

Abstract
c-Myc is frequently overexpressed in tumors and plays an important role in the regulation of cancer metabolism. Hypoxia-inducible factor-1 (HIF1), the master regulator of the hypoxic response, enhances tumorigenesis and influences metabolism via upregulation of the glycolytic pathway and suppression of mitochondrial respiration. Together, deregulated Myc and HIF1 cooperate to lend metabolic advantages to proliferating cancer cells and contribute to the Warburg Effect. Here we show that overexpression of Myc significantly stabilizes the alpha subunit of HIF1 (HIF1alpha) under normoxic conditions and enhances HIF1alpha accumulation under hypoxic conditions in cells. Post-transcriptional regulation of HIF1α by Myc led to the induction of HIF1α gene targets. Normoxic HIF1α protein expression was also dependent on Myc. Functionally; HIF1α expression was required for Myc-induced anchorage-independent growth and cell proliferation. Myc-dependent stabilization of HIF1α involved either disruption of binding to the VHL complex or post-translational protein modifications. Taken together, our findings uncover a previously uncharacterized regulatory relationship between Myc and HIF1 that has important implications for cancer metabolism and development.
INTRODUCTION
Breast cancer is the most common malignancy of women worldwide and targets 13% of women in the United States (NCI). The c-myc oncogene, which encodes the transcription factor c-Myc, contributes to many forms of cancer and was one of the first genes found to be amplified in breast cancer (1). Studies suggest that at least 15–20% of breast cancers present with significant amplification of c-myc and that overexpression of c-Myc likely occurs in an even greater number of tumors through other mechanisms (2). Animal model studies have confirmed that constitutive Myc expression in the mammary gland is oncogenic in transgenic mice (3). Microarray studies have exposed Myc as a weak yet pleiotropic transcription factor, possessing the ability to activate or repress 5–10% of all genes by 1.5-fold or more (4). While these target genes vary greatly depending on the cellular context, commonly targeted processes include the cell cycle, metabolism, and apoptosis (5).
In transformed cells, Myc promotes glycolysis by activating glycolytic enzymes (6, 7). Tumor cells often exhibit increased rates of glycolysis even in the presence of adequate oxygen concentrations (the Warburg Effect), although increased rates of glycolysis normally occur under hypoxic conditions (8, 9). HIF1 is the key mediator of the hypoxic response in cellular tissues, comprised of a constitutively expressed HIF1β subunit and an oxygen-responsive HIF1α subunit (10, 11). In oxygenated tissues, HIF1α has a very short half-life (10, 12). Rapid turnover is mediated through the hydroxylation of two prolines at amino acids 402 and 564, performed by oxygen-dependent prolyl hydroxylases (13, 14, 15 (reviewed in {Kaelin, 2005 #118)). Prolyl-hydroxylation of HIF1α leads to ubiquitination by the Von Hippel-Lindau (VHL) E3 ubiquitin ligase and degradation via the 26S proteasome (reviewed in (16, 17)). In a hypoxic environment HIF1α becomes stabilized (18, 19), although in some instances HIF1α can be stabilized under normoxia as well (reviewed in (20, 21)). HIF1α is commonly expressed in cancers, as tumor microenvironments suffer from hypoxia owing to an incomplete vasculature.
Studies have reported HIF1-induced opposition of Myc under hypoxia when Myc is expressed at non-tumorigenic levels. Studies have also revealed the cooperation of HIF1 and deregulated Myc under hypoxia (22–25). However, our data show that Myc regulates HIF1α expression under both normoxia and hypoxia, and that HIF1α expression represents an important component of Myc function. Our results demonstrate a novel and complex relationship between Myc, HIF1α, and tumor development.
MATERIALS AND METHODS
Cell culture, transfection, and retroviral infection
Retroviral-producing PhoeNX cells and breast cancer lines MCF7 and T47D were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS. IMEC epithelial cells were cultured in 1:1 DMEM/F12 medium supplemented with 5% FBS and other factors (DiRenzo et al., 2002). Retroviral infection was performed using PhoeNX cells. Briefly, PhoeNX cells were transfected with 7μg DNA using LipoD293 (Signagen) according to manufacturer's instructions. Virus was harvested from the culture medium two days later and used to infect recipient IMECs. Cells were selected two days later and maintained in 150μg/mL hygromycin (Calbiochem) to select for LXSH vectors (LXSH, LXSH-Myc). Hypoxic conditions were induced by culturing cells for two hours in a sealed hypoxia chamber (Billups Rothenberg) after flushing with a mixture of 1% O2, 5% CO2, and 94% N2. Hypoxic conditions were also induced by the addition of CoCl2 (Sigma) at a concentration of 200μM for 3 hours. We refer to the ambient oxygen concentration as normoxic (95% air, 5% CO2). Although the ambient oxygen concentration is not the physiological normoxic state, this is a frequently used convention in the literature.
Cell counts
Cells were plated at 1 × 104 per well in 6-well plates on Day 0. Cells were maintained as above. Cells were harvested each day for four days and counted using a hemacytometer. At least 100 cells were counted. The experiment was performed in duplicate on two separate occasions. Knockdown cell growth curves were achieved by plating cells at 1 × 104 per well, transfecting with HIF1α and non-targeting control siRNA 24 hours later (Day 1), and counting cells for three days post-transfection. The experiment was performed in duplicate on two separate occasions.
Soft agar assay
Assays were performed in 6-well plates in duplicate on two separate occasions. The lower layer of agar consisted of 2mL IMEC medium containing 10% FBS and 0.6% Noble Agar (USB). The upper layer consisted of 2mL IMEC medium containing 10% FBS, 0.3% Noble Agar, and 4 × 104 cells. Agar at 50°C was mixed with medium at 37°C, plated, and left to set for 10 min. Plates were fed every other day with 250μl IMEC medium containing 10% FBS. After 10 days, undivided cells and colonies were scored using a reticle, measuring the greatest diameter. 100 cells were counted per well.
Real-Time (RT) PCR
RNA was extracted from log phase cells using Trizol (Invitrogen) according to manufacturer's instructions. RNA was converted into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad) and PCR was performed using iQ SYBR Green Supermix and the CFX96 Real-Time PCR Detection System (Bio-Rad). Primers used were: GAPDH GCTCAGACACCATGGGGAAGGTGAAG and GCTGATGATCTTGAGGCTGTTGTCATAC; Myc CGAGGAGGAGAACTTCTACCAGC and CGAGAAGCCGCTCCACATACAGTCC; HIF1α ACAGTATTCCAGCAGACTCAA and CCTACTGCTTGAAAAAGTGAA; HK2 GGATGATTGCCTCGCATCTGC and GGAACTCTCCGTGTTCTGTCC; ALDOA CCATGCCCTACCAATATCCAGC and GGTGGTAGTCTCGCCATTTGTCC; eNOS GGAGAGATCCACCTCACTGTAGCand CCTTTGCTCTCAATGTCATGC; BGN GGAACATGAACTGCATCGAGATGG and CGATCATCCTGATCTGGTTGTGG; VHL CCTTTGGCTCTTCAGAGATGC and GGTCTTTCTGCACATTTGGGTGG; CUL2 GCAGAAGATTCAACACTACACTTGC and CCTTGCTGTATTCTTCCCAGTACC; RBX1 GCAGCGATGGATGTGGATACC and CCTGTCGTGTTTTGAGCCAGC; Elongin B CCACAAGACCACCATCTTCACG and CGTGCTGTTTGACTGGTGAAGC; Elongin C CCTATGGTGGCTGTGAAGGACC and GCAGTTCCAGTGCAATTTCAGG.
Western blots
Cells were lysed in F-Buffer and cell extracts were normalized for protein content. Proteins were resolved by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibody in 5% milk. Antibodies used include: monoclonal anti-HIF1α (Novus Biologicals), polyclonal anti-HIF1α (Novus Biologicals), monoclonal anti-VHL (BD Biosciences), polyclonal anti-VHL (Novus Biologicals), anti-hydroxyproline-564-specific-HIF1α (kindly provided by R. Freeman), monoclonal anti-c-Myc antibody (C-33; Santa Cruz), polyclonal anti-c-Myc antibody (N262; Santa Cruz), anti-β-tubulin (Santa Cruz), monoclonal anti-CUL2 (Santa Cruz), polyclonal anti-CUL2 (Abcam), polyclonal anti-Rbx1 (Santa Cruz), polyclonal anti-Elongin B (Santa Cruz), monoclonal anti-Elongin C (BD Biosciences), and polyclonal anti-Elongin C (Novus Biologicals). Immunoprecipitations were performed by complexing Protein A/G-Plus Agarose beads with 1μg of antibody for one hour and then mixing the beads with cell extract overnight. ImageJ (NCBI) was used to quantitate protein expression levels.
Cycloheximide assay
Cells were plated in 6-well plates and treated with cycloheximide (CHX) 24 hours post-plating. CHX was added at a concentration of 50μg/mL to the cells at 20-minute intervals. Hypoxic extracts were made by treating cells with 200μM CoCl2 for 3 hours prior to CHX treatment. Protein extracts were prepared as previously described. HIF1α and Myc half-lives were calculated after quantitating protein and normalizing to β-tubulin.
siRNA
Experiments involving siRNA were performed by transfecting HIF1A, c-myc, or non-targeting siRNA (Dharmacon) at 2μM in 6-well plates according to manufacturer's instructions. Cells were replated 24 hours following transfection for soft agar analysis. RNA and protein was harvested for analysis 24 or 48 hours following transfection. siRNA growth curve experiments were performed by transfecting HIF1A and non-targeting siRNA at 2μM in 24-well plates according to manufacturer's instructions.
ON-TARGETplus Human HIF1A siRNA: J-004018-07-0010, J-004018-08-0010
ON-TARGETplus Human c-myc siRNA: J-003282-23-0005, J-003282-24-0005
ON-TARGETplus Non-targeting siRNA: D-001810-01-05
RESULTS
Myc enhances accumulation of hypoxia regulator HIF1α
We have previously shown that overexpression Myc in TERT-immortalized human mammary epithelial cells (IMECs) leads to a partial oncogenic transformation with an increased proliferation rate and anchorage-independent growth (26). It is important to note that Myc expression in the transduced IMECs is similar to Myc protein levels found in breast cancer cell lines T47D and MDA-231 and hence represents physiologically relevant expression (26). Whole genome expression analysis revealed that the most significantly enriched Myc-induced cluster corresponded to angiogenesis and vascularization (Supplemental Table; data available on request). Previous studies have linked angiogenesis to Myc in a transgenic model (27, 28). Furthermore, many genes involved in these pathways have been scored as potential direct Myc target genes in microarrays. However, the most prominent regulator of angiogenesis is Hypoxia Inducible Factor 1 (HIF1), which coordinately controls the expression of a large number of genes in the pathway. Therefore, we explored if the strong induction of an angiogenic program is due to activation of HIF1α instead of a direct effect of Myc.
To assess a potential role for HIF1 in the induction of an angiogenic gene response, we examined the levels of HIF1α protein in vector and Myc-overexpressing cells under normoxic (95% air, 5%CO2) or hypoxic conditions (1% O2, 5% CO2, 94% N2). As expected, there were very low levels of HIF1α in IMECs cultured under normoxic conditions, and the level was induced substantially under hypoxia. Surprisingly, we found that overexpression of Myc enhances the accumulation of HIF1α under normoxic conditions to a significant level. Moreover, overexpression of Myc exaggerates HIF1α accumulation under hypoxic conditions (Figure 1A). On average, HIF1α expression in Myc-overexpressing cells under normoxia is ten times more than in vector IMECs (Figure 1B). We determined whether the increase in protein expression is due to an increase in transcription of the HIF1α gene by performing quantitative real-time (RT) PCR under normoxic and hypoxic conditions. We found that HIF1α is not transcriptionally induced by hypoxia, as expected, or by Myc (Figure 1C). Myc transcription is also not affected by hypoxia. These data suggest that Myc-enhanced HIF1α expression is post-transcriptionally regulated.
Figure 1. Myc post-transcriptionally enhances accumulation of HIF1α under both normoxia and hypoxia.

A) Western blot analyses of IMECs were performed with equivalent amounts of protein using anti-HIF1α antibody, anti-hydroxyl P564-specific HIF1α antibody (OH-P564), anti-c-Myc antibody, and anti-β-tubulin antibody. Hypoxic cell extracts were made after incubating cells for two hours in a hypoxia chamber with 1% oxygen. Normoxic HIF1α blots are shown at a longer exposure than the hypoxic HIF1α blots. Norm= Normoxia, Hypo= Hypoxia. B) Average HIF1α protein expression, normalized to β-tubulin, was quantitated using three independent cell extract sets. Expression levels are normalized to normoxic vector. C) Real-time RT-PCR was performed using RNA harvested from IMECs and primers specific to HIF1A, c-myc, and GAPDH in triplicate. Average c-myc and HIF1A RNA levels are normalized to GAPDH and then normalized to their respective normoxic vectors. D) IMECs were treated with proteasome inhibitor MG132 under normoxic and hypoxic conditions. Western blot analyses were performed with equivalent amounts of protein using anti-HIF1α and anti-β-tubulin antibodies. E) After treatment with MG132, average hydroxy-P564 HIF1α protein expression was determined in comparison to total HIF1α protein expression under normoxia and hypoxia. F) Western blot analyses of myc−/− rat fibroblasts (vector or Myc-reconstituted) were performed as described above. G) Real-time RT-PCR was performed using RNA harvested from myc−/− rat fibroblasts and primers specific to HIF1A, c-myc, and GAPDH in triplicate. Average c-myc and HIF1A RNA levels are normalized to GAPDH. HIF1A is normalized to its normoxic vector. c-myc is normalized to normoxic myc−/−(Myc) since vector cells express no c-myc RNA. Error bars reflect the standard deviation.
We treated the vector control IMEC and IMEC-Myc cells with the proteasome inhibitor MG132 to block protein degradation and assess the total amount of HIF1α protein being translated under normoxia and hypoxia. As expected, the enhanced level of HIF1α under hypoxia is an effect of stabilization and not increased translation since protein levels become equal when degradation is inhibited (Figure 1D). We find that, even though normoxic HIF1α is stabilized significantly by Myc, treatment with MG132 leads to further stabilization indicating that there is some ongoing degradation in IMEC-Myc cells. In contrast, all of the HIF1α protein appears to be fully stabilized under hypoxia in Myc-overexpressing cells because MG132 treatment gives no further enhancement.
Since HIF1 protein levels are primarily controlled by proline hydroxylation and interaction with the VHL E3 ligase, we tested if the enhanced HIF1α expression in response to Myc under normoxia is due to the loss of proline hydroxylation. We used an antibody specific to the prolylhydroxylation site (hydroxy-P564 HIF1α) to measure the extent of modification relative to HIF1α protein levels. We confirmed that HIF1α is prolyl-hydroxylated in normoxic cells, and hydroxylation is suppressed under hypoxia (Figure 1A). To compare HIF1α modification with or without high levels of Myc, we treated cells with MG132 to prevent turnover of HIF1α and quantitated the level of prolyl-hydroxylation to total HIF1α levels. We find that the ratio of prolyl-hydroxylated to total HIF1α is identical with high and low Myc (Figure 1D), thus the elevated levels of HIF1α are not due to the suppression of proline hydroxylation. Furthermore, prolyl-hydroxylation is sensitive to reactive oxygen species (ROS), but quenching of ROS with N-acetylcysteine had no effect on the Myc-dependent accumulation of HIF1α (Supplemental Figure).
We wanted to explore if HIF1α protein expression is induced by Myc in other cell types. We measured protein levels in myc−/− Rat1 fibroblasts before and after stable reconstitution with mouse c-Myc (Figure 1F). As in IMECs, HIF1α protein levels are enhanced with Myc reconstitution under normoxic conditions, and Myc dramatically exaggerates HIF1α levels in hypoxia. As expected, there are no changes in HIF1α mRNA levels due to Myc expression (Figure 1G).
HIF1 induces its target gene expression
We next looked at the expression levels of HIF1α target genes to determine if there is a functional downstream effect of enhanced HIF1α by Myc under normoxia in IMECs. We transiently transfected vector and Myc-overexpressing IMECs with siRNAs against HIF1A, c-myc, or a non-targeting control and achieved depletion at the RNA level (Figure 2A). By RT-PCR, we found that glycolytic enzymes HK2 and ALDOA, nitric oxide synthase eNOS, and collagen fibril assembly proteoglycan BGN are all transcriptionally upregulated in Myc-overexpressing cells under normoxia (Figure 2B). Of these genes, HK2 is the only one known to be a direct binding target of both HIF1α and Myc. Consistent with this, these genes are downregulated in response to HIF1α depletion. These genes are also all downregulated in response to Myc knockdown, suggesting that there is indeed a functional downstream effect of enhanced HIF1α expression by Myc.
Figure 2. HIF1α gene targets are transcriptionally upregulated in IMEC-Myc cells.

A) IMECs were transfected with non-targeting siRNA or siRNAs (1 or 2) targeting HIF1A or c-myc. RNA was isolated 48 hrs post-transfection. Real-time RT-PCR was performed using primers specific to HIF1A, c-myc, and GAPDH in triplicate. Average RNA levels were normalized to GAPDH and then normalized to their respective normoxic vectors. B) Real-time RT-PCR was performed using primers specific to ALDOA, HK2, eNOS, BGN, and GAPDH in triplicate. Average RNA levels were normalized to GAPDH and then normalized to their respective normoxic vectors. Error bars reflect the standard deviation.
Myc induces HIF1 expression in breast cancer cells
We also explored whether HIF1α protein levels are dependent on Myc in human cancer cells. To this end, we examined HIF1α and Myc in the breast cancer cell lines T47D and MCF7 (Figure 3A). MCF7 cells have 3.5-fold higher Myc protein levels than T47D and have significant constitutive HIF1α protein under normoxic conditions, whereas T47D cells have virtually undetectable levels (Figure 3B). As in other cell types, Myc protein levels largely correspond to mRNA levels, whereas HIF1α mRNA levels are equivalent between cell lines (Figure 3C). To test if HIF1α protein levels are Myc-dependent in MCF7 cells, we used siRNA to deplete native Myc. We found that the loss of Myc led to a corresponding reduction in HIF1α, suggesting that HIF1α expression is dependent on Myc in MCF7 cells (Figure 3D).
Figure 3. Myc-dependent HIF1α expression in breast cancer cell lines.
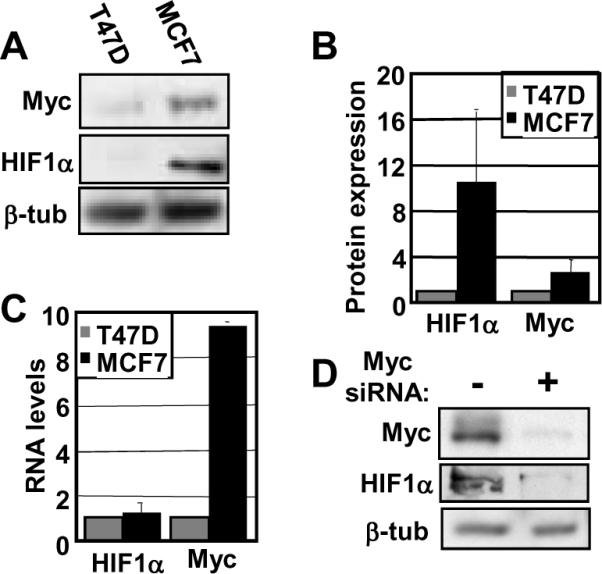
A) Western blot analyses of T47D and MCF7 breast cancer cell lines were performed with equivalent amounts of protein using anti-HIF1α, anti-c-Myc, and anti-β-tubulin antibodies. B) Average HIF1α and Myc protein expression, normalized to β-tubulin, were quantitated using three independent cell extract sets. Expression levels for HIF1α and Myc are normalized to T47D. C) Real-time RT-PCR was performed using RNA harvested from T47D and MCF7 cells and primers specific to HIF1A, c-myc, and GAPDH. Average HIF1A and c-myc RNA levels are normalized to GAPDH and then normalized T47D levels. D) MCF7 cells were transfected with siRNA against c-myc or non-targeting siRNA. Western blot analyses were performed after 24 hours with equivalent amounts of protein using anti-HIF1α, anti-c-Myc, and anti-β-tubulin antibodies. Error bars reflect the standard deviation.
Myc stabilizes HIF1α protein
Since we found elevated HIF1α protein with no change in mRNA levels (Figure 1), we hypothesized that the substantial accumulation of HIF1α in response to Myc under normoxia and hypoxia could be due to a decrease in its rate of degradation. We treated IMEC and IMEC-Myc cells with cycloheximide (CHX) to block protein synthesis and assessed HIF1α protein levels at 20 min intervals under both normoxic and hypoxic conditions (Figure 4). As previously described, HIF1α has an extremely short half-life under normoxia (t1/2<7 min) and is barely detectable in the vector cells. However, we found that overexpression of Myc under normoxic conditions significantly stabilizes HIF1α protein (t1/2=56 min). This stabilization can account for most or all of the enhanced HIF1α protein levels in IMEC-Myc. In contrast, Myc protein turnover is similar between control and Myc-overexpressing cells. Under hypoxia, HIF1α protein is stabilized (t1/2=42 min) as previously described (11, 29, 30). However, Myc-overexpression further stabilizes HIF1 (t1/2>90 min), which can account for the hyper-induction of HIF1α in hypoxic IMEC-Myc cells.
Figure 4. Expression of Myc stabilizes HIF1α protein.
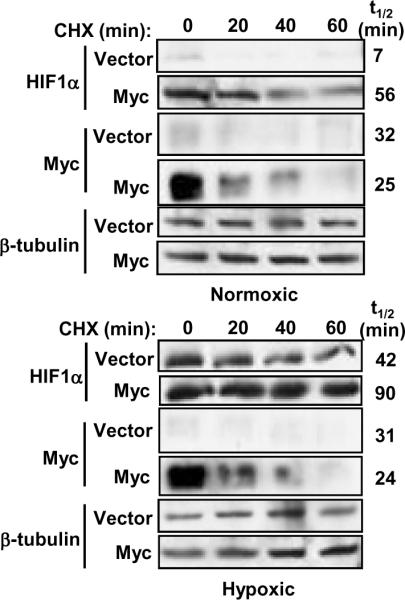
IMECs (vector or Myc-overexpressing) were treated with cycloheximide (CHX) for 20, 40, and 60 minutes. Hypoxic extracts were made by treating cells with 200μM CoCl2 for 3 hours prior to CHX treatment. Western blot analyses were performed with equivalent amounts of protein using anti-HIF1α, anti-c-Myc, and anti-β-tubulin antibodies. Half-lives of HIF1α and Myc were determined after normalization to β-tubulin. Normoxic HIF1α blots are shown at a longer exposure than the hypoxic HIF1α blots.
Myc enhances accumulation of VHL complex components
We next sought to determine other possible mechanisms that could account for Myc-induced stabilization of HIF1α protein. Since HIF1α prolyl-hydroxylation is maintained in IMEC-Myc cells (Figure 1E), we turned our focus to the next step in the normoxic regulation of HIF1α: the recognition and ubiquitination of prolyl-hydroxylated HIF1α by the VHL complex. We hypothesized a role for Myc in rendering the VHL complex less functional. We considered that Myc could be regulating expression levels of the components themselves, promoting protein modifications that could impact the formation of the VHL complex or affecting external cofactors that influence function. We first assessed protein levels to determine whether Myc might be downregulating one or more of the VHL components, which would de-stabilize complex formation. Quite unexpectedly, we found that Myc significantly enhances the accumulation of all of the VHL complex components under both normoxic and hypoxic conditions (Figure 5A). Furthermore, except for a two-fold induction of VHL RNA, the regulation appears to be entirely post-transcriptional (Figure 5B). As with HIF1α previously, we wanted to determine whether the expression of these VHL complex components is induced by Myc in other cell types. We again measured protein levels in myc−/− Rat1 fibroblasts before and after stable reconstitution with mouse c-Myc. As in IMECs, the protein levels of the VHL components are enhanced in response to elevated Myc expression under both normoxic and hypoxic conditions (Figure 5C). Of note, both VHL and Elongin B not only change in abundance but also shift in migration in SDS/PAGE (Figure 5A and 5C), suggesting that there may be Myc-induced changes in protein modification.
Figure 5. Myc enhances post-transcriptional accumulation of VHL complex components under both normoxia and hypoxia.

A) Western blot analyses of IMEC cells were performed with equivalent amounts of protein using anti-VHL, anti-CUL2, anti-RBX1, anti-Elongin B, anti-Elongin C, and anti-β-tubulin antibodies. B) Real-time RT-PCR was performed using RNA harvested from IMECs and primers specific to VHL, CUL2, RBX1, Elongin B (TCEB2), Elongin C (TCEB1), and GAPDH in triplicate. Average RNA levels are normalized to GAPDH and then normalized to their respective normoxic vectors. C) Western blot analyses of myc−/− rat fibroblasts (+/− reconstituted mouse c-Myc) were performed as described above. Error bars reflect the standard deviation.
HIF1α exhibits reduced binding to the VHL complex in Myc-overexpressing cells
Since we find that HIF1α is stabilized even in the presence of upregulated VHL complex components, we hypothesized that the VHL complex is less effective at recognizing and targeting HIF1α for degradation in Myc-overexpressing cells. To assess the association between the VHL complex and HIF1α, we performed immunoprecipitations with different components (VHL, CUL2, and Elongin C) and with HIF1α. Indeed, we find that there is less binding of HIF1α to VHL complexes in IMEC-Myc cells compared to vector cells under both normoxia and hypoxia (Figure 6A). Normoxic association between HIF1α and CUL2, Elongin C, and VHL are each reduced to only about 30% of the association seen in vector cells. This reduction may account for a significant portion of the HIF1α stabilization in IMEC-Myc cells. Furthermore, by performing immunoprecipitations between CUL2, Elongin C, and VHL, we find that there are also apparent changes in VHL complex component associations in IMEC-Myc cells compared to vector cells (Figure 6B). This suggests that, in addition to associating less with HIF1α, there may be fewer functional VHL E3 ligase complexes formed in Myc-overexpressing cells.
Figure 6. Reduced association of HIF1α with the VHL complex in Myc-overexpressing cells.

A) Immunoprecipitation and western blot analyses were performed with equivalent amounts of protein from normoxic and hypoxic IMEC extracts. Immunoprecipitations were quantitated using at least two independent cell extract sets. The HIF1α immunoprecipitation:WB CUL2 was normalized to total CUL2 protein, and all other immunoprecipitations were normalized to total HIF1α protein. Expression levels were normalized to normoxic vector. B) Quantitated immunoprecipitations between VHL complex components were performed as described above. Immunoprecipitations were normalized to total CUL2 protein. Error bars reflect the standard deviation.
HIF1α expression is critical for Myc-induced soft agar growth and proliferation
One of the most important questions we wanted to explore is whether or not HIF1α accumulation contributes to Myc-induced oncogenic transformation. To address this question we performed siRNA depletion experiments and assessed their effects on Myc-induced anchorage independent growth, which is an established hallmark of transformation. We transiently transfected IMEC and IMEC-Myc with siRNAs against HIF1A, c-myc, or a non-targeting control. Myc depletion served as a positive control because soft agar growth is Myc-dependent, and loss of Myc expression should ablate the ability of the cells to undergo anchorage-independent growth. We achieved depletion at the protein level for both Myc and HIF1α (Figure 7A and 7B). Consistent with the Myc-dependence of HIF1α accumulation, depleting Myc from the IMEC-Myc cells reduced the HIF1α protein level nearly back to that found in the parental IMECs. We plated transfected cells in soft agar 24 hours post-transfection with siRNA and scored for growth 10 days later. The IMEC-Myc cells transfected with the non-targeting control grew into colonies as expected (Figure 7C). Depletion of Myc predictably led to an almost complete loss of colony growth. Strikingly, the loss of HIF1α expression was equally as effective in blocking anchorage-independent growth, as HIF1α depletion almost completely eliminated soft agar colony formation (Figure 7C). Vector control IMECs do not grow in soft agar and are therefore not represented (26). These data show that expression of HIF1α is a critical component of Myc-induced anchorage independent growth. In support of these data, multiple studies have shown that the knockdown of HIF1α in tumor cells suppresses tumor growth (31–33).
Figure 7. HIF1α expression is affected by Myc levels and is necessary for anchorage-independent and log phase growth.

A, B) IMECs were transfected with non-targeting siRNA or siRNAs (1 or 2) targeting HIF1A or c-myc. Cell extracts were made 48 hrs post-transfection. Western blot analyses were performed with equivalent amounts of cell extracts using anti-HIF1α, anti-c-Myc, and anti-β-tubulin antibodies. Average Myc and HIF1α protein expression, normalized to β-tubulin, were quantitated using three independent cell extract sets. Expression levels for Myc and HIF1α protein were normalized to their respective vectors. C) IMECs transfected with non-targeting siRNA or siRNAs (1 or 2) targeting HIF1A or c-myc were plated in soft agar 24 hours post-transfection. The number of colonies per 100 plated Myc-overexpressing cells were counted and measured after 10 days in two independent experiments. Vector cells do not grow in soft agar and are therefore not included in the analysis. D) Cell proliferation rates were measured by counting equivalently plated cells that had been transfected at Day 1 with either HIF1A or non-targeting siRNA under normoxia. The graph shows the mean cell number for each day for duplicate experiments. Error bars reflect the standard deviation. Similar results were found for independent siRNAs.
Beyond anchorage-independent growth, we showed previously that Myc overexpression also enhances the overall proliferation rate of IMECs compared to vector control cells (26). Because of the importance of HIF1α expression for Myc-induced soft agar growth, we considered that HIF1α might also influence proliferation rates. We measured IMEC growth rates after transfection with HIF1A siRNA and compared them to IMECs transfected with non-targeting siRNA under normoxia (Figure 7D). Depletion of HIF1α expression greatly reduces the proliferation rate of Myc-overexpressing IMECs, down to the rate of vector control IMECs. HIF1A siRNA also had a smaller inhibitory effect on control IMECs. Thus, the enhanced growth rate of IMEC-Myc cells is highly dependent on elevated HIF1α levels even under normoxia, and the parental cells are also dependent on low basal HIF1α expression. Although our IMECs are non-transformed, these results are consistent with other studies that found that HIF1α knockdown significantly reduces the growth rates of glioma cells and prostate cancer cells, respectively (34, 35).
DISCUSSION
In this study we have explored a previously uncharacterized relationship between Myc and HIF1α expression under both normoxic and hypoxic conditions: Myc-dependent induction of HIF1 protein levels. Previous studies have shown interactions between Myc and HIF1 regulation and function, both synergistic and antagonistic. For example, in cells expressing physiologically normal levels of Myc, HIF1 has been found to functionally oppose Myc activity by disrupting Myc repression of CDKI p21 (36, 37). In addition to antagonizing Myc function through interaction, HIF1 also counters Myc-mediated regulation of mitochondrial biogenesis (4, 24, 38–40). While Myc itself promotes mitochondrial biogenesis and oxidative phosphorylation, HIF1 activates target genes that impair mitochondrial respiration. On the other hand, HIF1 and deregulated Myc cooperate to induce glycolysis in both normoxic and hypoxic tissues (22, 23, 25, 41). Studies suggest that Myc overexpression disrupts the sensitive stoichiometry normally governing HIF1 regulation of Myc. Normoxic cooperation of these transcription factors contributes to the Warburg Effect, a metabolic phenotype of aerobic glycolysis often seen in cancers.
We report that overexpression of Myc induces a dramatic enhancement in the stability of HIF1α protein under normoxia and also increases stability under hypoxia. A previous study reported that knockdown of Myc in a multiple myeloma cell line decreases HIF1α expression (42), but we provide the first evidence that overexpression of Myc in non-transformed epithelial and fibroblastic cells single-handedly induces the accumulation of HIF1α. Furthermore, we show that this induction is entirely post-transcriptional and that it results in the upregulation of HIF1α target genes. We show that Myc is also required for the post-transcriptional accumulation of HIF1α protein in MCF7 breast cancer cells. At the functional level, we show that HIF1α expression is essential for both Myc-induced anchorage-independent growth and Myc-induced proliferation.
The mechanism of Myc-induced HIF1α stabilization remains unresolved. Based on measurement of HIF1α hydroxy-P564 levels after blocking protein turnover, it appears that there is no reduction in hydroxylation with high Myc expression (Figure 1E). The primary effect we observed is that Myc post-transcriptionally upregulates the expression of VHL complex components. Furthermore, we find that HIF1α association with the VHL complex in cells overexpressing Myc is substantially reduced compared to vector control cells. The decreased interaction between HIF1α and the VHL complex likely contributes significantly to the enhanced accumulation of HIF1α protein. This could be due to an unknown modification of HIF1α or to inactivation of the VHL complex E3 ligase activity. We find evidence of disruption of VHL complex protein stoichiometry and possible post-translational modifications of VHL and Elongin B. Further characterization of changes in the VHL complex in response to high Myc expression will be required to resolve these questions.
Supplementary Material
ACKOWLEDGEMENTS
We thank Dr. Robert Freeman for the hydroxy-P564 HIF1α antibody. We thank members of the Cole lab for helpful discussions.
GRANT SUPPORT This work was supported by a grant from the National Cancer Institute (RO1CA055248).
REFERENCES
- 1.Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143–64. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]
- 2.Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83:1688–95. doi: 10.1054/bjoc.2000.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D'Cruz C, Gunther E, Boxer R, Hartman J, Sintasath L, Moody S, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–9. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 4.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J, Zeller K, Wang Y, Jegga A, Aronow B, O'Donnell K, et al. Evaluation of myc E-box phlyogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol. 2004;24:509–20. doi: 10.1128/MCB.24.13.5923-5936.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osthus R, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 8.Warburg O. On the Origin of Cancer Cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 9.Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg's effect. J Bioenerg Biomembr. 2007;39:223–29. doi: 10.1007/s10863-007-9080-3. [DOI] [PubMed] [Google Scholar]
- 10.Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular 02 tension. Proc Natl Acad Sci. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang E, Arany Z, Livingston D, Bunn H. Activation of Hypoxia-inducible Transcription Factor Depends Primarily upon Redox-sensitive Stabilization of Its a Subunit. J Biol Chem. 1996;271:32253–9. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 12.Kallio P, Pongratz I, Gradin K, McGuire J, Poellinger L. Activation of hypoxia-inducible factor 1α: Posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc Natl Acad Sci. 1997;94:5667–72. doi: 10.1073/pnas.94.11.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaakkola P, Mole D, Tian Y, Wilson M, Gielbert J, Gaskell S, et al. Targeting of HIF-α to the von Hippel Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 14.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFα Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 15.Masson N, Willam C, Maxwell P, Pugh C, Ratcliffe P. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung S, Ohh M. Playing Tag with HIF: The VHL Story. J Biomed Biotechnol. 2002;2:131–5. doi: 10.1155/S1110724302205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugh C, Ratcliffe P. The von Hippel–Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. 2003;13:83–9. doi: 10.1016/s1044-579x(02)00103-7. [DOI] [PubMed] [Google Scholar]
- 18.Sutter C, Laughner E, Semenza G. HIF-1α protein expression is controlled by oxygen-regulated ubiquitination that is disrupted by deletions and missense mutations. Proc Natl Acad Sci. 2000;97:4748–53. doi: 10.1073/pnas.080072497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahimi-Horn M, Pouyssegur J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bull Cancer. 2006;93:73–80. [PubMed] [Google Scholar]
- 20.Denko N. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 21.Semenza G. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–61. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Gao P, Liu Y, Semenza G, Dang C. HIF-1 and dysregulated c-Myc cooperatively induces VEGF and metabolic switches, HK2 and PDK1. Mol Cell Biol. 2007;27:7381–93. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J-W, Gao P, Liu Y-C, Semenza G, Dang C. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol Cell Biol. 2007;27:7381–93. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang C, Kim J-W, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–6. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 25.Yeung S, Pan J, Lee M-H. Roles of p53, Myc and HIF-1 in Regulating Glycolysis – the Seventh Hallmark of Cancer. Cell Mol Life Sci. 2008;65:3981–99. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cowling VH, D'Cruz CM, Chodosh LA, Cole MD. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol Cell Biol. 2007;27:5135–46. doi: 10.1128/MCB.02282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knies-Bamforth U, Fox S, Poulsom R, Evan G, Harris A. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004;64:6563–70. doi: 10.1158/0008-5472.CAN-03-3176. [DOI] [PubMed] [Google Scholar]
- 28.Shchors K, Shchors E, Rostker F, Lawlor E, Brown-Swigart L, Evan G. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527–38. doi: 10.1101/gad.1455706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moroz E, Carlin S, Dyomina K, Burke S, Thaler H, Blasberg R, et al. Real-time imaging of HIF-1alpha stabilization and degradation. PloS One. 2009;4:e5077. doi: 10.1371/journal.pone.0005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chua Y, Dufour E, Dassa E, Rustin P, Jacobs H, Taylor C, et al. Stabilization of Hypoxia-inducible Factor-1-alpha Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production. J Biol Chem. 2010;285:31277–84. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–8. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Shi M, Cao Y, Yuan W, Pang T, Li B, et al. Knockdown of hypoxia-inducible factor-1alpha in breast carcinoma MCF-7 cells results in reduced tumor growth and increased sensitivity to methotrexate. Biochem Biophys Res Commun. 2006;342:1341–51. doi: 10.1016/j.bbrc.2006.02.094. [DOI] [PubMed] [Google Scholar]
- 33.Méndez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang S, et al. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer. 2010;9 doi: 10.1186/1476-4598-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-Inducible Factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–13. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen N, Chen X, Huang R, Zeng H, Gong J, Meng W, et al. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha} J Biol Chem. 2009;284:10004–12. doi: 10.1074/jbc.M805997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koshiji M, To K, Hammer S, Kumamoto K, Harris A, Modrich P, et al. HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Kleine-Kohlbrecher D, Adhikary S, Eilers M. Mechanisms of transcriptional repression by Myc. Curr Top Microbiol Immunol. 2006;302:51–62. doi: 10.1007/3-540-32952-8_3. [DOI] [PubMed] [Google Scholar]
- 38.Li F, Wang Y, Zeller K, Potter J, Wonsey D, O'Donnell K, et al. Myc Stimulates Nuclearly Encoded Mitochondrial Genes and Mitochondrial Biogenesis. Mol Cell Biol. 2005;25:6225–34. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordan J, Thompson C, Simon M. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell. 2007;12:108–13. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutphin P, Giaccia A, Chan D. Energy Regulation: HIF MXIes It Up with the C-MYC Powerhouse. Dev Cell. 2007;12:845–6. doi: 10.1016/j.devcel.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 41.Dang C, Le A, Gao P. MYC-induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Sattler M, Tonon G, Grabher C, Lababidi S, Zimmerhackl A, et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009;69:5082–90. doi: 10.1158/0008-5472.CAN-08-4603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.