transcriptional memory. Biochim Biophys Acta. 2009;1790:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2009;66:596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. . Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. . Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colaneri A, Staffa N, Fargo DC, Gao Y, Wang T, Peddada SD, Birnbaumer L. Expanded methyl-sensitive cut counting reveals hypomethylation as an epigenetic state that highlights functional sequences of the genome. Proc Natl Acad Sci U S A. 2011;108:9715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. . The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandez AF, Assenov Y, Martin-Subero J, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC, Galm O, et al. . A DNA methylation fingerprint of 1628 human samples. Genome Res. Epub 2011 Jul 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang S, Lu Y, Jelinek J, Estecio M, Li H, Issa JP. Analysis of epigenetic modifications by next generation sequencing. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:6730. [DOI] [PubMed] [Google Scholar]
- 16. Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. . Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2011;28:1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sved J, Bird A. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci U S A. 1990;87:4692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalgo ML, Jones PA. Mutagenic and epigenetic effects of DNA methylation. Mutat Res. 1997;386:107–18. [DOI] [PubMed] [Google Scholar]
- 20. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. [DOI] [PubMed] [Google Scholar]
- 21. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr Biol. 1999;9:R661–7. [DOI] [PubMed] [Google Scholar]
- 23. Illingworth RS, Bird AP. CpG islands–'a rough guide'. FEBS Lett. 2009;583:1713–20. [DOI] [PubMed] [Google Scholar]
- 24. Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–71. [DOI] [PubMed] [Google Scholar]
- 25. Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. [DOI] [PubMed] [Google Scholar]
- 26. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- 27. Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A. 1983;80:5559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971–83. [DOI] [PubMed] [Google Scholar]
- 29. Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cravo M, Fidalgo P, Pereira AD, Gouveia-Oliveira A, Chaves P, Selhub J, Mason JB, Mira FC, Leitao CN. DNA methylation as an intermediate biomarker in colorectal cancer: modulation by folic acid supplementation. Eur J Cancer Prev. 1994;3:473–9. [DOI] [PubMed] [Google Scholar]
- 31. Smith AD, Kim YI, Refsum H. Is folic acid good for everyone? Am J Clin Nutr. 2008;87:517–33. [DOI] [PubMed] [Google Scholar]
- 32. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. [DOI] [PubMed] [Google Scholar]
- 33. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiss A, Keshet I, Razin A, Cedar H. DNA demethylation in vitro: involvement of RNA. Cell. 1996;86:709–18. [DOI] [PubMed] [Google Scholar]
- 35. Swisher JF, Rand E, Cedar H, Marie Pyle A. Analysis of putative RNase sensitivity and protease insensitivity of demethylation activity in extracts from rat myoblasts. Nucleic Acids Res. 1998;26:5573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–83. [DOI] [PubMed] [Google Scholar]
- 37. Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends Genet. 2009;25:82–90. [DOI] [PubMed] [Google Scholar]
- 38. Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, et al. . Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–5. [DOI] [PubMed] [Google Scholar]
- 39. Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin SG, Guo C, Pfeifer GP. GADD45A does not promote DNA demethylation. PLoS Genet. 2008;4:e1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. . Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Véron N, Peters AH. Epigenetics: Tet proteins in the limelight. Nature. 2011;473:293–4. [DOI] [PubMed] [Google Scholar]
- 44. Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. . Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. . Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;2011:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamagata K, Okada Y.. Understanding paternal genome demethylation through live-cell imaging and siRNA. Cell Mol Life Sci. 2011;2011:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. El-Maarri O, Becker T, Junen J, Manzoor SS, Diaz-Lacava A, Schwaab R, Wienker T, Oldenburg J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet. 2007;122:505–14. [DOI] [PubMed] [Google Scholar]
- 48. Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wallace K, Grau MV, Levine AJ, Shen LL, Hamdan R, Chen XL, Gui JA, Haile RW, Barry EL, Ahnen D, et al. . Association between Folate Levels and CpG Island Hypermethylation in Normal Colorectal Mucosa. Cancer Prev Res (Phila). 2010;3:1552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. . Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–31. [DOI] [PubMed] [Google Scholar]
- 52. Dhasarathy A, Wade PA. The MBD protein family-reading an epigenetic mark? Mutat Res. 2008;647:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001;15:827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. [DOI] [PubMed] [Google Scholar]
- 55. Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–90. [DOI] [PubMed] [Google Scholar]
- 56. Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Das R, Hampton DD, Jirtle RL. Imprinting evolution and human health. Mamm Genome. 2009;20:563–72. [DOI] [PubMed] [Google Scholar]
- 58. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. [DOI] [PubMed] [Google Scholar]
- 59. Ostertag EM, Kazazian HH Jr. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–38. [DOI] [PubMed] [Google Scholar]
- 60. Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23:8841–6. [DOI] [PubMed] [Google Scholar]
- 61. Jones PA, Gonzalgo ML. Altered DNA methylation and genome instability: a new pathway to cancer? Proc Natl Acad Sci U S A. 1997;94:2103–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–40. [DOI] [PubMed] [Google Scholar]
- 63. Razin A, Kafri T. DNA methylation from embryo to adult. Prog Nucleic Acid Res Mol Biol. 1994;48:53–81. [DOI] [PubMed] [Google Scholar]
- 64. Taberlay PC, Jones PA. DNA methylation and cancer. Prog Drug Res. 2011;67:1–23. [DOI] [PubMed] [Google Scholar]
- 65. Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002;101:335–41. [DOI] [PubMed] [Google Scholar]
- 66. Chen C, Yang MC, Yang TP. Evidence that silencing of the HPRT promoter by DNA methylation is mediated by critical CpG sites. J Biol Chem. 2001;276:320–8. [DOI] [PubMed] [Google Scholar]
- 67. Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–75. [DOI] [PubMed] [Google Scholar]
- 68. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. . Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. . Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Weber M, Schubeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–80. [DOI] [PubMed] [Google Scholar]
- 71. Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–72. [DOI] [PubMed] [Google Scholar]
- 72. Hornstra IK, Yang TP. High-resolution methylation analysis of the human hypoxanthine phosphoribosyltransferase gene 5′ region on the active and inactive X chromosomes: correlation with binding sites for transcription factors. Mol Cell Biol. 1994;14:1419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. [DOI] [PubMed] [Google Scholar]
- 74. Hirasawa R, Feil R. Genomic imprinting and human disease. Essays Biochem. 2010;48:187–200. [DOI] [PubMed] [Google Scholar]
- 75. Hudson QJ, Kulinski TM, Huetter SP, Barlow DP. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity. 2010;105:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. El-Maarri O, Buiting K, Peery EG, Kroisel PM, Balaban B, Wagner K, Urman B, Heyd J, Lich C, Brannan CI, et al. . Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet. 2001;27:341–4. [DOI] [PubMed] [Google Scholar]
- 77. Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Moore GE, Oakey R. The role of imprinted genes in humans. Genome Biol. 2011;12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morcos L, Ge B, Koka V, Lam KC, Pokholok DK, Gunderson KL, Montpetit A, Verlaan DJ, Pastinen T. Genome-wide assessment of imprinted expression in human cells. Genome Biol. 2011;12:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 2010;329:682–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. [DOI] [PubMed] [Google Scholar]
- 82. Szabó PE, Hübner K, Schüler H, Mann JR. Allele-specific expression of imprinted genes in mouse migratory primordial germ cells. Mech Dev. 2002;115:157–60. [DOI] [PubMed] [Google Scholar]
- 83. Maatouk DM, Kellam LD, Mann MR, Lei H, Li E, Bartolomei MS, Resnick JL. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development. 2006;133:3411–8. [DOI] [PubMed] [Google Scholar]
- 84. Lees-Murdock DJ, De Felici M, Walsh CP. Methylation dynamics of repetitive DNA elements in the mouse germ cell lineage. Genomics. 2003;82:230–7. [DOI] [PubMed] [Google Scholar]
- 85. Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet. 2008;9:129–40. [DOI] [PubMed] [Google Scholar]
- 86. Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Waterland RA. Early environmental effects on epigenetic regulation in humans. Epigenetics. 2009;4:523–5. [DOI] [PubMed] [Google Scholar]
- 88. Waterland RA, Kellermayer R, Rached MT, Tatevian N, Gomes MV, Zhang J, Zhang L, Chakravarty A, Zhu W, Laritsky E, et al. . Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum Mol Genet. 2009;18:3026–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fulka H, Mrazek M, Tepla O, Fulka J Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction. 2004;128:703–8. [DOI] [PubMed] [Google Scholar]
- 90. Feil R. Epigenetic asymmetry in the zygote and mammalian development. Int J Dev Biol. 2009;53:191–201. [DOI] [PubMed] [Google Scholar]
- 91. Dolinoy DC, Das R, Weidman JR, Jirtle RL. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res. 2007;61:30R–7R. [DOI] [PubMed] [Google Scholar]
- 92. Swanson JM, Entringer S, Buss C, Wadhwa PD. Developmental origins of health and disease: environmental exposures. Semin Reprod Med. 2009;27:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bailey RL, Mills JL, Yetley EA, Gahche JJ, Pfeiffer CM, Dwyer JT, Dodd KW, Sempos CT, Betz JM, Picciano MF. Unmetabolized serum folic acid and its relation to folic acid intake from diet and supplements in a nationally representative sample of adults aged > or =60 y in the United States. Am J Clin Nutr. 2010;92:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Stover PJ. One-carbon metabolism-genome interactions in folate-associated pathologies. J Nutr. 2009;139:2402–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Winter-Vann AM, Kamen BA, Bergo MO, Young SG, Melnyk S, James SJ, Casey PJ. Targeting Ras signaling through inhibition of carboxyl methylation: an unexpected property of methotrexate. Proc Natl Acad Sci U S A. 2003;100:6529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stead LM, Jacobs RL, Brosnan ME, Brosnan JT. Methylation demand and homocysteine metabolism. Adv Enzyme Regul. 2004;44:321–33. [DOI] [PubMed] [Google Scholar]
- 98. Shin W, Yan J, Abratte CM, Vermeylen F, Caudill MA. Choline intake exceeding current dietary recommendations preserves markers of cellular methylation in a genetic subgroup of folate-compromised men. J Nutr. 2010;140:975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bailey LB. editor Folate in health and disease. Boca Raton, FL: Taylor and Francis; 2009. [Google Scholar]
- 100. Hoffman DR, Marion DW, Cornatzer WE, Duerre JA. S-Adenosylmethionine and S-adenosylhomocystein metabolism in isolated rat liver. Effects of L-methionine, L-homocystein, and adenosine. J Biol Chem. 1980;255:10822–7. [PubMed] [Google Scholar]
- 101. James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr. 2002;132:2361S–6. [DOI] [PubMed] [Google Scholar]
- 102. Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and kymphocyte DNA hypomethylation. J Biol Chem. 2000;102:29318–23. [DOI] [PubMed] [Google Scholar]
- 103. Erickson JD. Folic acid and prevention of spina bifida and anencephaly. 10 years after the U.S. Public Health Service recommendation. MMWR Recomm Rep. 2002;51:1–3. [PubMed] [Google Scholar]
- 104. Lamprecht SA, Lipkin M. Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nat Rev Cancer. 2003;3:601–14. [DOI] [PubMed] [Google Scholar]
- 105. Holmquist C, Larsson S, Wolk A, de Faire U. Multivitamin supplements are inversely associated with risk of myocardial infarction in men and women–Stockholm Heart Epidemiology Program (SHEEP). J Nutr. 2003;133:2650–4. [DOI] [PubMed] [Google Scholar]
- 106. Kimura M, Umegaki K, Higuchi M, Thomas P, Fenech M. Methylenetetrahydrofolate reductase C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in cultured human lymphocytes. J Nutr. 2004;134:48–56. [DOI] [PubMed] [Google Scholar]
- 107. Issa J-PJ. DNA Methylation as a Therapeutic Target in Cancer. 2008. p. 643–9. [DOI] [PubMed] [Google Scholar]
- 108. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, et al. . Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8. [DOI] [PubMed] [Google Scholar]
- 110. Herman JG, Baylin SB. Promoter-region hypermethylation and gene silencing in human cancer. Curr Top Microbiol Immunol. 2000;249:35–54. [DOI] [PubMed] [Google Scholar]
- 111. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. [DOI] [PubMed] [Google Scholar]
- 112. Zhu ZZ, Sparrow D, Hou L, Tarantini L, Bollati V, Litonjua AA, Zanobetti A, Vokonas P, Wright RO, Baccarelli A, et al. . Repetitive element hypomethylation in blood leukocyte DNA and cancer incidence, prevalence, and mortality in elderly individuals: the Normative Aging Study. Cancer Causes Control. 2011;22:437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. . Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011 Mar 23;3:75ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, Kim DH, Son HJ, Rhee PL, Kim JJ, et al. . Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol. 2011. ;2011:2338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nagothu KK, Jaszewski R, Moragoda L, Rishi AK, Finkenauer R, Tobi M, Naumoff JA, Dhar R, Ehrinpreis M, Kucuk O, et al. . Folic acid mediated attenuation of loss of heterozygosity of DCC tumor suppressor gene in the colonic mucosa of patients with colorectal adenomas. Cancer Detect Prev. 2003;27:297–304. [DOI] [PubMed] [Google Scholar]
- 116. Wasson GR, McGlynn AP, McNulty H, O'Reilly SL, McKelvey-Martin VJ, McKerr G, Strain JJ, Scott J, Downes CS. Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J Nutr. 2006;136:2748–53. [DOI] [PubMed] [Google Scholar]
- 117. Jhaveri MS, Wagner C, Trepel JB. Impact of extracellular folate levels on global gene expression. Mol Pharmacol. 2001;60:1288–95. [DOI] [PubMed] [Google Scholar]
- 118. Cravo ML, Pinto AG, Chaves P, Cruz JA, Lage P, Nobre Leitao C, Costa Mira F. Effect of folate supplementation on DNA methylation of rectal mucosa in patients with colonic adenomas: correlation with nutrient intake. Clin Nutr. 1998;17:45–9. [DOI] [PubMed] [Google Scholar]
- 119. Pufulete M, Al-Ghnaniem R, Khushal A, Appleby P, Harris N, Gout S, Emery PW, Sanders TAB. Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut. 2005;54:648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cravo M, Pinto R, Fidalgo P, Chaves P, Gloria L, Nobre-Leitao C, Costa Mira F. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut. 1996;39:434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pufulete M, Al-Ghnaniem R, Leather AJ, Appleby P, Gout S, Terry C, Emery PW, Sanders TA. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: a case control study. Gastroenterology. 2003;124:1240–8. [DOI] [PubMed] [Google Scholar]
- 122. Piyathilake CJ, Azrad M, Jhala D, Macaluso M, Kabagambe EK, Brill I, Niveleau A, Jhala N, Grizzle WE. Mandatory fortification with folic acid in the United States is not associated with changes in the degree or the pattern of global DNA methylation in cells involved in cervical carcinogenesis. Cancer Biomark. 2006;2:259–66. [DOI] [PubMed] [Google Scholar]
- 123. Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, Kelsey KT. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16:108–14. [DOI] [PubMed] [Google Scholar]
- 124. Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, Garcia-Closas R, Chanock S, Tardon A, Serra C, et al. . Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol. 2008;9:359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Piyathilake CJ, Macaluso M, Alvarez RD, Chen M, Badiga S, Siddiqui NR, Edberg JC, Partridge EE, Johanning GL. A higher degree of LINE-1 methylation in peripheral blood mononuclear cells, a one-carbon nutrient related epigenetic alteration, is associated with a lower risk of developing cervical intraepithelial neoplasia. Nutrition. 2011;27:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pufulete M, Al-Ghnaniem R, Rennie JA, Appleby P, Harris N, Gout S, Emery PW, Sanders TA. Influence of folate status on genomic DNA methylation in colonic mucosa of subjects without colorectal adenoma or cancer. Br J Cancer. 2005;92:838–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wu HC, Delgado-Cruzata L, Flom JD, Kappil M, Ferris JS, Liao Y, Santella RM, Terry MB. Global methylation profiles in DNA from different blood cell types. Epigenetics. 2011;6:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int J Cancer. 2009;125:723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nephew KP, Balch C, Skalnik DG. Methyl group acceptance assay for the determination of global DNA methylation levels. Methods Mol Biol. 2009;507:35–41. [DOI] [PubMed] [Google Scholar]
- 130. Levine JJ, Stimson-Crider KM, Vertino PM. Effects of methylation on expression of TMS1/ASC in human breast cancer cells. Oncogene. 2003;22:3475–88. [DOI] [PubMed] [Google Scholar]
- 131. Christensen BC, Kelsey KT, Zheng SC, Houseman EA, Marsit CJ, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Kushi LH, et al. . Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6:e100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim JW, Park HM, Choi YK, Chong SY, Oh D, Kim NK. Polymorphisms in genes involved in folate metabolism and plasma DNA methylation in colorectal cancer patients. Oncol Rep. 2011;25:167–72. [PubMed] [Google Scholar]
- 133. Issa JP. Opinion: CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. [DOI] [PubMed] [Google Scholar]
- 134. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa J-PJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Rampersaud GC, Kauwell GP, Hutson AD, Cerda JJ, Bailey LB. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr. 2000;72:998–1003. [DOI] [PubMed] [Google Scholar]
- 136. Jacob RA, Gretz DM, Taylor PC, James SJ, Pogribny IP, Miller BJ, Henning SM, Swendseid ME. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr. 1998;128:1204–12. [DOI] [PubMed] [Google Scholar]
- 137. Shelnutt KP, Kauwell GP, Gregory JF 3rd, Maneval DR, Quinlivan EP, Theriaque DW, Henderson GN, Bailey LB. Methylenetetrahydrofolate reductase 677C→T polymorphism affects DNA methylation in response to controlled folate intake in young women. J Nutr Biochem. 2004;15:554–60. [DOI] [PubMed] [Google Scholar]
- 138. Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. The methylenetetrahydrofolate reductase 677TT genotype and folate intake interact to lower global leukocyte DNA methylation in young Mexican American women. Nutr Res. 2007;27:1365–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, et al. . A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99:5606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ingrosso D, Cimmino A, Perna AF, Masella L, De Santo NG, De Bonis ML, Vacca M, D'Esposito M, D'Urso M, Galletti P, et al. . Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003;361:1693–9. [DOI] [PubMed] [Google Scholar]
- 141. Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, Tarantini L, Schwartz J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119: 977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, et al. . Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–80. [DOI] [PubMed] [Google Scholar]
- 143. Friso S, Lamon-Fava S, Jang H, Schaefer EJ, Corrocher R, Choi SW. Oestrogen replacement therapy reduces total plasma homocysteine and enhances genomic DNA methylation in postmenopausal women. Br J Nutr. 2007;97:617–21. [DOI] [PubMed] [Google Scholar]
- 144. Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–6. [DOI] [PubMed] [Google Scholar]
- 145. Barker DJ. A new model for the origins of chronic disease. Med Health Care Philos. 2001;4:31–5. [DOI] [PubMed] [Google Scholar]
- 146. Waterland RA. Do Maternal Methyl Supplements in Mice Affect DNA Methylation of Offspring? J Nutr. 2003;133:238. [DOI] [PubMed] [Google Scholar]
- 147. Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, Zhang W, Torskaya MS, Zhang J, Shen L, et al. . Season of conception in rural Gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, et al. . Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–6. [DOI] [PubMed] [Google Scholar]
- 151. Gong L, Pan YX, Chen H. Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics. 2010;5:619–26. [DOI] [PubMed] [Google Scholar]
- 152. Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lumey LH, Stein AD, Kahn HS, Romijn JA. Lipid profiles in middle-aged men and women after famine exposure during gestation: the Dutch Hunger Winter Families Study. Am J Clin Nutr. 2009;89:1737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kahn HS, Graff M, Stein AD, Lumey LH. A fingerprint marker from early gestation associated with diabetes in middle age: the Dutch Hunger Winter Families Study. Int J Epidemiol. 2009;38:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. de Rooij SR, Painter RC, Phillips DI, Osmond C, Michels RP, Godsland IF, Bossuyt PM, Bleker OP, Roseboom TJ. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care. 2006;29:1897–901. [DOI] [PubMed] [Google Scholar]
- 157. Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. 2006;82:485–91. [DOI] [PubMed] [Google Scholar]
- 158. Lumey LH, Stein AD, Susser E. Prenatal famine and adult health. Annu Rev Public Health. 2011;32:237–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS ONE. 2009;4:e7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, Bao Y, Li Y, Wu L, Zheng X, et al. . Tissue-specific distribution of aberrant DNA methylation associated with maternal low-folate status in human neural tube defects. J Nutr Biochem. Epub 2011 Feb 16. [DOI] [PubMed] [Google Scholar]
- 162. Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics. 2009;4:394–8. [DOI] [PubMed] [Google Scholar]
- 163. Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, Farrell WE. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics. Jan 1;6:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ba Y, Yu H, Liu F, Geng X, Zhu C, Zhu Q, Zheng T, Ma S, Wang G, Li Z, et al. . Relationship of folate, vitamin B(12) and methylation of insulin-like growth factor-II in maternal and cord blood. Eur J Clin Nutr. 2011;65:480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Hoyo C, Murtha AP, Schildkraut JM, Jirtle RL, Demark-Wahnefried W, Forman MR, Iversen ES, Kurtzberg J, Overcash F, Huang Z, et al. . Methylation variation at IGF2 differentially methylated regions and maternal folic acid use before and during pregnancy. Epigenetics. 2011;6:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Berry RJ, Li Z, Erickson JD, Li S, Moore CA, Wang H, Mulinare J, Zhao P, Wong LY, Gindler J, et al. . Prevention of neural-tube defects with folic acid in China. China-U.S. Collaborative Project for Neural Tube Defect Prevention. N Engl J Med. 1999;341:1485–90. [DOI] [PubMed] [Google Scholar]
- 167. Painter RC, de Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, Bleker OP, Roseboom TJ. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2006;84:322–7, quiz 466–7. [DOI] [PubMed] [Google Scholar]
- 168. Ulrich CM, Potter JD. Folate and cancer–timing is everything. JAMA. 2007;297:2408–9. [DOI] [PubMed] [Google Scholar]
- 169. Kim YI. Folic acid supplementation and cancer risk: point. Cancer Epidemiol Biomarkers Prev. 2008;17:2220–5. [DOI] [PubMed] [Google Scholar]
- 170. Rauch TA, Zhong X, Wu X, Wang M, Kernstine KH, Wang Z, Riggs AD, Pfeifer GP. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci U S A. 2008;105:252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tellez CS, Shen L, Estecio MR, Jelinek J, Gershenwald JE, Issa JP. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009;19:146–55. [DOI] [PubMed] [Google Scholar]
- 172. Warren ST. The epigenetics of fragile X syndrome. Cell Stem Cell. 2007;1:488–9. [DOI] [PubMed] [Google Scholar]
- 173. Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. [DOI] [PubMed] [Google Scholar]
- 174. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. [DOI] [PubMed] [Google Scholar]
- 175. Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Huevel LP, et al. . A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–3. [DOI] [PubMed] [Google Scholar]
- 176. Green R, Miller JW. Folate deficiency beyond megaloblastic anemia: hyperhomocysteinemia and other manifestations of dysfunctional folate status. Semin Hematol. 1999;36:47–64. [PubMed] [Google Scholar]
- 177. Hoffbrand AV, Jackson BF. Correction of the DNA synthesis defect in vitamin B12 deficiency by tetrahydrofolate: evidence in favour of the methyl-folate trap hypothesis as the cause of megaloblastic anaemia in vitamin B12 deficiency. Br J Haematol. 1993;83:643–7. [DOI] [PubMed] [Google Scholar]
- 178. Quinlivan EP, Davis SR, Shelnutt KP, Henderson GN, Ghandour H, Shane B, Selhub J, Bailey LB, Stacpoole PW, Gregory JF III. Methylenetetrahydrofolate reductase 677C->T polymorphism and folate status affect one-carbon incorporation into human DNA deoxynucleosides. J Nutr. 2005;135:389–96. [DOI] [PubMed] [Google Scholar]
- 179. Stimson KM, Vertino PM. Methylation-mediated silencing of TMS1/ASC is accompanied by histone hypoacetylation and CpG island-localized changes in chromatin architecture. J Biol Chem. 2002;277:4951–8. [DOI] [PubMed] [Google Scholar]
- 180. Jackson K, Yu MC, Arakawa K, Fiala E, Youn B, Fiegl H, Muller-Holzner E, Widschwendter M, Ehrlich M. DNA hypomethylation is prevalent even in low-grade breast cancers. Cancer Biol Ther. 2004;3:1225–31. [DOI] [PubMed] [Google Scholar]
- 181. Narayan A, Ji W, Zhang XY, Marrogi A, Graff JR, Baylin SB, Ehrlich M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer. 1998;77:833–8. [DOI] [PubMed] [Google Scholar]
- 182. Frigola J, Sole X, Paz MF, Moreno V, Esteller M, Capella G, Peinado MA. Differential DNA hypermethylation and hypomethylation signatures in colorectal cancer. Hum Mol Genet. 2005;145:319–26. [DOI] [PubMed] [Google Scholar]
- 183. Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kota SK, Feil R. Epigenetic transitions in germ cell development and meiosis. Dev Cell. 2010;19:675–86. [DOI] [PubMed] [Google Scholar]
- 185. Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, Andrews TD, Howe JK, Otto T, Olek A, Howe KL, et al. . DNA methylation profiling of the human major histocompatibility complex: a pilot study for the Human Epigenome Project. PLoS Biol. 2004;2:e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Armstrong KM, Bermingham EN, Bassett SA, Treloar BP, Roy NC, Barnett MP. Global DNA methylation measurement by HPLC using low amounts of DNA. Biotechnol J. 2011;6:113–7. [DOI] [PubMed] [Google Scholar]
- 187. Balaghi M, Wagner C. DNA methylation in folate deficiency: use of CpG methylase. Biochem Biophys Res Commun. 1993;193:1184–90. [DOI] [PubMed] [Google Scholar]
- 188. Wolf SF, Migeon BR. Studies of X chromosome DNA methylation in normal human cells. Nature. 1982;295:667–71. [DOI] [PubMed] [Google Scholar]
- 189. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007;2:2265–75. [DOI] [PubMed] [Google Scholar]
- 191. Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, et al. . High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–62. [DOI] [PubMed] [Google Scholar]
- 194. Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MW, Clark SJ. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics. 2011;6:34–44. [DOI] [PubMed] [Google Scholar]
- 195. Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–47. [DOI] [PubMed] [Google Scholar]
- 196. Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6:468–81. [DOI] [PubMed] [Google Scholar]
- 197. Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, Wen B, Feinberg AP. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008;18:780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, Figueroa ME, Glass JL, Chen Q, Montagna C, et al. . Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Timing of DNA Methylation Patterning. The DNA methylation patterns in an individual are determined during critical windows during development. Many of the details of DNA methylation patterning during human development remain unknown. The purpose of this figure is to provide a very broad overview and illustrate (1) the times during development that DNA methylation patterns are set, and thus times that may be particularly sensitive and (2) the possible tissues impacted at specific developmental windows and the distance in time between exposures of interest and the impact on an individual. A. Imprinting patterns are set in the gamete of the parent, starting during the parents embryonic period; the precise timing for each loci is still under investigation (184). The DNA methylation patterns at imprinted loci may reflect exposures experienced by the grandmother when the parent was in utero through the gametogenesis period until fertilization, if the changes resulting from an exposure are inherited like a somatic mutation, this is under active investigation. B. The DNA methylation patterns of the majority of the genome are re-patterned in the early embryo (89). Exposures during this small window of time in the days after conception may be reflected in many tissues depending on the exact timing and affected cell lineages. C. Throughout development, a limited subset of loci change methylation patterns as cells differentiate (88, 185). Exposures later in development would only affect those limited loci that are differentiating in the affected tissues.
Studies in humans that have examined the association between folate/folic acid intake or blood folate levels and DNA methylation