

1. Introduction
Cyclopropane, epoxide, and aziridine groups are three-membered ring structural elements found in a wide variety of natural products, some of which are depicted in Figure 1.1 The antibiotic and antitumor properties of many of these compounds, including duocarmycin (1),2 dynemicin (2),3 epothilone (3),4 mitomycin (4),5–8 and azinomycin (5)9–11 are well known. The pharmacological activities of others are more diverse. For example, pentalenolactone P (6)12,13 has antineoplastic and antiviral activities,14 scopolamine (7) has a subduing effect on the central nervous system,15 azicemicin (8) is active against Gram-negative bacteria and mycobacteria,16 and ficellomycin (9) shows inhibitory activities towards Gram-positive bacteria.17,18
Figure 1.

Representative three membered ring-containing natural products.
The inherent ring strain present in the small ring moiety is frequently responsible for the biological activities of these compounds, many of which are potent alkylation agents.1,19,20 For example, upon sequence specific binding to the minor groove of double stranded DNA, a twist around the amide bond of duocarmycin (1) activates the cyclopropyl ring towards alkylation by a suitably positioned adenine residue to give 10 as an adduct (Scheme 1A).21,22 Opening of the oxiranyl ring in the reduced dynemicin A (11) is known to trigger the rearrangement of the enediyne group to a 1,4-dehydrobenzene biradical (11→12) that initiates DNA degradation (Scheme 1B).23,24 The reductive activation of mitomycin C (4) involves opening of the aziridine ring (13→14), which unmasks the electrophilic site at C-1 and results in DNA alkylation (14→15→16, Scheme 1C).5–7 In other examples, such as the tubulin-binding cytotoxin epothilone (3, Figure 1), the epoxide ring introduces a rigid structural element into the parent compound. While derivatives lacking the epoxide ring exhibit similar activities as the parent compound,25 the epoxide moiety may be important for the directing/binding of epothilone to its biological target in the cell.4,26,27
Scheme 1.

Mechanism of action of (A) duocarmycin, (B) dynemicin, and (C) mitomycin.
Despite the recent advances in our understanding of the biosynthesis of many different types of natural products, the specific enzymes responsible for making these strained, 3-membered rings have only been identified in a few cases, and many of the mechanistic details regarding small ring closure remain obscure. Due to the impressive array of diverse biological activities exhibited by these small-ring containing compounds and their potential applications as therapeutic agents as well as mechanistic probes for studying enzyme catalysis,1,19,20,28–30 a thorough understanding of their biosynthesis is warranted. These studies are a crucial first step in maximizing the potential of these compounds as general mechanistic probes, or as specific tools in the rational design of drugs with optimal in vivo specificity. In this review, we will illustrate the established biosynthetic strategies for construction of cyclopropane, epoxide, and aziridine rings, focusing primarily on studies that were performed in the last decade. Only those pathways that have been genetically and/or biochemically verified will be discussed. Special attention will be directed to the prototypes of chemical transformations observed in small-ring biosynthesis and, when applicable, mechanistic details of the enzymes involved. In addition, we will highlight the occurrence of several poorly understood, but potentially novel modes of enzyme-catalyzed small-ring biosynthesis that merit further investigation.
2. Cyclopropane Biosynthesis
Several reviews on the biosynthesis and occurrence of the cyclopropane moiety in natural products have been published.31–36 The present review will focus on established chemical mechanisms of cyclopropane biosynthetic enzymes, some of which have been characterized only recently. For more comprehensive surveys of the occurrence of cyclopropane rings in natural compounds, the reader is directed to these earlier contributions. The biosynthetic enzymes that construct cyclopropane rings can be broadly divided into two classes: those whose mechanisms involve carbocationic intermediates (or transition states) and those that proceed through carbanionic intermediates (or transition states) to form the cyclopropane moiety.
2.1. Cyclopropane Biosynthesis via Inter- and Intramolecular Electrophilic Addition
Most cyclopropane containing natural products belong to the terpenoid (or isoprenoid) family of natural products, a large and structurally diverse class of compounds derived from the two ubiquitous isoprene units, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) (17 and 18, respectively, Scheme 2).37,38 The core carbon skeletons of most isoprenoids are assembled by two classes of structurally and mechanistically related isoprenoid biosynthetic enzymes. First, prenyltransferase enzymes, such as farnesyl pyrophosphate (FPP, 20) synthase, link together linear prenyl diphosphates of various lengths to form a variety of long chain isoprenoids (Scheme 2).39,40 FPP synthase catalyzes the formation of 1′-4 linkages (as in 19 and 20) between linear prenyl diphosphates, but other types of prenyltransferase-catalyzed linkages, such as the cyclopropyl 1′-2–3 linkage catalyzed by chrysanthemyl pyrophosphate (CPP, 21) synthase (Scheme 2), are also observed in nature.41 The higher order linear isoprenoids (i.e., those isoprenoids comprised of two or more isoprene units) can then be intramolecularly cyclized by a group of enzymes called terpene synthases (or terpene cyclases), such as carene synthase, which cyclizes the monoterpene geranyl pyrophosphate (GPP, 19) into carene (22, Scheme 2).42–44 Together, the prenyltransferases and terpene synthases are responsible for the construction of the majority of the cyclopropane moieties observed in isoprenoid compounds.40,45–48
Scheme 2.

Representative examples of prenyltransferase- and terpene synthase-catalyzed reactions.
Despite low overall amino acid sequence similarity, prenyltransferases and terpene synthases adopt similar 3-dimensional folds (termed the class I terpene cyclase fold46), characterized by a highly α-helical region surrounding a deep hydrophobic cleft where the isoprene substrate binds. The prenyltransferases and terpene synthases are mechanistically linked by a common early step in their catalyzed reactions (Scheme 3). Namely, both groups of enzymes employ a divalent metal ion (coordinated by a conserved DDXXD/E motif) to facilitate cleavage of the pyrophosphate bond of an allylic diphosphate substrate.46 The resulting allylic carbocation intermediate (such as 23) can then undergo a variety of chemical transformations to achieve the vast number of isomeric carbon skeletons observed in the isoprenoid family of natural products. Typically, the allylic carbocation undergoes an intermolecular (in the case of the prenyltransferases) or an intramolecular electrophilic alkylation (in the case of the terpene synthases). This may be followed by a series of rearrangements. Eventually, the enzyme-bound carbocation intermediate is quenched, usually by stereospecific deprotonation (e.g., 24→20) or hydration (e.g., 25→26) to terminate the reaction sequence. Thus, both prenyltransferase and terpene cyclase enzymes function by first generating a reactive carbocation intermediate. This intermediate is then manipulated in ways specific to the particular isoprenoid biosynthetic enzyme in order to direct it to specific final products. For more detailed information on the structure, chemistry, and evolution of these remarkable and versatile enzymes, the reader is directed to several excellent reviews.39–41,45–50
Scheme 3.

Mechanistic link between prenyltransferases and terpene synthases. Both families of enzymes catalyze C-C bond formation via reactive carbocation intermediates (such as 23, 24 and 25).
One of the best studied cyclopropane-forming isoprenoid biosynthetic enzymes is squalene synthase (SQS, Scheme 4), a prenyltransferase enzyme that catalyzes formation of the irregular 1′-1 linkage between two molecules of farnesyl pyrophosphate (20) to form squalene (33), a key triterpenoid intermediate in cholesterol biosynthesis.51,52 In the first step of SQS catalysis, a tyrosine residue may assist in the protonation of the pyrophosphate leaving group of one FPP molecule, generating an allylic cation (27) that then alkylates the other FPP molecule bound in the same active site.51,53 Deprotonation of the resulting carbocation (either 28 or 29) yields the cyclopropane-containing intermediate, presqualene pyrophosphate (PSPP, 30), which can be released from the enzyme and isolated from reaction mixtures lacking NADPH.54 In the second step of SQS catalysis, the pyrophosphate moiety of PSPP is cleaved to generate the primary cyclopropylcarbinyl cation (31), which undergoes alkyl rearrangement to the more stable tertiary carbocation, 32, prior to stereospecific reduction by NADPH to complete the reaction cycle (32 →33).54,55 Formation of the highly-reactive cation 31 may be facilitated by electrostatic interactions with the pyrophosphate leaving group. Evidence for the presence of cyclopropylcarbinyl cation 32 in the SQS reaction cycle was obtained from mechanistic studies of SQS performed in the presence of NADPH3 (34) – an NADPH analogue that is not capable of reducing 32 to form squlene (33). In these reaction mixtures, a series of products (35–37) consistent with either hydration or deprotonation of 32 were obtained.
Scheme 4.

Chemical mechanism of squalene synthase, which proceeds through the stable cyclopropyl intermediate, presqualene pyrophosphate (30).
Chrysanthemyl pyrophosphate (CPP) synthase is another prenyltransferase enzyme that catalyzes formation of a cyclopropyl 1′-2–3 linkage between two molecules of DMAPP (18) to form (1R,3R)-chrysanthemyl pyrophosphate (21, Scheme 5),56,57 the monoterpene precursor of the pyrethrins (such as pyrethrin I, 38) - a commercially important class of neurotoxic insecticides. Once again, the CPP synthase-catalyzed reaction is triggered by the cleavage of the pyrophosphate moiety of DMAPP to yield an allylic carbocation (39) that then couples with the other molecule of DMAPP bound in the active site, forming either a tertiary carbocation (40) or a protonated cyclopropane intermediate (41). Stereospecific deprotonation at C1′ forms the cyclopropnae moeity and gives CPP (21) as the major product. In addition to 21, a variety of minor products (42–44) are also isolated from CPP synthase reaction mixtures. These side products are likely derived from enzymatic processing of a C3-centered tertiary carbocation intermediate (40).
Scheme 5.

Proposed chemical mechanism for chrysanthemyl pyrophosphate (CPP) synthase, which catalyzes the first step in the biosynthesis of the pyrethrin class of insecticides.
The exact nature of the carbocation intermediate(s) formed in the initial electrophilic alkylation steps of the SQS and CPP synthase-catalyzed reactions (compounds 28/29 and 40/41, respectively) and of the transition states leading to cyclopropane ring formation by these enzymes are not known. Based on the side products observed in the CPP synthase-catalyzed reaction, it is possible that the tertiary carbocation intermediates (28 and 40) form first, and that the protonated cyclopropane species (29 and 41) are transition states along the reaction coordinates leading to cyclopropane ring formation. While the tertiary carbocations (28 and 40) would be the more thermodynamically stable species, protonated cyclopropanes (such as 29 and 41) are also well documented in studies of carbocation rearrangement reactions in solution58 and from computational studies.59 Furthermore, the energetic differences between isomeric tertiary and protonated cyclopropyl carbocations are relatively modest. For example, the barrier for the 1,2-methyl shift observed in the rearrangement of the tertiary heptyl cation (45, Scheme 6), which likely proceeds through the protonated cyclopropane transition state (46), has been estimated to be approximately 2 – 6 kcal/mol.58 A similar barrier height for the interconversion of 28/29 and 40/41 by SQS and CPP synthase, respectively, may be expected due to the similar level of alkyl substitution on the carbon atoms of the nacent cyclopropane moeities in these reactions. A barrier height of this magnitude should be surmountable by the stabilizing interactions (hydrogen bonding, electrostatic interactions, etc.) typically provided by enzyme active sites during catalysis.
Scheme 6.
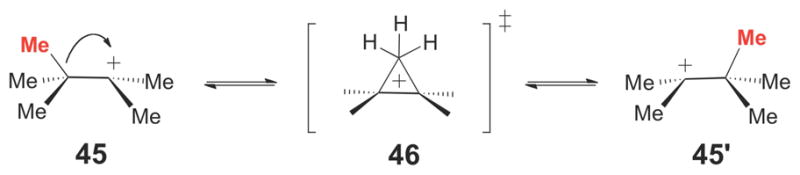
Rearrangement of the tertiary heptyl cation (45) likely involves a protonated cyclopropane transition state (46).
A number of enzymes in the terpenoid synthase family that catalyze cyclopropane formation have also been biochemically and mechanistically characterized. For example, enzymes responsible for the production of the bicyclic plant monoterpene (+)-3-carene (22, Scheme 7),60,61 have been isolated from several sources.43,44 Some carene derivatives are known to have strong local anesthetic, anti-inflammatory, and antifungal activities.62,63 In the initial step of the 3-carene synthase-catalyzed reaction, geranyl pyrophosphate (GPP, 19) undergoes a syn-1,3-isomerization to linalyl pyrophosphate (LPP, 47a). The LPP intermediate can have either R- or S- configuration at C3 depending on the particular monoterpene synthase involved and the GPP binding mode in the enzyme active site.64,65 This step is important since formation of 47a facilitates rotation around the newly formed C2–C3 single bond, placing the C1–C2 double bond into a cisoid conformation (47b) that is poised for intramolecular cyclization. Kinetic studies with chemically synthesized LPP (47) have shown that the kcat/Km values for 47 often exceed those for 19 for any given monoterpene cyclase.66 This finding suggests that 47 is likely a true reaction intermediate for this class of enzymes. The LPP intermediate then undergoes an intramolecular rearrangement to form the cyclohexene ring and the 4-α-terpinyl cation (48) – a key intermediate in all monoterpene cyclases. Stereochemical studies using a (5S)-[3H1]-GPP analogue suggested that the cyclopropane moiety in (+)-3-carene (22) is derived from the half-chair conformer of 48 by direct elimination of the pro-R hydrogen at C5 in an anti-1,3-elimination reaction.42
Scheme 7.

Monoterpene synthase-catalyzed formation of cyclopropane rings in the carene (22), sabinene (51), and thujene (52) families of monoterpenoids.
Another group of plant monoterpene synthases, the sabinene synthases, catalyze a similar reaction, except that the 4-α-terpinyl cation (48) undergoes a 1,2-hydride shift to generate the tertiary carbocation (49), prior to intramolecular electrophilic alkylation to form the cyclopropane ring in 50.67–69 Regiospecific deprotonation of 50 then yields the sabinene- or thujene-type monoterpenoid skeletons (51 and 52, respectively).
At least one diterpene synthase, casbene synthase, has been shown to cyclize geranylgeranyl pyrophosphate (53) into casbene (55, Scheme 8),70–73 which is believed to be the precursor for a variety of diterpenoid natural products, many of which [such as the ingenanes (56), tiglianes (57), and lathyranes (58)] retain the cyclopropane ring in their final structures.73 The majority of naturally occurring diterpenoid compounds are found in the latex of plants, primarily those species in the Euphorbiaceae family.74 Several of these diterpenes, such as the latent HIV-1 activator, prostratin (59),75,76 and the anti-cancer compound, ingenol-3-angelate (60),77 are of pharmaceutical interest. Though biochemical details on the casbene synthase-catalyzed reaction are sparse, this enzyme conceivably employs a mechanism similar to the other class I terpenoid cyclase enzymes discussed above (including texadiene synthase whose structure was recently determined78), where divalent metal ion-dependent cleavage of the allylic pyrophosphate moiety of GGPP (53) leads to an allylic carbocation (54) that triggers an intramolecular alkylation and deprotonation to generate the cyclopropane moiety in 55.
Scheme 8.

Casbene (55), a diterpene produced from geranylgeranyl pyrophosphate (53) by casbene synthase, is the likely intermediate for the ingenane (56), tigliane (57), and lathyrane (58) families of diterpenoid natural products.
Another well-studied isoprenoid biosynthetic enzyme is cycloartenol synthase,79,80 which converts the triterpene (S)-2,3-oxidosqualene (61) into cycloartenol (64), the sterol precursor in plants and some bacteria (Scheme 9). Interestingly, in animals and fungi, the sterol precursor is lanosterol (65), which is also generated from 61 by lanosterol synthase81 (which shares 40% amino acid sequence identity with plant cycloartenol synthases).82 Both of these enzymes are members of the class II terpene cyclase superfamily, enzymes that generate carbocation intermediates by protonation of their substrates (rather than by divalent metal ion-dependent cleavage of the pyrophosphate bond as seen in the class I terpenoid cyclase superfamily).46,50 Both cycloartenol synthase and lanosterol synthase begin their catalytic cycles by protonating the epoxide moiety of 61. This triggers a cyclization cascade that converts 61 into 62, which then undergoes a series of superfacial 1,2-methyl and 1,3-hydride shifts to give the tertiary carbocation intermediate, 63. At this point, the cycloartenol synthase catalyzes a 1,2-hydride shift and a deprotonation of the C19 methyl group in sequence to form the cyclopropyl group in 64 (path A). In contrast, lanosterol synthase catalyzes elimination of the C9 proton of 63 to form the Δ8,9-alkene moiety in 65 (path B).
Scheme 9.

Proposed chemical mechanisms for cycloartenol (64) and lanosterol (65) synthases.
Based on homology modeling and site-directed mutagenesis, several residues required for directing cyclopropane formation in Arabidopsis thaliana cycloartenol synthase have been identified.50,83,84 The C19 methyl group of 63 is believed to be deprotonated by a conserved Tyr410 residue that forms a hydrogen bond with a conserved His477 residue. Mutation of either Tyr410 or His477 allows cycloartenol synthase to produce lanosterol. Quite impressively, the H477N/I481V double mutation converted cycloartenol synthase into an efficient lanosterol synthase (where 99% of the detectable product was lanosterol), providing convincing support for the proposed role of these residues in mediating formation of the cyclopropane moiety in 64.83 The isoleucine residue likely promotes cycloartenol formation by sterically hindering the movement of 62 in the enzyme active site.83
2.2. Cyclopropane Biosynthesis via S-adenosyl-methionine (SAM)-dependent Methylation
Formation of cyclopropanes via carbocationic intermediates is not limited to isoprenoid biosynthesis. Cyclopropane fatty acids (CFAs) are common constituents of bacterial and plant cell membranes and they are also found in the cyclopropane mycolic acid (CMA)-containing cell walls of Mycobacterium tuberculosis.85–90 The exact function of CMAs in M. tuberculosis is unknown, but CMA synthase activity appears to be correlated with the virulence and persistence of this pathogen,91–93 making the CMA synthases potential drug targets. To date, the X-ray crystal structures of three CMA synthases from M. tuberculosis have been reported,94 but due to difficulties in obtaining the corresponding mycolic acid substrates, mechanistic studies have not yet been conducted. However, the E. coli CFA synthase, which shares a high degree of amino acid sequence identity with the CMA synthases,88,89 can be expressed and purified in workable quantities, and its unsaturated phospholipid substrates are commercially available.
In E. coli, CFAs are synthesized by addition of a methylene unit derived from S-adenosyl-Lmethionine (SAM, 66) across the double bond of unsaturated acyl chains (67) to generate the corresponding cylcopropane fatty acid (72) and S-adenosylhomocysteine (68) as the products (Scheme 10). Mechanistic studies on E. coli cyclopropane fatty acid synthase (CFAS) using SAM analogues containing selenium or telurium in place of the sulfur atom have shown that the rate-determining step in this transformation involves nucleophilic attack of the alkene on the SAM methyl group to generate a carbocation intermediate (perhaps the corner protonated species, 69).95 In addition, by using isotope-labeled SAM, it was recently demonstrated that deprotonation of 69 is also at least partially rate limiting.96 The cationic nature of the transition state in the CFAS-catalyzed reaction is supported by kinetic studies with 3-palmitoyl-2-(9-/10-fluorooleoyl)phosphatidylethanolamine (PFOPE, 70), which is a competitive inhibitor of CFAS. The fluorine substitution at the vinyl position of 70 likely deactivates the olefin and destabilizes the carbocation intermediate/transition state.97,98
Scheme 10.

SAM-dependent cyclopropane biosynthesis catalyzed by CFA and CMA synthases.
The X-ray crystal structures of the highly homologous CMA synthases suggest that this putative carbocation intermediate (or transition state) may be stabilized through cation-π interactions with several conserved active site tyrosine residues.94 Interestingly, a bicarbonate ion (71) was also bound in the active site of the CMA synthases in the crystal structure.94 Using an enzymatic system to degrade the bicarbonate that co-purifies with the enzyme, Booker and coworkers demonstrated that the bicarbonate ion is required for E. coli CFAS catalysis in vitro,99 and subsequent mutagenesis of the putative bicarbonate ligands led to drastic reductions in the catalytic efficiency of CFAS.100 The exact role of the bicarbonate ion is unclear, but it could serve as the base to deprotonate the carbocation intermediate (69). Interestingly, in the structurally homologous M. tuberculosis enzyme Hma, which catalyzes SAM-dependent methylation and hydration of the alkene moieties in mycolic acids to form 73 (Scheme 10), the bicarbonate ion is absent.101 Thus, an alternative role for bicarbonate in CFA and CMA synthases could be simply to exclude water from binding to the active site, where it could potentially add as a nucleophile to the incipient carbocation intermediate and disfavor cyclopropane formation.
Several natural products also contain cyclopropane moieties that are likely derived from insertion of a methylene group from SAM into double bonds. These include the antifungal compound FR-900848 (74, Scheme 11) isolated from Streptoverticillium fervens,102–104 and the cholesterol ester transfer protein inhibitor, U-106305 (75), isolated from Streptomyces sp. UC 11136.105 In the case of the latter compound, isotope tracer experiments in the producing organism using [1-13C]- and [2-13C]-acetate (76 and 77, respectively) established that the hydrophobic chain is derived from head-to-tail condensation of nine acetate units and is likely of fatty acid synthase or polyketide synthase origin. In addition, feeding experiments using [methyl-13C]-L-methionine (78) established that the methylene carbon of each of the six cyclopropane moieties is most likely supplied by SAM. The authors proposed that a highly unsaturated polyketide intermediate might serve as a precursor in the biosynthesis of this compound. To date, however, the putative cyclopropane synthase enzymes have not been discovered. In addition to these two unusual bacterial cyclopropane fatty acid natural products, several sterols produced by marine microorganisms also contain cyclopropane moieties that are likely constructed in a similar SAM-dependent manner.32,33
Scheme 11.

The cyclopropane-containing natural products FR-900848 (74) and U-106305 (75) are likely biosynthesized by a series of insertion of a SAM-derived methylene group into each of the reacting alkene moiety.
2.3. Cyclopropane Biosynthesis via Carbanionic Intermediates
Another strategy for the biosynthetic formation of cyclopropane moieties is by the displacement of a good leaving group by intramolecular attack of a carbanion using SN2-type chemistry. This type of cyclopropane-forming reaction was first demonstrated in the biosynthesis of 1-aminocyclopropane-1-carboxylate (ACC, 79, Scheme 12) - a cyclic amino acid precursor to ethylene. The latter is a plant hormone that is involved in leaf senescence, fruit ripening, seed germination, wound healing, responses to environmental stress, and many other biological processes.106,107 ACC synthase is a pyridoxal-5′-phosphate (PLP)-dependent enzyme that catalyzes the formation of ACC from SAM (66).108–111 Following formation of the external aldimine (80) between the PLP coenzyme and the amino group of SAM, the pKa of Cα is significantly reduced, allowing proton abstraction by the now liberated Schiff base-forming lysine residue to form the resonance-stabilized Cα-anion intermediate (81). Intramolecular nucleophilic attack by the Cα-anion on Cγ displaces the methylthioadenosine leaving group (82) and generates the ACC-PLP external aldimine adduct (83). Aldimine exchange then releases ACC (79) and regenerates the internal aldimine resting form of the enzyme (84). Support for the SN2-type α,γ-elimination to form the cyclopropane moiety is derived from studies using stereospecifically deuterated SAM analogues. The results indicated that the Cβ protons are not exchanged during turnover and that the reaction proceeds with stereochemical inversion at Cγ.108
Scheme 12.

ACC synthase-catalyzed cyclopropane formation, which proceeds from the resonance-stabilized carbanion (81) via an intramolecular SN2 reaction.
Several interesting variations of this nucleophilic displacement mechanism for cyclopropane ring formation have been recently unraveled during the biosynthesis of nonribosomal peptides112,113 and hybrid nonribosomal peptide-polyketide compounds (Scheme 13).114,115 In each case, a chlorinated, carrier protein-linked intermediate (85, 89, or 94) is converted into a cyclopropane containing intermediate (87, 91, and 96, respectively) by an intramolecular nucleophilic displacement mechanism. Interestingly, the triggering mechanism for cyclopropane ring formation is distinct in each pathway. In the biosynthesis of the phytotoxin coronatine (88),115 the Zn2+-dependent enzyme CmaC deprotonates Cα of 85. The Zn2+ ion likely stabilizes the enolate intermediate (86), which is then used to displace the chloride ion at Cγ, forming the coronamic acid moiety (see 87) that is eventually incorporated into coronatine (88).
Scheme 13.


Halogenated carrier protein-linked intermediates serve as the substrates for SN2-like cyclopropane ring formation in the biosynthesis of coronatine (88), kutzneride 2 (92), and curacin A (97). The triggering mechanism for cyclopropane formation in each case is believed to involve a carbanionic intermediate (or transition state).
In the biosynthetic studies of non-ribosomally produced peptides by Kutzneria sp.,113 a similar γ-chloro-L-isoleucine unit (89) is used to form allo-coronamic acid (see 91) via an enolate intermediate (90). In this case, however, the enzyme that catalyzes the cyclopropanation (KtzA) is an FAD-dependent enzyme that shares homology with the acyl-CoA dehydrogenases (ACADs). In the generalized mechanism of ACADs (Scheme 14),116–118 an acyl-CoA substrate (98) is deprotonated at Cα to form an enolate (99), which then transfers a hydride equivalent from Cβ; to the oxidized flavin to generate the two-electron reduced flavin and the α,α-unsaturated acyl thioester product (100). The two-electron oxidation of 99 could potentially occur via either hydride transfer to the oxidized flavin (path a) or through sequential one-electron transfer steps (path b).116–121 Regardless of the nature of the electron transfer steps, a key feature of this mechanism is the ability of the transient enolate (99) to form a charge-transfer interaction with the oxidized flavin, which, in combination with other electrostatic and hydrogen bonding interactions in the active site, is believed to significantly lower the pKa of Cα proton. In KtzA catalysis, the oxidized FAD may play a similar role in lowering the pKa of the Cα proton in 89. In the case of KtzA catalysis, however, no hydride equivalent is transferred to the oxidized flavin. Instead, the two-electron oxidation of the substrate is provided by the departure of the chloride leaving group, and the flavin coenzyme undergoes no net redox change. Interestingly, the allo-CMA amino acid (see 91) produced by the ktz genes in vitro differs from the 2-(1- methylcyclopropyl)glycine moitey (93) expected from the structure of the natural product (e.g., see kutzneride 2, 92). Further studies will be required to determine if 93 is somehow derived from 91, or whether it is biosynthesized by a different set of genes. Finally, during the biosynthesis of the anticancer cytotoxin curacin A (97),114 NADPH-dependent reduction at Cβ of the chlorinated ACP-linked intermediate (94) by an enoyl reductase module (encoded by curF) triggers formation of the Cα anion (95), that then attacks Cγ to form the cyclopropane ring in 96.
Scheme 14.

Generalized chemical mechanism for acyl-CoA dehydrogenases.
The biosynthetic gene cluster for hormaomycin (101, Scheme 15), a non-ribosomally synthesized peptide produced by Steptomyces griseoflavus that functions as a bacterial hormone and a narrow spectrum antibiotic, was recently sequenced.122 This compound contains two highly unusual 3-(trans-2′-nitrocyclopropyl)alanine moieites (see 102) that are biosynthetically derived from L-lysine.123 The exact biosynthetic mechanism for construction of the cyclopropane moiety in this compound is unknown, and an enzymatic route to synthesize 102 from lysine is not obvious based on the identities of the genes encoded in the cluster. The pathway may involve oxidation of the Nε and Cγ positions of lysine to form 103, followed by lactonization to 104, deprotonation and intramolecular nucleophilic attack to give 105, and oxidation to give 102.122
Scheme 15.

Putative mechanism for cyclopropane formation in hormaomycin (101).
3. Epoxide Biosynthesis
Most of the biosynthetic enzymes used to form epoxide moieties require O2, either as a source for the epoxide oxygen atom and/or as an oxidizing agent. Despite the exothermic nature of O-O bond cleavage, the triplet ground state of O2 presents a significant kinetic barrier to reaction with most organic compounds - the vast majority of which have singlet ground states. Thus, in order to react with most biological molecules, O2 must be converted to a more reactive, singlet state. In enzymes, the conversion of molecular oxygen to form the reactive oxygen species is usually achieved by electron transfer from redox active transition metal ions (such as Fe, Cu, or Mn) or from organic cofactors or substrates that have stable radical states (such as flavin and pterin coenzymes).124–127
3.1. P450-dependent Epoxidases
The best characterized group of epoxide forming enzymes are the P450 epoxidases, which use a heme coenzyme to insert one atom of oxygen from O2 into the double bond of an alkene. In a typical P450 reaction cycle (Scheme 16), substrate binding converts the six-coordinate, low spin (S = 3/2) ferric resting state (FeIII-H2O, 106) to a five-coordinate high spin (S = 5/2) ferric state (107). The iron center is then reduced to its ferrous state (108) by a single electron supplied by an external reductase protein (typically an NAD(P)H-dependent P450 reductase). Upon O2 binding, an electron is transferred from the coenzyme to O2, generating a ferric superoxo [FeIIIOO·] species (109). This species is then reduced by a second external electron and is protonated to form the ferric hydroperoxide (FeIII-OOH, 110), also known as compound 0 (Cpd 0). Protonation of Cpd 0 leads to cleavage of the O-O bond to form a water molecule and the hypervalent oxy-ferryl porphyrin cation radical species (Por+•FeIV=O, 111), also known as Cpd I, which is believed to be the active oxidant in P450 enzymes. Two-electron oxidation of the substrate (depicted here as epoxidation of an alkene) could occur in a concerted (111→113) or stepwise fashion (111→112→113) via a Cpd II species (112). Product dissociation and coordination of a water molecule to the ferric iron center regenerates the resting form of the enzyme for another round of catalysis. Detailed descriptions of the structural, mechanistic, and biochemical evidence supporting the general P450 reaction cycle shown in Scheme 16 have been thoroughly reviewed in several recent, excellent articles.128–132
Scheme 16.

Typical catalytic cycle for a P450-dependent epoxidase.
The mechanism of oxygen insertion across the double bond (and of other oxidation reactions catalyzed by P450-dependent enzymes) is still under debate. The key unresolved issues for P450 epoxidases are the exact nature of the oxidizing species and whether the two C-O bond forming steps occur in stepwise fashion via a discreet radical intermediate (111→112→113), or whether oxygen insertion is concerted (111→113). The fact that most P450 epoxidases catalyze reactions with retention of stereochemistry implies a concerted mechanism for oxygen insertion. However, examples of P450-catalyzed epoxidation reactions with stereochemical inversion are known, suggesting the existence of an oxidized substrate intermediate in some cases.133–136
Recent computational work has provided a potential explanation for these observations. Using a variety of quantum chemical calculations, Shaik and coworkers have proposed a twostate reactivity model for P450 enzymes (Scheme 17), in which Cpd I exists in energetically similar low spin (S = ½, 111) and high spin (S = 3/2, 111b) states, both of which are catalytically competent towards alkene epoxidation.130,131,137,138 Their calculations indicate that, following formation of the Cpd II-substrate radical state (112 or 112b), oxygen rebound along the low spin reaction coordinate is barrierless, making this reaction effectively concerted and leading to retention of configuration in the epoxide product (112→113). In contrast, a significant kinetic barrier to rebound exists for the high spin Cpd II-substrate radical species (112b). This would result in a substrate radical intermediate with a finite lifetime and, in cases where the carbon substituents are sufficiently small, rotation around the C-C single bond would lead to inversion of stereochemistry upon epoxide formation (112b→112c→113c). This two state reactivity model also provides a satisfactory explanation for the reactivity patterns observed in other P450- catalyzed reactions.129–131,138,139
Scheme 17.

Two-state reactivity model for P450 enzymes.
To date, the epoxidase activity of several natural product biosynthetic P450 enzymes has been verified biochemically or through gene disruption experiments.140 These include EpoK, which installs the epoxide moiety of the anticancer agent epothilone (3, Figure 2),141 a hybrid non-ribosomal peptide-polyketide compound produced by Sorangium cellulosum,142–144 MycG, which is involved in the biosynthesis of mycinamicin (114), a macrolide antibiotic produced by Micromonospora griseorubida,145 GfsF, which catalyzes epoxidation in the biosynthesis of FD- 891 (115), a macrolide antibiotic isolated from Streptomyces graminofaciens A-8890,146 and PimD, which is involved in the biosynthesis of the potent antifungal polyene compound pimaricin (116), produced by Streptomyces natalensis.147–149 Interestingly, both MycG and GfsF are dual-function P450 monooxygenases that catalyze not only the epoxidation reaction but also the α-hydroxylation step in their respective biosynthetic pathways (Figure 2). In the case of MycG (see 114), C-14 hydroxylation occurs prior to Δ12,13-epoxidation.145 In contrast, Δ8,9- epoxidation occurs first followed by C-10 hydroxylation in GfsF-catalyzed reaction (see 115).146 In addition, putative P450 epoxidase genes are present in the biosynthetic gene clusters of griseorhodin A (117),150 a human telomerase and retroviral reverse transcriptase inhibitor,150 hedamycin (118), an antitumor compound produced by Streptomyces griseoruber,151 maduropeptin (119), an enediyne antitumor antibiotic compound found in Actinomadura madurae,152 and FR901464 (120) a hybrid non-ribosomal peptide/polyketide compound with anticancer activity produced by Psedomonas sp. No. 2663.153
Figure 2.

Representative epoxide-containing natural products whose biosynthetic gene clusters contain genes encoding putative P450 epoxidases.
In the case of PimD, recent structural studies provided an interesting twist to our understanding of the mechanism of P450 epoxidases.149 Namely, in the co-crystal structure of PimD solved in the presence of its substrate under anaerobic conditions, the p-orbitals of the double bond to be oxidized are oriented nearly perpendicular to the plane of the π-electron system of the heme coenzyme. As such, the substrate does not appear to be oriented properly for oxygen insertion via a Cpd I species. Furthermore, based on steric constraints imposed by the PimD active site, the authors argue that substantial changes in the observed substrate binding mode are unlikely. In addition, a conserved serine residue, which was expected to participate in Cpd I formation by protonating the Cpd 0 species (110→111, Scheme 16) is oriented away from the O2 binding site, where it hydrogen bonds with the backbone carbonyl group of a highlyconserved alanine residue. The authors suggested that in PimD, formation of Cpd I may not be possible due to this disrupted proton transfer network, and that the reactive oxidant could instead be a ferric hydroperoxide species (Cpd 0), which could approach the alkene moiety of the substrate in an optimal configuration to undergo a concerted six-electron rearrangement to form the epoxide moiety (110→121, Scheme 18). However, from computational studies,131 Cpd 0 is thought to be a sluggish oxidant towards alkene epoxidation, with a calculated barrier to oxygen insertion of > 36 kcal/mol (versus 14–15 kcal/mol for a Cpd I species). In addition, these calculations suggested that Cpd 0 reacts with alkenes to generate a Cpd II-substrate radical complex (112), which has a calculated barrier of 6 kcal/mol for radical-radical recombination to form the ferric glycol species (122) (path A) and a barrier of 20 kcal/mol to form the corresponding epoxide complex (121) (path B). Thus, according to these calculations, if Cpd 0 were the reactive oxidant in P450 epoxidases, one would expect to observe predominantly diol products, which were not reported in the biochemical studies of PimD.148–149 Additional biochemical and computational studies of the PimD-catalyzed reaction could help to resolve these ambiguities, and may provide insight into the mechanism of this interesting class of epoxidases.
Scheme 18.

Alternative mechanism of epoxidation by P450 enzymes involving a Cpd 0 species (110).
Transformation of fatty acid hydroperoxides (123) to allene oxides (125) represents a novel epoxide formation reaction (Scheme 19). Fatty acid hydroperoxides, which are biosynthesized from polyunsaturated fatty acids by hemoprotein dioxygenases and/or non-heme iron lipoxygenases, are precursors of a diverse spectrum of signaling molecules, such as leukotrienes, thromboxane, prostacyclin and jasmonate.154–157 Further metabolism of these peroxides to allene oxides is commonly catalyzed by cytochrome P450s or by catalase-related enzymes, which also possess a heme coenzyme.157–160 As shown in Scheme 19, the reaction is believed to be initiated by a homolytic cleavage of the hydroperoxyl O-O bond of the fatty acid peroxide (123) followed by cyclization to generate an epoxyallylic radical intermediate (124). Subsequent electron transfer followed by proton abstraction would complete the formation of the allene oxide (125).161 In a recent study of allene oxide formation catalyzed by a catalase-like synthase from Acaryochloris marina, a cis-epoxyalcohol derivative (126) was detected.162 This observation is consistent with the intermediacy of a carbocation species in the reaction pathway that can be trapped by hydration at C-13.
Scheme 19.

Radical and ionic pathways to allene oxide (125) catalyzed by a catalase-related allene oxide synthase from the cyanobacterium Acaryochloris marina.
3.2. Flavin-dependent Epoxidases
Like the cytochrome P450 epoxidases, the flavin-dependent epoxidases are monooxygenases that insert one atom of oxygen from O2 into an alkene moiety. These enzymes are believed to adopt a mechanism similar to other flavin-dependent monooxygenases (Scheme 20),163,164 where the anionic reduced flavin (127) first transfers a single electron to O2, forming the caged semiquinone-superoxide radical pair (128), which subsequently undergoes radical recombination and protonation to form the flavin C4a-hydroperoxide intermediate (129). The distal oxygen atom of the flavin C4a-hydroperoxide is electrophilic and, as such, can serve as a suitable oxidant for oxygen insertion into the alkene moiety. As with the P450 epoxidases, the exact mechanism of oxygen insertion is unknown, but following epoxidation of the alkene, a flavin C4a-hydroxy species (130) is likely generated at some point along the reaction coordinate. This species can then spontaneously decay to produce a water molecule and the two-electron oxidized flavin (131). A reductase enzyme can then catalyze the NAD(P)H-dependent two-electron reduction of the flavin to regenerate the catalytically competent form of the epoxidase. In support of this mechanism for flavin-dependent epoxidation, the flavin C4a-hydroperoxy intermediate (129), which has a unique UV-visible absorption profile, has been detected in the reaction catalyzed by StyA, a microbial enzyme that catalyzes the stereospecific epoxidation of styrene during the catabolism of this compound.165,166 To date, StyA is the best-characterized flavin-dependent epoxidase.165–169
Scheme 20.

Generalized chemical mechanism for flavin dependent epoxidases.
Recent bioinformatic studies suggest that flavin-dependent epoxidases are potentially widespread and are found in a number of natural product biosynthetic gene clusters. Several recent examples include the biosynthetic gene clusters for the enediyne compounds neocarzinostatin170 (132, Figure 3) and dynemicin171 (2), anticancer agents produced by Streptomyces carzinostaticus and Micromonospora chersina, respectively, and the polyketide antitumor compound hedamycin (118, Figure 2).151 Interestingly, as noted above, the hedamycin cluster also encodes a putative P450 epoxidase. It is currently not known whether the two epoxide moieties of this compound are installed by the same or different enzymes. The putative flavin-dependent epoxidase (Hpm7) in the biosynthesis of the fungal polyketide hypothemycin (133, Figure 3) in Hypomyces subiculosus – a compound that irreversibly alkylates a conserved cysteine residue in certain human kinases172 – has also been purified and shown to catalyze Δ1′,2′- epoxidation of substrate analogues in vitro.173
Figure 3.

Representative natural products whose epoxide moieties are likely installed by flavindependent epoxidases. See also hedamycin (118) in Figure 2.
Very recently, the biosynthetic gene cluster for asukamycin A1 (134, Figure 3), a manumycin-type compound with a variety of biological activities,174–176 was cloned from Streptomycesnodosus subsp. asukaensis, sequenced, and heterologously expressed in Streptomyces lividans.176 The activity of the putative flavin-dependent epoxidase (AsuE3) was verified by gene disruption experiments, which led to the accumulation of 4- hydroxyprotoasukamycin D (135) as the major product.176 When compound 135 was fed to an S. lividans strain expressing the asuE3 gene, asukamycin A1 production was restored. Although AsuE3 has not been purified and biochemically characterized, its assignment as a monooxygenase is consistent with earlier 18O2 feeding studies of structurally related manumycin compounds, which demonstrated that the epoxide oxygen atom in this group of natural products is derived from O2.177 A similar mode of epoxidation is likely operative in other members of the manumycin-type compounds.174
Putative flavin-dependent epoxidase genes have also been found in the biosynthetic gene clusters of a number of different polyether compounds (Figure 4), including those leading to production of monensin (136),178 lasalocid (137),179,180 nanchangmycin (138),181 nigericin (139),182 and tetronomycin (140).183 In the biosynthesis of these compounds (as shown for monensin in Scheme 21), the flavin-dependent epoxidase (MonCI in the case of monensin) is believed to introduce multiple epoxide moieties into a polyunsaturated biosynthetic intermediate (141). A conserved epoxide hydrolase (encoded by each of the polyether biosynthetic gene clusters that have been sequenced, e.g., MonBI/MonBII in the case of monesin184) is thought to generate the polycyclic ether ring system by initiating a cyclization cascade,185 likely in a stepwise manner,186 from the polyepoxy intermediate (142) using acid/base catalysis.185–187 Support for this biosynthetic mechanism has been derived from studies of monensin biosynthesis, where disruption of the putative flavin epoxidase gene led to accumulation of a polyene precursor (141, Scheme 21).188 Recently, using a chemically synthesized substrate, the activity of the epoxide hydrolase, Lsa19, which catalyzes polyether formation by a cascade of epoxide opening reactions in the biosynthesis of lasalocid (137) was demonstrated in vitro (Scheme 21).189 Thus, the epoxide moiety, while absent in the final structure of the polyether natural products, serves a critical role in the biosynthesis of these compounds by priming a biosynthetic intermediate for the key cyclization step. The epoxide moiety in 2,3-oxidosqualene (61, Scheme 9), which serves a similar biosynthetic function, is also installed by a flavin-dependent enzyme, squalene epoxidase.190,191
Figure 4.

Chemical structures of several polyether natural products whose biosynthetic gene clusters have been sequenced.
Scheme 21.

Involvement of epoxide intermediate in the biosynthesis of polyether natural products.
3.3. Cofactor Independent Epoxidation
Several dioxygenase enzymes (that incorporate both oxygen atoms of O2 into their products) catalyze epoxidation reactions using O2 as the oxygen source without the assistance of any redox active metal ions or organic coenzymes.127,140 In these unusual enzymes, the initial 1e- reduction of O2 (required to convert O2 to its reactive singlet state) is believed to be carried out by the substrate which, for all known dioxygenases in this class, is a phenolic compound that can likely access a stable radical state.140 Biochemically well characterized examples of epoxidases from this class of enzymes include the vitamin K-dependent glutamate carboxylase, an enzyme that couples the post-translational carboxylation of glutamate residues in certain proteins to the epoxidation of vitamin K,192–195 and the dihydroxyacetanilide epoxidases, DHAE I and DHAE II, which respectively catalyze the key steps in the biosynthesis of the epoxyquinone compounds LL-C10037α (143) and MM14201 (144) (Scheme 22). The former is an antitumor agent produced by Streptomyces LL-C10037, and the latter is an anticancer compound isolated from Streptomyces MPP 3051.196–198 Studies on DHAE I have shown that this enzyme converts dihydroxyacetanilide (145) to the (5R,6S)-epoxyquinone (147), with quantitative incorporation of 18O (derived from 18O2) into the epoxide.198 Control experiments demonstrated that the oxygen atom at C4 exchanges quickly with solvent. However, when an NADPH-dependent ketoreductase (AEBQOR I) from the same organism was included in the DHAE I reaction mixture, 18O label from 18O2 was also incorporated into C4, forming the final product, LLC10037α (143), and suggesting that DHAE I is a dioxygenase. Based on this observation, a plausible chemical mechanism involving a dioxetane intermediate (146) has been proposed for DHAE (Scheme 22).198
Scheme 22.

Cofactor-independent epoxidation catalyzed by DHAE I.
3.4. Non-heme Iron-dependent Epoxidases
While molecular oxygen serves as the oxygen source for most natural product epoxidases, several other modes of enzyme-catalyzed epoxidation have recently been discovered and mechanistically characterized. One of these unusual modes of epoxidation is present in the biosynthesis of fosfomycin (149, Scheme 23), a small molecule antibiotic produced by several species of Streptomyces and Pseudomonas that irreversibly inhibits peptidoglycan biosynthesis in Gram-positive bacteria by covalent modification of the MurA (UDP-GlcNAc enolpyruvyl transferase) active site.199,200 The epoxidation step in fosfomycin biosynthesis is catalyzed by (S)- 2-hydroxypropylphosphonate (HPP) epoxidase (HppE), a mononuclear non-heme irondependent enzyme that catalyzes the two-electron oxidation of (S)-HPP (148) to fosfomycin (149).201,202 The HppE-catalyzed reaction requires FeII, O2, NADPH, and an electron mediator (such as flavin or an enzymatic reductase) for activity.203–205 Early studies demonstrated that the oxygen atom in the epoixde ring of fosfomycin is derived from the C2 hydroxyl group of (S)- HPP, rather than from O2,203,206 and that the pro-R hydrogen atom at C1 is stereospecifically removed during turnover.207,208 Thus, the HppE-catalyzed reaction is effectively a two-electron dehydrogenation of a secondary alcohol to from an epoxide – a highly unusual mode of C-O bond formation that is thought to occur in only few other non-heme iron dependent enzymes.209- 211
Scheme 23.

The epoxidation reaction catalyzed by HppE involves the two-electron oxidation of a secondary alcohol.
The high resolution X-ray crystal structure of the active, ferrous form of HppE, solved anaerobically in the presence and absence of (S)-HPP (148), provided important insights into the mechanism of HppE.212 HppE is a member of the cupin superfamily and it employs a 2-His-1- carboxylate facial triad (formed by His138, His180, and Glu142 in Streptomyces wedmorensis HppE) to bind an iron atom.213 Upon binding, (S)-HPP forms a bidentate interaction with the metal center, leaving the last coordination site on the iron center available for O2 binding.212 The crystal structures determined in the presence of the dioxygen mimic nitric oxide (NO) revealed that NO is indeed bound to the open iron coordination site.214 Interestingly, when the dioxygen is bound to the iron center as predicted, it would only be able to access the pro-R hydrogen at C1,213,214 consistent with the pro-R stereospecificity of C1 H-abstraction.207,208 EPR analysis of anaerobic, enzyme-bound FeII-NO complexes in the presence and absence of (S)-HPP and various substrate analogues confirmed that both the C2-OH and phosphonate functional groups of (S)-HPP were required for bidentate binding.204,215 The direct coordination of the negatively charged phosphonate group to the iron center in this binding mode likely helps activate FeII towards O2 binding and may also assist in the formation of higher iron oxidation states for substrate oxidation.216 Overall, the crystallographic and EPR studies shed light on how HppE is able to bind HPP, prime iron for O2 binding, and shield reactive intermediates from solvent.
A plausible chemical mechanism for the conversion of (S)-HPP (148) to fosfomycin (149) involves bidentate binding of the substrate to the ferrous iron center, which triggers O2 binding and single electron transfer to generate the ferric superoxide species (150, Scheme 24). Stereospecific hydrogen atom abstraction from C1 by a reactive oxygen species (150, 151, or 152), followed by epoxide ring formation and electron transfer to the iron center (via 150→153→154→149, 151→154→149, or 152→155→149) complete catalysis. The two reducing equivalents required to complete the catalytic cycle are supplied by an NADPH-dependent reductase protein in vivo. Evidence for the involvement of a substrate radical intermediate during HppE catalysis was obtained from studies using (R)-HPP (156) and (R)-1,1-difluoro-HPP (158), both of which are converted to 2-keto products (157 and 159, respectively), perhaps via hydrogen atom abstraction at C2 to form a ketyl radical species (Scheme 25).217
Scheme 24.

Proposed chemical mechanism for HppE.
Scheme 25.

HppE-catalyzed conversion of substrate analogues to the corresponding ketones likely proceeds via hydrogen atom abstraction and ketyl radical intermediates.
To investigate the mechanism of regiospecificity of HppE, four X-ray structures were recently determined: two of the R-stereoisomer of substrate [(R)-HPP (156)] with the inert cobalt-containing enzyme (Co(II)-HppE); one of (R)-HPP with the active iron-containing enzyme (Fe(II)-HppE) under anaerobic conditions; and one of (S)-HPP-Fe(II)-HppE in complex with the dioxygen mimic nitric oxide (NO).214 The structural data suggest that the formation of different products from (S)- and (R)-HPP (149 and 157, respectively) is not due to the presence of different binding sites for the two HPP enantiomers in the active site, but rather to the distinct orientation of the labile C-H bonds relative to the O2 binding site on the iron center. Specifically, both enantiomers of substrate bind in a bidentate mode to iron such that a single hydrogen atom is accessible for abstraction by a reactive iron-oxygen intermediate (C1-H in the case of (S)-HPP and C2-H in the case of (R)-HPP).214
To probe the chemical mechanism of HppE more closely, several kinetic isotope effect (KIE) studies were performed.218,219 Experiments using appropriately deuterated (S)- and (R)-HPP compounds revealed a small, primary isotope effect on (S)-HPP turnover (Dkcat= 1.5) and no isotope effect on (R)-HPP (Dkcat= 1.0), suggesting that hydrogen atom abstraction is at least partially rate limiting for (S)-HPP turnover.218 This difference in KIE between (S)- and (R)-HPP could be due to the slightly different strengths of the C1-H and C2-H bonds, which were calculated to have bond dissociation energies of 96.5 and 89.0 kcal/mol, respectively.218 The reactive oxygen species is likely either 150, 151, or 152 (Scheme 24), all of which could potentially abstract a hydrogen atom from C1 of (S)-HPP or from C2 of (R)-HPP. To study the reactive oxygen species more closely, the 18O KIE on kcat/Km(O2) for the reaction with native substrate, (S)-HPP, was measured to be 1.0120, the magnitude of which is consistent with the formation of an [FeIII-OOH] species in the rate-determining step.219 Formation of the FeIII-OOH species could involve direct hydrogen atom abstraction from the substrate (150→153). It may also be generated via a rate-limiting proton coupled electron transfer to form a ferric peroxy intermediate (151) that is then used to oxidize substrate directly (151→154),220 or to form an [FeIV=O] species which oxidizes substrate (151→152→155).216 Mechanistic studies to further characterize the unusual mode of epoxide formation catalyzed by HppE are currently in progress.
A similar mode of epoxide ring formation is believed to be catalyzed by hyoscyamine 6β-hydroxylase (H6H) in the biosynthesis of the plant tropane alkaloid, scopolamine (162) isolated from Hyoscyamus niger (Scheme 26).221 Although H6H is a mononuclear non-heme irondependent oxidase,222–224 it is distinct from HppE in that it requires α-ketoglutarate (α-KG) for catalysis. H6H is a highly versatile enzyme that catalyzes hydroxylation (160→161), dehydrogenation (161→162), and epoxidation (163→162) reactions (Scheme 26).225,226 The reactions catalyzed by Fe(II)/α-KG-dependent enzymes are characterized by a two-stage catalytic cycle, where the first stage involves the reaction of α-KG with O2 to generate a high valent iron-oxo species (164), which then oxidizes the substrate (RH) in the second half of the reaction (Scheme 27).216,227 The hydroxylation of hyoscyamine (160) to form the 6β-hydroxylated product (161) is predicted to follow a similar path. The same iron-oxo species (164) is also expected to play a key role in the epoxidation reaction (161→165→162, Scheme 27). However, detailed biochemical, mechanistic, and structural studies on H6H have not yet been conducted.
Scheme 26.

The epoxidation reaction catalyzed by H6H is α-KG-dependent and involves the two-electron oxidation of a secondary alcohol.
Scheme 27.

Oxygen activation catalyzed by Fe(II)/α-KG-dependent enzymes and the proposed mechanism of H6H catalyzed two-electron oxidation of a secondary alcohol to generate scopolamine (162).
More recently, several other Fe(II)/α-KG-dependent enzymes have been shown to epoxidize alkene moieties. DdaC was found to catalyze the epoxidation of Nβ-fumaramoyl-L-2,3- diaminopropionyl-S-DdaD (166) to Nβ-epoxysuccinamoyl-L-2,3-diaminopropionyl-S-DdaD (167), a key step in the biosynthesis of the dapdiamide-typeantibiotic Nβ-epoxysuccinamoyl- DAP-Val (168) in Pantoea agglomerans (Scheme 28).211 The epoxy oxygen in 167 was shown to be derived from molecular oxygen since incubation in the presence of 18O2 led to 18O incorporation in the product. Finally, the functions of PenD and PntD, two Fe(II)/α-KG-dependent epoxidases involved in pentalenolactone biosynthesis (169, Scheme 28) in Streptomyces exfoliatus and Streptomyces arenae, respectively, have recently been characterized.228 Interestingly, the purified PenD and PntD proteins were shown to catalyze two sequential two-electron oxidation reactions that convert pentalenolactone D (170) to pentalenolactone F (171) via the unsaturated intermediate pentalenolactone E (172). In addition, Streptomyces knockout strains harboring deletions of the penD or pntD genes accumulated pentalenolactone D with no detectable formation of 169, 171, or 172, strongly suggesting that these enzymes indeed convert 170→171 in vivo. As with other Fe(II)/α-KG-dependent enzymes, the key catalytic oxidant in the PenD- and PntD-catalyzed desaturation (170→172) and epoxidation (172→171) reactions is likely a high valent iron-oxo species.
Scheme 28.

Epoxidation reactions catalyzed by the Fe(II)/α-KG dependent enzymes DdaC and PenD/PntD during Nβ-epoxysuccinamoyl-DAP-Val (168) and pentalenolactone (169) biosynthesis, respectively.
3.5. Epoxide Formation via Intramolecular Nucleophilic Substitution
Unlike the mechanisms for epoxide formation discussed above, the haloalcohol (or halohydrin) dehalogenases catalyze epoxide formation without oxidizing their substrates.229,230 Instead, these enzymes use acid/base chemistry to catalyze a reversible SN2-like reaction, where a 1,2-haloalcohol (such as 173, Scheme 29) is converted to an epoxide (174). These enzymes are involved in the catabolism of organohalide pollutants by certain soil bacteria, and they have attracted attention as useful synthetic catalysts because of their broad substrate specificity and their potential for catalyzing stereoselective epoxidation, as well as regio- and stereoselective epoxide ring opening using a variety of alternative nucleophiles in place of the halide.231–234
Scheme 29.

Structure-based mechanism proposed for the haloalcohol dehydrogenase, HheC, which likely catalyzes formation of an epoxide from a 1,2-haloalcohol via an SN2-type reaction.
The X-ray crystal structure of HheC from Agrobacterium radiobacter revealed that the haloalcohol dehalogenases belong to the short chain dehydrogenase/reductase (SDR) family of enzymes, a group of enzymes that typically employ a reduced nicotinamide coenzyme to catalyze a transient two-electron oxidation of their substrates. The haloalcohol dehalogenases, however, have lost the ability to bind NAD(P)H and, instead, have evolved a different set of residues to bind to the halide moiety of their substrates. The typical SDR catalytic triad (Ser- Tyr-Arg/Lys) is conserved in the dehalogenases.235 The pKa of the conserved Tyr145 residue (in HheC from A. radiobacter) is likely lowered by its interaction with the conserved Arg149, enabling Tyr145 to deprotonate the alcohol moiety to initiate the intramolecular SN2-type transformation (Scheme 29). The conserved Ser132 residue likely helps bind substrate and stabilize the transition state for epoxide formation by hydrogen bonding to the developing oxyanion. The backbone amides of Tyr177 and Leu178 are believed to facilitate the displacement reaction by stabilizing the free halide. Recent computation studies, guided by the available crystal structures of HheC, suggest that a concerted SN2-type mode of epoxide formation is energetically feasible for this class of dehalogenases.236
4. Aziridine Biosynthesis
While our understanding of the biosynthetic mechanisms for cyclopropane and epoxide formation is steadily improving, the biosynthetic strategies for aziridine formation remain elusive. To date, only a handful of aziridine-containing natural products are known,237 and the biosynthetic gene clusters for only three of these have been published: the mitomycin (4) cluster from Streptomyces lavendulae,238 the azinomycin (5) cluster from Streptomyces sahachiroi,239 and the azicemicin (8) cluster from Kibdelosporangium sp. MJ126-NF4 (see Figure 1 for structures).240 The gene cluster for the enediyne compound maduropeptin (119, Scheme 30) has also been sequenced.152 However, due to its inherent instability, 119 has never been isolated from the producing organism, Actinomadura madurae. Instead, the presence of the aziridine moiety in 119 has been inferred from the structures of derivatives of 119 (such as 175, Scheme 30) that are released upon denaturation of the maduropeptin binding protein (MdpA, which forms a 1:1 complex with 119)241 and from the reactivity patterns of maduropeptin analogues.242
Scheme 30.

The major compound (175) isolated from the alcohol extract of the tightly bound maduropeptin (119) from MdpA.
Comparison of these gene clusters revealed that they likely employ distinct strategies to generate the aziridine unit. Early isotope-tracer experiments indicated that glucosamine (176, Scheme 31A)243–245 is the precursor of the aziridine moiety and 3-amino-5-hydroxylbenzoic acid (AHBA, 177) is likely the precursor of the quinone ring246 in the biosynthesis of mitomycin (4). Indeed, a set of genes required for AHBA biosynthesis and a gene encoding chitinase (MmcY), which may be needed to hydrolyze suitable glycans to provide glucosamine, are located in the mitomycin gene cluster.238 As depicted in Scheme 31A, coupling of glucosamine (176) to AHBA (177) could lead to a bicyclic intermediate 178, formation of which is possibly catalyzed by a glycosyltransferase (MitB) and a plant berberine bridge C-C bond-forming enzyme (MitR).247 Since a similar compound 182 (Scheme 31B) is believed to be an intermediate generated in the reductive activation of the DNA alkylating agent FR66979 (181) (and related compounds) via a mitomycin-like species (183),248,249 compound 178 (or 180) is thus likely a biosynthetic intermediate in the pathway en route to mitomycin (4). However, details of the initial coupling steps and the subsequent transformations of 178 to 4 remain obscure. In fact, except for the enzymes involved in the self-resistance mechanisms (Mcr, Mrd, Mct)250–252 and the final Omethylation step (MitM),253 functions of most genes in the mitomycin cluster have not been biochemically characterized. Interestingly, formation of the aziridine ring has been proposed to proceed through an SN2 mechanism involving nucleophilic displacement of the C-3 hydroxyl group (or its derivative) by the C-2 amino group of the glucosamine moiety (see 179→180 in Scheme 31A).237
Scheme 31.

(A) Proposed biosynthetic pathway of mitomycin C starting from glucosamine (176) and AHBA (177). (B) Proposed mechanism of reductive activation of FR66979 (181) via 183 to crosslink DNA.
For the biosynthesis of azinomycin (5), previous isotope-feeding experiments revealed efficient incorporation of 13C-labeled acetate into 5 suggesting that the azabicyclic fragment is in part derived from α-ketoglutarate (184, Scheme 32).254 In a more recent study using a protein cell-free system of the producer, S. sahachiroi, the incorporation of valine, glycine and threonine into the tripeptidyl backbone was demonstrated.255 Interestingly, ornithine (185) was also found to be an essential component for the in vitro reconstitution of azinomycin biosynthesis (Scheme 32).255 These results provided valuable information regarding the possible building blocks used in the azinomycin pathway. On the basis of the newly identified azinomycin gene cluster, a reaction sequence for the construction of the azabicyclic moiety (193, Scheme 33) starting from the peptidyl carrier protein (PCP)-linked glutamic acid (186) was proposed.239 As shown in Scheme 33, a two-carbon chain extension (187→188) catalyzed by a transketolase (AziC5/C6) helps to fully assemble the azabicyclic backbone. Following transamination (188→189), a dehydrogenation (189→190) catalyzed by AziC8 may trigger the subsequent cyclization of the first ring (190→191). A P450 catalyzed hydroxylation by AziC9 and a deacylation, catalyzed by AziC10 are likely the final steps to make 193, but how the aziridine ring in 192 is constructed remains uncertain.
Scheme 32.

Incorporation of various precursors into the azabicyclic fragment (circled in red) of azinomycin.
Scheme 33.

Proposed biosynthetic pathway for the azabicyclic moiety of azinomycin.
Feeding experiments using deuterium-labeled amino acids revealed that aspartic acid is the precursor of the aziridine moiety in azicemicin (8, Scheme 34).240 On the basis of the assigned functions of genes in the azicemicin gene cluster, a possible pathway for aziridine ring formation in the azicemicins was proposed (Scheme 34).240 The reaction is likely initiated by AzicMcatalyzed activation of the β-carboxylate group of either D- or L-aspartate (194) as an adenylate, which then serves as the aminoacyl donor for the carrier protein AzicP (196). The AzicP-linked aspartate (197), with a free α-carboxylate group, is the substrate for the PLP-dependent decarboxylase, AzicN. After decarboxylation, the resulting AzicP-tethered β-alanine (198) is oxidized by a P450 (AzicO2) or flavin-dependent (AzicO1 or O6) monooxygenase to the AzicPtethered isoserine (199). Cyclization of the resulting isoserine, which has a β-aminoalcohol moiety, may occur through an intramolecular SN2 reaction leading to the displacement of the hydroxyl group (or its derivative) by the amino group to generate the aziridine carboxyl starter unit (200). However, the identity of the enzyme that catalyzes the last cyclization step is not immediately apparent. The timing of aziridine ring formation, which may occur prior to or after the assembly of the angucycline core, is also uncertain. To obtain support for the proposed biosynthetic pathway, two genes encoding adenylyltransferases (AzicM and AzicV) were overexpressed and the resulting proteins were purified. Enzyme assays showed that one of the adenylyltransferases (AzicM) specifically recognizes aspartic acid, providing strong evidence that aspartate is the precursor of the aziridine moiety.
Scheme 34.

Proposed biosynthetic pathway for aziridine ring formation in the azicemicins.
One interesting possible general mechanism for aziridine formation in biological compounds was suggested by Bicker and Fischer.256 In this work, compound 201 was incubated with homogenized rat liver extract (Scheme 35). In the presence of both ATP and sulfate, 201 was converted to an aziridine compound (203). The authors proposed that sulfate was enzymatically converted to 3′-phophoadenosine 5′-phosphosulfate (PAPS, 204), a common intermediate in sulfur assimilation pathways. PAPS is formed from ATP and sulfate in two steps: a sulfate adenylyltransferase generates adenosine 5′-phosphosulfate (APS, 205), and an APS kinase generates PAPS (204). PAPS can then be enzymatically linked by sulfotransferases to a variety of acceptor groups, presumably including the hydroxyl group of 201. The sulfated hydroxyl group of 202 would then become a good leaving group, and an intramolecular nucleophilic attack by the vicinal secondary amine could form the aziridine (203). Interestingly, putative prokaryotic heterodimeric sulfate adenylyltransferase genes are present in both the mitomycin and azinomycin clusters (mmcU/mmcV and aziH1/aziH2, respectively). In addition, the azinomycin cluster also encodes a putative APS kinase (aziH3). The recently sequenced azicemicin and maduropeptin clusters, however, lack both of these genes, suggesting that other mechanisms likely exist for aziridine biosynthesis. Thus, it remains unclear whether a sulfation/intramolecular cyclization reaction sequence is a general strategy for aziridine biosynthesis, or whether other biosynthetic mechanisms are employed.
Scheme 35.

A potential biosynthetic route to the aziridine moiety.
5. Concluding Remarks
Numerous enzymatic strategies have evolved for small-ring biosynthesis, clearly underscoring the importance of these functional groups in biological systems. As more of these small-ring containing natural products are discovered and their biosynthetic gene clusters are identified, additional mechanisms for epoxide, cyclopropane, and aziridine ring formation may be unveiled. While chemical intuition and preliminary biochemical studies have enabled reasonable catalytic mechanisms to be proposed for many of these enzymes, detailed characterization of the transition states leading to ring closure merit further investigation. The interesting, diverse, and energetically difficult chemistry catalyzed by these enzymes should inspire attempts to mimic and/or manipulate their activities either for synthetic purposes or for enzymatic engineering of natural product structures.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (GM40541) and the Welch Foundation (F-1511).
Biographies

Christopher James Thibodeaux was born in 1980 and spent the majority of his childhood in Sulphur, Louisiana. He attended the Louisiana State University, where he graduated valedictorian in 2002 with degrees in Biochemistry, Plant Biology, and Chemistry. He joined the research group of Proffessor Hung-wen Liu at the University of Texas, Austin, in the summer of 2004, where he earned his Ph.D. in Cellular and Molecular Biology in 2010. His research in graduate school focused on elucidating the chemical and kinetic mechanisms of enzyme catalysis. In 2010, he began his post-doctoral work with Professor Taekjip Ha at the University of Illinois, Urbana-Champaign, as an Institute for Genomic Biology Fellow, where he applies cellular and molecular imaging techniques to understand enzyme function and cancer biology. His passions include spending time with his family, reading about history, fishing and hunting, and watching LSU Tiger football games.

Wei-chen Chang, a graduate student in the Medicinal Chemistry Division of the College of Pharmacy at the University of Texas at Austin, was born in Chai-yi, Taiwan, in 1980. He attended the National Taiwan University for his undergraduate studies where he worked with Professor Tien-Yau Luh on the development and synthesis of oligo aryl systems containing heteroatoms. After receiving his B.S. in Agricultural Chemistry in 2003, and completing his obligatory military service in 2005, he moved to the University of Texas at Austin to conduct his graduate research under the supervision of Professor Hung-wen Liu. His current research employs synthetic and biochemical methodologies to understand the chemical principles that underly biological processes and enzymatic reaction mechanisms. His specific areas of interest include mechanistic studies of 2-hydroxypropylphosphonate epoxidase (HppE) and enzymes involved in isoprenoid biosynthesis.

Hung-wen (Ben) Liu was born in Taipei, Taiwan (ROC). He graduated with a B.S. degree in Chemistry from Tunghai University, Taichung, in 1974. After completing his military service, he earned his Ph.D. at Columbia University, where he studied the additivity relation in exciton-split circular dichroism curves and its application to structural studies of oligosaccharides under Professor Koji Nakanishi. In 1981, his research interests turned to mechanistic enzymology, when he then joined the laboratory of Professor Christopher Walsh at Massachusetts Institute of Technology as a post-doctoral fellow. In 1984, he embarked on his independent career in the Chemistry Department at the University of Minnesota, Minneapolis, where he was promoted to the rank of Full Professor in 1994, and to Distinguished McKnight University Professor in 1999. In 2000, he moved to the University of Texas, Austin, where he is now the George H. Hitchings Regents Chair in Drug Design and Professor of Medicinal Chemistry, Chemistry, and Biochemistry. His research lies at the crossroads of organic and biological chemistry, with a particular emphasis on enzymatic reaction mechanisms, natural product biosynthesis, protein function regulation, inhibitor design and synthesis, and metabolic pathway engineering. In his spare time, he enjoys reading, music, and travels.
References
- 1.Steglich W, Fugmann B, Lang-Fugmann S, editors. Römpp Encyclopedia Natural Products. Georg Thieme Verlag; Stuttgart, Germany: 2000. [Google Scholar]
- 2.Takahashi I, Takahashi K, Ichinura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. J Antibiot. 1988;41:1915. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 3.Konichi M, Ohkuma H, Matusumoto K, Tsuno T, Kamei H, Miyaki T, Oki T, Kawaguchi H, Van Duyne GD, Clardy J. J Antibiot. 1989;42:1449. doi: 10.7164/antibiotics.42.1449. [DOI] [PubMed] [Google Scholar]
- 4.Hofle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem Int Ed. 1996;35:1567. [Google Scholar]
- 5.Tomasz M, Palom Y. Pharmacol Therap. 1997;76:73. doi: 10.1016/s0163-7258(97)00088-0. [DOI] [PubMed] [Google Scholar]
- 6.Rajski SR, Williams RM. Chem Rev. 1998;98:2723. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 7.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 8.Tomasz M, Lipman R, Chowdary D, Pawlak J, Verdine GL, Nakanishi K. Science. 1987;235:1204. doi: 10.1126/science.3103215. [DOI] [PubMed] [Google Scholar]
- 9.Salvati ME, Nguyen M, Armstrong RW. J Am Chem Soc. 1992;114:3144. [Google Scholar]
- 10.Hodgkinson TJ, Shipman M. Tetrahedron. 2001;57:4467. [Google Scholar]
- 11.Casely-Hayford M, Searcey M. DNA and RNA Binders, From Small Molecules to Drugs. Wiley-VCH; Weinheim: 2002. p. 676. [Google Scholar]
- 12.Seto H, Sasaki T, Yonehara H, Takahashi S, Takeuchi M, Kuwano H, Arai M. J Antibiot. 1984;37:1076. doi: 10.7164/antibiotics.37.1076. [DOI] [PubMed] [Google Scholar]
- 13.Cane DE, Oliver JS, Harrison PHM, Abell C, Hubbard BR, Kane CT, Lattman R. J Am Chem Soc. 1990;112:4513. [Google Scholar]
- 14.Nakagawa A, Tomoda H, Hao MV, Okano K, Iwai Y, Omura S. J Antibiot. 1985;38:1114. doi: 10.7164/antibiotics.38.1114. [DOI] [PubMed] [Google Scholar]
- 15.Foldor G, Dharanipragada R. Nat Prod Rep. 1994;11:443. doi: 10.1039/np9941100443. [DOI] [PubMed] [Google Scholar]
- 16.Tsuchida T, Iinuma H, Kinoshita N, Ikeda T, Sawa T, Hamada M, Takeuchi T. J Antibiot. 1995;48:217. doi: 10.7164/antibiotics.48.1104. [DOI] [PubMed] [Google Scholar]
- 17.Argoudelis AD, Reusser F, Whaley HA, Baczynskyj L, Mizsak SA, Wnuk RJ. J Antibiot. 1976;29:1001. doi: 10.7164/antibiotics.29.1001. [DOI] [PubMed] [Google Scholar]
- 18.Kuo MS, Yurek DA, Mitsak SA. J Antibiot. 1989;42:357. doi: 10.7164/antibiotics.42.357. [DOI] [PubMed] [Google Scholar]
- 19.Salaün J. Topics in Current Chemistry. Vol. 207. Springer-Verlag; Berlin: 2000. pp. 1–67. [Google Scholar]
- 20.Silverman RB. The Organic Chemistry of Drug Design and Drug Action. 2. Elsevier; London: 2004. [Google Scholar]
- 21.Boger DL, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645. [Google Scholar]
- 22.Boger DL, Garbaccio RM. Bioorg Med Chem. 1997;5:576. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 23.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043. [Google Scholar]
- 24.Searcey M. Curr Pharm Design. 2002;8:1375. doi: 10.2174/1381612023394539. [DOI] [PubMed] [Google Scholar]
- 25.Nicolaou KC, Winssinger N, Pastor J, Ninkovic S, Sarabia F, He Y, Vourloumis D, Yang Z, Li T, Glannakakou P, Hamel E. Nature. 1997;387:268. doi: 10.1038/387268a0. [DOI] [PubMed] [Google Scholar]
- 26.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325. [PubMed] [Google Scholar]
- 27.Kowalski RJ, Giannakakou P, Hamel E. J Biol Chem. 1997;272:2534. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 28.Suckling CJ. Angew Chem Int Ed. 1988;27:537. [Google Scholar]
- 29.Armstrong RN. In: Comprehensive Natural Products Chemistry. Barton D, Nakanishi K, Meth-Cohn O, editors. Vol. 5. Pergamon; New York: 1999. pp. 51–70. [Google Scholar]
- 30.Walsh CT. Tetrahedron. 1982;38:871. [Google Scholar]
- 31.Law JH. Acc Chem Res. 1971;4:199. [Google Scholar]
- 32.Liu H-w, Walsh CT. In: The Chemistry of the Cyclopropyl Group. Rappoport Z, editor. Vol. 2. John Wiley & Sons; New York: 1987. pp. 959–1025. [Google Scholar]
- 33.Djerassi C, Silva CJ. Acc Chem Res. 1991;24:371. [Google Scholar]
- 34.Baker BJ, Kerr RG. Topics in Current Chemistry. Vol. 167. Springer-Verlag; Berlin: 1993. pp. 1–32. [Google Scholar]
- 35.Moore BS, Floss HG. In: Comprehensive Chemistry of Natural Products Chemistry. Barton D, Nakanishi K, Meth-Cohn O, editors. Vol. 1. Pergamon; New York: 1999. pp. 61–82. [Google Scholar]
- 36.Wessjohann LA, Brandt W, Thiemann T. Chem Rev. 2003;103:1625. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- 37.Kuzuyama T, Hemmi H, Takahashi S. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 493–516. [Google Scholar]
- 38.Rohmer M. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 517–556. [Google Scholar]
- 39.Kellogg BA, Poulter CD. Curr Opin Chem Biol. 1997;1:570. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 40.Kurokawa H, Koyama T. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 557–584. [Google Scholar]
- 41.Poulter CD. Acc Chem Res. 1990;23:70. [Google Scholar]
- 42.Savage TJ, Croteau R. Arch Biochem Biophys. 1993;305:581. doi: 10.1006/abbi.1993.1464. [DOI] [PubMed] [Google Scholar]
- 43.Hoelscher DJ, Williams DC, Wildung MR, Croteau R. Phytochemistry. 2003;62:1081. doi: 10.1016/s0031-9422(02)00674-x. [DOI] [PubMed] [Google Scholar]
- 44.Faldt J, Martin D, Miller B, Rawat S, Bohlmann J. Plant Mol Biol. 2003;51:119. doi: 10.1023/a:1020714403780. [DOI] [PubMed] [Google Scholar]
- 45.Croteau R. Chem Rev. 1987;87:929. [Google Scholar]
- 46.Christianson DW. Chem Rev. 2006;106:3412. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 47.Allemann RK. Pure Appl Chem. 2008;80:1791. [Google Scholar]
- 48.Degenhardt J, Köllner TG, Gershenzon J. Phytochemistry. 2009;70:1621. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 49.Croteau R, Alonso WR, Koepp AE, Shim JH, Cane DE. Arch Biochem Biophys. 1993;307:397. doi: 10.1006/abbi.1993.1606. [DOI] [PubMed] [Google Scholar]
- 50.Wendt KU, Schulz GE. Structure. 1998;6:127. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 51.Poulter CD, Rilling HC. In: Biosynthesis of Isoprenoid Compounds. Porter JW, Spurgeon SL, editors. Vol. 1. Wiley; New York: 1981. pp. 413–441. [Google Scholar]
- 52.Abe I. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 709–732. [Google Scholar]
- 53.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr J Biol Chem. 2000;275:30610. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 54.Zhang D-l, Poulter CD. J Am Chem Soc. 1995;117:1641. [Google Scholar]
- 55.Jarstfer MB, Blagg BSJ, Rogers DH, Poulter CD. J Am Chem Soc. 1996;118:13089. [Google Scholar]
- 56.Erickson HK, Poulter CD. J Am Chem Soc. 2003;125:6886. doi: 10.1021/ja034520g. [DOI] [PubMed] [Google Scholar]
- 57.Thulasiram HV, Erickson HK, Poulter CD. Science. 2007;316:73. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 58.Saunders M, Vogel P, Hagen EL, Rosenfeld J. Acc Chem Res. 1973;6:53. [Google Scholar]
- 59.Dewar MJS, Healy EF, Ruiz JM. J Chem Soc, Chem Comm. 1987;12:943. [Google Scholar]
- 60.Norin T. Phytochemistry. 1972;11:1231. [Google Scholar]
- 61.Rudloff EV. Biochem Systemat Ecol. 1975;2:131. [Google Scholar]
- 62.Librowski T, Czarnecki R, Lochynski S, Frackowiak B, Pasenkiewicz-Gierula M, Grochowski J, Serda P. Pol J Pharmacol. 2001;53:535. [PubMed] [Google Scholar]
- 63.Nikitina LE, Startseva VA, Dorofeeva LY, Artemova NP, Kuznetsov IV, Lisovskaya SA, Glushko NP. Chem Nat Cmpds. 2010;46:28. [Google Scholar]
- 64.Savage TJ, Croteau R. Arch Biochem Biophys. 1993;305:581. doi: 10.1006/abbi.1993.1464. [DOI] [PubMed] [Google Scholar]
- 65.Wise ML, Croteau R. In: Comprehensive Natural Products Chemistry: Isoprenoids Including Carotenoids and Steroids. Cane DE, editor. Elsevier; Oxford: 1999. pp. 97–153. [Google Scholar]
- 66.Hallahan TW, Croteau R. Arch Biochem Biophys. 1988;264:618. doi: 10.1016/0003-9861(88)90328-1. [DOI] [PubMed] [Google Scholar]
- 67.Hallahan TW, Croteau R. Arch Biochem Biophys. 1989;269:313. doi: 10.1016/0003-9861(89)90113-6. [DOI] [PubMed] [Google Scholar]
- 68.Wise ML, Savage TJ, Katahira E, Croteau R. J Biol Chem. 1998;273:14891. doi: 10.1074/jbc.273.24.14891. [DOI] [PubMed] [Google Scholar]
- 69.Kampranis SC, Ioannidis D, Purvis A, Mahrez W, Ninga E, Katerelos NA, Anssour S, Dunwell JM, Degenhardt J, Makris AM, Goodenough PW, Johnsona CB. Plant Cell. 2007;19:1994. doi: 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dueber MT, Adolf W, West CA. Plant Physiol. 1978;62:598. doi: 10.1104/pp.62.4.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mau CJD, West CA. Proc Natl Acad Sci USA. 1994;91:8497. doi: 10.1073/pnas.91.18.8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang K-x, Huang Q-l, Scott AI. Arch Biochem Biophys. 1998;352:144. doi: 10.1006/abbi.1998.0578. [DOI] [PubMed] [Google Scholar]
- 73.Kirby JMN, Park JG, Withers ST, Nowroozi F, Behrendt D, Rutledge EJG, Fortman JL, Johnson HE, Anderson JV, Keasling JD. Phytochemistry. 2010;71:1466. doi: 10.1016/j.phytochem.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 74.Shi QW, Su XH, Kiyota H. Chem Rev. 2008;108:4295. doi: 10.1021/cr078350s. [DOI] [PubMed] [Google Scholar]
- 75.Kulkosky J, Culnan DM, Roman J, Dornadula G, Schnell M, Boyd MR, Pomerantz RJ. Blood. 2001;98:3006. doi: 10.1182/blood.v98.10.3006. [DOI] [PubMed] [Google Scholar]
- 76.Wender PA, Kee JM, Warrington JM. Science. 2008;320:649. doi: 10.1126/science.1154690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kedei N, Lundberg DJ, Toth A, Welburn P, Garfield SH, Blumberg PM. Cancer Res. 2004;64:3243. doi: 10.1158/0008-5472.can-03-3403. [DOI] [PubMed] [Google Scholar]
- 78.Köksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Nature. 2011;469:116. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Corey EJ, Matsuda SP, Bartel B. Proc Natl Acad Sci USA. 1993;90:11628. doi: 10.1073/pnas.90.24.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaller H. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 755–788. [Google Scholar]
- 81.Corey EJ, Matsuda SP, Bartel B. Proc Natl Acad Sci USA. 1994;91:2211. doi: 10.1073/pnas.91.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi Z, Buntel CJ, Griffin JH. Proc Natl Acad Sci USA. 1994;91:7370. doi: 10.1073/pnas.91.15.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lodeiro S, Schulz-Gasch T, Matsuda SPT. J Am Chem Soc. 2005;127:14132. doi: 10.1021/ja053791j. [DOI] [PubMed] [Google Scholar]
- 84.Wu TK, Chang CH, Liu YT, Wang TT. Chem Rec. 2008;8:302. doi: 10.1002/tcr.20157. [DOI] [PubMed] [Google Scholar]
- 85.Cronan JE, Jr, Nunn WD, Batchelor JG. Biochim Biophys Acta. 1974;348:63. doi: 10.1016/0005-2760(74)90093-9. [DOI] [PubMed] [Google Scholar]
- 86.Yuan Y, Lee RE, Besra GS, Belisle JT, Barry CE., III Proc Natl Acad Sci USA. 1995;92:6630. doi: 10.1073/pnas.92.14.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.George KM, Yuan Y, Sherman DR, Barry CE., III J Biol Chem. 1995;270:27292. doi: 10.1074/jbc.270.45.27292. [DOI] [PubMed] [Google Scholar]
- 88.Grogan DW, Cronan JE., Jr Microbiol Mol Biol Rev. 1997;61:429. doi: 10.1128/mmbr.61.4.429-441.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barry CE, III, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Prog Lipid Res. 1998;37:143. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 90.Bao X, Katz S, Pollard M, Ohlrogge J. Proc Natl Acad Sci USA. 2002;99:7172. doi: 10.1073/pnas.092152999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kastrinsky DB, McBride NS, Backus KM, LeBlanc JJ, Barry CE., III . In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 1. Elsevier; Oxford: 2010. pp. 65–146. [Google Scholar]
- 92.Glickman MS, Cox JS, Jacobs WR., Jr Mol Cell. 2000;5:717. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 93.Rao V, Fujiwara N, Porcelli SA, Glickman MS. J Exp Med. 2005;201:535. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang C-c, Smith CV, Glickman MS, Jacobs WR, Jr, Sacchettini JC. J Biol Chem. 2002;277:11559. doi: 10.1074/jbc.M111698200. [DOI] [PubMed] [Google Scholar]
- 95.Iwig DF, Grippe AT, McIntyre TA, Booker SJ. Biochemistry. 2004;43:13510. doi: 10.1021/bi048692h. [DOI] [PubMed] [Google Scholar]
- 96.Guangqi E, Lesage D, Ploux O. Biochimie. 2010;92:1454. doi: 10.1016/j.biochi.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 97.Molitor EJ, Paschal BM, Liu H-w. Chembiochem. 2003;4:1352. doi: 10.1002/cbic.200300767. [DOI] [PubMed] [Google Scholar]
- 98.Pongdee R, Liu H-w. Bioorg Chem. 2004;32:393. doi: 10.1016/j.bioorg.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 99.Iwig DF, Uchida A, Stromberg JA, Booker SJ. J Am Chem Soc. 2005;127:11612. doi: 10.1021/ja053899z. [DOI] [PubMed] [Google Scholar]
- 100.Courtois F, Ploux O. Biochemistry. 2005;44:13583. doi: 10.1021/bi051159x. [DOI] [PubMed] [Google Scholar]
- 101.Boissier F, Bardou F, Guillet V, Uttenweiler-Joseph S, Daffe M, Quemard A, Mourey L. J Biol Chem. 2006;281:4434. doi: 10.1074/jbc.M510250200. [DOI] [PubMed] [Google Scholar]
- 102.Yoshida M, Ezaki M, Hashimoto M, Yamashita M, Shigematsu N, Okuhara M, Kohsaka M, Horikoshi K. J Antibiot. 1990;43:748. doi: 10.7164/antibiotics.43.748. [DOI] [PubMed] [Google Scholar]
- 103.Watanabe H, Tokiwano T, Oikawa H. Tetrahedron Lett. 2006;47:1399. [Google Scholar]
- 104.Tokiwano T, Watanabe H, Seo T, Oikawa H. J Chem Soc, Chem Comm. 2008;45:6016. doi: 10.1039/b809610d. [DOI] [PubMed] [Google Scholar]
- 105.Kuo MS, Zielinski RJ, Cialdella JI, Marschke CK, Dupuis MJ, Li GP, Kloosterman DA, Spilman CH, Marshall VP. J Am Chem Soc. 1995;117:10629. [Google Scholar]
- 106.Abeles FB, Morgan PW, Saltveit ME., Jr . Ethylene in Plant Biology. 2. Academic; New York: 1992. [Google Scholar]
- 107.Kende H. Annu Rev Plant Physoiol Plant Mol Biol. 1993;44:283. [Google Scholar]
- 108.Ramalingam K, Lee K-M, Woodard RW, Bleecker AB, Kende H. Proc Natl Acad Sci USA. 1985;82:7820. doi: 10.1073/pnas.82.23.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White MF, Vasquez J, Yang SF, Kirsch JF. Proc Natl Acad Sci USA. 1994;91:12428. doi: 10.1073/pnas.91.26.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Y, Feng L, Kirsch JF. Biochemistry. 1997;36:15477. doi: 10.1021/bi971625l. [DOI] [PubMed] [Google Scholar]
- 111.Capitani G, Hohenester E, Feng L, Storici P, Kirsch JF, Jansonius JN. J Mol Biol. 1999;294:745. doi: 10.1006/jmbi.1999.3255. [DOI] [PubMed] [Google Scholar]
- 112.Frédéric H, Vaillancourt EY, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 113.Neumann CS, Walsh CT. J Am Chem Soc. 2008;130:14022. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelly WL, Boyne MT, Yeh E, Vosburg DA, Galoni DP, Kelleher NL, Walsh CT. Biochemistry. 2007;46:359. doi: 10.1021/bi061930j. [DOI] [PubMed] [Google Scholar]
- 116.Ghisla S, Thorpe C, Massey V. Biochemistry. 1984;23:3154. doi: 10.1021/bi00309a008. [DOI] [PubMed] [Google Scholar]
- 117.Kim JJ, Wang M, Paschke R. Proc Natl Acad Sci USA. 1993;90:7523. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ghisla S, Thorpe C. Eur J Biochem. 2004;271:494. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 119.Li D, Zhou HQ, Dakoji S, Shin I, Oh E, Liu H-w. J Am Chem Soc. 1998;120:2008. [Google Scholar]
- 120.Lis L, Koltun ES, Liu H-w, Kass SR. J Am Chem Soc. 2002;124:1276. doi: 10.1021/ja0114862. [DOI] [PubMed] [Google Scholar]
- 121.Mansoorabadi SO, Thibodeaux CJ, Liu H-w. J Org Chem. 2007;72:6329. doi: 10.1021/jo0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Höfer I, Crüsemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J. Chem Biol. 2011;18:381. doi: 10.1016/j.chembiol.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 123.Brandl M, Kozhushkov SI, Zlatopolskiy BD, Alvermann P, Geers B, Zeeck A, de Meijere A. Eur J Org Chem. 2005;1:123. [Google Scholar]
- 124.Nelson MJ, Seitz S. In: Active Oxygen in Biochemistry. Valentine JS, Foote CS, Greenberg A, Liebman JF, editors. Chapman & Hall; Glasgow, U.K: 1995. pp. 276–312. [Google Scholar]
- 125.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 126.Que L, Jr, Ho RYN. Chem Rev. 1996;96:2607. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 127.Bugg TDH. Tetrahedron. 2003;59:7075. [Google Scholar]
- 128.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 129.Ortiz de Montellano PR, De Voss JJ. Nat Prod Rep. 2002;19:477. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 130.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 131.Shaik S, Hirao H, Kumar K. Nat Prod Rep. 2007;24:533. doi: 10.1039/b604192m. [DOI] [PubMed] [Google Scholar]
- 132.Zhu Y, Silverman RB. Biochemistry. 2008;47:2231. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 133.Kumar D, de Visser SP, Shaik S. Chem Eur J. 2005;11:2825. doi: 10.1002/chem.200401044. [DOI] [PubMed] [Google Scholar]
- 134.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 135.Hirao H, Kumar D, Shaik S. J Inorg Biochem. 2006;100:2054. doi: 10.1016/j.jinorgbio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 136.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2010;110:949. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 137.de Visser SP, Ogliaro F, Harris N, Shaik S. J Am Chem Soc. 2001;123:3037. doi: 10.1021/ja003544+. [DOI] [PubMed] [Google Scholar]
- 138.Shaik S, de Visser SP, Ogliaro F, Schwartz H, Schroder D. Curr Opin Chem Biol. 2002;6:556. doi: 10.1016/s1367-5931(02)00363-0. [DOI] [PubMed] [Google Scholar]
- 139.Newcomb M, Toy PH. Acc Chem Res. 2000;33:449. doi: 10.1021/ar960058b. [DOI] [PubMed] [Google Scholar]
- 140.Gruschow S, Sherman DH. In: Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. Wiley-VCH; Weinheim: 2006. pp. 349–398. [Google Scholar]
- 141.Hőfle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem Int Ed. 1996;35:1567. [Google Scholar]
- 142.Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Science. 2000;287:640. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- 143.Nagano S, Li H, Shimizu H, Nishida C, Ogura H, Ortiz de Montellano PR, Poulos TL. J Biol Chem. 2003;278:44886. doi: 10.1074/jbc.M308115200. [DOI] [PubMed] [Google Scholar]
- 144.Ogura H, Nishida CR, Hoch UR, Perera R, Dawson JH, de Montellano PRO. Biochemistry. 2004;43:14712. doi: 10.1021/bi048980d. [DOI] [PubMed] [Google Scholar]
- 145.Anzai Y, Li S, Chaulagain MR, Kinoshita K, Kato F, Montgomery J, Sherman DH. Chem Biol. 2008;15:950. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kudo F, Motegi A, Mizoue K, Eguchi T. Chembiochem. 2010;11:1574. doi: 10.1002/cbic.201000214. [DOI] [PubMed] [Google Scholar]
- 147.Mendes MV, Recio E, Fouces R, Luiten R, Martin JF, Aparicio JF. Chem Biol. 2001;8:635. doi: 10.1016/s1074-5521(01)00033-3. [DOI] [PubMed] [Google Scholar]
- 148.Mendes MV, Anton N, Martin JF, Aparicio JF. Biochem J. 2005;386:57. doi: 10.1042/BJ20040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kells PM, Ouellet H, Santos-Aberturas J, Aparicio JF, Podust LM. Chem Biol. 2010;17:841. doi: 10.1016/j.chembiol.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li A, Piel J. Chem Biol. 2002;9:1017. doi: 10.1016/s1074-5521(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 151.Bililign T, Hyun CG, Williams JS, Czisny AM, Thorson JS. Chem Biol. 2004;11:959. doi: 10.1016/j.chembiol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 152.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. J Am Chem Soc. 2007;129:13082. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang F, He H-Y, Tang M-C, Tang Y-M, Zhou Q, Tang G-L. J Am Chem Soc. 2011;133:2452. doi: 10.1021/ja105649g. [DOI] [PubMed] [Google Scholar]
- 154.Ullrich V, Brugger R. Angew Chem Int Ed. 1994;33:1911. [Google Scholar]
- 155.Senger T, Wichard T, Kunze S, Gobel C, Lerchl J, Pohnert G, Feussner I. J Biol Chem. 2005;280:7588. doi: 10.1074/jbc.M411738200. [DOI] [PubMed] [Google Scholar]
- 156.Radmark O, Werz O, Steinhilber D, Samuelsson B. Trends Biochem Sci. 2007;32:332. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 157.Stumpe MFI. Phytochem Rev. 2006;5:347. [Google Scholar]
- 158.Koljak R, Boutaud O, Shieh BH, Samel N, Brash AR. Science. 1997;277:1994. doi: 10.1126/science.277.5334.1994. [DOI] [PubMed] [Google Scholar]
- 159.Oldham ML, Brash AR, Newcomer ME. Proc Natl Acad Sci USA. 2005;102:297. doi: 10.1073/pnas.0406352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee DS, Nioche P, Hamberg M, Raman CS. Nature. 2008;455:363. doi: 10.1038/nature07307. [DOI] [PubMed] [Google Scholar]
- 161.Ullrich V. Arch Biochem Biophys. 2003;409:45. doi: 10.1016/s0003-9861(02)00410-1. [DOI] [PubMed] [Google Scholar]
- 162.Gao B, Boeglin WE, Zheng Y, Schneider C, Brash AR. J Biol Chem. 2009;284:22087. doi: 10.1074/jbc.M109.013151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fagan RL, Palfey BA. In: Comprehensive Natural Products II, Chemistry and Biology. Mander L, Liu H-w, editors. Vol. 7. Elsevier; Oxford: 2010. pp. 37–113. [Google Scholar]
- 164.Palfey BA, McDonald CA. Arch Biochem Biophys. 2010;493:26. doi: 10.1016/j.abb.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 165.Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT. Arch Biochem Biophys. 2005;442:102. doi: 10.1016/j.abb.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 166.Kantz A, Gassner GT. Biochemistry. 2011;50:523. doi: 10.1021/bi101328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hollmann F, Lin PC, Witholt B, Schmid A. J Am Chem Soc. 2003;125:8209. doi: 10.1021/ja034119u. [DOI] [PubMed] [Google Scholar]
- 168.Otto K, Hofstetter K, Rothlisberger M, Witholt B, Schmid A. J Bacteriol. 2004;186:5292. doi: 10.1128/JB.186.16.5292-5302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ukaegbu UE, Kantz A, Beaton M, Gassner GT, Rosenzweig AC. Biochemistry. 2010;49:1678. doi: 10.1021/bi901693u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu W, Nonaka K, Nie L, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. Chem Biol. 2005;12:293. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 171.Gao Q, Thorson JS. FEMS Microbiol Lett. 2008;282:105. doi: 10.1111/j.1574-6968.2008.01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Proc Natl Acad Sci USA. 2006;103:4234. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reeves CD, Hu Z, Reid R, Kealey JT. Appl Environ Microbiol. 2008;74:5121. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sattler I, Thiericke R, Zeeck A. Nat Prod Rep. 1998;221 doi: 10.1039/a815221y. [DOI] [PubMed] [Google Scholar]
- 175.Shipley PR, Donnelly CCA, Le CH, Bernauer AD, Klegeris A. Int J Mol Med. 2009;25:711. doi: 10.3892/ijmm_00000283. [DOI] [PubMed] [Google Scholar]
- 176.Rui Z, Petríckova K, Skanta F, Pospisil S, Yang Y, Chen C-Y, Tsai S-F, Floss HG, Petrícek M, Yu T-W. J Biol Chem. 2010;285:24915. doi: 10.1074/jbc.M110.128850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Thiericke R, Zeeck A, Robinson JA, Beale JM, Floss HG. J Chem Soc, Chem Comm. 1989;7:402. [Google Scholar]
- 178.Oliynyk M, Stark CB, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF. Mol Microbiol. 2003;49:1179. doi: 10.1046/j.1365-2958.2003.03571.x. [DOI] [PubMed] [Google Scholar]
- 179.Smith L, Hong H, Spencer JB, Leadlay PF. Chembiochem. 2008;15:2967. doi: 10.1002/cbic.200800585. [DOI] [PubMed] [Google Scholar]
- 180.Migita A, Watanabe M, Hirose Y, Watanabe K, Tokiwano T, Kinashi H, Oikawa H. Biosci Biotechnol Biochem. 2009;73:169. doi: 10.1271/bbb.80631. [DOI] [PubMed] [Google Scholar]
- 181.Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, Deng Z. Chem Biol. 2003;10:431. doi: 10.1016/s1074-5521(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 182.Harvey BM, Mironenko T, Sun Y, Hong H, Deng Z, Leadlay PF, Weissman KJ, Haydock SF. Chem Biol. 2007;14:703. doi: 10.1016/j.chembiol.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 183.Demydchuk Y, Sun Y, Hong H, Staunton J, Spencer JB, Leadlay PF. Chembiochem. 2008;9:1136. doi: 10.1002/cbic.200700715. [DOI] [PubMed] [Google Scholar]
- 184.Gallimore AR, Stark CB, Bhatt A, Harvey BM, Demydchuk Y, Bolanos-Garcia V, Fowler DJ, Staunton J, Leadlay PF, Spencer JB. Chem Biol. 2006;13:453. doi: 10.1016/j.chembiol.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 185.Liu T, Cane DE, Deng Z. Methods Enzymol. 2009;459:187. doi: 10.1016/S0076-6879(09)04609-6. [DOI] [PubMed] [Google Scholar]
- 186.Oliynyk M, Stark CB, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF. Mol Microbiol. 2003;49:1179. doi: 10.1046/j.1365-2958.2003.03571.x. [DOI] [PubMed] [Google Scholar]
- 187.Gallimore AR. Nat Prod Rep. 2009;26:266. doi: 10.1039/b807902c. [DOI] [PubMed] [Google Scholar]
- 188.Bhatt A, Stark C, Harvey BM, Gallimore AR, Demydchuk YA, Spencer JB, Staunton J, Leadlay PF. Angew Chem Int Ed. 2005;44:7075. doi: 10.1002/anie.200501757. [DOI] [PubMed] [Google Scholar]
- 189.Shichijo Y, Migita A, Oguri H, Watanabe M, Tokiwano T, Watanabe K, Oikawa H. J Am Chem Soc. 2008;130:12230. doi: 10.1021/ja8040543. [DOI] [PubMed] [Google Scholar]
- 190.Ono T, Bloch K. J Biol Chem. 1975;250:1571. [PubMed] [Google Scholar]
- 191.Abe I, Prestwich GD. In: Squalene epoxidase and oxidosqualene: lanosterol cyclase. Key enzymes in cholesterol biosynthesis. Barton DHR, Nakanishi K, editors. Elsevier; Oxford, UK: 1999. [Google Scholar]
- 192.Kuliopulos A, Hubbard GR, Lam Z, Koski IJ, Furie B, Furie BC, Walsh CT. Biochemistry. 1992;31:7722. doi: 10.1021/bi00148a037. [DOI] [PubMed] [Google Scholar]
- 193.Dowd P, Ham S-W, Hershline R. J Am Chem Soc. 1992;114:7613. [Google Scholar]
- 194.Naganathan S, Hershline R, Ham SW, Dowd P. J Am Chem Soc. 1994;116:9831. [Google Scholar]
- 195.Dowd P, Hershline R, Ham SW, Naganathan S. Science. 1995;269:1684. doi: 10.1126/science.7569894. [DOI] [PubMed] [Google Scholar]
- 196.Shen B, Gould SJ. J Am Chem Soc. 1991;30:8936. doi: 10.1021/bi00101a004. [DOI] [PubMed] [Google Scholar]
- 197.Gould SJ, Shen B. J Am Chem Soc. 1991;113:684. [Google Scholar]
- 198.Gould SJ, Kirchmeier MJ, LaFever RE. J Am Chem Soc. 1996;118:7663. [Google Scholar]
- 199.Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, Walsh CT. Biochemistry. 1994;33:10646. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- 200.Skarzynski T, Kim DH, Lees WJ, Walsh CT, Duncan K. Biochemistry. 1998;37:2572. doi: 10.1021/bi9722608. [DOI] [PubMed] [Google Scholar]
- 201.Seto H, Kuzuyama T. Nat Prod Rep. 1999;16:589. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 202.Metcalf WW, Van der Donk WA. Annu Rev Biochem. 2009;78:65. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liu P, Murakami K, Seki T, He X, Yeung S-M, Kuzuyama T, Seto H, Liu H-w. J Am Chem Soc. 2001;123:4619. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 204.Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu H-w. Biochemistry. 2003;42:11577. doi: 10.1021/bi030140w. [DOI] [PubMed] [Google Scholar]
- 205.Yan F, Munos JW, Liu P, Liu H-w. Biochemistry. 2006;45:11473. doi: 10.1021/bi060839c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hammerschmidt F, Bovermann G, Bayer K. Liebigs Annalen Der Chemie. 1990:1055. [Google Scholar]
- 207.Hammerschmidt F, Kahlig H. J Org Chem. 1991;56:2364. [Google Scholar]
- 208.Woschek A, Wuggenig F, Peti W, Hammerschmidt F. ChemBioChem. 2002;3:829. doi: 10.1002/1439-7633(20020902)3:9<829::AID-CBIC829>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 209.Hashimoto T, Matsuda J, Yamada Y. FEBS Lett. 1993;329:35. doi: 10.1016/0014-5793(93)80187-y. [DOI] [PubMed] [Google Scholar]
- 210.Busby RW, Townsend CA. Bioorg Med Chem. 1996;4:1059. doi: 10.1016/0968-0896(96)00088-0. [DOI] [PubMed] [Google Scholar]
- 211.Hollenhorst MA, Bumpus SB, Matthews ML, Bollinger JM, Jr, Kelleher NL, Walsh CT. J Am Chem Soc. 2010;132:15773. doi: 10.1021/ja1072367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Higgins LJ, Yan F, Liu P, Liu H-w, Drennan CL. Nature. 2005;437:838. doi: 10.1038/nature03924. [DOI] [PubMed] [Google Scholar]
- 213.Yan F, Li T, Lipscomb JD, Liu A, Liu H-w. Arch Biochem Biophys. 2005;442:82. doi: 10.1016/j.abb.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 214.Yun D, Higgins LJ, Yan F, Liu H-w, Drennan CL. J Am Chem Soc. 2011;133:11262. doi: 10.1021/ja2025728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yan F, Moon S-J, Liu P, Zhao Z, Lipscomb JD, Liu A, Liu H-w. Biochemistry. 2007;46:12628. doi: 10.1021/bi701370e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 217.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu H-w. Angew Chem Int Ed. 2002;41:4529. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 218.Munos JW, Moon S-J, Mansoorabadi SO, Chang W, Hong L, Yan F, Liu A, Liu H-w. Biochemistry. 2008;47:8726. doi: 10.1021/bi800877v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mirica LM, McCusker KP, Munos JW, Liu H-w, Klinman JP. J Am Chem Soc. 2008;130:8122. doi: 10.1021/ja800265s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.van der Donk W, Krebs C, Bolinger JM., Jr Curr Opin Struc Biol. 2010;20:673. doi: 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Evans WC. In: The Biology and Taxonomy of the Solanaceae. Hawkes JG, Lester RN, Skelding AD, editors. Academic Press; London: 1979. pp. 241–254. [Google Scholar]
- 222.Hashimoto T, Yamada Y. Eur J Biochem. 1987;164:277. doi: 10.1111/j.1432-1033.1987.tb11055.x. [DOI] [PubMed] [Google Scholar]
- 223.Matsuda J, Okabe S, Hashimoto T, Yamada Y. J Biol Chem. 1991;266:9460. [PubMed] [Google Scholar]
- 224.Liu T, Zhu P, Cheng K-d, Meng C, He H-x. Planta Med. 2005;71:249. doi: 10.1055/s-2005-837825. [DOI] [PubMed] [Google Scholar]
- 225.Hashimoto T, Kohno J, Yamada Y. Plant Physiol. 1987;84:144. doi: 10.1104/pp.84.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hashimoto T, Matsuda J, Yamada Y. FEBS Lett. 1993;329:35. doi: 10.1016/0014-5793(93)80187-y. [DOI] [PubMed] [Google Scholar]
- 227.Prescott AG, Lloyd MD. Nat Prod Rep. 2000;17:367. doi: 10.1039/a902197c. [DOI] [PubMed] [Google Scholar]
- 228.Seo M-J, Zhu D, Endo S, Ikeda H, Cane DE. Biochemistry. 2011;50:1739. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fetzner S. Appl Microbiol Biotechnol. 1998;50:633. doi: 10.1007/s002530051346. [DOI] [PubMed] [Google Scholar]
- 230.de Jong RM, Dijkstra BW. Curr Opin Struct Biol. 2003;13:722. doi: 10.1016/j.sbi.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 231.LutjeSpelberg JH, van Hylckama Vlieg JET, Tang L, Janssen DB, Kellogg RM. Org Lett. 2001;3:41. doi: 10.1021/ol0067540. [DOI] [PubMed] [Google Scholar]
- 232.de Jong RM, Tiesinga JJW, Villa A, Tang L, Janssen DB, Dijkstra BW. J Am Chem Soc. 2005;127:13338. doi: 10.1021/ja0531733. [DOI] [PubMed] [Google Scholar]
- 233.Haak RM, Tarabiono C, Janssen DB, Minnaard AJ, de Vries JG, Feringa BL. Org Biomol Chem. 2007;5:318. doi: 10.1039/b613937j. [DOI] [PubMed] [Google Scholar]
- 234.Hopmann KH, Himo F. Biochemistry. 2008;47:4973. doi: 10.1021/bi800001r. [DOI] [PubMed] [Google Scholar]
- 235.de Jong RM, Tiessinga JJW, Rozeboom HJ, Kalk KH, Tnag L, Janssen DB, Dijkstra BW. EMBO J. 2003;22:4933. doi: 10.1093/emboj/cdg479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hopmann KH, Himo F. J Chem Theory Comput. 2008;4:1129. doi: 10.1021/ct8000443. [DOI] [PubMed] [Google Scholar]
- 237.Lowden PAS. In: Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. Wiley-VCH; Weinheim: 2006. pp. 399–442. [Google Scholar]
- 238.Mao Y, Varoglu M, Sherman DH. Chem Biol. 1999;6:251. doi: 10.1016/S1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- 239.Zhao Q, He Q, Ding W, Tang M, Kang Q, Yu Y, Deng W, Zhang Q, Fang J, Tang G, Liu W. Chem Biol. 2008;15:693. doi: 10.1016/j.chembiol.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 240.Ogasawara Y, Liu H-w. J Am Chem Soc. 2009;131:18066. doi: 10.1021/ja907307h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Schroeder DR, Colson KL, Klohr SE, Zein N, Langley DR, Lee MS, Matson JA, Doyle TW. J Am Chem Soc. 1994;116:9351. [Google Scholar]
- 242.Norizuki Y, Komano K, Sato I, Hirama M. J Chem Soc, Chem Comm. 2008;42:5372. doi: 10.1039/b811355f. [DOI] [PubMed] [Google Scholar]
- 243.Hornemann U, Cloyd LC. J Chem Soc D: Chem Comm. 1971;7:301. [Google Scholar]
- 244.Hornemann U, Aikman MJ. J Chem Soc, Chem Comm. 1973;3:88. [Google Scholar]
- 245.Hornemann U, Kehrer JP, Nunez CS, Ranieri RL. J Am Chem Soc. 1974;96:320. doi: 10.1021/ja00808a087. [DOI] [PubMed] [Google Scholar]
- 246.Anderson MG, Kibby JJ, Rickards RW, Rothschild JM. J Chem Soc, Chem Comm. 1980;24:1277. [Google Scholar]
- 247.Fukuyama T, Xu L, Goto S. J Am Chem Soc. 1992;114:383. [Google Scholar]
- 248.Huang H, Pratum TK, Hopkins PB. J Am Chem Soc. 1994;116:2703. [Google Scholar]
- 249.Williams RM, Rajski SR, Rollins SB. Chem Biol. 1997;4:127. doi: 10.1016/s1074-5521(97)90256-8. [DOI] [PubMed] [Google Scholar]
- 250.Johnson DA, August PR, Shackleton C, Liu H-w, Sherman DH. J Am Chem Soc. 1997;119:2576. [Google Scholar]
- 251.Sheldon PJ, Johnson DA, August PR, Liu H-w, Sherman DH. J Bacteriol. 1997;179:1796. doi: 10.1128/jb.179.5.1796-1804.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sheldon PJ, Mao Y, He M, Sherman DH. J Bacteriol. 1999;181:2507. doi: 10.1128/jb.181.8.2507-2512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Varoglu M, Mao Y, Sherman DH. J Am Chem Soc. 2001;123:6712. doi: 10.1021/ja015646l. [DOI] [PubMed] [Google Scholar]
- 254.Corre C, Lowden PAS. J Chem Soc, Chem Comm. 2004:990. doi: 10.1039/b400093e. [DOI] [PubMed] [Google Scholar]
- 255.Liu C, Kelly GT, Watanabe CM. Org Lett. 2006;8:1065. doi: 10.1021/ol052987l. [DOI] [PubMed] [Google Scholar]
- 256.Bicker U, Fischer W. Nature. 1974;249:344. doi: 10.1038/249344a0. [DOI] [PubMed] [Google Scholar]