

Abstract
Although adenophostin A (AdA), the most potent agonist of d-myo-inositol 1,4,5-trisphosphate receptors (IP3R), is thought to mimic IP3, the relative roles of the different phosphate groups and the adenosine motif have not been established. We synthesized all three possible bisphosphate analogues of AdA and glucose 3,4-bisphosphate (7, AdA lacking the 2′-AMP). 2′-Dephospho-AdA (6) was prepared via a novel regioselective dephosphorylation strategy. Assessment of the abilities of these bisphosphates to stimulate intracellular Ca2+ release using recombinant rat type 1 IP3R (IP3R1) revealed that 6, a mimic of Ins(4,5)P2, is only 4-fold less potent than IP3, while 7 is some 400-fold weaker and even 3″-dephospho-AdA (5) is measurably active, despite missing one of the vicinal bisphosphate groups normally thought to be crucial for IP3-like activity. Compound 6 is the most potent bisphosphate yet discovered with activity at IP3R. Thus, adenosine has a direct role independent of the 2′-phosphate group in contributing toward the potency of adenophostins, the vicinal bisphosphate motif is not essential for activity at the IP3R, as always thought, and it is possible to design potent agonists with just two of the three phosphates. A model with a possible adenine–R504 interaction supports the activity of 5 and 6 and also allows a reappraisal of the unexpected activity previously reported for the AdA regioisomer 2″-phospho-3″-dephospho-AdA 40.
Introduction
d-myo-Inositol 1,4,5-trisphosphate receptors (IP3Rs) are Ca2+ channels located on the endoplasmic reticulum.1 IP3 [Ins(1,4,5)P3, 1] is a second messenger produced by the action of phospholipase C on phosphatidylinositol 4,5-bisphosphate in response to various extracellular signals. IP3 binds to its receptor and opens its intrinsic Ca2+ channel, allowing Ca2+ to leak into the cytosol and so cause the increase in cytosolic [Ca2+] that regulates many cellular events.2 To study the interaction of 1 with its receptor and to understand structure–activity relationships (SARs), many synthetic analogues of IP3 have been synthesized.3 These studies revealed that the 4,5-bisphosphate functionality and 6-OH group are apparently crucial for activity, while the 1-phosphate has a supplementary role, and the 2-OH and 3-OH groups are much less important.4 Although the relative importance of all of the hydroxyl and phosphate groups has been established, none of the synthetic analogues has proven to be more potent than the natural ligand. In 1993, two potent agonists of IP3 receptors, adenophostin A (3a, Figure 1) and adenophostin B (3b), were isolated from culture broths of Penicillium brevicompactum.5 Both 3a and 3b bind to IP3 receptors with much greater affinity than IP3 and are 10–100 times more potent than IP3 in evoking Ca2+ release.6 This finding has stimulated many syntheses of various analogues of the adenophostins and studies of their SARs.7−10
Figure 1.

Structures of IP3 (1), Ins(4,5)P2 (2), and adenophostins (3a and 3b).
SAR studies with synthetic analogues of adenophostin A (AdA) with and without a purine ring established that the presence of the adenine ring (or an aromatic surrogate) is crucial for enhanced affinity.10 This suggests that the adenosine moiety either disposes the 2′-phosphate in a special spatial arrangement to strengthen its interactions with the receptor (indirect role)11 or that the adenine moiety itself is involved in supplementary binding interactions with a nearby region of the binding site (direct role).12 It has been shown that the glucose 2″-OH group (analogous to 6-OH in IP3) is important, while the glucose 5″-CH2OH (analogous to the 3-OH in IP3) and ribose 4′-CH2OH are less important for AdA activity.13,14 The roles of the phosphate groups of IP3 seem to be established,4,14 but their relative importance in the adenophostins has not been examined systematically using synthetic chemistry. We have proposed a model for AdA binding to the IP3-binding core (IBC) of the receptor in which the glucose 2″-OH and 3″,4″-bisphosphate triad mimic the 6-OH and 4,5-bisphosphate triad of IP3, while the adenine engages in a cation-π interaction with Arg504 adjacent to the binding site. In this model, AdA takes on a 2′-endo anti extended binding conformation.7 In addition, the ribose 2′-phosphate may have a stabilizing helix–dipole interaction with the N terminus of an α-helix in the IBC.15 We recently reviewed aspects of the chemistry and biology of AdA in its interaction with IP3R16 and examined the thermodynamics of both IP3 and AdA binding to both the N terminus and IBC of the IP3R.17 AdA binds to both proteins with significantly higher affinity than does IP3.
Bisphosphate 6 (Figure 2) was previously made by enzymatic hydrolysis of AdA using alkaline phosphatase and was reported to be 1800 times weaker than AdA.18 This implied a critical role for the 2′-phosphate group and led some workers to conclude that the adenine had only an indirect role, by enhancing the positioning of the 2′-phosphate group. However, this finding seems incompatible with our suggestion based on detailed SAR and molecular modeling7,19 that the adenine may form its own interactions with the IP3R. Furthermore, inframolecular protonation studies of AdA suggest that, at least in solution, the 2′-P is close to the glucose ring and the three phosphate groups behave similarly to those in IP3.20 Hence, we were curious to re-examine the biological activity of 6 with a chemically synthesized sample and to compare its activity with a minimal motif of AdA, glucose 3,4-bisphosphate [Gluc(3,4)P2,7]. Quantitative insight into the contribution of the adenosine moiety might be obtained by comparison of the activities of 6 and 7, which differ only by the presence of the adenosine moiety.
Figure 2.

Bisphosphate analogues of AdA.
Thus, to explore our model further and to investigate whether the extra binding motif of AdA as compared to IP3 might compensate for removal of a phosphate interaction, we report the synthesis and biological evaluation of all three possible bisphosphates of AdA. A preliminary communication on this work has appeared,21 and we recently reported the ability of 6 to evoke Ca2+ release via recombinant and mutant IP3Rs.19
Results and Discussion
For the synthesis of adenophostin analogues, an ideal and economical strategy would be the Vorbrüggen condensation of a silylated purine with an appropriately protected disaccharide, followed by deprotection of hydroxyl groups (to be later phosphorylated), phosphorylation, and final deprotection.22,23 It is a prerequisite to have ester functionalities at 1-O- and 2-O-positions of the disaccharide for the Vorbrüggen condensation.24 Other hydroxyl groups on the disaccharide have to be protected with orthogonal protecting groups, such that the hydroxyls to be phosphorylated are protected with an easily removable temporary protecting group and the other hydroxyls are protected with a stable protecting group that can be cleaved at the final step. Because the nucleoside after Vorbrüggen condensation will have an ester protecting group at the 2′-O-position (which has to be phosphorylated in 4 and 5) and the fact that this ester can be removed under milder conditions, it is ideal to protect the other hydroxyl to be phosphorylated as a similar ester. Benzyl is the protecting group of choice for all other hydroxyl groups as it is stable to various conditions and also due to the convenience in its deprotection along with other benzyl protecting groups on phosphate triesters in a single final step.
The synthesis of 4 started from the cheaply available penta-O-acetyl-β-d-glucose (8, Scheme 1). The glycosyl donor 15 was prepared from 8 by a series of protecting group manipulations. The butane 2,3-diacetal (BDA) derivative 9 was obtained from 8 as reported previously.25 Diol 9 on benzylation gave the dibenzyl ether 10 in excellent yield. The acid-labile BDA protecting group was removed by hydrolysis with 80% acetic acid. Various conditions were tried to achieve selective benzylation at the 2-OH in 11. Stannylene-mediated benzylation was sluggish and gave a 1:1 mixture of isomeric tribenzyl ethers 12 (27%) and 13 (23%) along with unreacted 11 (36%). Sodium hydride-mediated benzylation also gave a 1:1 mixture of tribenzyl ethers 12 (18%) and 13 (16%) in very low yield along with fully benzylated 14 (32%) and starting material 11 (24%). Fortunately, benzylation under biphasic conditions with tetrabutylammonium hydrogen sulfate as phase transfer catalyst gave predominantly the tribenzyl ether 12 (74%) along with minor amounts of 13 (8%) and 14 (10%). Acetylation of 12 provided the acetate 15 in 95% yield. N-Iodosuccinimide (NIS)-mediated glycosylation of 5-O-benzyl-1,2-O-isopropylidene-α-d-ribofuranose (16), which was prepared from 1,2-O-isopropylidene-α-d-xylofuranose by a known procedure26 with glycosyl donor 15, gave the α-glycoside 17 selectively. No detectable amount of the β-isomer was obtained. Such a high degree of selectivity has been observed before when structurally similar compounds were used.25,27,28 To make the disaccharide donor for Vorbrüggen glycosylation, the isopropylidene group in 17 was hydrolyzed with 90% trifluoroacetic acid (TFA), and the resulting mixture of diols (anomeric mixture) was acetylated under standard conditions to produce a chromatographically inseparable α/β mixture of triacetates 18. Vorbrüggen condensation of 18 with silylated 6-chloropurine gave the β-nucleoside 19 as the exclusive product. The yield of the condensation depended on the nature of the silylating agents used for the persilylation of purine. Among the various silylating agents used, N,O-bis-trimethylsilyl-acetamide (BSA) gave a very good yield for the subsequent same pot glycosylation. Removal of the acetate protecting groups in nucleoside 19 and substitution of chlorine with an amino group were achieved concomitantly by ammonolysis. The diol 20 was then selectively phosphitylated with dibenzyl N,N-diisopropyl phosphoramidite in the presence of imidazolium triflate as catalyst,29 and the resulting bisphosphite was oxidized in situ to bisphosphate 21. Finally, all of the benzyl protecting groups (on both the phosphate and the sugar) were removed by transfer hydrogenolysis30 to provide the bisphosphate 4, which was purified by ion exchange chromatography.
Scheme 1. Synthesis of 4″-Dephospho-adenophostin A (4).

Reagents and conditions: (a) BnBr, NaH, DMF, 0 °C–room temperature, 2 h. (b) 80% aqueous HOAc, reflux, 1 h. (c) Bu2SnO, BnBr, Bu4NBr, MeCN, 3 Å molecular sieves, reflux, 24 h. (d) Bu4NHSO4, BnBr, DCM: 5% aqueous NaOH (1:1), room temperature. (e) Ac2O, pyr, room temperature, 12 h. (f) Compound 16, NIS, TMSOTf, dioxane:toluene (3:1 v/v), 3 Å molecular sieves, room temperature, 30 min. (g) 90% TFA, room temperature, 15 min. (h) Ac2O, pyr, room temperature, 3 h. (i) 6-Chloropurine, BSA, TMSOTf, MeCN, 70 °C, overnight. (j) NH3, EtOH, 70 °C, 5 days. (k) (1) (BnO)2PN(iPr)2, ImOTf, DCM, room temperature, 30 min; (2) mCPBA, −78 °C–room temperature, 1 h. (l) Cyclohexene, Pd(OH)2, MeOH, H2O, 80 °C, overnight.
Synthesis of bisphosphate 5 started with commercially available tetra-acetate 22 (Scheme 2). The acetate protecting groups on 22 were removed by methanolysis with a catalytic amount of sodium methoxide in methanol, and the resulting crude tetrol was converted into diol 23 by benzylidenation using benzaldehyde dimethyl acetal and camphorsulphonic acid (CSA). Crude 23 was then benzylated with an excess of benzyl bromide and sodium hydride to afford the dibenzyl ether 24. Reductive opening of the benzylidene acetal by following a known procedure31 gave the tribenzyl ether 25(32) selectively. Glycosylation of ribose derivative 16 with the glycosyl donor 26, obtained by acetylation of the tribenzyl ether 25, gave α-glycoside 27 selectively. As in the previous case, no detectable amount of β-isomer was obtained. Disaccharide 27 was converted into the triacetate 28 (α- and β-anomer) in a one pot reaction of hydrolysis of acetonide with TFA followed by acetylation. Vorbrüggen condensation with 6-chloropurine gave the nucleoside 29, which on ammonolysis provided the diol 30. Chemoselective phosphitylation followed by in situ oxidation afforded the fully protected bisphosphate 31. Transfer hydrogenolysis removed all of the benzyl protecting groups to provide the bisphosphate 5, which was purified by ion exchange chromatography.
Scheme 2. Synthesis of 3″-Dephospho-adenophostin A (5).

Reagents and conditions: (a) MeOH, NaOMe, room temperature, 30 min. (b) Dowex-H+. (c) PhCH(OMe)2, CSA, DMF, 70 °C. (d) BnBr, NaH, DMF, room temperature, 2 h. (e) TFA, Et3SiH, DCM, 0 °C. (f) Ac2O, pyr, room temperature, 3 h. (g) Compound 16, NIS, TMSOTf, dioxane:toluene, 3 Å molecular sieves, room temperature, 30 min. (h) 90% TFA, room temperature, 10 min. (i) 6-Chloropurine, BSA, TMSOTf, MeCN, 70 °C, overnight. (j) NH3, EtOH, 74 °C, 5 days. (k) (1) (BnO)2PN(iPr)2, ImOTf, DCM, room temperature, 30 min; (2) mCPBA, −78 °C–room temperature, 1 h. (l) Cyclohexene, Pd(OH)2, MeOH, H2O, 80 °C, overnight.
The requirement that the 1-O- and 2-O-positions of the disaccharide should be protected as esters for Vorbrüggen condensation leaves an ester group at the 2′-O-position of nucleosides 19 and 29. With the next step being the substitution of 6-Cl with NH2 and deprotection of the hydroxyl groups to be phosphorylated, the ester functionality at 2′-O- position is advantageous, as treatment with NH3 effects both of the transformations. The other hydroxyl group to be phosphorylated (3″ in 18 and 4″ in 28) was also protected as its acetate for convenient one-step deprotection. This strategy offered an economical synthesis of 4 and 5. However, it cannot be applied to the synthesis of bisphosphate 6 as there is no phosphate at the 2′-O-position. This prompted us to explore other possibilities.
During our synthesis of the AdA analogue, guanophostin,15 several attempts to substitute the chlorine in the intermediate 32 with different oxygen nucleophiles under basic conditions [4 M NaOH, dioxane; 3-hydroxypropiononitrile, 1,8-diaza-bicycloundec-7-ene (DBU), dichloromethane (DCM);33 3-hydroxypropiononitrile, NaH, tetrahydrofuran (THF); BnOH, K2CO334] were marred by accompanied dephosphorylation. For instance, when 3-hydroxypropiononitrile in the presence of a base (DBU or NaH) was used, it was observed that at first all of the benzyl groups were displaced by the 2-cyanoethoxy group, which underwent β-elimination to give water-soluble phosphate monoesters. Prolonged treatment with the oxygen nucleophile resulted in P–O–sugar cleavage, also giving all of the possible mono-, bis-, and tris-phosphates. Possible transesterification pathways for a protected sugar phosphate are shown in Figure 4. Because the P–O–sugar also underwent cleavage, we were curious to investigate the possible selective dephosphorylation at the 2′-position from a protected trisphosphate such as 33 (Scheme 3) via transesterification with an alcohol. We envisioned that if we used the incoming nucleophile (RO–), the same as the protecting group on the phosphorus (benzyl), the unwanted loss of protecting group on phosphorus can be prevented. Thus, benzyl alcohol was our agent of interest for transesterification.
Figure 4.

Various possible transesterification mechanisms for phosphate triesters. Only the first step is shown. The intermediates can undergo further transesterification.
Scheme 3. Synthesis of 2′-Dephospho-adenophostin A (6).

Reagents and conditions: (a) BnOH, NaH, room temperature, 30 min. (b) BnOH, K2CO3, 70 °C, overnight. (c) Pd(OH)2, cyclohexene, MeOH, H2O, 80 °C, overnight. (d) R–NH2 (R = H, Bu, Pr), EtOH, room temperature.
Figure 3.

Base-mediated dephosphorylation.
Trisphosphate 33 was made in 12 steps from 8 by following the reported procedure.35 Trisphosphate 33 on treatment with in situ-generated sodium benzoxide [benzyl alcohol (5 equiv) and NaH (2 equiv)] in N,N-dimethylformamide (DMF) at room temperature for 30 min gave a bisphosphate as major product with minor amounts of other byproducts. Fortunately, the use of the milder base potassium carbonate and BnOH as solvent resulted in the exclusive formation of this bisphosphate in excellent yield. A detailed spectral analysis revealed that the bisphosphate is the expected 34. The fact that the 2′-phosphate is comparatively less crowded and hence more prone to nucleophilic attack than those in the 3″,4″ vicinal bisphosphate functionality could be the reason for this high selectivity. This assumption is supported by the fact that triacetate 35 on aminolysis with various amines gives the 2′-O-deacetylated derivative 36 exclusively. Finally, the benzyl protecting groups on both sugar and phosphorus were removed by transfer hydrogenolysis and purification of the product by ion exchange column chromatography provided bisphosphate 6 quantitatively.
For biological comparison with 6, the previously unknown glucose 3,4-bisphosphate 7 was synthesized from allyl glycoside 37 (Scheme 4).36 Phosphorylation of 37 using standard phosphoramidite methodology37gave the fully protected bisphosphate 38, and the allyl protecting group was removed using PdCl2 in methanol38 to give bisphosphate 39 as a mixture of α- and β-anomers. Hydrogenolysis of 39 then provided the bisphosphate 7, and 1H NMR and 31P NMR spectroscopy showed this to exist as a mixture of α- and β-anomers in aqueous or methanolic solution. The relative proportions of the two anomers were dependent on salt form and pH.
Scheme 4. Synthesis of Glucose 3,4-Bisphosphate (7).

Reagents and conditions: (a) (1) (BnO)2PN(iPr)2, 1H-tetrazole, DCM, room temperature, 1 h; (2) mCPBA, −78 °C, 10 min, 92%. (b) PdCl2, MeOH, 81%. (c) H2, Pd(OH)2, MeOH, H2O, 85%.
The ability of bisphosphates 4–7, IP3 (1), AdA (3a), and d-myo-inositol 4,5-bisphosphate [Ins(4,5)P2 (2)] to stimulate the IP3R of intracellular Ca2+ stores was measured using a low-affinity Ca2+ indicator trapped within the intracellular stores of permeabilized DT40 cells expressing only recombinant rat IP3R1, as previously reported.19 The amount of Ca2+ released by each of these compounds was expressed as a fraction of the total Ca2+ content of the endoplasmic reticulum (ER) as assessed by the addition of ionomycin. Results are shown in Table 1.
Table 1. Ca2+ Release by IP3R1 Mediated by Compounds 1–7a.
| ligand | EC50 (nM) | nHill | % release |
|---|---|---|---|
| IP3 (1) | 23 ± 4 | 1.00 ± 0.12 | 74 ± 2 |
| AdA (3a) | 2.8 ± 0.4 | 0.87 ± 0.09 | 73 ± 2 |
| 2′-dephospho AdA (6) | 107 ± 15 | 1.79 ± 0.41 | 67 ± 3 |
| 3″-dephospho AdA (5) | 53030 ± 6249* | 1.31 ± 0.36 | 53 ± 8 at 50 μM |
| 4″-dephospho AdA (4) | ND | ND | 8% release at 50 μM |
| Gluc(3,4)P2 (7) | 25190 ± 4782 | 1.04 ± 0.17 | 72 ± 4 |
| Ins(4,5)P2 (2) | 8634 ± 1005 | 0.86 ± 0.07 | 73 ± 10 |
Results show the half-maximal effective concentration of each ligand (EC50), the Hill coefficient (nHill), and the percentage of the intracellular stores released by a maximal concentration of each ligand. Results are means ± SEMs from eight independent experiments. The EC50 for 5, where even the maximal practicable concentration (50 μM) failed fully to release the IP3-sensitive Ca2+ stores, was calculated by assuming that it was a full agonist capable of releasing the entire IP3-sensitive Ca2+ store.
Among the bisphosphates, 6 is the most potent; it releases the same fraction of the Ca2+ stores as other full agonists, and it is only 40 times less potent than AdA and only four times less potent than IP3. This contradicts an earlier report18 where 6 was found to be 1800-fold less potent than AdA, although this is now thought to be in error (see ref (19) for discussion). Bisphosphate 4 released only 8% of the Ca2+ stores at a concentration of 50 μM. It was impracticable to assess the activity at higher concentrations and so impossible to resolve whether 4 is a full agonist with very low affinity or a partial agonist. Bisphosphate 5 had some activity, but even at 50 μM, it released only 53% of the intracellular stores. Again, it was impracticable to increase the concentration of 5 sufficiently to assess whether it is a full agonist, capable at a high enough concentration, of mimicking the response to a maximal concentration of AdA or IP3. Assuming 5 to be a full agonist, then it is 2300 times less potent than IP3. Gluc(3,4)P2 (7) is approximately 1000 times less potent than IP3. This is broadly in agreement with an earlier report,39 where Ins(4,5)P2 was 460-fold less potent than IP3 in evoking Ca2+ release from rat brain microsomes. When compared in permeabilized DT40 cells (Table 1), synthetic Ins(4,5)P2 from the present study is ca. 375-fold less potent than IP3 but ca. 3-fold more potent than 7. The marginally weaker activity of 7 as compared to Ins(4,5)P2 is not surprising, given the existence of 7 as a mixture of anomers. The 7-β isomer mimics four of the IP3 carbon centers retaining the important triad binding motif (4,5-bisphosphate and 6-OH); hence, 7-β might be more or less active as IP2 (Figure 5). However, 7-α can orient in two different ways (A and B; Figure 5), mimicking the orientation of IP3. Although orientation A has the binding motif in full, it mimics only three carbons of IP3, while orientation B mimics four centers of IP3 but lacks the 6-OH mimic of the binding motif. While it is reasonable to expect that the mode with maximum resemblance to IP3 (B) would be the preferred fit, the association constant could be less due to the lack of 6-OH interaction with the receptor. Competition between these two orientations (A and B) for receptor binding could reduce the overall potency. The ratio of 7-α to 7-β at the receptor is unknown.
Figure 5.

Structural comparison of Ins(4,5)P2 (2), 7, and IP3.
AdA binds to a site that at least substantially overlaps the IP3-binding site and in a manner that is thought to be broadly similar to IP3.10 The crystal structure41 of IP3 bound to the IBC (residues 224–604) of IP3R1 and our model7 for AdA binding together allow a rationalization of the biological activities of the bisphosphates of adenophostin A. The IBC comprises an α-domain and β-domain, with IP3 sandwiched between the two domains (Figure 6).
Figure 6.

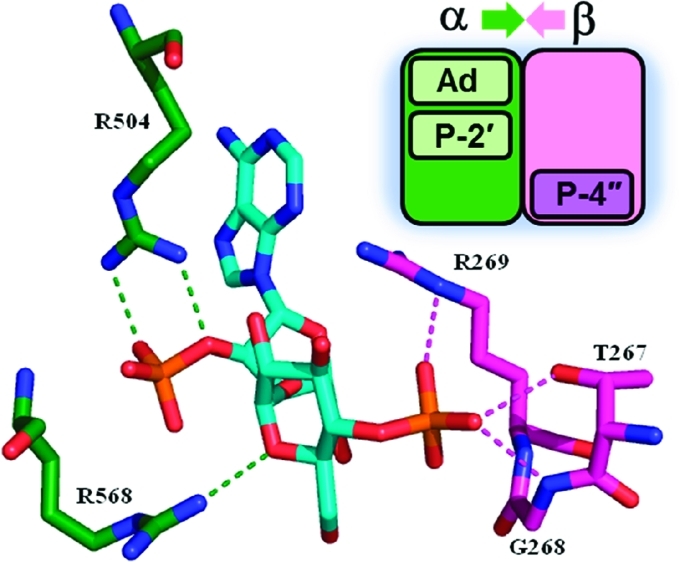
Possible hydrogen bonds between IP3 and IP3R in the crystal structure (PDB: 1N4K) of the IBC of IP3R1. Waters not shown. Green carbons, α-domain; purple carbons, β-domain.
The 4,5-bisphosphate motif of IP3 makes more contacts with the receptor than the more exposed 1-phosphate. The 1-phosphate interacts only with R568 and the backbone NH of K569 in the α-domain, while the 4-phosphate interacts mainly with residues in the β-domain (R265, R269, and T267), and the 5-phosphate interacts predominantly with amino acids in the α-domain (R504, K508, R511, and Y567). We and others have speculated1,42 that binding of IP3 pulls the domains together and closes the “clam-like” binding core, in a manner similar to glutamate binding to ionotropic glutamate receptors.43 This is illustrated schematically in Figure 7 (top left).
Figure 7.

Schematic representation of IP3R activation by inositol phosphates and adenophostin analogues. IP3 (1) and Ins(4,5)P2 (2) activate IP3R by engaging residues in the α- (green) and β- (pink) domains of the IBC, stabilizing a closed conformation that favors opening of the C-terminal Ca2+ channel.45 Phosphate groups may have strong (dark green) or weaker (light green) interactions with the α-domain. Molecular modeling suggests that Ada (3a) has additional interactions (light green) with the α-domain of the IBC, accounting for the greater potency of AdA. 4″-Dephospho-AdA (4) is essentially inactive because it cannot form effective β-domain interactions. However, 3″-dephospho-Ada (5) and 2′-dephospho-AdA (6) retain activity because they can effectively engage both domains, even though 5 does not contain a vicinal bisphosphate pair. The previously unexplained and relatively potent activity of AdA regiosomer 40 can now be explained by analogy with 5. P = phosphate group, and Ad = adenine.
Recently published X-ray structures of the N-terminal ligand-binding domain (LBD)44 and the N-terminal domain of rat IP3R1 (residues 1–604) with and without IP3 bound support the idea that IP3 causes domain closure within the IBC.45 As IP3 binds, side chains of nine residues within the α- and β-domains of the IBC become organized around IP3, causing the clam-like structure to partially close, reducing the angle between the two domains by ∼8°. Unfortunately, the resolution of these structures is not sufficient to provide further clues about how AdA and its analogues might bind. The 4,5-bisphosphate of IP3 clearly plays a major role in cross-linking the two domains of the IBC, and the 1-phosphate exerts its enhancing effect by providing an additional, weaker interaction with the α-domain, accounting for the greater potency of IP3 relative to Ins(4,5)P2 (Figure 7). The known inactivity of d-myo-inositol 1,4-bisphosphate [Ins(1,4)P2] suggests that the 1-P alone cannot interact strongly enough with the α-domain to pull the two domains together. Our model for AdA binding7 shows how the 3″,4″-bisphosphate of AdA can mimic the 4,5-bisphosphate of IP3, while both its 2′-phosphate group and adenine moiety have additional interactions with the IBC (Figures 7 and 8A). Specifically, we have proposed that while the 2′-phosphate of AdA essentially mimics the 1-phosphate of IP3, the adenine can engage in a cation-π interaction with the guanidinium side chain of R504 in the α-domain.
Figure 8.

Interactions of AdA (A), compound 5 (B), and compound 6 (C) with the IBC of IP3R as predicted by molecular docking experiments. Waters not shown. Green carbons, α-domain; purple carbons, β-domain. See the Experimental Section for details.
The present study shows that bisphosphate analogues of AdA lacking the 3″- or 4″-phosphate (4 and 5) are very weak agonists, confirming the important role for the 3″,4″-vicinal bisphosphate in AdA.40 However, our finding that 4 and 5 differ in their potency suggests unequal contributions from the two phosphates (4″-P and 3″-P). While bisphosphate 4 is almost inactive, 5 is a weak agonist of IP3R, being some 2300-fold weaker than IP3. This shows that between the vicinal bisphosphates, the 3″-phosphate in AdA is less important than the 4″-phosphate. Loss of activity after deletion of the 4″-phosphate from AdA is consistent with it providing the major contact with the β-domain, which may be essential for “clam” closure. Thus, 4 and 5 can be considered as analogues of d-myo-inositol 1,5-bisphosphate [Ins(1,5)P2] and Ins(1,4)P2, respectively, and the fact that 5 is a weak agonist while its inositol equivalent Ins(1,4)P2 is inactive can be explained in terms of our model by the enhancing role of the adenine in 5, strengthening its α-domain interactions (Figures 7 and 8B).
2′-Dephospho-AdA (6) differs from Gluc(3,4)P2 (7) only in having an adenosine moiety; yet, it is 200-fold more potent than 7 (Table 1). This 200-fold enhancement of activity by the adenosine moiety in the absence of a 2′-phosphate establishes unequivocally that the adenosine moiety contributes directly to enhanced activity independent of any effect on positioning of that phosphate. In a recent report,19 we demonstrated, in a study linking chemical modification with receptor mutagenesis, that removal of the 2′-phosphate from AdA to give 6 has significantly lesser effects on affinity for the IBC than did removal of the 1-phosphate from IP3 to give Ins(4,5)P2. A cell line was established that expresses only IP3R1, and mutation of R504 more profoundly reduces the affinity of IP3R for AdA (353-fold) than for IP3 (13-fold). The activities of other adenophostin analogues and the corresponding IP3 analogues were also comparable. Thus, when an amino acid thought to be responsible for engaging the adenine unit (R504) is mutated, the enhanced activity of adenophostin ligands disappears.
Bisphosphate 6 is the most potent known agonist of the IP3R with only two phosphates, suggesting that it may be possible to develop high-affinity ligands with fewer phosphates or even nonphosphate moieties. The possibility to develop less charged agonists or antagonists would be particularly attractive for cellular or in vivo chemical biological studies. Even though 5 is a weak agonist, its measurable activity suggests that a vicinal bisphosphate is not absolutely required for a ligand to activate IP3R. This now explains an apparent anomaly observed in previous work. The regioisomeric AdA analogue 2″-phospho-3″-dephospho-AdA 40 (Figure 9) with the 3″-phosphate group of AdA transposed to the 2″-position was previously shown to possess surprisingly potent activity that did not fit established SAR considerations.
Figure 9.

Structural comparison of IP3R ligands lacking vicinal bisphosphates with IP3.
Compound 40, although lacking the vicinal 3″,4″-bisphosphate, is only 12–20 times less potent than IP3 in liver flux and binding assays, respectively.46 Interestingly, d-myo-inositol 1,4,6-trisphosphate (41), which may be pictured as having a similar arrangement of phosphate groups to 40 (Figure 9), is also an agonist, only 2–3 times weaker than IP3.47 However, 41 may be able to present an IP3-like arrangement of phosphate groups to the IBC simply by binding in an alternative orientation (41b, Figure 9). Similar reasoning was used to explain the unexpected activity of 40 in the original study, where it was suggested that it too may be able to adopt an inverted binding orientation.40 This may seem improbable given the size and complexity of 40 as compared to 41, but the alternative explanation, namely, that a vicinal bisphosphate is not required, would have been totally unprecedented. Given the new finding of activity for 5, which cannot present a vicinal bisphosphate in any binding orientation, we can now suggest a simpler explanation for the activity of both 5 and 40 that is also consistent with more recent information on the structure of the IBC and with the concept of a direct role for the adenine. We propose that, contrary to previous assumptions, a vicinal bisphosphate motif is not absolutely essential for activation of IP3R, given sufficiently strong compensating contributions from other components of the ligand. Thus, we suggest that for bisphosphate 5, interactions of the adenine with the α-domain partially compensate the loss of one phosphate group in the vicinal pair, while in 40, these interactions are supplemented by additional contributions from the 2″-phosphate, presumably also with the α-domain, strengthening binding still further (Figure 7). Thus, it appears that two or more weak interactions with the α-domain can, to some extent, compensate for the loss of a strong interaction.
Conclusion
In summary, as a continuation of our efforts to understand SARs of adenophostins at the IP3 receptor and to gain further insight into the molecular mechanism of IP3-mediated signal transduction pathways in general, we have synthesized all of the bisphosphate analogues of AdA, excising one of its three phosphates at a time. Bisphosphates 4 and 5 were prepared in a series of reactions involving α-selective glycosylation of an appropriately protected glucose (glycosyl donor) and protected ribose acceptor, Vorbrüggen condensation of an appropriately protected disaccharide with 6-chloropurine, followed by further manipulation and chemoselective phosphitylation. A novel strategy for regioselective dephosphorylation by transesterification has been introduced for the efficient synthesis of 6. The glucose bisphosphate 7 was synthesized as a control. The ability of these novel bisphosphates to stimulate IP3R-mediated Ca2+ release was measured using rat type I IP3R expressed in chicken DT40 cells and compared to that of glucose 3,4-bisphosphate (7) and Ins(4,5)P2. This study reveals that although the 3″,4″-bisphosphate functionality is important for high affinity binding and channel opening, it is still possible for molecules lacking this feature to stimulate the IP3R, albeit to a lesser extent. Such knowledge is important in designing novel ligands as tools for pharmacological intervention. We conclude that a vicinal bisphosphate moiety is not essential for IP3R activation. P-4 of IP3, which contacts the β-domain of the IBC, is required, but P-5, which contacts the α-domain, can be replaced by a cation-π interaction between the adenine of AdA and R504 in the α-domain. The same interaction can substantially compensate the loss of P-1 to provide the potent agonist 6 with only two phosphate groups. Inositol polyphosphates often bind to sites rich in Arg and Lys residues, and replacing such interactions with a polar phosphate by a cation-π motif could have more general applications in the chemical biology of inositol phosphate signaling and probably also in other fields.
Glucose-3,4-bisphosphate essentially mimicks Ins(4,5)P2 in activity as expected. A 200-fold enhancement in activity is found in going from the simple glucose bisphosphate 7 to the related 6 bearing an adenosyl moiety, establishing a direct role for the adenosine of AdA in increasing affinity for the receptor. Compound 6, the most potent among the bisphosphates synthesized, approaches the potency of IP3. This is the first report of a relatively potent agonist of IP3R devoid of one phosphate group from the natural ligand and suggests the possibility to develop other ligands having a fewer number of phosphates. Such ligands, with less charge, could be useful for pharmacological intervention in this cellular signaling system and, at their simplest, might comprise two motifs that interact with the IBC domains linked by a suitable spacer. The reappraisal here of the relatively potent activity and surprising activity of 40, published earlier, in the light of these new results and particularly the activity of 5, begins to demonstrate the potential of this approach, suggesting that it should be possible in principle to develop IP3R ligands without a vicinal bisphosphate moiety.
Experimental Section
General
Chemicals were purchased from Aldrich, Sigma, and Fluka. All anhydrous solvents were purchased from Aldrich or Fluka. TLC was performed on precoated plates (Merck Aluminum sheets silica 60 F254, art No. 5554). Chromatograms were visualized under UV light and by dipping plates into either phosphomolybdic acid in MeOH or anisaldehyde in ethanol, followed by heating. Ion exchange chromatography was performed on an LKB-Pharmacia Gradifrac medium pressure ion-exchange chromatograph using MP1 AG ion-exchange resin and a gradient of 0–100% 150 mM TFA as eluent or Q Sepharose Fast Flow resin and a gradient of 0–100% 1.0 M triethylammonium bicarbonate (TEAB). 1H NMR, COSY, NOESY, HMBC, and HMQC spectra were recorded on Varian EX-400 (400 MHz) and Bruker Avance III (400 and 500 MHz) spectrometers. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.0 ppm) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ 7.26 ppm; D2O, δ 4.79 ppm). Data are reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m)], integration, coupling constants [Hz], annotation). 13C and DEPT spectra were recorded on Varian EX-400 (100 MHz) and Bruker Avance III (100 and 126 MHz) spectrometers with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with the respective solvent resonance as the internal standard (CDCl3, δ 77.0 ppm). 31P NMR spectra were recorded on Varian EX-400 (162 MHz) or Bruker Avance III (162 MHz) spectrometers with complete proton decoupling. Phosphorus chemical shifts are reported in ppm (δ) relative to an 85% H3PO4 external standard (H3PO4, δ 0.0 ppm). All NMR data were collected at 25 °C. Melting points were determined using a Reichert-Jung Thermo Galen Kofler block and are uncorrected. Microanalysis was carried out at the University of Bath microanalysis service. Mass spectra were recorded at the SERC Mass Spectrometry Service Centre, Swansea, and at the University of Bath on VG Autospec or MicroTOF instruments. The method of ionization is given in parentheses. Optical rotations were measured at ambient temperature using an Optical Activity Ltd. AA-10 polarimeter in a cell volume of 5 cm3, and specific rotation is given in 10–1 deg cm3 g–1. UV spectra were recorded using a Perkin-Elmer Lambda EZ201 spectrometer. The purities of final compounds were determined to be greater than 98% by HPLC analysis. HPLC analysis was carried out on a Hewlett-Packard series chromatograph with a strong anion-exchange resin (MP1 AG, column size 3 mm × 150 mm). A linear gradient of 0–50% 150 mM TFA was used as eluent at 1 cm3/min over 60 min, with the UV detector set at 254 nm. Synthetic phosphates were assayed using an adaptation of the modified Brigg's phosphate assay48 and/or Ames phosphate assay.49 Flash column chromatography was performed using Silica Gel 60 A (32–63 μm). All reactions were carried out under argon or nitrogen atmosphere employing oven-dried glassware unless stated otherwise. Usual work up refers to taking up the crude material in an organic solvent (ethyl acetate, DCM, or CHCl3) followed by washing successively with water, cold diluted HCl, saturated NaHCO3 solution, and brine and drying over anhydrous MgSO4.
3′-O-Acetyl-5,2′,4′,6′-tetra-O-benzyl-3-O-(α-d-glucopyranosyl)-1,2-O-isopropylidene-α-d-ribo-furanoside (17)
Both donor 15 (1.925 g, 3.587 mmol) and acceptor 16 (981 mg, 3.5 mmol) were dissolved in toluene–dioxane (10 mL: 30 mL), and the mixture was stirred with powdered 3 Å molecular sieves for 15 min. NIS (866 mg, 3.85 mmol) was then added, and when it had dissolved, TMSOTf (100 μL, 0.55 mmol) was added dropwise, and the mixture was stirred at room temperature. TLC after 30 min showed disappearance of the starting material. NaHCO3 solution was added to quench the acidic residue, and the mixture was diluted with ethyl acetate and washed with saturated Na2S2O3 solution and then with brine. The solution was dried over MgSO4 and concentrated under reduced pressure. The residue thus obtained was chromatographed using 15% ethyl acetate–hexane to afford the α-glycoside 17 (2 g, 76%) as a colorless crystalline solid; mp 112 °C; [α]D +120.8 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3): 1.35 (s, 3H, CH3 isopropylidene), 1.50 (s, 3H, CH3 isopropylidene), 1.96 (s, 3H, COCH3), 3.46 (dd, 1H, 10.87 Hz, 1.00 Hz, H-6A′), 3.51 (dd, 1H, 9.88 Hz, 3.95 Hz, H-2′), 3.59 (dd, 1H, 10.87 Hz, 2.47 Hz, H-6B′), 3.67 (dd, 1H, 8.40 Hz, 2.97 Hz, H-5A), 3.67–3.74 (m, 2H, H-4′ and H-5′), 3.79 (dd, 1H, 11.61 Hz, 1.73 Hz, H-5B), 4.15 (dd, 1H, 9.14 Hz, 3.95 Hz, H-3), 4.29 (ddd, 1H, 9.14 Hz, 3.21 Hz, 1.73 Hz, H-4), 4.44 (AB q, 2H, 25.69 Hz, 11.36 Hz, CH2Ph), 4.47 (AB q, 2H, 82.02 Hz, 11.86 Hz, CH2Ph), 4.53 (AB q, 2H, 46.44 Hz, 11.86 Hz, CH2Ph), 4.64 (AB q, 2H, 50.89 Hz, 12.35 Hz, CH2Ph), 4.71 (t, 1H, 3.95 Hz, H-2), 5.23 (d, 1H, 3.95 Hz, H-1′), 5.45 (t, 1H, 9.39 Hz, H-3′), 5.83 (d, 1H, 3.95 Hz, H-1), 7.11–7.35 (m, 20 H, 4 × C6H5). Elemental analysis calcd for C44H50O11: C, 70.01; H, 6.68. Found: C, 69.8; H, 6.69. m/z (ES+) = 777.4 [(M + Na)+, 100%]. HRMS: mass calcd for C44H54O11N1 [M + NH4]+, 772.3691; found, 772.3691.
2′,3″-Di-O-acetyl-5′,2″,4″,6″-tetra-O-benzyl-6-chloro-3′-O-(α-d-glucopyranosyl)-9-β-d-ribo-furanosylpurine (19)
Disaccharide 17 (128 mg, 0.17 mmol) was stirred with 90% TFA (0.5 mL) for 10–15 min at room temperature. The TFA was then evaporated off, and the residue was coevaporated several times with toluene to remove traces of water. The residue was then dissolved in pyridine (2 mL), Ac2O (0.5 mL, 5.3 mmol) was then added, and the mixture was stirred for 4 h at room temperature. Work up was as usual, and the crude product was chromatographed to afford an inseparable mixture of the α- and β-triacetates 18 (120.6 mg, 89%). A suspension of triacetate 18 (120 mg, 0.15 mmol), 6-chloropurine (46 mg, 0.3 mmol), and BSA (182 μL, 0.74 mmol) in MeCN (5 mL) was refluxed until the solution became clear. The mixture was cooled to room temperature, and then, TMSOTf (60 μL, 0.33 mmol) was added, and the solution was stirred at 70 °C overnight. Workup was in ethyl acetate, and the crude product was chromatographed to afford the nucleoside 19 (103 mg, 77%) as a colorless gum. [α]D +47.5 (c 1.6, CHCl3). 1H NMR (400 MHz, CDCl3): 1.84 (s, 3H, COCH3), 1.86 (s, 3H, COCH3), 3.41 (dd, 1H, 10.14 Hz, 3.48 Hz, H-2″), 3.44 (dd, 1H, 8.99 Hz, 1.74 Hz, HA-6″), 3.56 (dd, 1H, 10.72 Hz, 3.19 Hz, HB-6″), 3.56–3.61 (m, 1H, HA-5′), 3.60 (t, 1H, 9.27 Hz, H-4″), 3.70–3.80 (m, 2H, H-5″, HB-5′), 4.40 (AB q, 2H, 33.62 Hz, 11.01 Hz, PhCH2), 4.41 (AB q, 2H, 78.84 Hz, 11.88 Hz, PhCH2), 4.42–4.46 (m, 1H, H-4′), 4.47 (AB q, 2H, 14.49 Hz, 11.59 Hz, PhCH2), 4.49 (AB q, 2H, 64.92 Hz, 12.17 Hz, PhCH2), 4.68 (t, 4.93 Hz, H-3′), 4.95 (d, 1H, 3.48 Hz, H-1″), 5.45 (dd, 1H, 10.14 Hz, 9.28 Hz, H-3″), 5.68 (dd, 1H, 5.22 Hz, 4.64 Hz, H-2′), 6.30 (d, 1H, 4.64 Hz, H-1′), 7.00–7.30 (m, 20H, 4 × Ph), 8.45 (s, 1H, H-8), 8.66 (s, 1H, H-2). m/z (ES+) = 915.44 [(M + Na)+, 100%]. HRMS: mass calcd for C48H50ClN4O11 [M + H]+, 893.3159; found, 893.3160.
6-Amino-5′,2″,4″,6″-tetra-O-benzyl-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (20)
A solution of chloro-nucleoside 19 (100 mg, 0.112 mmol) in ethanol (10 mL) in a pressure tube was saturated with NH3 (bubbled at 0 °C for 10–15 min). The tube was then closed tightly and was heated at 74 °C for 5 days. Solvents were evaporated off in vacuo, and the crude product was chromatographed with 4% MeOH–CHCl3 to afford the diol 20 (86 mg, 97%) as a colorless gum. 1H NMR (400 MHz, CDCl3): 3.51 (dd, 1H, 9.78 Hz, 3.52 Hz, H-2″), 3.56 (dd, 1H, 10.17 Hz, 9.39 Hz, H-4″), 3.55–3.61 (m, 2H, HA-5′ and HA-6″), 3.66 (dd, 2H, 10.57 Hz, 3.52 Hz, HB-5′ and HB-6″), 3.82 (ddd, 1H, 10.17 Hz, 3.13 Hz, 1.96 Hz, H-5″), 4.04 (br, 1H, OH), 4.20 (dd, 1H, 9.39 Hz, 9.78 Hz, H-3″), 4.28 (dd, 1H, 6.26 Hz, 3.13 Hz, H-4′), 4.41 (dd, 1H, 5.87 Hz, 3.52 Hz, H-3′), 4.48–4.52 (br, 1H, OH), 4.48 (s, 2H, PhCH2), 4.51 (AB q, 2H, 43.82 Hz, 12.13 Hz, PhCH2), 4.67 (dd, 11.74 Hz, 5.87 Hz, H-2′), 4.69 (AB q, 2H, 116.21 Hz, 11.35 Hz, PhCH2), 4.80 (AB q, 2H, 17.61 Hz, 11.74 Hz, PhCH2), 4.96 (d, 1H, 3.91 Hz, H-1″), 5.98 (d, 1H, 5.87 Hz, H-1′), 6.05 (br s, 2H, NH2), 7.15–7.35 (m, 20H, 4 × Ph), 7.98 (s, 1H, H-8), 8.39 (s, 1H, H-2). m/z (ES+) = 812.48 [(M + Na)+, 100%]. HRMS: mass calcd for C44H48O9N5 [M + H]+, 790.3447; found, 790.3449.
6-Amino-5′,2″,4″,6″-tetra-O-benzyl-2′,3″-bis-O-(dibenzyloxyphosphoryl)-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (21)
To a solution of diol 20 (123 mg, 0.156 mmol) and dibenzyl-N,N-di-isopropyl phosphoramidite (113 mg, 0.327 mmol) in DCM (5 mL) was added imidazolium triflate (75 mg, 0.343 mmol), and the solution was stirred at room temperature for 30 min. When TLC showed the disappearance of diol 20, the temperature was reduced to −78 °C, and then, mCPBA (170 mg, 60–70%) was added, and the solution was stirred for 1 h, gradually allowing the temperature to attain room temperature. Workup was in ethyl acetate, and the crude product was chromatographed with ethyl acetate:hexane:Et3N, 77:20:3 (v/v/v), to get pure bisphosphate 21 (165 mg, 81%) as a colorless oil. [α]D +22.6 (c 1.15, CHCl3). 1H NMR (400 MHz, CDCl3): 3.46 (dd, 1H, H-6A″), 3.56 (dd, 1H, H-6B″), 3.58–3.64 (m, 2H, H-2″, H-5A′), 3.67–3.76 (m, 3H, H-4″, H-5″, H-5B′), 4.35–4.54 (m, 7H, H-4′, 3 × CH2Ph), 4.62–4.77 (m, 5H, H-3′, 2 × CH2Ph), 4.79–4.98 (m, 7H, H-3″, 3 × CH2Ph), 5.40 (d, 1H, 3.52 Hz, H-1″), 5.66 (ddd, 1H, 8.22 Hz, 5.48 Hz, 4.90 Hz, H-2′), 5.70–6.00 (br, 2H, NH2), 6.34 (d, 1H, 5.48 Hz, H-1′), 7.00–7.42 (m, 40H, 8 × Ph), 7.93 (s, 1H, H-8), 8.27 (s, 1H, H-2). 31P NMR (161.94 MHz, CDCl3): −1.625, −1.101. Elemental analysis calcd for C72H73N5O15P2 C, 66.00; H, 5.62; N, 5.34. Found: C, 65.8; H, 5.94; N, 5.21. m/z (ES+) = 1334.29 [(M + Na)+, 100%], 1311.38 [(M + 1)+, 100%]. HRMS (FAB, CsI/glycerol): mass calcd for C72H73O15N5P2 [M]+, 1309.4573; found, 1309.4585.
3′-O-(α-d-Glucopyranosyl)-adenosine-2′,3″-bisphosphate (4)
A suspension of bisphosphate 21 (130 mg, 0.1 mmol) and 20% Pd(OH)2 on carbon (300 mg) in a mixture of cyclohexene (4 mL), MeOH (10.5 mL), and H2O (0.75 mL) was refluxed at 80 °C overnight. After filtration through a membrane filter, the filtrate was concentrated in vacuo. Purification by an AG column with 0–100% gradient elution using 150 mM TFA as an eluent gave pure bisphosphate 4 (53 mg, 91%). 1H NMR (400 MHz, D2O): 3.58 (t, 1H, 8.5 Hz, H-4″), 3.66–3.76 (m, 3H, H-2″, H-5″, H-6A″), 3.80–3.90 (m, 3H, H-5A′, H-5B′, H-6B″), 4.34 (ddd, 1H, 9.45 Hz, 8.50 Hz, 8.50 Hz, H-3″), 4.43 (dd, 1H, 7.09 Hz, 3.78 Hz, H-4′), 4.63 (dd, 1H, 5.20 Hz, 3.31 Hz, H-3′), 5.24–5.29 (m, 1H, H-2′), 5.27 (d, 1H, 3.78 Hz, H-1″), 6.35 (d, 1H, 5.61 Hz, H-1′), 8.40 (s, 1H, H-2), 8.50 (s, 1H, H-8). 31P NMR (161.94 MHz, D2O with excess of TEA): 4.25, 3.35. m/z (ES+) = 590.1 [(M + H)+, 90%]; 612.1 [(M + Na)+, 100%]. m/z (ES−) 588.2 [(M – H)+, 100%]. HRMS: mass calcd for C16H26O15N5P2 [M + H]+, 590.0895; found, 590.0895.
3-O-(4′-O-Acetyl-2′,3′,6′-tri-O-benzyl-α-d-glucopyranosyl)-5-O-benzyl-1,2-O-isopropylidene-α-d-ribofuranoside (27)
To a solution of donor acetate 26 (prepared from the known dibenzyl ether 24(50)) (1.007 g, 1.876 mmol), acceptor 16 (527 mg, 1.88 mmol), and N-iodosuccinimide (640 mg, 2.845 mmol) in a mixture of toluene (14 mL) and dioxane (19 mL) were added powdered 3 Å molecular sieves, and the mixture was stirred for 15 min under argon atmosphere. TMSOTf (100 μL, 0.55 mmol) was then added dropwise, and the mixture was stirred at room temperature for 30 min. When TLC showed disappearance of the starting material, the reaction mixture was filtered through a small pad of Celite, and the filtrate was diluted with ethyl acetate and washed successively with saturated Na2S2O3 solution, NaHCO3 solution, and brine. After it was dried over MgSO4, chromatography using ethyl acetate–hexane gave pure α-disaccharide 27 (1 g, 71%) as a colorless gum. [α]D +82.6 (c 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): 1.38 (s, 3H, CH3 isopropylidene), 1.60 (s, 3H, CH3 isopropylidene), 1.85 (s, 3H, COCH3), 3.33 (dd, 1H, 10.59 Hz, 4.33 Hz, H-6A′), 3.39 (dd, 1H, 10.59 Hz, 2.89 Hz, H-6B′), 3.65 (dd, 1H, 9.63 Hz, 3.85 Hz, H-2′), 3.69 (dd, 1H, 11.55 Hz, 3.85 Hz, H-5A), 3.77 (ddd, 1H, 10.11 Hz, 3.85 Hz, 3.85 Hz, H-5′), 3.80 (dd, 1H, 11.55 Hz, 1.93 Hz, H-5B), 3.88 (t, 1H, 9.63 Hz, H-3′), 4.15 (dd, 1H, 9.63 Hz, 4.33 Hz, H-3), 4.32–4.38 (m, 1H, H-4), 4.43 (AB q, 2H, 47.18 Hz, 12.04 Hz, CH2Ph), 4.57 (AB q, 2H, 47.66 Hz, 12.04 Hz, CH2Ph), 4.72 (AB q, 2H, 32.98 Hz, 11.80 Hz, CH2Ph), 4.73 (dd, 1H, 4.70 Hz, 1.17 Hz, H-2), 4.75 (AB q, 2H, 119.15 Hz, 11.31 Hz, CH2Ph), 5.09 (dd, 1H, 10.11 Hz, 9.63 Hz, H-4′), 5.21 (d, 1H, 3.37 Hz, H-1′), 5.83 (d, 1H, 3.85 Hz, H-1), 7.20–7.40 (m, 20 H, 4 × C6H5). Elemental analysis calcd for C44H50O11: C, 70.00; H, 6.68. Found: C, 69.79; H, 6.69. m/z (ES+) = 777.47 [(M + Na)+, 100%]. HRMS: mass calcd for C44H54O11N [M + NH4]+, 772.3691; found, 772.3694.
3-O-(4′-O-Acetyl-2′,3′,6′-tri-O-benzyl-α-d-glucopyranosyl)-5-O-benzyl-1,2-di-O-acetyl-α/β-d-ribofuranoside (28)
Disaccharide 27 (270 mg, 0.358 mmol) was treated with 90% TFA (2 mL) at room temperature for 15 min. TFA was evaporated off, and the residue was coevaporated with toluene several times. The residue thus obtained was dissolved in pyridine (5 mL), added Ac2O (2 mL, 21.2 mmol), and stirred at room temperature for 3 h. Workup was as usual, and the product was crystallized from ethyl acetate–hexane to afford pure crystals of α-triacetate 28-α (100 mg). The mother liquor was concentrated and chromatographed using 25% ethyl acetate–hexane to give an inseparable α/β mixture of the triacetate 28 (149 mg, total yield = 249 mg, 87%). Data for 28-α: mp 127–128 °C; [α]D +46 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3): 1.80 (s, 3H, CH3), 1.89 (s, 3H, CH3), 1.93 (s, 3H, CH3), 3.31 (dd, 1H, 10.77 Hz, 4.72 Hz, H-6A′), 3.38 (dd, 1H, 10.77 Hz, 2.94 Hz, H-6B′), 3.55 (dd, 1H, 9.78 Hz, 3.52 Hz, H-2′), 3.60 (dd, 1H, 11.16 Hz, 4.11 Hz, H-5A), 3.69 (dd, 1H, 11.35 Hz, 3.13 Hz, H-5B), 3.81 (ddd, 1H, 10.17 Hz, 4.70 Hz, 2.94 Hz, H-5′), 3.89 (t, 1H, 9.78 Hz, H-3′), 4.39 (AB q, 2H, 41.87 Hz, 11.74 Hz, CH2Ph), 4.40 (t, 1H, 3.52 Hz, H-4), 4.50 (AB q, 2H, 14.87 Hz, 11.74 Hz, CH2Ph), 4.60 (t, 1H, 5.87 Hz, H-3), 4.65 (s, 2H, CH2Ph), 4.73 (AB q, 2H, 94.68 Hz, 11.74 Hz, CH2Ph), 4.95 (d, 1H, 3.52 Hz, H-1′), 5.02 (dd, 1H, 10.17 Hz, 9.40 Hz, H-4′), 5.31 (dd, 1H, 4.70 Hz, 1.17 Hz, H-2), 6.12 (d, 1H, 1.17 Hz, H-1), 7.22–7.33 (m, 20 H, 4 × C6H5). Elemental analysis calcd for C45H50O13: C, 67.66; H, 6.31. Found: C, 67.5; H, 6.34. m/z (ES+) = 821.73 [(M + Na)+, 100%]. HRMS: mass calcd for C45H54O13N [M + NH4]+, 816.3590; found, 816.3592.
2′,4″-Di-O-acetyl-5′,2″,3″,6″-tetra-O-benzyl-6-chloro-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (29)
A suspension of triacetate 28 (234 mg, 0.293 mmol), 6-chloropurine (91 mg, 0.589 mmol), and BSA (356 μL, 1.46 mmol) in acetonitrile (9 mL) was refluxed until the solution became clear. After it was cooled and the addition of TMSOTf (115 μL, 0.635 mmol), the solution was heated to 70 °C, stirred overnight, and quenched with a saturated solution of NaHCO3, and the mixture worked up in ethyl acetate. The residue was chromatographed with 35% ethyl acetate–hexane to yield the chloronucleoside 29 (241 mg, 92%) as a colorless gum. [α]D +14.1 (c 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): 2.16 (s, 3H, COCH3), 2.20 (s, 3H, COCH3), 3.67–3.75 (m, 2H, HA-6″ and HB-6″), 3.80–3.92 (m, 3H, H-2″, HA-5′, HB-5′), 4.01 (m, 1H, H-5″), 4.27 (t, 1H, 9.50 Hz, H-3″), 4.72 (AB q, 2H, 29.72 Hz, 11.64 Hz, PhCH2), 4.80 (AB q, 2H, PhCH2), 4.91 (q, 1H, H-4′), 4.93–5.00 (m, 3H, H-3′, PhCH2), 5.10 (AB q, 2H, 87.33 Hz, 11.34 Hz, PhCH2), 5.23 (d, 1H, 3.68 Hz, H-1″), 5.29 (dd, 1H, 10.11 Hz, 9.50 Hz, H-4″), 5.95 (dd, 1H, 6.44 Hz, 5.52 Hz, H-2′), 6.79 (d, 1H, 6.44 Hz, H-1′), 7.50–7.70 (m, 20H, 4 × Ph), 8.79 (s, 1H, H-8), 9.06 (s, 1H, H-2). m/z (ES+) = 915.4 [(M + Na)+, 100%]. HRMS: mass calcd for C48H49O11N4Cl1Na1 [M + Na]+, 915.2979; found, 915.2984.
6-Amino-5′,2″,3″,6″-tetra-O-benzyl-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (30)
A solution of chloronucleoside 29 (360 mg, 0.403 mmol) in ethanol was saturated with ammonia and heated at 74 °C in a sealed pressure tube for 5 days. The solvents were evaporated off, and the residue was dissolved in DCM and washed with water and then with brine. The organic layer was dried over MgSO4, and the solvents were evaporated under reduced pressure to afford the pure diol 30 (318 mg, 100%) as a colorless gum. [α]D +6 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3): 3.54 (dd, 1H, 9.39 Hz, 3.91 Hz, H-2″), 3.57–3.68 (m, 5H, H-4″, H-5A′, H-5B′, H-6A″ and H-6B″), 3.76–3.90 (m, 2H, H-3″, H-5″), 4.28–4.38 (m, 1H, H-4′), 4.38–4.52 (m, 5H, H-3′, 2 × CH2Ph), 4.64–4.72 (m, 1H, H-2′), 4.72 (ABq, 2H, 46.17 Hz, 11.74 Hz, CH2Ph), 4.86 (d, 3.52 Hz, H-1″), 4.89 (ABq, 2H, 66.52 Hz, 11.35 Hz, CH2Ph), 5.74–5.90 (br, 2H, NH2), 6.03 (d, 1H, 5.48 Hz, H-1′), 7.20–7.38 (m, 20 H, 4 × C6H5), 7.98 (s, 1H, H-8), 8.30 (s, 1H, H-2). m/z (ES+) = 812.70 [(M + Na)+, 100%]. HRMS: mass calcd for C28H42O7NS [M + H]+, 790.3447; found, 790.3454.
6-Amino-5′,2″,3″,6″-tetra-O-benzyl-2′,4″-bis-O-(dibenzyloxyphosphoryl)-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (31)
To a solution of 30 (165.5 mg, 0.21 mmol) and dibenzyl-N,N-di-isopropyl phosphoramidite (152 mg, 0.44 mmol) in DCM (5 mL) was added imidazolium triflate (102 mg, 0.468 mmol), and the solution was stirred at room temperature for 30 min. When TLC showed disappearance of the starting material, the temperature was reduced to −78 °C, mCPBA (170 mg) was added, and the mixture was stirred for 30 min, allowing the temperature to attain room temperature. The mixture was taken up in ethyl acetate, and the solution was washed successively with Na2SO3 solution, water, and brine. The solution was dried over MgSO4, and the residue after evaporation was chromatographed to yield bisphosphate 31 (233 mg, 85%) as a colorless oil.
3′-O-(α-d-Glucopyranosyl)-adenosine-2′,4″-bisphosphate (5)
A suspension of bisphosphate 31 (155 mg, 0.118 mmol) and 20% Pd(OH)2 on carbon (400 mg) in a mixture of cyclohexene (5.5 mL), MeOH (10.5 mL), and H2O (0.75 mL) was heated at 80 °C overnight. After filtration through a membrane filter, the evaporated filtrate was purified on an AG column using 0–100% gradient elution using 150 mM TFA as eluent to give pure bisphosphate 5 (62 mg, 89%). 1H NMR (400 MHz, D2O): 3.52 (dd, 1H, 9.78 Hz, 3.91 Hz, H-2″), 3.60–3.79 (m, 5H, H-5″, H-5A′, H-5B′ H-6A″, H-6B″), 3.82 (dd, like a t, 9.29 Hz, 8.80 Hz, H-3″), 3.89 (ddd, like a q, 1H, 9.29 Hz, 8.80 Hz, 8.80 Hz, H-4″), 4.31 (dd, 1H, 6.85 Hz, 3.42 Hz, H-4′), 4.52 (dd, 1H, 4.89 Hz, 3.42 Hz, H-3′), 5.11 (d, 1H, 3.91 Hz, H-1″), 5.18 (ddd, 1H, 9.78 Hz, 5.87 Hz, 5.38 Hz, H-2′), 6.23 (d, 1H, 6.36 Hz, H-1′), 8.28 (s, 1H, H-2), 8.39 (s, 1H, H-8). 31P NMR (161.94 MHz, D2O with excess of TEA): 4.22, 3.13. m/z (ES−) = 588.1 [(M – H), 50%]. HRMS (ES−): mass calcd for C16H24O15N5P2 [M – H], 588.0750; found, 588.0751.
6-Amino-5′,2″,6″-tri-O-benzyl-3″,4″-bis-O-(dibenzyloxyphosphoryl)-3′-O-(α-d-glucopyranosyl)-9-β-d-ribofuranosylpurine (34)
To a solution of trisphosphate 33 (60 mg, 0.0405 mmol) in BnOH (2 mL) was added anhydrous K2CO3 (25 mg, 0.18 mmol), and the mixture was stirred at 70 °C overnight. Chromatography with 30% ethyl acetate–hexane removed the benzyl alcohol, and then, elution with 5% MeOH–CHCl3 gave the bisphosphate 34 (46 mg, 93%) as a colorless gum. 1H NMR (400 MHz, CDCl3): 3.46 (dd, 1H, 10.70 Hz, 3.30 Hz, H-5A′), 3.50 (dd, 1H, 9.78 Hz, 3.70 Hz, H-2″), 3.54 (dd, 1H, 10.84 Hz, 2.38 Hz, H-5B′), 3.54–3.62 (m, 2H, H-6A″ and H-6B″), 3.88 (ddd, 1H, 10.04 Hz, 3.83 Hz, 2.78 Hz, H-5″), 4.24–4.30 (m, 2H, H-3′ and H-4′), 4.27 (AB q, 2H, 47.56 Hz, 11.63 Hz, C–O–CH2Ph), 4.35 (s, 2H, C–O–CH2Ph), 4.49 (ddd, like a q, 9.78 Hz, 9.51 Hz, 9.51 Hz, H-4″), 4.51 (AB q, 2H, 40.83 Hz, 12.03 Hz, C–O–CH2Ph), 4.58–4.66 (m, 1H, H-2′), 4.72 (d, 1H, 3.44 Hz, H-1″), 4.82–5.06 (m, 9H, 4 × P–O-CH2Ph, H-3″), 5.81 (br, 2H, NH2), 5.99 (d, 1H, 5.02 Hz, H-1′), 7.10–7.26 (m, 35 H, 7 × Ph), 7.91 (s, 1H, H-8), 8.17 (s, 1H, H-2). 31P NMR (161.94 MHz, CDCl3): −1.967, −1.223. m/z (ES+) = 1243.16 [(M + Na)+, 100%], (ES−) 1218.64 [(M – H)−, 100%]. HRMS (FAB, CsI/glycerol): mass calcd for C65H67O15N5P2 [M]+, 1219.4103; found, 1219.4096.
3′-O-(α-d-Glucopyranosyl)-adenosine-3″,4″-bisphosphate (6)
A suspension of bisphosphate 34 (45 mg, 0.037 mmol) and 20% Pd(OH)2 on carbon (120 mg) in a mixture of cyclohexene (1.6 mL), MeOH (3 mL), and H2O (0.22 mL) was heated at 80 °C overnight. After filtration through a membrane filter, the evaporated filtrate was purified on an AG column using 0–100% gradient elution using 150 mM TFA as eluent to give pure bisphosphate 6 (20 mg, 92%). 1H NMR (400 MHz, D2O): 3.74–3.90 (m, 6H, H-2″, H-5″, H-5A′, H-5B′ H-6A″, H-6B″), 4.10 (ddd, like a q, 1H, 9.66 Hz, 9.66 Hz, 9.66 Hz, H-4″), 4.42 (dd, 1H, 7.24 Hz, 3.86 Hz, H-4′), 4.47–4.55 (m, 2H, H-3′, H-3″), 4.87 (dd, like a t, 1H, 5.80 Hz, 5.31 Hz, H-2′), 5.19 (d, 1H, 3.38 Hz, H-1″), 6.19 (d, 1H, 5.80 Hz, H-1′), 8.40 (s, 1H, H-2), 8.50 (s, 1H, H-8). 31P NMR (161.94 MHz, D2O with excess of TEA): 4.50, 3.62. m/z (ES+) = 590.2 [(M + H)+, 100%]; 612.1 [(M + Na)+, 100%]; m/z (ES−) 588.2 [(M – H)+, 100%]. HRMS: mass calcd for C16H26O15N5P2 [M + H]+, 590.0895; found, 590.0895.
Allyl 2,6-Di-O-benzyl-3,4-bis-O-(dibenzyloxyphosphoryl)-α-d-glucopyranoside (38)
To a stirred solution of allyl 2,6-di-O-benzyl-α-d-glucopyranoside (37) (400 mg, 1.00 mmol) and 1H-tetrazole (280 mg, 4.00 mmol) in dry CH2Cl2 (5 mL) at room temperature was added dibenzyl-N,N-diisopropylphosphoramidite (1.0 mL, 3.0 mmol). After 1 h, the mixture was cooled to −78 °C, and mCPBA (1.0 g, 57%, 3.3 mmol) was added in portions over 1 min. After a further 10 min at −78 °C, a solution of Na2SO3 (50 mL, 10% W/V) was added, and the mixture was stirred vigorously for 2–3 min, until the mixture began to freeze. The cooling bath was then removed, and the mixture was allowed to reach room temperature and diluted with CH2Cl2 (50 mL). The organic layer was separated, washed with a saturated solution of NaHCO3 (50 mL), dried over MgSO4, and concentrated. Purification of the residue by flash chromatography (EtOAc/hexane 1:2 then 1:1) gave 38 as a colorless oil (846 mg, 0.919 mmol, 92%); [α]D +15.9 (c 2.3, CHCl3). 1H NMR (400 MHz, CDCl3): 3.57 (dd, 1H, 9.7 Hz, 3.6 Hz, H-2), 3.72 (dd, 1H, 10.9 Hz, 2.1 Hz, H-6A), 3.76 (dd, 1H. 10.9 Hz, 4.4 Hz, H-6B), 3.84–3.90 (m, 2H, H-5 and OCH2CH=CH2), 4.07–4.12 (m, 1H, one proton of OCH2CH=CH2), 4.38, 4.51 (ABq, 2H, JAB = 12.1 Hz, OCH2Ph), 4.47, 4.72 (ABq, 2H, JAB = 12.1 Hz, OCH2Ph), 4.61 (ddd, 1H, 9.8 Hz, 9.4 Hz, 9.4 Hz, H-4), 4.72 (d, 1H, 3.6 Hz, H-1), 4.90–5.08 (m, 9H, 4 × POCH2Ph and H-3), 5.16–5.21 (m, 1H, OCH2CH=CHH cis), 5.26–5.32 (m, 1H, OCH2CH=CHH trans), 5.82–5.93 (m, 1H, OCH2CH=CH2), 7.15–7.30 (m, 28H, Ph), 7.31–7.35 (m, 2H, Ph). 31P NMR (162 MHz, CDCl3): −2.28, −1.89. MS m/z (ES–); 919 [(M – H)−, 45%], 829 [(M – C7H7)−, 100%]. HRMS: mass calcd for C51H54O12P2, 943.2983 [M + Na]+; found, 943.2954. Elemental analysis calcd for C51H54O12P2 (920.91): C, 66.51; H, 5.91. Found: C, 66.6; H, 5.97.
2,6-Di-O-benzyl-3,4-bis-O-(dibenzyloxyphosphoryl)-d-glucopyranose (39)
To a solution of 38 (400 mg, 0.434 mmol) in dry methanol (5 mL) was added PdCl2 (20 mg, 0.11 mmol). The mixture was stirred vigorously in a flask fitted with a drying tube (air is required for the reaction) for 3 h, after which time TLC (ethyl acetate/hexane 1:1) showed the reaction to be essentially complete, with conversion of 38 (Rf 0.36) into two products (Rf 0.10 and 0.16). The acidic solution was neutralized by stirring with excess NaHCO3 for 5 min, then filtered through Celite, and concentrated. Purification by flash chromatography (EtOAc/hexane 2:3, then EtOAc) gave 39 (mixture of α and β anomers) as a colorless oil (311 mg, 0.353 mmol, 81%); [α]D +6.4 (c 1.1, CHCl3). 1H NMR (400 MHz, CDCl3): 3.46 (dd, 0.25H*, 8.9 Hz, 7.7 Hz, H-2 in β-anomer), 3.54, (dd, 0.75H, 9.5 Hz, 3.4 Hz, H-2 in α-anomer), 3.56 (m, 0.25H, buried, H-5 in β-anomer), 3.64–3.79 (m, 2H, CH2-6 in α- and β-anomers), 4.09–4.14 (m, 1.5H, OH-1 and H-5 in α-anomer), 4.32–4.66 (m, 5.25H, H-4 and 2 × OCH2Ph in α- and β-anomers, H-3 in β-anomer), 4.69–4.76 (m, 0.75H, H-1 and POCH2Ph in β-anomer), 4.87–5.07 (m, 8.25H, H-3 and 4 × POCH2Ph in α-anomer, 3 × POCH2Ph in β-anomer), 5.13 (dd, 0.75H, 3.2 Hz, 3.2 Hz, H-1 in α-anomer), 7.02–7.35 (m, 30H, Ph). 31P NMR (162 MHz, CDCl3): −2.27 (0.75P), −2.24 (0.25P), −1.99 (0.25P), −1.72 (0.75P). m/z (FAB+) 881 [(M + H)+, 90%], 91 [C7H7+, 100%]. HRMS: mass calcd for C48H50O12P2, 903.2670 [M + Na]+. Found, 903.2649. Elemental analysis calcd for C48H50O12P2 (880.85): C, 65.45; H, 5.72. Found C, 65.4; H, 5.70. *Approximately 3:1 mixture of α- and β-anomers; integrals are therefore approximate.
d-Glucopyranose 3,4-Bisphosphate (7)
To a solution of 39 (225 mg, 0.255 mmol) in MeOH (20 mL) and water (5 mL) was added Pd(OH)2–C (20%, 50% water, 600 mg). The mixture was shaken in a Parr hydrogenator under H2 (50 psi) for 24 h. The catalyst was removed by filtration through a PTFE syringe filter, and 1.0 M TEAB (1 mL) was added. The solvents were removed by evaporation under reduced pressure, and the residue was purified by ion-exchange chromatography on Q-Sepharose Fast Flow resin eluting with a gradient of triethylammonium bicarbonate (0–1 M). Fractions containing the target compound were identified by a modification of the Briggs phosphate test.48 The combined fractions were concentrated by evaporation in vacuo, and methanol was repeatedly added and evaporated, eventually leaving the triethylammonium salt of 7 as a colorless glass (0.216 mmol, 85%); [α]D +19 (c 1.5, MeOH). 1H NMR (400 MHz, TEA+ salt, D2O, approximately 1:1 mixture of α- and β-anomers): 3.41 (dd, 0.5H, 8.9 Hz, 8.5 Hz, H-2 in β-anomer), 3.55–3.59 (m, 0.5H, H-5 in β-anomer), 3.70 (dd, 0.5H, 9.6 Hz, 3.8 Hz, H-2 in α-anomer), 3.77–3.88 (m, 2H, H-6A and H-6B in α- and β-anomers), 3.92–3.96 (m, 0.5H, H-5 in α-anomer), 4.00–4.09 (m, 1H, H-4 in α- and β-anomers), 4.23 (ddd, 0.5H, 9.0 Hz, 9.0 Hz, 9.0 Hz, H-3 in β-anomer), 4.41 (ddd, 0.5 H, 9.0 Hz, 8.6 Hz, 8.6 Hz, H-3 in α-anomer), 4.70 (d, 0.5H, 8.0 Hz, H-1 in β-anomer), 5.25 (d, 0.5H, 3.8 Hz, H-1 in α-anomer). 31P NMR (162 MHz, TEA+ salt, CD3OD, TEA added, approximately 1:1 mixture of α- and β-anomers): 2.22 (0.5P), 2.63 (0.5P), 2.79 (0.5P), 3.04 (0.5P) m/z (FAB–) 338.9 [M–, 100%]. HRMS: mass calcd for C6H13O12P2, 338.9888 [M–]; found, 338.9894.
d-myo-Inositol 4,5-Bisphosphate (2)
A sample of d-2,3,6-tri-O-benzyl-myo-inositol 4,5-bis-O-(dibenzylphosphate)51 (94 mg, 0.10 mmol) was subjected to hydrogenolytic deprotection as described for 39, above. Purification of the product by ion-exchange chromatography on Q-Sepharose Fast Flow resin, as before, gave the triethylammonium salt of 2 as a colorless glass (0.076 mmol, 76%); [α]D −17 (c 1.0, MeOH), Lit.52 −15.4 (c 0.6, H2O, cyclohexylammonium salt); Lit.39 −10 (c 1, H2O, tetrapotassium salt); Lit.53 −4.4 (c 0.3, H2O, pH 6); Lit.53 +7.9 (c 1.2, H2O, free acid). 1H NMR (500 MHz, TEA+ salt, D2O): 3.60 (dd, 1H, 10.0 Hz, 2.8 Hz, H-1), 3.70 (dd, 1H, 9.8 Hz, 2.8 Hz, H-3), 3.81 (dd, 1H, 9.7 Hz, 9.5 Hz, H-6), 3.99 (ddd, 9.0 Hz, 9.0 Hz, 9.0 Hz, H-5), 4.07 (dd, 1H, 2.8 Hz, 2.8 Hz, H-2), 4.27 (dd, 1H, 9.3 Hz, 9.2 Hz, 9.2 Hz, H-4). 31P NMR (162 MHz, D2O, TEA added): 4.50 (1P), 4.64 (1P). 31P NMR (162 MHz, TEA+ salt, D2O, TEA added): 4.50 (1P), 4.64 (1P). HRMS: mass calcd for C6H13O12P2, 338.9888 [M–]; found, 338.9896.
Measurement of Ca2+ Release from Permeabilized Cells
The effects of AdA and its analogues on intracellular Ca2+ stores were measured using a low-affinity Ca2+-indicator trapped within the intracellular stores of permeabilized cells. DT40 cells stably expressing only rat type 1 IP3R (DT40-IP3R1) were harvested by centrifugation (650g; 2 min) and resuspended [(2–3) × 107 cells/mL] in hepes-buffered saline (HBS: 135 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.5 mM CaCl2, 11.6 mM hepes, and 11.5 mM d-glucose, pH 7.3) supplemented with Mag-fluo-4AM (20 μM), Pluronic F-127 (0.02%), and bovine serum albumin (1 mg/mL). After 1 h at 20 °C in the dark, the Mag-fluo-4-loaded cells were harvested (650g; 2 min) and resuspended (∼2 × 106 cells/mL) in Ca2+-free cytosolic-like medium (CLM: 140 mM KCl, 20 mM NaCl, 2 mM MgCl2, 1 mM EGTA, and 20 mM pipes, pH 7.0). The cells were permeabilized by incubation with saponin (10 μg/mL, 4 min at 37 °C), harvested (650g; 2 min), and resuspended in Mg2+-free CLM (140 mM KCl, 20 mM NaCl, 1 mM EGTA, 375 μM CaCl2 (∼200 nM free [Ca2+]), and 20 mM pipes, pH 7.0). The permeabilized cells (with Mag-fluo-4 trapped within the lumen of the ER) were then attached to 96-well plates (∼8 × 105 cells/well) coated with poly-l-lysine (0.01%) and centrifuged onto the plate (300g; 2 min). Immediately before an experiment, the cells were washed twice in Mg2+-free CLM to remove cytosolic Mag-fluo-4, and the plates were then mounted in a Flexstation fluorescence plate reader (Molecular Devices, Sunnyvale, CA), which allows automated additions to the sample wells while recording fluorescence. Mag-fluo-4 fluorescence was monitored by excitation at 485 nm with emission detected at 520 nm. Active Ca2+ uptake into the ER was initiated by the addition of Mg-ATP (1.5 mM), and after 150 s, when the stores had loaded to a steady-state Ca2+ content, AdA or its analogues were added. The amount of Ca2+ released was expressed as a fraction of the total Ca2+ content of the ER as assessed by addition of 1 μM ionomycin. Data are presented as means ± SEMs from at least three independent experiments, each performed in triplicate. Concentration–effect relationships were fitted to four-parameter logistic equations using nonlinear curve-fitting procedures (GraphPad Prism, San Diego, CA).
Molecular Modeling Methods
The 1N4K structure41 was used in this work. The docking of AdA into IP3R has been described previously.7 In the present work, Schrödinger software running under Maestro version 9.2.112 was used unless otherwise stated. Compounds 5, 6, and 40 were built and minimized. The 1N4K crystal structure and the model of adenophostin A docked into IP3R were prepared using the Protein Preparation Wizard, with exhaustive sampling, including sampling the water orientations, for the hydrogen bond assignment, and minimization of the resulting structures. GOLD version 5.1 was used for docking experiments. In the structure with AdA, the phosphates and the hexose were removed and replaced with hydroxyls. The resulting adenine-ribose was used as a template when docking compounds 5, 6, and 40. The compounds were docked 25 times, with the five water molecules in the binding site retaining their position and orientation. The highest scoring solution had a sensible pose, and the protein–ligand complex was run through the Protein Preparation Wizard to optimize hydrogen bonding, with minimization of the resulting structure. To better enable comparison of the structures, the five protein–ligand complexes were superimposed using the Protein Structure Alignment tool. Ligand interaction diagrams were prepared for all five ligands (IP3, adenophostins A, and compounds 5, 6, and 40) with IP3R (see the Supporting Information). All other figures were prepared using PyMOL (DeLano Scientific LLC).
Acknowledgments
We thank the Wellcome Trust for Programme Grant Support (060544 to B.V.L.P., 082837 to B.V.L.P. and A.M.R., and 085295 to C.W.T.) and the EPSRC Mass Spectrometry Service Centre, Swansea, for high-resolution accurate mass spectra.
Glossary
Abbreviations Used
- AdA
adenophostin A
- IP3
d-myo-inositol 1,4,5-trisphosphate
- IP3R
d-myo-inositol 1,4,5-trisphosphate receptor
- 2′-AMP
2′-adenosine monophosphate
- SAR
structure–activity relationship
- IBC
IP3-binding core
- BDA
butane 2,3-diacetal
- NIS
N-iodosuccinimide
- BSA
N,O-bissilylacetamide
- CSA
camphorsulphonic acid
- TFA
trifluoroacetic acid
- DBU
1,8-diaza-bicycloundec-7-ene
- DCM
dichloromethane
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- Ins(4,5)P2
d-myo-inositol 4,5-bisphosphate
- Ins(1,5)P2
d-myo-inositol 1,5-bisphosphate
- Ins(1,4)P2
d-myo-inositol 1,4-bisphosphate
- ER
endoplasmic reticulum
Supporting Information Available
Full synthetic details for compounds 10–12, 15, and 24–26; 13C NMR data for all compounds; overlays, molecular docking, and ligand interaction diagrams for IP3, Ada, 5, 6, and 40 (SI-1); and spectral charts of all compounds (SI-2). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ School of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Kerala-695016, India.
Supplementary Material
References
- Taylor C. W.; da Fonseca P. C. A.; Morris E. P. IP3 Receptors: The Search for Structure. Trends Biochem. Sci. 2004, 29, 210–219. [DOI] [PubMed] [Google Scholar]
- Berridge M. J.; Lipp P.; Bootman M. D. The Versatility and Universality of Calcium Signalling. Nature Rev. Mol. Cell Biol. 2000, 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Potter B. V. L; Lampe D. Chemistry of Inositol Lipid Mediated Cellular Signaling. Angew. Chem., Int. Ed. Engl. 1995, 34, 1933–1972. [Google Scholar]
- Nerou E. P.; Riley A. M.; Potter B. V. L; Taylor C. W. Selective Recognition of Inositol Phosphates by Subtypes of the Inositol Phosphate Receptors. Biochem. J. 2001, 355, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M.; Kagasaki T.; Hosoya T.; Takahashi S. Adenophostins A and B: Potent Agonists of Inositol-1,4,5-Trisphophate Receptor. J. Antibiot. 1993, 46, 1643–1647. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Tanzawa K.; Takahashi S. Adenophostins, Newly Discovered Metabolites of Penicillium brevicompactum, Act as Potent Agonists of the Inositol 1,4,5-Trisphosphate Receptor Interaction. J. Biol. Chem. 1994, 269, 369–372. [PubMed] [Google Scholar]
- Rosenberg H. J.; Riley A. M.; Laude A. J.; Taylor C. W.; Potter B. V. L. Synthesis and Ca2+ Mobilizing Activity of Purine Modified Mimics of Adenophostin A: A Model for the Adenophostin-Ins(1,4,5)P3 Receptor. J. Med. Chem. 2003, 46, 4860–4871. [DOI] [PubMed] [Google Scholar]
- Chrètien F.; Moitessier N.; Roussel F.; Mauger J.-P.; Chapleur Y. Carbohydrate Based Mimics of D-myo-Inositol 1,4,5-Trisphosphate. Curr. Org. Chem. 2000, 4, 513–534. [Google Scholar]
- Hotoda H.; Murayama K.; Miyamoto S.; Iwata Y.; Takahashi M.; Kawase Y.; Tanzawa K.; Kaneko M. Molecular Recognition of Adenophostin, a Very Potent Ca2+ Inducer at the D-myo-Inositol 1,4,5-Trisphosphate Receptor. Biochemistry 1999, 38, 9234–9241. [DOI] [PubMed] [Google Scholar]
- Correa V. A.; Riley A. M.; Shuto S.; Horne G.; Nerou E. P.; Marwood R. D.; Potter B. V. L.; Taylor C. W. Structural Determinants of Adenophostin A Activity at Inositol Trisphosphate Receptors. Mol. Pharmacol. 2001, 59, 1206–1215. [DOI] [PubMed] [Google Scholar]
- Wilcox R. A.; Erneux C.; Primrose W. U.; Gigg R.; Nahorski S. R. 2-Hydroxyethyl α-D-Glucopyranoside 2,3′,4′-Trisphosphate, a Novel Metabolically Resistant Adenophostin A and myo-Inositol 1,4,5-Trisphosphate Analog, Potently Interacts with the myo-Inositol 1,4,5-Trisphosphate Receptor. Mol. Pharmacol. 1995, 47, 1204–1211. [PubMed] [Google Scholar]
- Riley A. M.; Correa V.; Mahon F.; Taylor C. W.; Potter B. V. L. Bicyclic Analogs of D-myo-Inositol 1,4,5-Trisphosphate Related to Adenophostin A: Synthesis and Biological Activity. J. Med. Chem. 2001, 44, 2108–2117. [DOI] [PubMed] [Google Scholar]
- Rosenberg H. J; Riley A. M.; Marwood R. D.; Correa V.; Taylor C. W.; Potter B. V. L. Xylopyranoside-Based Agonists of D-myo-Inositol 1,4,5-Trisphosphate Receptors: Synthesis and Effect of Stereochemistry on Biological Activity. Carbohydr. Res. 2001, (332), 53–66. [DOI] [PubMed] [Google Scholar]
- Roussel F.; Moitessier N.; Hilly M.; Chrètien F.; Mauger J.-P.; Chapleur Y. D-myo-Inositol-1,4,5-Trisphosphate and Adenophostin Mimics: Importance of the Spatial Orientation of a Phosphate Group on the Biological Activity. Bioorg. Med. Chem. 2002, 10, 759–768. [DOI] [PubMed] [Google Scholar]
- Sureshan K. M.; Trusselle M.; Tovey S. C.; Taylor C. W.; Potter B. V. L. Guanophostin A: Synthesis and Evaluation of a High Affinity Agonist of the d-myo-Inositol 1,4,5-Trisphosphate Receptor. Chem. Commun. 2006, 2015–2017. [DOI] [PubMed] [Google Scholar]
- Rossi A. M.; Riley A. M.; Potter B. V. L.; Taylor C. W.. Adenophostins: High-affinity Agonists of IP3 Receptors. In Current Topics in Membranes; Serysheva I., Ed.; Academic Press: Burlington, 2010; Vol. 66, pp 209–233. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Rossi A. M.; Riley A. M.; Rahman T.; Potter B. V. L; Taylor C. W. Binding of IP3 and Adenophostin A to the N-terminal Region of the IP3 Receptor: Thermodynamic Analysis Using Fluorescence Polarization with a Novel IP3 Receptor Ligand. Mol. Pharmacol. 2010, 77, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S.; Kinoshita T.; Takahashi M. Adenophostins A and B: Potent Agonists of Inositol 1,4,5-Trisphosphate Receptor Produced by Penicillium brevicompactum. Structure Elucidation. J. Antibiot. 1994, 47, 95–100. [DOI] [PubMed] [Google Scholar]
- Rossi A. M.; Sureshan K. M.; Riley A. M.; Potter B. V. L.; Taylor C. W. Selective Determinants of IP3 and Adenophostin A Interactions with Type 1 IP3 Receptors. Br. J. Pharmacol. 2010, 161, 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felemez M.; Marwood R. D.; Potter B. V. L.; Spiess B. Inframolecular Studies of the Protonation of Adenophostin A: Comparison with 1-D-myo-Inositol 1,4,5-Trisphosphate. Biochem. Biophys. Res. Commun. 1999, 266, 334–340. [DOI] [PubMed] [Google Scholar]
- Sureshan K. M.; Riley A. M.; Rossi A. M.; Tovey S. C.; Dedos S. G.; Taylor C. W.; Potter B. V. L. Activation of IP3 Receptors by Synthetic Bisphosphate Ligands. Chem. Commun. 2009, 1204–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwood R. D.; Jenkins D. J.; Correa. V.; Taylor C. W.; Potter B. V. L. Contribution of the Adenine Base to the Activity of Adenophostin A Investigated Using a Base Replacement Strategy. J. Med. Chem. 2000, 43, 4278–4287. [DOI] [PubMed] [Google Scholar]
- Shuto S.; Horne G.; Marwood R. D.; Potter B. V. L. Total Synthesis of Nucleobase-Modified Adenophostin A Mimics. Chem. Eur. J. 2001, 7, 4937–4946. [DOI] [PubMed] [Google Scholar]
- Vorbrüggen H.; Krolikiewicz K.; Bennua B. Nucleoside Synthesis with Trimethyl Triflate and Perchlorate as the Catalyst. Chem. Ber. 1981, 114, 1234–1255. [Google Scholar]
- de Kort M.; Correa V.; Valentijn R. P. M; van der Marel G. A.; Potter B. V. L.; Taylor C. W.; van Boom J. H. Synthesis of Potent Agonists of the D-myo-Inositol 1,4,5-Trisphosphate Receptor Based on Clustered Disaccharide Polyphosphate Analogs of Adenophostin A. J. Med. Chem. 2000, 43, 3295–3303. [DOI] [PubMed] [Google Scholar]
- Alper P. B.; Hendrix M.; Sears P.; Wong C.-H. Probing the Specificity of Aminoglycoside Ribosomal RNA Interaction with Designed Synthetic Analogs. J. Am. Chem. Soc. 1998, 120, 1965–1978. [Google Scholar]
- de Kort M.; Regenbogen A. D.; Overkleeft H. S.; Challiss R. A. J.; Iwata Y.; Miyamoto S.; van der Marel G. A.; van Boom J. H. Synthesis and Biological Evaluation of Cyclophostin: A 5′,6″-Tethered Analog of Adenophostin A. Tetrahedron 2000, 56, 5915–5928. [Google Scholar]
- de Kort M.; Luijendijk J.; van der Marel G. A.; van Boom J. H. Synthesis of Photoaffinity Derivatives of Adenophostin A. Eur. J. Org. Chem. 2000, 3085–3092. [Google Scholar]
- Hayakawa Y.; Kataoka M. Facile Synthesis of Oligodeoxy Ribo Nucleotides via the Phosphoramidite Method Without Nucleoside Base Protection. J. Am. Chem. Soc. 1998, 120, 12395–12401. [Google Scholar]
- Hanessian S.; Liak T. J.; Vanasse B. Facile Cleavage of Benzyl Ethers by Catalytic Transfer Hydrogenation. Synthesis 1981, 396–397. [Google Scholar]
- Arasappan A.; Fraser-Reid B. n-Pentenyl Glycoside Methodology in the Stereoselective Construction of the Tetrasaccharyl Cap Portion of Leishmania Lipophosphoglycan. J. Org. Chem. 1996, 61, 2401–2406. [Google Scholar]
- van Steijn A. M. P.; Kamerling J. P.; Vliegenthart J. F. G. Synthesis of Trisaccharide Methyl Glycosides Related to Fragments of the Capsular Polysaccharides of Streptococcus pneumoniae type 18C. Carbohydr. Res. 1992, 225, 229–245. [DOI] [PubMed] [Google Scholar]
- Ashwell M.; Bleasdale C.; Golding B. T.; O'Neill I. K. An Improved Route to Guanines Substituted at N-9. J. Chem. Soc. Chem. Commun. 1990, 955–956. [Google Scholar]
- Chen X.; Kern E. R.; Drach J. C.; Gullen E.; Cheng Y.-C.; Zemlicka J. Structure Activity Relationships of (S,Z)-2-Aminopurine Methylene Cyclopropane Analogs of Nucleosides. Variation of Purine-6-Substituents and Activity Against Herpes Viruses and Hepatitis B Viruses. J. Med. Chem. 2003, 46, 1531–1537. [DOI] [PubMed] [Google Scholar]
- Borissow C. N.; Black S. J.; Paul M.; Tovey S. C.; Dedos S. G.; Taylor C. W.; Potter B. V. L. Adenophostin A and Analogs Modified at the Adenine Moiety: Synthesis, Conformational Analysis and Biological Activity. Org. Biomol. Chem. 2005, 3, 245–252. [DOI] [PubMed] [Google Scholar]
- Rosenberg H.; Riley A. M.; Correa V.; Taylor C. W.; Potter B. V. L. C-Glycoside Based Mimics of d-myo-Inositol 1,4,5-Trisphosphate. Carbohydr. Res. 2000, 329, 7–16. [DOI] [PubMed] [Google Scholar]
- Fraser-Reid B.; Yu K. L. A Novel Reagent for the Synthesis of myo-Inositol Phosphates: N,N-Diisopropyl Dibenzyl Phosphoramidite. Tetrahedron Lett. 1988, 29, 979–982. [Google Scholar]
- Liao W.; Lu D. Synthesis of a Hexasaccharide Acceptor Corresponding to the Reducing Terminus of Mycobacterial 3-O-Methylmannose Polysaccharide (MMP). Carbohydr. Res. 1996, 296, 171–182. [DOI] [PubMed] [Google Scholar]
- Lu P.-J.; Gou D.-M.; Shieh W.-R.; Chen C.-S. Molecular Interactions of Endogenous D-myo-Inositol Phosphates with the Intracellular D-myo-Inositol 1,4,5-Trisphosphate Recognition Site. Biochemistry 1994, 33, 11586–11597. [DOI] [PubMed] [Google Scholar]
- Murphy C. T.; Riley A. M.; Lindley C. J.; Jenkins D. J.; Westwick J.; Potter B. V. L Structural Analogs of D-myo-Inositol-1,4,5-Trisphosphate and Adenophostin A: Recognition by Cerebellar and Platelet Inositol-1,4,5-Trisphosphate Receptors. Mol. Pharmacol. 1997, 52, 741–748. [DOI] [PubMed] [Google Scholar]
- Bosanac I.; Alattia J.-R.; Mal T. K.; Chan J.; Talarico S.; Tong F. K.; Tong K. I.; Yoshikawa F.; Furuichi T.; Iwai M.; Michikawa T.; Mikoshiba K.; Mitsuhiko I. Structure of the Inositol 1,4,5-Trisphosphate Receptor Binding Core in Complex with its Ligand. Nature 2002, 420, 696–700. [DOI] [PubMed] [Google Scholar]
- Bosanac I.; Michikawa T.; Mikoshiba K.; Ikura M. Structural Insights into the Regulatory Mechanism of IP3 Receptor. Biochim. Biophys. Acta 2004, 1742, 89–102. [DOI] [PubMed] [Google Scholar]
- Madden D. R. The Structure and Function of Glutamate Receptor Ion Channels. Nat. Rev. Neurosci. 2002, 3, 91–101. [DOI] [PubMed] [Google Scholar]
- Lin C.-C.; Back K.; Lu Z. Apo- and InsP3-bound Crystal Structures of the Ligand-binding Domain of an InsP3 Receptor. Nat. Struct. Mol. Biol. 2011, 18, 1172–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M.-D.; Velamakanni S.; Ishiyama N.; Stathopulos P. B.; Rossi A. M.; Kahn S. A.; Dale P.; Li C.; Ames J. B.; Ikura M.; Taylor C. W.. Structural and Functional Conservation of Key Domains in InsP3 and Ryanodine Receptors. Nature 2012, in press. [DOI] [PMC free article] [PubMed]
- Beecroft M. D.; Marchant J. S.; Riley A. M.; van Straten N. C. R.; van der Marel G. J.; van Boom J. H.; Potter B. V. L.; Taylor C. W. Acyclophostin: A Ribose-Modified Analog of Adenophostin A with High Affinity for Inositol 1,4,5-Trisphosphate Receptors and pH-Dependent Efficacy. Mol. Pharmacol. 1999, 1, 109–117. [DOI] [PubMed] [Google Scholar]
- Murphy C. T.; Bullock A. J.; Lindley C. J.; Mills S. J.; Riley A. M.; Potter B. V. L.; Westwick J. Enantiomers of myo-Inositol-1,3,4-Trisphosphate and myo-Inositol-1,4,6-Trisphosphate: Stereospecific Recognition by Cerebellar and Platelet myo-Inositol-1,4,5-Trisphosphate Receptors. Mol. Phramacol. 1996, 50, 1223–1230. [PubMed] [Google Scholar]
- Lampe D.; Liu C.; Potter B. V. L. Synthesis of Selective Non-Ca2+ Mobilizing Inhibitors of D-myo-Inositol 1,4,5-Trisphosphate 5-Phosphatase. J. Med. Chem. 1994, 37, 907–912. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Dubin D. T. The Role of Polyamines in the Neutralization of Bacteriophage Deoxyribonucleic Acid. J. Biol. Chem. 1960, 235, 769–775. [PubMed] [Google Scholar]
- Alpe M.; Oscarson S. Synthesis of Oligosaccharides Corresponding to Streptococcus pneumoniae Type 9 Capsular Polysaccharide Structures. Carbohydr. Res. 2002, 337, 1715–1722. [DOI] [PubMed] [Google Scholar]
- Desai T.; Gigg J.; Gigg R.; Martín-Zamora E. The Preparation of Intermediates for the Synthesis of 1d-myo-Inositol 1,4,5- and 2,4,5-Trisphosphates, 1,4-Bisphosphate 5-Phosphorothioate, and 4,5-Bisphosphate 1-Phosphorothioate from 1d-3,6-di-O-Benzyl-1,2-O-Isopropylidene-myo-Inositol. Carbohydr. Res. 1994, 262, 59–77. [DOI] [PubMed] [Google Scholar]
- Krylova V. N.; Kobelkova N. I.; Oleinik G. F.; Shvets V. I. Study of the Derivatives of Asymmetrically Substituted myo-Inositol. 27. Synthesis of myo-Inositol Diphosphates. Zh. Org. Khim. 1980, 16, 62–68. [Google Scholar]
- Podeschwa M. A. L.; Plettenburg O.; Altenbach H.-J. Flexible Stereo- and Regioselective Synthesis of myo-Inositol Phosphates (Part 1): Via Symmetrical Conduritol B Derivatives. Eur. J. Org. Chem. 2005, 3101–3115. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.