

Short abstract
In silico and experimental approaches have been used to identify the non-long terminal repeat retrotransposons of the urochordate Ciona intestinalis providing valuable data for understanding the evolution of early chordate genomes.
Abstract
Background
Non-long terminal repeat (non-LTR) retrotransposons have contributed to shaping the structure and function of genomes. In silico and experimental approaches have been used to identify the non-LTR elements of the urochordate Ciona intestinalis. Knowledge of the types and abundance of non-LTR elements in urochordates is a key step in understanding their contribution to the structure and function of vertebrate genomes.
Results
Consensus elements phylogenetically related to the I, LINE1, LINE2, LOA and R2 elements of the 14 eukaryotic non-LTR clades are described from C. intestinalis. The ascidian elements showed conservation of both the reverse transcriptase coding sequence and the overall structural organization seen in each clade. The apurinic/apyrimidinic endonuclease and nucleic-acid-binding domains encoded upstream of the reverse transcriptase, and the RNase H and the restriction enzyme-like endonuclease motifs encoded downstream of the reverse transcriptase were identified in the corresponding Ciona families.
Conclusions
The genome of C. intestinalis harbors representatives of at least five clades of non-LTR retrotransposons. The copy number per haploid genome of each element is low, less than 100, far below the values reported for vertebrate counterparts but within the range for protostomes. Genomic and sequence analysis shows that the ascidian non-LTR elements are unmethylated and flanked by genomic segments with a gene density lower than average for the genome. The analysis provides valuable data for understanding the evolution of early chordate genomes and enlarges the view on the distribution of the non-LTR retrotransposons in eukaryotes.
Background
The ascidian Ciona intestinalis has joined the select group of fully sequenced genomes [1]. The draft sequence shows interesting features of an invertebrate chordate: a genome size of 153-159 megabases (Mb); base composition of 65% AT; 15,852 predicted transcripts; and a gene density of one per 7.5 kilobases (kb). Ciona genome organization lies between that of protostomes (most animals other than echinoderms and chordates) and vertebrates. The released sequence allows new approaches to study the structure of the still poorly characterized repetitive DNA fraction, which accounts for 30-35% of the urochordate genome [2]. Although rRNA and tRNA families have been described, the different classes of transposable elements were not surveyed. Indeed, current information about ascidian transposable elements is limited to only 1 Mb of genomic sequences [3]. These elements are, however, invariably found in eukaryotes and most probably have contributed greatly to shaping the structure and function of vertebrate genomes [4].
Transposable elements are grouped into two major classes - class I and class II - depending on the mechanism of transposition [5,6]. Class I elements can be further classified into three categories: short interspersed nucleotide elements (SINEs); long terminal repeat (LTR) retrotransposons; and non-LTR retrotransposons (also termed LINE-like elements or retroposons). Although elements in the last category are among the most abundant, frequency estimates vary greatly depending on the species and the DNA segment considered, as most copies are 5'-truncated. Full-length non-LTR elements contain either one or two open reading frames (ORFs), all of them encode a reverse transcriptase, and some have additional motifs [7-10]. On the basis of the reverse transcriptase, non-LTR retrotransposons have been clustered into 14 different clades, the L1, L2, CR1, Rex1, RTE and R4 clades being the six major lineages present in vertebrates [11-15]. In contrast to vertebrates, our knowledge of LINE-like elements in other chordates is scanty: Cili-1 and Cili-2 [3] and BfCR1 [16] are the only non-LTR elements reported in nonvertebrate chordates. If, however, non-LTR clades originated before the divergence of the major animal phyla [11], urochordate and cephalochordate genomes should harbor representatives of these clades.
We have conducted an exhaustive search for non-LTR elements, initially on raw data and more recently on the draft genome, of the urochordate C. intestinalis. Phylogenetic analysis based on the reverse transcriptase domain showed that the ascidian elements grouped within five non-LTR clades. The structural features of the non-LTR elements, copy number, genome distribution and methylation status have been analyzed and inferences on the evolution of chordate genomes are presented.
Results
Non-LTR elements in the ascidian genome
Five consensus non-LTR retrotransposons, termed CiI, CiL1, CiL2, CiLOA and CiR2, were derived from five, five, six, five and five C. intestinalis scaffolds, respectively (Figures 1, 2 and see Additional data file 1). TBLASTX comparisons showed that the ascidian elements belonged to the I, LINE1, LINE2, LOA and R2 clades (E-values: 4e-69 with Biomphalaria glabrata (snail) BGR, 2e-89 with Nycticebus coucang (slow loris) L1, 2e-50 with Danio rerio CR1Dr2, e-146 with Aedes aegypti Lian, and e-106 with Drosophila melanogaster R2, respectively). CiL1.2 (Figure 2b), derived from five scaffolds, was another ascidian LINE1 element. It showed homology with the Xenopus laevis Tx1 retroelement (E-value: 2e-40) but was not further analyzed because it was significantly shorter than CiL1 (CiL1.2 only encompassed the reverse transcriptase region).
Figure 1.
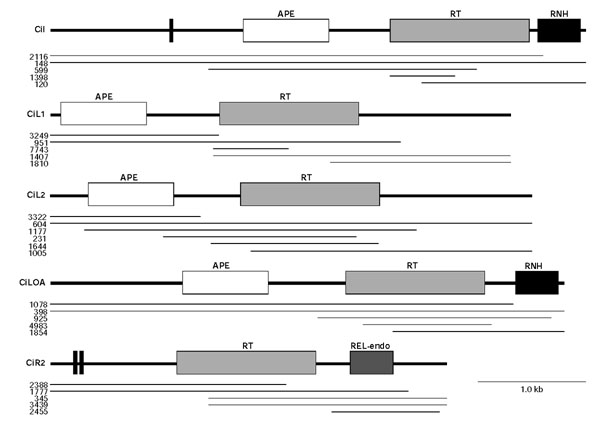
Schematic representation of the ascidian non-LTR retrotransposons. The conserved domains are depicted on the sequence derived from the Ciona scaffolds (thick line). Thin lines correspond to the physical segments covered by the scaffolds, which are numbered on the left. For clarity, indels are not shown. APE, apurinic/apyrimidinic endonuclease; REL-endo, restricted enzyme-like endonuclease; RNH, RNase H domain; RT, reverse transcriptase. Vertical bars indicate the location of cysteine-histidine motifs typical of nucleic acid-binding domains.
Figure 2.

Multiple-sequence alignments of the Ciona non-LTR retrotransposons. (a) The APE region. Only blocks of highly similar residues are shown. The Roman numerals above the alignments correspond to those defined by Tu et al. [17]. Highly conserved residues, as convenient landmarks, are shaded. (b) Reverse transcriptase sequences. Numbers above the sequences and the black-shaded residues refer to the conserved peaks described by Malik et al. [11]. Gray-shaded residues correspond to the CRE/R2/R4/L1/RTE and Tad/R1/LOA/Jockey/CR1/I subgroups described in [11]. (c) CiR2 domains. The CCHH and c-Myb DNA-binding motifs are shown in the amino-terminal domain and the REL-endo domain with the CCHC and KPDI motifs in the carboxy-terminal domain [18]. (d) CiLOA and CiI RNH domains. Only the highly conserved regions of the RNase H domain of the blocks defined by Tu et al. [17] are depicted. The three amino-acid residues identified in the active site of E. coli RNase H are shaded. (e) CCHC motif of the putative CiI-ORF1 as defined by Fawcett et al. [32].
All the Ciona elements encoded the conserved reverse transcriptase with the distinctive structural hallmarks defined as block 0, 1, 2, 2a, 3, 4, 5, 6 and 7 [11] (Figure 2b). Although conservation of the thumb region (block 8 and 9) was weak, preservation of the ascidian sequences defining the CRE/R2/R4/L1/RTE subgroup was still found. The apurinic/apyrimidic endonuclease (APE) region was clearly identified in CiI, CiL1, CiL2 and CiLOA (Figure 2a) on the basis of the reported domains I to VII [17]. CiI and CiLOA also contained the RNaseH (RNH) domain at the carboxylic end (Figures 1, 2d). Concerning ORF1, partial sequences were assembled for CiI and CiLOA, but a CCHC motif (single-letter amino-acid code) in this region was only identified in the CiI element (Figure 2e). Finally, for CiR2, a restriction enzyme-like endonuclease (REL-endo) containing the CCHC and KPDI motifs [18] was found in the carboxy-terminal region, and a CCHH domain and a putative c-Myb DNA-binding motif were identified at the amino terminus (Figure 2c). Overall, the structure and organization of the ascidian non-LTR retrotransposons is consistent with those reported for each non-LTR clade.
Phylogenetic relationships of Ciona retrotransposons
The reverse transcriptase domain of non-LTR elements was used to establish the phylogenetic relationships of the ascidian elements and the 14 reported non-LTR clades. In the neighbor-joining tree (Figure 3), all clades were supported with significant bootstrap values (≥70%), except clade I, with the lowest bootstrap value (67%) in agreement with previous analyses [11,12,14]. Therefore, ascidian sequences clustered within five distinct clades: I, L1, L2, LOA and R2 (bootstraps: 67%, 70%, 97%, 100% and 100%, respectively), as a result of which they were recorded as new members of such groups.
Figure 3.

Phylogenetic tree of non-LTR elements based on the reverse transcriptase sequence. The elements identified in C. intestinalis are indicated with an asterisk. The number next to each node of the 14 clades indicates the bootstrap value as the percentage out of 1,000 replicates. The name of each non-LTR element and the species harboring it is listed to the right of the figure, shaded in light gray (protostomes) or dark gray (deuterostomes).
Copy number and genomic features
Fragments of about 300 nucleotides of the reverse transcriptase domain of each ascidian element were PCR amplified, cloned, sequenced and used for copy-number estimations and methylation analyses. To quantify the copy number for each element, two independent experimental approaches - slot blot analysis and genomic library screenings (Figure 4a,b) - were combined with in silico scores on the number of Ciona scaffold-containing elements (Table 1). When the reverse transcriptase was considered, the data from the different approaches were consistent and mean values for each element were in the range 3-7, far below the copy numbers of the vertebrate counterparts (Table 2). CiR2 slot-blot analysis did not give a signal, in agreement with the low estimates obtained after in silico searches and library screenings. Indeed, full-length copies could not be assembled for any of the families after database searches. In silico estimates with sequences that also encompassed the 5' and 3' sequences of reverse transcriptase increased the numbers slightly: 9, 22, 24, 69 and 13 for CiI, CiL1, CiL2, CiLOA and CiR2, respectively (Table 1).
Figure 4.
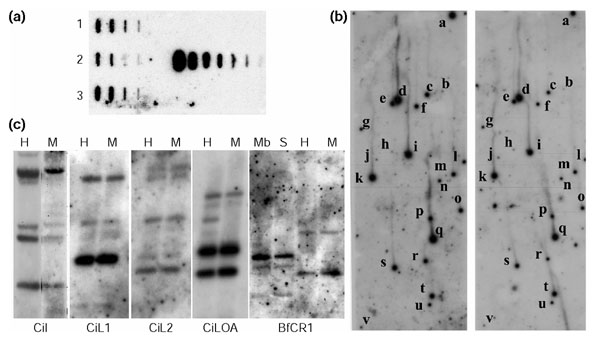
Slot-blot analysis, library screening and Southern blot of ascidian non-LTR elements. (a) On the left is shown a representative experiment of slot-blot analysis of CiLOA elements in three specimens with (from left to right) 500 ng, 250 ng, 50 ng and 25 ng of EcoRI-digested C. intestinalis genomic DNA. On the right is shown slot-blot analysis of serial dilutions of plasmid containing CiLOA which has been EcoRI-restricted and mixed with 1 μg mouse DNA. (b) Hybridization of a C. intestinalis genomic library screened with CiLOA. Positive signals have been depicted (from a to v) in the original (left) and its duplicate (right). (c) The first four panels show Southern analyses of 10 μg C. intestinalis genomic DNA digested with HpaII (H) or MspI (M) and probed with the non-LTR element indicated under each panel; the fifth panel shows 10 μg Branchiostoma floridae (amphioxus) genomic DNA digested with HpaII (H), MboI (Mb), MspI (M) or Sau3A (S) and probed with BfCR1.
Table 1.
Copy number of the Ciona non-LTR elements
| Total number* | Number based on reverse transcriptase | ||||
| Screening | Slot-blot | Databank | Average | ||
| CiI | 9 | 7.5 | 5 | 6 | 6 |
| CiL1 | 22 | 3 | 3 | 6 | 4 |
| CiL2 | 24 | 3 | 5 | 4 | 4 |
| CiLOA | 69 | 6 | 5 | 9 | 7 |
| CiR2 | 13 | 3 | - | 3 | 3 |
*In silico estimates based on all the sequence available.
Table 2.
Non-LTR retrotransposons in Ciona
| Deuterostomes | Protostomes | Other organisms‡ | ||
| Non-LTR clade | Ciona copy number* | Vertebrates (copy number in Fugu [15]) | Copy number in D. melanogaster [30] and A. gambiae [31]† | |
| CR1 | + (NF§) | + (NF, 152) | ||
| CRE | + (NF, NF) | |||
| I | + (6-9) | + (67, 19) | ||
| Jockey | + (392, 28) | |||
| L1 | + (4-22) | + (500) | +¶ | + |
| L2 | + (4-24) | + (6,500) | + (NF, NF) | |
| LOA | + (7-69) | + (18, 19) | ||
| NeSL1 | + (NF, NF) | |||
| R1 | + (130, 3¥) | |||
| R2 | + (3-13) | + (0, NF) | ||
| R4 | + (1,000) | + (NF, 2) | ||
| Rex1 | + (2,000) | + | ||
| RTE | + (2,300) | + (NF, 167) | ||
| Tad1 | + | |||
| Total copy number | 24-137 | 12,300 | 607, 390 | |
| Clade complexity** | Five in Ciona | Five in Fugu | Five in D. melanogaster and seven in A. gambiae | |
Gene density and GC content in the surrounding retrotransposon sequences was estimated from 26 10-kb regions flanking CiI, CiL1, CiL2 and CiLOA elements of 17 scaffolds. Overall, 16.5 genes were found in the 260 kb analyzed. Therefore, the average gene density (1 gene per 15.8 kb) was lower than that of the whole genome (1 gene per 7.5 kb) [1]. However, no differences were observed when the GC content of those segments (35.7%) was compared with the overall genomic value (35%). Concerning CiR2, our data confirmed the target specificity for rRNA genes associated with the REL-endo domain: 9 out of 13 CiR2s were indeed linked to rRNA sequences.
Finally, the methylation status of the genomic regions containing the elements was investigated by comparing the hybridization patterns of genomic DNA restricted with the methylation-sensitive enzyme HpaII, and the methylation-insensitive isoschizomer MspI. The identical HpaII and MspI patterns obtained for all the elements (except for CiR2, which gave no signal) supported the location of the ascidian elements in unmethylated genomic segments (Figure 4c).
Discussion
Ciona non-LTR retrotransposons
The analysis of non-LTR elements in the urochordate Ciona provides valuable data for understanding the evolution of early chordate genomes and enlarges the view of the distribution of the non-LTR clades in eukaryotes. The Ciona genome harbors: I, LOA and R2 elements, hitherto restricted to protostomes; L1 elements, formerly uncharacterized in invertebrates; and L2 elements, previously described in protostomes and vertebrates.
Clade I was the least supported branch of our analysis (bootstrap value, 67%). However, ascription of CiI to this clade was unambiguous as it shares with the other I elements the CCHC motif and the APE, reverse transcriptase and RNH domains (Figure 1), and also because it clearly clustered with the I non-LTR retrotransposon BGR of the snail Biomphalaria glabrata (bootstrap 93%). In regard to the LOA clade, representatives in urochordates had previously been identified after BLASTN and BLASTX comparisons. The ascidian Cili-2 retrotransposon gives the closest match with the RNH domain of the mosquito Lian element [3]. We have now derived CiLOA, an element that encodes APE, reverse transcriptase and RNH. The phylogenetic analysis of the reverse transcriptase domain together with the other structural hallmarks improved the assignment of the Ciona sequence to the LOA clade (bootstrap 100%). Finally, the phylogenetic analysis and structural features clearly placed CiR2 within the R2 clade (Figures 1-3). As well as the reverse transcriptase, the preservation in the deuterostome lineage of the distinctive R2 structural hallmarks, such as the REL-endo domain and the 5' CCHH and c-Myb DNA-binding motifs, indicate the ancient structural organization of this clade. Additionally, insertions of the element near the Ciona rRNA genes suggest that target specificity through the REL-endo mechanism has been preserved. Overall, not only does the analysis of CiI, CiLOA and CiR2 agree with the origin of these retrotransposons in the Precambrian era [11], but the fact that the urochordate elements resemble the protostome counterparts points to their ancient structural organization.
We derived CiL1 and CiL1.2, whose structural organization and phylogenetic relationship made them cluster within the L1 clade (bootstrap 70%) and supported a previous BLAST analysis of two short Ciona sequences [3]. Our data allowed the first structural characterization of the L1 clade in invertebrates. CiL2 clustered within the L2 clade (bootstrap 97%), a novel group of non-LTR retrotransposons closely related to the CR1 and Rex1 clades [14], which includes members previously described in the protostome and deuterostome lineages (Table 2). Interestingly, the CR1 and RTE clades, which are also shared by protostomes and deuterostomes, have not been identified in Ciona. Whether these clades were lost in the whole urochordate subphylum needs further investigation.
Retrotransposon frequency, genomic features and genome evolution
Sequence analysis of the scaffolds harboring the non-LTR elements revealed that ascidian transposable elements are flanked by regions of low gene density. However, no differences in GC content with respect to the average genome value were found when comparing these genomic segments. Moreover, Southern analysis showed that ascidian non-LTR retrotransposons are unmethylated. Overall, these data suggest that mobile elements, gene density and methylation status have not influenced the nucleotide composition in urochordates.
Copy-number estimates of the non-LTR elements in the Ciona genome suffer from slight inaccuracies due to the hybridization reaction, which disregards highly divergent elements, and to the fact that computational estimates only refer to the available 90% of the genome. However, the agreement between the in silico and experimental estimations indicates that, in this case, the biases have been minimized. The data show a low copy number per haploid genome of the different ascidian elements: from 9 to 69 copies, which decreased to three to seven copies when estimates were based on the reverse transcriptase domain only. These values are far below the vertebrate counterparts, but similar to numbers reported for protostome genomes. This also seems to apply to another lower chordate genome. In amphioxus (subphylum Cephalochordata) a low copy number has been reported for a non-LTR retrotransposon, BfCR1, [16] and for ATE1, a class II transposable element [19].
The factors involved in retrotransposon control are still an open question. The view that methylation evolved to suppress the activity of transposable elements in vertebrates [20] pointed to DNA methylation as a good candidate for transposition control in lower chordates. However, ascidian transposable elements are clearly unmethylated ([21] and this study) and, hence, the genome-defense model cannot be extended to urochordates, and perhaps not to cephalochordates, as the amphioxus BfCR1 element also belongs to the unmethylated genomic fraction (Figure 4c). Among other mechanisms, if required at all for retrotransposon control in lower chordates, co-suppression, which operates on I elements in Drosophila [22,23] and in transposable element silencing in plant genomes [24], is a possibility.
Conclusions
In summary, ascidian and amphioxus genomes do not harbor high copy numbers of retrotransposons. If this reflects the condition of the pre-duplicative genome of the ancestor of the vertebrates, substantial increases in the number of transposons in vertebrates could only have been attained after the large-scale duplications that provided the raw material to buffer the transposable element-induced genome rearrangements, and after the recruitment of methylation to control transposable element mobility. Therefore, beyond the extensive duplications occurring at the origin of the vertebrates, expansion of mobile elements linked to new roles for DNA methylation would have to be considered as significant factors in the modeling of the highly complex genomes.
Materials and methods
Non-LTR retrotransposons in the Ciona database
The C. intestinalis non-LTRs were identified through a TBLASTX [25] search on the Ciona genome draft deposited in the JGI database [26]. The following non-LTR retrotransposons were used as queries: CRE1 from Crithidia fasciculata (accession number M33009), CZAR from Trypanosoma cruzi (M62862), Dong from Bombyx mori (L08889), L1 from Rattus norvegicus (U83119), RTE1 from Caenorhabditis elegans (AF025462), Tad1 from Neurospora (L25662), R1 from D. melanogaster (X51968), Jockey from D. melanogaster (M22874), L1Tc from T. cruzi (X83098), R2 from Porcellio scaber (AF015818), LOA from Drosophila silvestris (X60177), Rex3 from Tetraodon nigroviridis (AJ312226), NeSL-1 from C. elegans (Z82058), CR1 from Gallus gallus (AAC60281) and Maui from Takifugu rubripes (AF086712). The retrieved Ciona sequences were aligned by eye on the basis of the DotPlot comparisons of the MegAlign program from the DNASTAR package, and a consensus composite was assembled. Sequence differences between scaffolds due to nucleotide substitutions or indels were analyzed and the sequence maximizing the similarity to reported elements was selected. The non-LTR nature of each composite sequence was further verified through a TBLASTX search against the GenBank database. The consensus sequence was named after the defined non-LTR clade to which it belonged. The CiI consensus sequence was derived from scaffolds 120, 148, 599, 1398 and 2116; CiL1 from 951, 1407, 1810, 3249 and 7743; CiL1.2 from 388, 890, 1138, 2278 and 2648; CiL2 from 231, 604, 1005, 1177, 1644 and 3322; CiLOA from 398, 925, 1078, 1854 and 4983; CiR2 from 345, 1777, 2388, 2455 and 3439.
The in silico copy number of each repetitive DNA element was estimated from the Ciona database. Two types of search were performed. First, all the derived sequences were used to retrieve scaffold-containing elements that matched with a BLAST expect value of <10-3 [15]. To discard wrongly assigned elements, a threshold was defined at the score value of the first match of an element that belonged to another clade. Second, for the sake of comparison with experimental data, only the consensus reverse transcriptase region was used for the search and the scaffolds showing a minimum match of 300 nucleotides with the same BLAST expect value were considered.
PCR amplification, cloning and sequence of non-LTR elements
PCR amplifications with primers designed from the consensus reverse transcriptase sequence of each identified ascidian non-LTR elements were performed with 250 pg of genomic DNA and 1 U Taq DNA polymerase (BioTherm) in 25 μl of reaction volume containing 0.2 μM for each primer, 32 μM each dNTP and 2 mM MgCl2. The sequences of the primers were: CiL1-F (forward): 5'-AACTAGTGATACCGCGCC-3', CiL1-R (reverse): 5'-ACACCTCGTTTGATCGG-3', CiL2-F: 5'-GTTGAGGTAAATGGCGC-3', CiL2-R: 5'-CGTTCGTCATTATCTGGG-3', CiR2-F: 5'-TTCCGCAAGGTCGATG-3', CiR2-R: 5'-CAGATAGGGCCCAATCC-3', CiI-F: 5'-CGATCTACCACCGACCAC-3', CiI-R: 5'-GCTTGTCACAGGCAGTTG-3', CiLOA-F: 5'-AACTGCGGAGATCCATGG-3' and CiLOA-R: 5'-GTCGCAGTCTTGATGCGG-3'. PCR conditions were as follows: the initial denaturation step at 94°C for 2 min was followed by 40 cycles at 94°C for 45 sec, 53°C for 30 sec and 72°C for 30 sec and a final extension step at 72°C for 5 min. In each PCR assay, a fragment of approximately 300 bp was amplified and then cloned in a pUC18 plasmid and sequenced using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) in a 3730 DNA Analyzer (Applied Biosystems).
Genomic library screenings, slot and Southern blot analyses
A C. intestinalis λZapII genomic library (kindly provided by M. Levine) was screened with each of the fragments of the identified Ciona non-LTR elements. The probes were labeled with [α-32P]dCTP by random-hexamer priming and hybridized to phage DNA transferred on Hybond-N nylon filters (Amersham Pharmacia Biotech, Uppsala, Sweden) in duplicate. Approximately 70,000 phages were screened. Hybridizations were performed in phosphate-SDS solution [27] at 65°C overnight. Two 15-min washes were performed at 65°C in 2 × SSC, 0.1% SDS, 2 × 15 min at 65°C in 1 × SSC, 0.1% SDS and 1 × 15 min at 65°C in 0.2 × SSC, 0.1% SDS. Hybridization signals were detected by autoradiography. Only the signals present in the original and duplicated filters were considered.
For quantitative slot-blot analysis, 500 ng, 250 ng, 50 ng and 25 ng of EcoRI-digested C. intestinalis genomic DNA and serial dilutions of each plasmid-containing probe, EcoRI-restricted and mixed with 1 μg mouse genomic DNA (as nonspecific DNA) were denatured with 0.4 M NaOH and 25 mM EDTA in a final volume of 200 μl and blotted on Hybond-N nylon filters (Amersham Pharmacia Biotech) with a slot-blot device (Minifold II, Schleicher & Schuell, Dassel, Germany). Three genomic DNA replicates of isolated animals were performed. Before sample loading, the membrane was soaked in water and then neutralized with 2 M sodium acetate, pH 5.4 and fixed with UV light. Membranes were hybridized with the same probes used for library screening at the same hybridization and washing conditions. The slot-blot signal was quantified with the GS525 Molecular Imager System (Bio-Rad, Hercules, CA).
For Southern analyses 10 μg of C. intestinalis genomic DNA digested with HpaII or MspI was resolved on 0.8% agarose gels and transferred to nylon membranes. Southern blots were hybridized with the same non-LTR probes used for library screening at identical hybridization and washing conditions.
Sequence and phylogenetic analyses
Gene density and GC content of the retrotransposon insertion sites was assessed. Only the scaffolds that expanded at least 10 kb upstream or downstream from an element were considered. Gene density in the 10-kb flanking regions was calculated by scoring the predicted genes according the Ciona gene model v1.0. When the gene sequence was only partially contained in the region analyzed, it was scored as 0.5.
For phylogenetic analysis the C. intestinalis sequences were added to a previous alignment by Malik [11], updated by adding the NeSL-1 (C. elegans, Z82058), LINE2 (Patella, X77618; Danio rerio, AL591210; Oryzias, AB054295; Fugu, AF086712) and Rex1 (Xiphophorus, AF155728; Batrachocottus, AAA83744) clades. The new alignment was generated using Clustal X [28], maintaining the same pairwise gap penalties and multiple alignment parameters, and adjusted by eye (see Additional data files 2 and 3). Phylogenetic analyses were performed using the neighbor-joining method, rooted with the reverse transcriptase sequence of Neurospora organellar group II intron (accession number S07649) and drawn with the TreeViewPPC program [29]. Confidence in each node was assessed by 1,000 bootstrap replicates.
Additional data files
The consensus DNA sequences of each derived Ciona non-LTR retrotransposon (Additional data file 1) and the reverse transcriptase alignment used to reconstruct the phylogenetic relationship with the non-LTR clades (Additional data file 2, Additional data file 3) are available with the online version of this article.
Supplementary Material
The consensus DNA sequences of each derived Ciona non-LTR retrotransposon
The reverse transcriptase alignment used to reconstruct the phylogenetic relationship with the non-LTR clades
The reverse transcriptase alignment used to reconstruct the phylogenetic relationship with the non-LTR clades
Acknowledgments
Acknowledgements
We thank G. Marfany for helpful discussion and R. Rycroft for revising the English. We also thank the Serveis Científico-Tècnics (UB) for DNA sequencing. This study was supported by the Ministerio de Educación y Cultura (grant BMC2000-0536 and BMC 2003-05211). J.P. was the recipient of an fellowship from the Universitat de Barcelona.
References
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002;298:2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- Schmidtke J, Epplen JT, Engel W. Genome analysis of Amphioxus and speculation as to the origin of contrasting vertebrate genome organization patterns. Comp Biochem Physiol. 1979;63:455–458. doi: 10.1016/0300-9629(79)90171-3. [DOI] [PubMed] [Google Scholar]
- Simmen MW, Bird A. Sequence analysis of transposable elements in the sea squirt, Ciona intestinalis. Mol Biol Evol. 2000;17:1685–1694. doi: 10.1093/oxfordjournals.molbev.a026267. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–220. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- Finnegan DJ. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989;5:103–107. doi: 10.1016/0168-9525(89)90039-5. [DOI] [PubMed] [Google Scholar]
- Finnegan DJ. Transposable elements. Curr Opin Genet Dev. 1992;2:861–867. doi: 10.1016/s0959-437x(05)80108-x. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990;9:3353–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Burch JB, Davis DL, Haas NB. Chicken repeat 1 elements contain a pol-like open reading frame and belong to the non-long terminal repeat class of retrotransposons. Proc Natl Acad Sci USA. 1993;90:8199–8203. doi: 10.1073/pnas.90.17.8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín F, Olivares M, López MC. Do non-long terminal repeat retrotransposons have nuclease activity? Trends Biochem Sci. 1996;21:283–285. doi: 10.1016/0968-0004(96)80839-5. [DOI] [PubMed] [Google Scholar]
- Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- Malik HS, Eickbush TH. NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics. 2000;154:193–203. doi: 10.1093/genetics/154.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff JN, Korting C, Schartl M. Multiple lineages of the non-LTR retrotransposon Rex1 with varying success in invading fish genomes. Mol Biol Evol. 2000;17:1673–1684. doi: 10.1093/oxfordjournals.molbev.a026266. [DOI] [PubMed] [Google Scholar]
- Lovsin N, Gubensek F, Kordi D. Evolutionary dynamics in a novel L2 clade of non-LTR retrotransposons in Deuterostomia. Mol Biol Evol. 2001;18:2213–2224. doi: 10.1093/oxfordjournals.molbev.a003768. [DOI] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Albalat R, Permanyer J, Cañestro C, Martínez-Mir A, Gonzàlez-Angulo O, Gonzàlez-Duarte R. The first non-LTR retrotransposon characterised in the cephalochordate amphioxus, BfCR1, shows similarities to CR1-like elements. Cell Mol Life Sci. 2003;60:803–809. doi: 10.1007/s00018-003-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Isoe J, Guzova JA. Structural, genomic, and phylogenetic analysis of Lian, a novel family of non-LTR retrotransposons in the yellow fever mosquito, Aedes aegypti. Mol Biol Evol. 1998;15:837–853. doi: 10.1093/oxfordjournals.molbev.a025989. [DOI] [PubMed] [Google Scholar]
- Burke WD, Malik HS, Jones JP, Eickbush TH. The domain structure and retrotransposition mechanism of R2 elements are conserved throughout arthropods. Mol Biol Evol. 1999;16:502–511. doi: 10.1093/oxfordjournals.molbev.a026132. [DOI] [PubMed] [Google Scholar]
- Cañestro C, Albalat R, Gonzàlez-Duarte R. Isolation and characterization of the first non-autonomous transposable element in amphioxus, ATE-1. Gene. [DOI] [PubMed]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/S0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Simmen MW, Leitgeb S, Charlton J, Jones SJ, Harris BR, Clark VH, Bird A. Nonmethylated transposable elements and methylated genes in a chordate genome. Science. 1999;283:1164–1167. doi: 10.1126/science.283.5405.1164. [DOI] [PubMed] [Google Scholar]
- Chaboissier MC, Bucheton A, Finnegan DJ. Copy number control of a transposable element, the I factor, a LINE-like element in Drosophila. Proc Natl Acad Sci USA. 1998;95:11781–11785. doi: 10.1073/pnas.95.20.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S, Gassama MP, Heidmann T. Taming of transposable elements by homology-dependent gene silencing. Nat Genet. 1999;21:209–212. doi: 10.1038/5997. [DOI] [PubMed] [Google Scholar]
- Flavell RB. Inactivation of gene expression in plants as a consequence of specific sequence duplication. Proc Natl Acad Sci USA. 1994;91:3490–3496. doi: 10.1073/pnas.91.9.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOE Joint Genome Institute http://www.jgi.doe.gov
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Bucheton A, Busseau I. A search for reverse transcriptase-coding sequences reveals new non-LTR retrotransposons in the genome of Drosophila melanogaster. Genome Biol. 2000;1:research0012.1–0012.15. doi: 10.1186/gb-2000-1-6-research0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Fawcett DH, Lister CK, Kellett E, Finnegan DJ. Transposable elements controlling I-R hybrid dysgenesis in D. melanogaster are similar to mammalian LINEs. Cell. 1986;47:1007–1015. doi: 10.1016/0092-8674(86)90815-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The consensus DNA sequences of each derived Ciona non-LTR retrotransposon
The reverse transcriptase alignment used to reconstruct the phylogenetic relationship with the non-LTR clades
The reverse transcriptase alignment used to reconstruct the phylogenetic relationship with the non-LTR clades