

Abstract
Previously, mutational analyses identified six disease-causing mutations in 24 amelogenesis imperfecta (AI) kindreds. We have since expanded the number of AI kindreds to 39, and performed mutation analyses covering the coding exons and adjoining intron sequences for the six proven AI candidate genes [amelogenin (AMELX), enamelin (ENAM), family with sequence similarity 83, member H (FAM83H), WD repeat containing domain 72 (WDR72), enamelysin (MMP20), and kallikrein-related peptidase 4 (KLK4)] and for ameloblastin (AMBN) (a suspected candidate gene). All four of the X-linked AI families (100%) had disease-causing mutations in AMELX, suggesting that AMELX is the only gene involved in the aetiology of X-linked AI. Eighteen families showed an autosomal-dominant pattern of inheritance. Disease-causing mutations were identified in 12 (67%): eight in FAM83H, and four in ENAM. No FAM83H coding-region or splice-junction mutations were identified in three probands with autosomal-dominant hypocalcification AI (ADHCAI), suggesting that a second gene may contribute to the aetiology of ADHCAI. Six families showed an autosomal-recessive pattern of inheritance, and disease-causing mutations were identified in three (50%): two in MMP20, and one in WDR72. No disease-causing mutations were found in 11 families with only one affected member. We conclude that mutation analyses of the current candidate genes for AI have about a 50% chance of identifying the disease-causing mutation in a given kindred.
Keywords: ameloblastin, amelogenin, enamelin, enamelysin, kallikrein-related peptidase 4
Amelogenesis imperfecta (AI) is a group of hereditary conditions featuring the pathological formation of dental enamel. By the narrow definition used here, the phenotype is limited to the dental apparatus and is not associated with more generalized defects (1). Enamel malformations are categorized as hypoplastic, hypocalcified, or hypomaturation types (2, 3), which reflect the timing of the developmental disturbance. During the secretory stage of amelogenesis, the enamel layer achieves its final dimensions by the lengthening of enamel crystals (4). Hypoplastic (thin) enamel results from secretory-stage pathologies. During the maturation stage the enamel layer hardens by the widening and thickening of the crystals deposited during the secretory stage (5). Hypomaturation enamel has normal thickness, but is soft and contains residual protein as a result of maturation-stage disturbances. Hypocalcified AI is the most severe form of AI and may be the result of pathology starting in the secretory stage and continuing through maturation. When mode of inheritance is included in the classification, 14 subtypes of AI are recognized (2, 3). The complexity of the AI aetiology is suggested by the diversity of its phenotypes and its multiple patterns of inheritance.
Dental enamel formation is a specialized process. Unravelling the mechanisms of dental enamel formation requires knowledge of all critical molecular participants. Determining the genes that cause syndromic and isolated AI provides this information. Identifying the genes that cause isolated AI narrows the focus to components that are most specialized for dental enamel formation. At the time of writing there were six proven candidate genes for AI: amelogenin (AMELX, Xp22.3-p22.1); enamelin (ENAM, 4q21); WD repeat containing domain 72 (WDR72, 15q21.3); family with sequence similarity 83, member H (FAM83H, 8q24.3); enamelysin (MMP20, 11q22.3–q23); and kallikrein-related peptidase 4 (KLK4, 19q13.4). The major secretory-stage enamel constituents (amelogenin, enamelin, ameloblastin, and MMP20) are thought to be specialized for tooth formation, as these genes degenerate in mammals that have lost, during evolution, the ability to make teeth or dental enamel (6–10).
At the Enamel VII conference (in 2005), we presented a paper that summarized the results of mutational analyses of 24 kindreds with isolated AI (11). At that time, only six of their disease-causing mutations had been identified. Since that report, new candidate genes for AI have been identified, and additional AI kindreds have been recruited. We now know the disease-causing mutations for 12 of the original 24 families and have ruled out the presence of coding region and splice junction mutations in the six known AI candidate genes plus ameloblastin (AMBN) in the families where the genetic cause could not be determined. Here we report the results of mutation analyses in the original 24 and 15 additional AI kindreds and discuss them in the context of new information concerning normal and pathological enamel formation.
Material and methods
The human study protocol and patient consents were reviewed and approved by the Institution Review Boards at the University of Michigan.
Fifteen families with isolated enamel defects were recruited for genetic studies. Mutational analyses for AMBN were conducted in all families. Based upon the enamel phenotype and pattern of inheritance, the most likely of the six proven candidate genes for AI (AMELX, ENAM, FAM83H, WDR72, MMP20, and KLK4) was selected and the coding exons and nearby intron sequences were amplified, using genomic DNA from each proband as the template. The amplification products were characterized by DNA sequencing and then other family members were tested to determine their genotype with respect to each sequence variation; however, typically there were too few people in each family for haplotype analyses. All subjects received an oral examination, and intra-oral photographs and dental radiographs were obtained.
In most cases, 10 ml of peripheral whole blood was obtained from participating family members. Alternatively, buccal swabs were performed. Genomic DNA was isolated using the QIAamp DNA Blood Maxi Kit and protocol (Qiagen, Valencia, CA, USA). Genomic DNA (50 ng) from affected individuals was amplified using the Platinum PCR Supermix (Invitrogen, Carlsbad, CA, USA), and the amplification products were purified using the QIAquick PCR Purification Kit and protocol (Invitrogen). The concentration of purified amplimer was estimated by the intensity of its ethidium bromide-stained band on a 1% agarose gel. The DNA-sequencing reactions used 1.0 pmol/µl of oligonucleotide primer and 3 ng/µl for each 1000 bp of amplification product, and were analyzed using an ABI Model 3700 DNA sequencer (Applied Biosystems, Foster City, CA, USA) at the University of Michigan DNA sequencing core. The primer pairs and PCR conditions for the amplification of the coding regions were as previously described for AMBN (11), AMELX (12), ENAM (13), FAM83H (14), WDR72 (15), KLK4 (16), and MMP20 (17).
Results
Original 24 AI kindreds
Disease-causing mutations in 12 of our original 24 AI kindreds (11) have been identified (Table 1). Two families showed an X-linked pattern of inheritance and both had AMELX mutations (12). Eleven families showed a dominant pattern of inheritance. Three of these families had ENAM mutations, and five had FAM83H defects. Five families showed recessive transmission. Six families had only a single affected individual (simplex pedigrees), so the pattern of transmission could not be determined. Simplex cases are usually recessive, but they can be the manifestations of de novo mutations with any inheritance pattern possible. Among the five families showing recessive transmission, we identified one disease-causing mutation in both alleles of MMP20 and one in both alleles of WDR72. Among the six simplex cases, a disease-causing mutation was identified in a single allele of FAM83H, so the inheritance pattern in this simplex case was actually autosomal dominant. In addition to the six proven AI candidate genes, mutational analyses for AMBN were performed for all of the probands, but no disease-causing mutations were identified in this gene.
Table 1.
Patterns of inheritance and amelogenesis imperfecta (AI) aetiologies of the 39 AI kindreds
| Not identified | Mutation identified | ||||||
|---|---|---|---|---|---|---|---|
| No. | Pattern | No. | Pattern | Gene | cDNA | Protein | References |
| Original 24 AI kindreds | |||||||
| 4 | Simplex | 1 | X-linked | AMELX | c.2T > C | p.M1T | 12 |
| 5 | Simplex | 2 | X-linked | AMELX | c.11G > C | p.W4S | 12 |
| 6 | ADAI | 3 | ARAI | WDR72 | c.1467_1468delAT | p.V491fsX497 | 15 |
| 9 | Simplex | 7 | ADAI | FAM83H | c.1330C > T | p.Q444X | 19 |
| 10 | ADAI | 8 | ADAI | FAM83H | c.1243G > T | p.E415X | 64 |
| 11 | ADAI | 12 | Simplex/AD | FAM83H | c.891T > A | p.Y297X | 64 |
| 14 | Simplex | 13 | ADAI | ENAM | c.(583−588 + 1)delG | p.R179-N196del | 13 |
| 16 | ARAI | 15 | ARAI | MMP20 | c.954−2A > T | IVS6-2A > T | 17 |
| 18 | ARAI | 17 | ADAI | ENAM | c.211−2A > C | p.M71_Q157del | 13 |
| 20 | Simplex | 19 | ADAI | ENAM | c.157A > T | p.K53X | 11 |
| 22 | ARAI | 21 | ADAI | FAM83H | c.1380G > A | p.W460X | 64 |
| 23 | ADAI | 24 | ADAI | FAM83H | c.2029C > T | pQ677X | 64 |
| Additional 15 kindreds | |||||||
| 25 | ADAI | ||||||
| 26 | ADAI | 33 | Simplex/AR | MMP20 | c.102G > A | p.W34X | 18 |
| 27 | Simplex | 34 | X-linked | AMELX | Gene deleted | p.0 | (here) |
| 28 | Simplex | 35 | ADAI | FAM83H | c.1192C > T | p.Q398X | 19 |
| 29 | Simplex | 36 | ADAI | ENAM | c.647G > T | p.S216L | 20 |
| 30 | Simplex | 37 | X-linked | AMELX | c.208C > A | p.P70T | (here) |
| 31 | Simplex | 38 | ADAI | FAM83H | c.2029C > T | p.Q677X | (here) |
| 32 | Simplex | 39 | ADAI | FAM83H | c.1354C > T | pQ452X | (here) |
A simplex inheritance pattern is when only one person in a kindred is affected, so the inheritance pattern appears to be recessive but could be a de novo or sporadic mutation. Note that one simplex case was found to be dominant and another was found to be recessive once the disease-causing mutation was identified.
AD, autosomal dominant; ADAI, autosomal-dominant amelogenesis imperfecta; AMELX, amelogenin; AR, autosomal recessive; ARAI, autosomal-recessive amelogenesis imperfecta; ENAM, enamelin; FAM83H, family with sequence similarity, member H; MMP20, enamelysin; WDR72, WD repeat containing domain 72.
Success in determining the causative mutation in these AI kindreds varied, depending upon the pattern of transmission. The disease-causing mutations were identified in both cases showing X-linked inheritance (100%). Among the 12 kindreds with dominant conditions (11 with a dominant pattern of inheritance and a dominant de novo mutation), disease mutations were characterized in eight (67%). Among the five recessive conditions, only two (40%) of the causative mutations were identified.
Additional AI kindreds
Fifteen additional AI kindreds were recruited and extensively, but not exhaustively, characterized. Of these additional kindreds, two were X-linked, six were dominant, and seven were simplex. Mutational analyses for the six AI candidate genes were prioritized based upon previously established genotype–phenotype correlations, and their transmission patterns were deduced from the pedigrees. Using this approach we were not able to identify a disease-causing mutation in eight of the 15 probands (Fig. 1). Among the seven remaining probands, four had novel mutations (Fig. 2), which included a nonsense mutation (p.W34X) in both alleles of MMP20 (18), a complete deletion of AMELX (H-C. Chan, unpublished data), a FAM83H nonsense (p.Q398X) mutation (19), and an ENAM compound heterozygote (p.S216L/p.422FsX448) (20). The final three AI kindreds in the new group were found to have previously identified disease-causing mutations: two had FAM83H nonsense mutations [p.Q677X (Fig. 3) and p.Q452X (Figs 4 and S1)], and one had an AMELX missense mutation [p.P70T (Figs 5, S2, and S3)]. Thus, in the 15 additional AI kindreds, both X-linked mutations (100%), four of the six dominant mutations (67%), and one of the seven simplex mutations (14%) were identified (Table 1).
Fig. 1.

Oral photographs of probands and pedigrees of eight amelogenesis imperfecta (AI) families of unknown aetiology. A dot on the pedigree marks subjects recruited in the study. Numbering starts with 25, as a continuation of previous work reporting mutational analyses of 24 AI kindreds (11). Pedigree analyses showed that families 25 and 26 have a dominant pattern of inheritance. Families 27–32 have either a recessive pattern of transmission or are de novo mutations.
Fig. 2.

Oral photographs of probands and pedigrees of four amelogenesis imperfecta (AI) families of known genetic aetiology. The proband of family 33 has autosomal-recessive hypomaturation AI, resulting from a p.W34X mutation in both enamelysin (MMP20) alleles (18). The proband of family 34 has X-linked dominant hypoplastic hypomaturation AI, resulting from a complete deletion of amelogenin (AMELX) (H-C. Chan, unpublished data). The proband of family 35 has autosomal-dominant hypocalcified AI caused by a p.Q398X mutation in one allele of family with sequence similarity 83, member H (FAM83H) (19). The proband of family 36 has a severe form of hypoplastic AI resulting from different mutations (p.422FsX448 and p.S216L) in both enamelin (ENAM) alleles (20). The father (p.422FsX448) and mother (p.S216L) had only one affected ENAM allele and both showed a very mild, but detectable, enamel phenotype.
Fig. 3.

Pedigree and sequencing chromatograms for family 37 [with the amelogenin (AMELX) missense mutation, p.P70T] and oral photographs of the proband’s affected mother. The pedigree is consistent with an X-linked pattern of inheritance. The AMELX DNA sequencing chromatogram shows a doublet of C and A (c.208C > A; arrowhead). (A) Maxillary occlusal, (B) mandibular occlusal, (C) frontal, (D) right buccal, (E) left buccal, (F) frontal/buccal views of mandibular teeth, and (G) occlusal/incisal views of mandibular teeth.
Fig. 4.

Pedigree and sequencing chromatograms for family 38 (FAM83H, p.Q677X) and oral photographs and radiographs of the proband. This 3-yr-old Caucasian boy showed hypocalcified amelogenesis imperfecta (AI) in his primary dentition. (A) Maxillary occlusal, (B) mandibular occlusal, (C) frontal, (D) left buccal, (E) right buccal, (F) radiographs, (G) DNA-sequencing chromatograms showing a C/T doublet demonstrating the c.2029C > T mutation in the proband, but not in the unaffected mother, and (H) a pedigree showing the autosomal-dominant pattern of inheritance (a dot marks each person recruited in the study).
Fig. 5.

Pedigree and DNA-sequencing chromatograms for family 39 (FAM83H, p.Q452X) and oral photographs of the 8-yr-old proband. The sequencing chromatogram shows a C/T doublet demonstrating the c.1354C > T mutation in one FAM83H allele. (A) maxillary occlusal, (B) mandibular occlusal, (C) frontal, (D) left buccal, and (E) right buccal. The phenotype in this Caucasian family is representative of autosomal-dominant hypocalcified amelogenesis imperfecta (ADHCAI).
Among the 39 AI kindreds characterized, disease-causing mutations were identified in 19, so screening a given AI kindred for mutations in the six proven candidate genes has about a 50% chance of success. AMELX mutations were identified in all four of the X-linked families (100%). Four ENAM and eight FAM83H mutations were identified among the 18 autosomal-dominant families (67%). One WDR72 and two MMP20 mutations were found among the six AI kindreds with known recessive conditions (50%). In addition, there were 11 simplex cases in which the disease-causing mutation could not be identified by mutational analyses of the known candidate genes.
Discussion
Screening the six proven AI candidate genes in kindreds with isolated AI has about a 50% chance of identifying the causative mutation, but the odds vary depending upon the pattern of inheritance. Next we discuss the findings of this study in the light of recent advances in our understanding of each form of AI according to its transmission pattern.
X-linked AI
About 5–10% of all AI cases are X-linked (21). Currently, 16 different AMELX mutations have been reported in subjects with X-linked AI (Table 2). If the AI is X-linked, a mutation in AMELX is predictably found, suggesting that no other genes on the X-chromosome are involved in its aetiology. In all 16 AMELX mutations, the phenotype was limited to the enamel layer, excepting an open bite, which is probably a secondary effect as it is observed in all forms of AI, regardless of which gene is defective.
Table 2.
Amelogenin (AMELX) disease-causing mutations
 | ||||
|---|---|---|---|---|
| Mutation no. | Gene | cDNA | Protein | References |
| 1 | g.1378T > C | c.2T > C | p.M1T | 12 |
| 2 | g.1387G > C | c.11G > C | p.W4S | 12 |
| 3 | g.1387G > A | c.11G > A | p.W4X | 65 |
| 4 | g.1390_1398del9 | c.14_22del9 | p.I5-A8delinsT | 66 |
| 5 | g.2525_7247del4723 | c.55-842_665del4723 | No protein | 67, 68 |
| 6 | g.4831C > T | c.152C > T | p.T51I | 67 |
| 7 | g.(4832_4834)delC | c.(152_155)delC | p.P52LfsX53 | 68, 69 |
| 8 | g.4834G > C | c.155C > G | p.P52R | 32, 70 |
| 9 | g.5157C > A | c.208C > A | p.P70T | 29–32 (here) |
| 10 | g.5179 | c.230A > T | p.H77L | 71 |
| 11 | g.(5333_5335)delC | c.(384_385)delC | p.H129fsX187 | 72 |
| 12 | g.(5367_5369)delC | c.(418_420)delC | p.Y141fsX187 | 73 |
| 13 | g.(5421_5422)delC | c.(472_473)delC | p.P158HfsX187 | 32, 67 |
| 14 | g.(5466_5477)delC | c.(517_518)delC | p.P173LfsX16 | 74 |
| 15 | g.(5487_5490)delC | c.(538_541)delC | p.L181CfsX187 | 71, 75 |
| 16 | g.5520G > T | c.571G > T | p.E191X. | 67 |
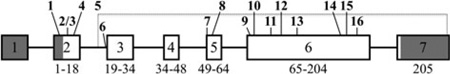
AMELX gene structure: numbered boxes indicate exons; introns are lines connecting the exons. Shaded exon regions are non-coding. The numbers below each exon show the range of amino acids encoded by it. Bold numbers indicate AMELX mutations. Mutation 5 is the deletion indicated by dashed lines. The gene numbers start from the first nucleotide of the AMELX reference sequence NG_012040.1. The cDNA numbers start from the translation initiation site of AMELX reference sequence NM_182680.1.
None of the reports that describe the dental phenotype resulting from defined AMELX mutations detected developmental abnormalities in the periodontium (cementum, periodontal ligament, and alveolar bone), despite the fact that such abnormalities would certainly have been diagnosed if they were part of the dental phenotype, given that dental examinations and radiographs are routinely used to characterize the dentition in patients with AI. In addition to there being no reports of developmental periodontal defects in individuals with AMELX mutations, there are solid data showing no, or only trace expression of amelogenin along developing tooth roots (22–24). These findings undermine the conclusions of an extensive and growing literature that describes amelogenin as a signalling molecule which serves important functions in the formation and healing of the periodontium.
As a rule, enamel malformations in X-linked AI are more severe in men than in women (who have a second copy of AMELX). Women often show vertical grooves in their enamel crowns. These are believed to result from alternating bands of ameloblasts secreting normal and defective amelogenin during amelogenesis, depending upon which X-chromosome had been inactivated by the individual cells that later developed into ameloblasts (2, 25). The severity of the enamel phenotype in women can be influenced by skewed X-inactivation, where a disproportionate number of cells inactivate the X-chromosome carrying the mutant, or alternatively, the normal allele (26). The enamel phenotype in subjects with AMELX defects generally correlates with the nature of the AMELX mutation (27, 28). The phenotype in our family with the p.P70T defect in AMELX is consistent with previous descriptions of hypomineralization/hypomaturation enamel with brown discolouration (Figs 5, S1, and S2) (29–32).
Although amelogenins comprise 90% of the enamel matrix, only 5–10% of cases AI are X-linked. Abundance of the protein product is not a factor in determining the relative importance of AMELX in the aetiology of AI, which is a collection of many diseases exhibiting isolated enamel malformations. The number of AI-causing genes relates to the number of genes specialized for dental enamel formation (i.e. a loss of function would result in isolated enamel defects) plus the number of genes that function in multiple places, but enamel formation is most sensitive to their absence or alteration. Even if we knew the number of genes involved in the aetiology of AI, their individual contributions (in percentages of the total number of cases) would vary significantly, depending upon the number of mutations in each gene that could cause AI, the chances of these mutations occurring, and whether the resulting phenotypes would be dominant or recessive. Identifying the pantheon of genes that cause AI, and understanding their pathogenesis and mode of inheritance will allow us to appreciate the contributions of individual genes to the aetiology of AI as a whole.
Autosomal-dominant AI
About 46% of our AI kindreds showed a dominant pattern of inheritance, which is a lower proportion than found in Sweden (~61%) (33). It is likely, however, that a few of the remaining 11 simplex cases will prove to be dominant mutations. Most of the autosomal-dominant AI (ADAI) in our kindreds (12 out of 18 cases) was caused by defects in ENAM or FAM83H.
To date, 11 different disease-causing ENAM mutations have been reported (Table 3). ENAM defects show a dose effect (34). When one allele is defective the phenotype can range from non-penetrant (35), to minor well-circumscribed pits or horizontal grooves (13, 20, 34, 36), to pronounced horizontal grooves (37) with generally very thin enamel (38, 39), to virtually no enamel (13). When both ENAM alleles are defective, there is extreme enamel hypoplasia or no enamel (20, 34, 35, 40). In all four reported cases where both ENAM alleles are defective, one or both alleles had the p.V422PfsX448 mutation. In one case where the enamel was extremely thin and smooth, there was also defective eruption and coronal resorption (40), which we hypothesize was caused by secondary pathology in the enamel organ. The ameloblast layer becomes increasingly pathological as amelogenesis progresses in the absence of a true enamel layer in the Enam null mice (41, 42).
Table 3.
Enamelin (ENAM) disease-causing mutations
 | ||||
|---|---|---|---|---|
| No. | Gene | cDNA | Protein | References |
| 1 | g.3139A > T | c.157A > T | p.K53X | 11, 37 |
| 2 | g.55636A > C | c.211−2A > C | p.M71_Q157del | 13 |
| 3 | g.7152G > A | c.534 + 1G > A | p.A158_Q178del | 76, 77 |
| 4 | g.9045A > G | c.535−2A > G | p.R179-N196del | 32 |
| 5 | g.9048G > T | c.536G > T | p.R179M | 78 |
| 6 | g.(9095–9101)delG | c.(583−588 + 1)delG | p.R179-N196del (or p.N197fsX277) | 13, 36, 38, 39) |
| 7 | g.13330 C > T | c.647G > T | p.S216L | 20 |
| 8 | g.13420C > A | c.737C > A | p.S246X | 34 |
| 9 | * | † | ‡ | 34 |
| 10 | g.13942_13943/insAG | c.1259_1230insAG | p.V422PfsX448 | 20, 34–36, 40, 79 |
| 11 | g.15674delT | c.2991delT | p.L998WfsX1062 | 35 |
| Both ENAM alleles affected | ||||
| No. | Allele 1 | Allele 2 | References | |
| 1 | p.S216L | p.V422PfsX448 | 20 | |
| 2 | p.V340_M341insSQYQYCV | p.V422PfsX448 | 34 | |
| 3 | p.V422PfsX448 | p.V422PfsX448 | 35, 40 | |
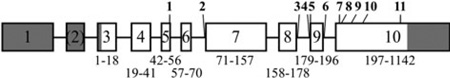
ENAM gene structure: numbered boxes indicate exons; introns are lines connecting the exons. The numbers below each exon show the range of amino acids encoded by it. Shaded exon regions are non-coding. Bold numbers indicate ENAM mutations. The gene numbers start from the first nucleotide of the ENAM reference sequence NG_013024.1. The cDNA numbers start from the translation initiation site of ENAM reference sequence NM_031889.2.
g.13703_13704insAGTCAGTACCAGTACTGTGTC;
c.1020_1021insAGTCAGTACCAGTACTGTGTC;
p.V340_M341insSQYQYCV
Although only recently discovered to be part of the aetiology of inherited enamel defects, FAM83H accounts for more cases of ADAI than any other gene. This was true for this study and also for another study that performed mutational analyses on 91 families with inherited enamel defects, 71 of which satisfied the classical criteria for non-syndromic AI (32). FAM83H encodes an intracellular protein of unknown function that appears to be associated with the Golgi apparatus or trans-Golgi network (19), and is most strongly expressed by pre-a-meloblasts (43). In the last few years, 18 different FAM83H disease-causing mutations have been reported (Table 4). All of the defects are missense or frameshift mutations located within a discrete region of the last exon that truncate the protein and apparently target it to the nucleus (44). The N-terminal region is the only part of the mutant proteins that is translated. This domain shares homology with the phospholipase D superfamily and is assumed to form dimers or to interact with another protein to cause dominant-negative effects in ameloblasts or their progenitors.
Table 4.
Family with sequence similarity 83, member H (FAM83H) disease-causing mutations
 | ||||
|---|---|---|---|---|
| No. | Gene | cDNA | Protein | References |
| 1 | g.5411C > A | c.860C > A | p.S287X | 80 |
| 2 | g. 5175T > A | c.891T > A | p.Y297X | 64 |
| 3 | g.5207_5208delTC | c.923_924delTC | p.L308fsX323 | 80 |
| 4 | g.5257C > T | c.973C > T | p.R325X | 14 |
| 5 | g.5476C > T | c.1192C > T | p.Q398X | 14, 19, 32, 81 |
| 6 | g.5528G > T | c.1243G > T | p.E415X | 64 |
| 7 | g.5573C > A | c.1289C > A | p.S430X | 32 |
| 8 | g.5614C > T | c.1330C > T | p.Q444X | 19, 81 |
| 9 | g.5638C > T | c.1354C > T | pQ452X | 82 (here) |
| 10 | g.5650C > T | c.1366C > T | p.Q456X | 81 |
| 11 | g.5658C > A | c.1374C > A | p.Y458X | 83 |
| 12 | g.5663G > A | c.1379G > A | p.W460X | 80 |
| 13 | g.5664G > A | c.1380G > A | p.W460X | 64 |
| 14 | g.5692G > A | c.1408C > T | p.Q470X | 80 |
| 15 | g.6156_6157delCC | c.1872_1873delCC | p.L625fsX703 | 80 |
| 16 | g.6277C > T | c.1993 C > T | p.Q665X | 44 |
| 17 | g.6313C > T | c.2029 C > T | p.Q677X | 32, 44, 64 (here) |
| 18 | g.6364G > T | c.2080G > T | p.E694X | 80 |
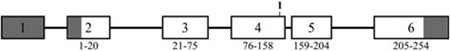
FAM83H gene structure: numbered boxes indicate exons; introns are lines connecting the exons. The numbers above each intron indicate the length of the intron in base pairs (bp). The numbers below each exon show the length of the exon in bp and below that the range of amino acids encoded by it. Shaded exon regions are non-coding. The 17 reported FAM83H missense or frameshift mutations are located between the sites marked 1 and 17 in bold. The gene numbers start from the first nucleotide of the FAM83H reference sequence NG_016652.1. The cDNA numbers start from the translation initiation site of FAM83H reference sequence NM_198488.3.
Defects in FAM83H cause autosomal-dominant hypocalcified AI (ADHCAI). The enamel layer has normal thickness in newly erupted teeth, but is rapidly lost by attrition following eruption and the teeth turn brown. Enamel that is not lost by attrition can sometimes persist indefinitely, such as islands of normal enamel. In this report we show the oral phenotypes of two new cases of hypocalcification AI caused by FAM83H defects. The first is the primary dentition of a Caucasian 3-yr-old boy with a p.Q677X mutation in FAM83H (Fig. 4). The second is a Caucasian family with a p.Q452X mutation (Figs 5, S2, and S3).
Some isolated cases of ADAI may come from defects in genes associated with syndromic AI. Collagen, type XVII, alpha 1 (COL17A1) contributes to the aetiology of junctional epidermolysis bullosa, a recessive disorder showing skin fragility, and AI (45). Mutations in a single allele of COL17A1 can cause isolated enamel defects (46, 47) or enamel defects combined with relatively minor skin fragility (48). Amelogenesis imperfecta can also occur along with nephrocalcinosis. Although nephrocalcinosis can be associated with impaired renal function, it is often asymptomatic and may go undiagnosed (49, 50). The genetic aetiology of this condition is currently unknown.
Autosomal-recessive AI
The diversity of enamel phenotypes in autosomal-recessive AI (ARAI) suggests that a large number of genes are involved in its aetiology (51). Only 10 mutations (in KLK4, MMP20, and WDR72) causing ARAI have been reported (Table 5). Consanguinity is a major contributor, as in all cases the same gene defect was observed in both alleles. Six of our 39 AI kindreds are known to have autosomal-recessive transmission patterns, and three of the causative mutations have been characterized in this group. We suspect that most of our simplex cases will turn out to be recessive conditions, as was determined for our kindred 33.
Table 5.
Autosomal-recessive amelogenesis imperfecta (ARAI)-causing mutations in kallikrein-related peptidase 4 (KLK4), enamelysin (MMP20), and WD repeat containing domain 72 (WDR72)
| No. | Gene | cDNA | Protein | References |
|---|---|---|---|---|
 | ||||
| KLK4 disease-causing mutation | ||||
| 1 | g.2143G > A | c.458G > A | p.W153X | 16, 32 |
 | ||||
| MMP20 disease-causing mutations | ||||
| 1 | g.115G > A | c.102G > A | p.W34X | 18 |
| 2 | g.16263T > A | c.678T > A | p.H226Q | 84 |
| 3 | g.18,755G > A | c.910G > A | p.A304T | 85 |
| 4 | g.30,574A > T | c.954-2A > T | IVS6-2ART | 17, 32 |
 | ||||
| WDR72 disease-causing mutations | ||||
| 1 | g.57,427_57,428delAT | c.1467_1468delAT | p.V491DfsX497 | 15, 32 |
| 2 | g.143805C > G | c.2348C > G | p.S783X | 59 |
| 3 | g.144143C > T | c.2686C > T | p.R897X | 86 |
| 4 | g.145982delA | c.2857delA | p.S976VX20 | 59 |
| 5 | g.150132G > A | c.2934G > A | p.W978X | 59 |
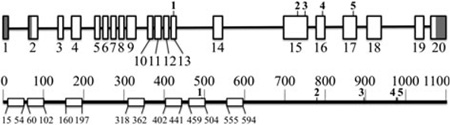
Exons are numbered boxes; introns are lines connecting the exons. The number below the intron indicates its length (in base pairs). The numbers below each exon show the range of amino acids encoded by it. Shaded exon regions are non-coding. Below the WDR72 gene diagram is the WDR72 protein (1104 amino acids) showing the seven WD repeats (boxes), with the range of amino acids indicated below each. Mutation sites are numbered in bold. The gene numbers start from the first nucleotide of the genomic reference sequences (KLK4, NG_012154.1; MMP20, NG_012151.1; WDR72, NG_017034.1). The cDNA numbers start from the translation initiation site (KLK4, NM_004917.3; MMP20, NM_004771.3; WDR72, NM_182758.2).
MMP20 is and KLK4 are secreted proteases that cleave amelogenin, ameloblastin, and enamelin in developing teeth (52). Cleavage of amelogenin reduces its affinity for hydroxyapatite (53). Mmp20 cleaves enamel proteins during the secrectory stage and the cleavage products accumulate in the matrix. Klk4 is expressed in the maturation stage and degrades the accumulated organic matrix to facilitate its reabsorption into ameloblasts (54). The enamel produced by Mmp20 and Klk4 null mice is soft and chips away after the teeth erupt into function (55–58). Based upon their known roles in enamel formation, it is not surprising that MMP20 and KLK4 defects cause AI.
WDR72 had no suspected role in enamel formation before it was shown, by genetic analyses, to cause AI (59). The gene is expressed by maturation-stage ameloblasts, and defects in both alleles cause hypomaturation AI. The teeth erupt with a creamy-brown colour, a rough texture, and reduced radiodensity when compared with normal enamel. Posteruptive changes vary, but the enamel can turn a deep orange-brown colour and undergo accelerated attrition, particularly on working surfaces. The five reported WDR72 disease-causing mutations were all truncation mutations in both alleles. Perhaps the deleted C-terminal domain performs an essential function in ameloblasts, although there are currently no data showing that the defective transcripts are translated into protein.
Aetiology of ARAI
The six proven candidate genes for AI account for about half of all AI cases (Table 1). Although the genes responsible for ADAI remain to be identified, less is known about the genetic aetiology of ARAI. So far, no AI-causing mutations have been identified in the genes encoding several proteins exported by ameloblasts, such as AMBN (60), amelotin (AMTN) (61), and odontogenic ameloblast-associated protein (ODAM) (62). We suspect that these genes are involved in ARAI. The characterization of large consanguineous AI kindreds will continue to yield discoveries, but the application of whole-exome sequencing, which involves fragmenting genomic DNA and capturing coding sequences by hybridization to a chip, followed by next-generation DNA sequencing, is likely to yield important information concerning the diverse genetic aetiology of ARAI. This technique has recently been used to discover that FAM20A is defective in subjects with AI and gingival hyperplasia syndrome (63).
Supplementary Material
Acknowledgements
We thank the family for their participation, and the Pediatric Dental Clinic at the University of Michigan for their cooperation. This investigation was supported by USPHS Research Grant DE015846 from the National Institute of Dental and Craniofacial Research (NIDCR), National Institutes of Health (NIH), Bethesda, MD 29892, and a Science Research Center grant to the Bone Metabolism Research Center (2010-0001741) funded by the Ministry of Education, Science, and Technology (MEST) of Korea.
Footnotes
Conflicts of interest – All authors declare that there are no potential conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Oral photographs and radiographs of the family 37 (AMELX, pP70T) proband at age 11.5.
Fig. S2. Oral photographs of the unaffected father (II:4) of family 37 (AMELX, pP70T).
Fig. S3. Oral photographs for family 39 (FAM83H, pQ452X).
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Witkop CJ., Jr Heterogeneity in inherited dental traits, gingival fibromatosis and amelogenesis imperfecta. South Med J. 1971;64 Suppl 1:16–25. doi: 10.1097/00007611-197102001-00005. [DOI] [PubMed] [Google Scholar]
- 2.Witkop CJ, Jr, Sauk JJ., Jr . Heritable defects of enamel. In: Stewart RE, Prescott GH, editors. Oral facial genetics. St. Louis: C.V. Mosby Co; 1976. pp. 151–226. [Google Scholar]
- 3.Witkop CJ., Jr Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1988;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 4.Simmer JP, Papagerakis P, Smith CE, Fisher DC, Rountrey AN, Zheng L, Hu JC. Regulation of dental enamel shape and hardness. J Dent Res. 2010;89:1024–1038. doi: 10.1177/0022034510375829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith CE. Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med. 1998;9:128–161. doi: 10.1177/10454411980090020101. [DOI] [PubMed] [Google Scholar]
- 6.Sire JY, Delgado SC, Girondot M. Hen’s teeth with enamel cap: from dream to impossibility. BMC Evol Biol. 2008;8:246. doi: 10.1186/1471-2148-8-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davit-Beal T, Tucker AS, Sire JY. Loss of teeth and enamel in tetrapods: fossil record, genetic data and morphological adaptations. J Anat. 2009;214:477–501. doi: 10.1111/j.1469-7580.2009.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meredith RW, Gatesy J, Murphy WJ, Ryder OA, Springer MS. Molecular decay of the tooth gene Enamelin (ENAM) mirrors the loss of enamel in the fossil record of placental mammals. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000634. e1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meredith RW, Gatesy J, Cheng J, Springer MS. Pseudogenization of the tooth gene enamelysin (MMP20) in the common ancestor of extant baleen whales. Proc Biol Sci. 2010;278:993–1002. doi: 10.1098/rspb.2010.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Hashimi N, Lafont AG, Delgado S, Kawasaki K, Sire JY. The enamelin genes in lizard, crocodile, and frog and the pseudogene in the chicken provide new insights on enamelin evolution in tetrapods. Mol Biol Evol. 2010;27:2078–2094. doi: 10.1093/molbev/msq098. [DOI] [PubMed] [Google Scholar]
- 11.Kim JW, Simmer JP, Lin BP, Seymen F, Bartlett JD, Hu JC. Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci. 2006;114 Suppl 1:3–12. doi: 10.1111/j.1600-0722.2006.00278.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim J-W, Simmer JP, Hu YY, Lin BP-L, Boyd C, Wright JT, Yamada CJM, Rayes SK, Feigal RJ, Hu JC-C. Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res. 2004;83:378–383. doi: 10.1177/154405910408300505. [DOI] [PubMed] [Google Scholar]
- 13.Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, Hu JC. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res. 2005;84:278–282. doi: 10.1177/154405910508400314. [DOI] [PubMed] [Google Scholar]
- 14.Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, Park JT, Seo BM, Hu JC, Simmer JP. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet. 2008;82:489–494. doi: 10.1016/j.ajhg.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SK, Seymen F, Lee KE, Kang HY, Yildirim M, Tuna EB, Gencay K, Hwang YH, Nam KH, De La Garza RJ, Hu JC, Simmer JP, Kim JW. Novel WDR72 mutation and cytoplasmic localization. J Dent Res. 2010;89:1378–1382. doi: 10.1177/0022034510382117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, Wright JT. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41:545–549. doi: 10.1136/jmg.2003.017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, Hu JC. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papagerakis P, Lin HK, Lee KY, Hu Y, Simmer JP, Bartlett JD, Hu JC. Premature stop codon in MMP20 causing amelogenesis imperfecta. J Dent Res. 2008;87:56–59. doi: 10.1177/154405910808700109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Y, Estrella MR, Hu YY, Chan HL, Zhang HD, Kim JW, Simmer JP, Hu JC. Fam83h is associated with intracellular vesicles and ADHCAI. J Dent Res. 2009;88:991–996. doi: 10.1177/0022034509349454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan HC, Mai L, Oikonomopoulou A, Chan HL, Richardson AS, Wang SK, Simmer JP, Hu JC. Altered enamelin phosphorylation site causes amelogenesis imperfecta. J Dent Res. 2010;89:695–699. doi: 10.1177/0022034510365662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backman B. Amelogenesis imperfecta – clinical manifestations in 51 families in a northern Swedish county. Scand J Dent Res. 1988;96:505–516. doi: 10.1111/j.1600-0722.1988.tb01590.x. [DOI] [PubMed] [Google Scholar]
- 22.Luo W, Slavkin HC, Snead ML. Cells from Hertwig’s epithelial root sheath do not transcribe amelogenin. J Periodontal Res. 1991;26:42–47. doi: 10.1111/j.1600-0765.1991.tb01624.x. [DOI] [PubMed] [Google Scholar]
- 23.Hu JC, Sun X, Zhang C, Simmer JP. A comparison of enamelin and amelogenin expression in developing mouse molars. Eur J Oral Sci. 2001;109:125–132. doi: 10.1034/j.1600-0722.2001.00998.x. [DOI] [PubMed] [Google Scholar]
- 24.Torres-Quintana MA, Gaete M, Hernandez M, Farias M, Lobos N. Ameloblastin and amelogenin expression in posnatal developing mouse molars. J Oral Sci. 2005;47:27–34. doi: 10.2334/josnusd.47.27. [DOI] [PubMed] [Google Scholar]
- 25.Berkman MD, Singer A. Demonstration of the lyon hypothesis in X-linked dominant hypoplastic amelogenesis imperfecta. Birth Defects Orig Artic Ser. 1971;7:204–209. [PubMed] [Google Scholar]
- 26.Hobson GM, Gibson CW, Aragon M, Yuan ZA, Davis-Williams A, Banser L, Kirkham J, Brook AH. A large X-chromosomal deletion is associated with microphthalmia with linear skin defects (MLS) and amelogenesis imperfecta (XAI) Am J Med Genet A. 2009;149A:1698–1705. doi: 10.1002/ajmg.a.32968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright JT, Hart PS, Aldred MJ, Seow K, Crawford PJ, Hong SP, Gibson CW, Hart TC. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 2003;44 Suppl 1:72–78. [PubMed] [Google Scholar]
- 28.Wright JT. The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am J Med Genet A. 2006;140:2547–2555. doi: 10.1002/ajmg.a.31358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collier PM, Sauk JJ, Rosenbloom SJ, Yuan ZA, Gibson CW. An amelogenin gene defect associated with human X-linked amelogenesis imperfecta. Arch Oral Biol. 1997;42:235–242. doi: 10.1016/s0003-9969(96)00099-4. [DOI] [PubMed] [Google Scholar]
- 30.Hart S, Hart T, Gibson C, Wright JT. Mutational analysis of X-linked amelogenesis imperfecta in multiple families. Arch Oral Biol. 2000;45:79–86. doi: 10.1016/s0003-9969(99)00106-5. [DOI] [PubMed] [Google Scholar]
- 31.Ravassipour DB, Hart PS, Hart TC, Ritter AV, Yamauchi M, Gibson C, Wright JT. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J Dent Res. 2000;79:1476–1481. [Google Scholar]
- 32.Wright JT, Torain M, Long K, Seow K, Crawford P, Aldred MJ, Hart PS, Hart TC. Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells Tissues Organs. 2011;194:279–283. doi: 10.1159/000324339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backman B, Holmgren G. Amelogenesis imperfecta: a genetic study. Hum Hered. 1988;38:189–206. doi: 10.1159/000153785. [DOI] [PubMed] [Google Scholar]
- 34.Ozdemir D, Hart PS, Firatli E, Aren G, Ryu OH, Hart TC. Phenotype of ENAM mutations is dosage-dependent. J Dent Res. 2005;84:1036–1041. doi: 10.1177/154405910508401113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang HY, Seymen F, Lee SK, Yildirim M, Tuna EB, Patir A, Lee KE, Kim JW. Candidate gene strategy reveals ENAM mutations. J Dent Res. 2009;88:266–269. doi: 10.1177/0022034509333180. [DOI] [PubMed] [Google Scholar]
- 36.Pavlic A, Petelin M, Battelino T. Phenotype and enamel ultrastructure characteristics in patients with ENAM gene mutations g.13185–13186insAG and 8344delG. Arch Oral Biol. 2007;52:209–217. doi: 10.1016/j.archoralbio.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Mårdh CK, Backman B, Holmgren G, Hu JC, Simmer JP, Forsman-Semb K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2) Hum Mol Genet. 2002;11:1069–1074. doi: 10.1093/hmg/11.9.1069. [DOI] [PubMed] [Google Scholar]
- 38.Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exonintron boundary. J Dent Res. 2002;81:738–742. doi: 10.1177/0810738. [DOI] [PubMed] [Google Scholar]
- 39.Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT. Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol. 2003;48:589–596. doi: 10.1016/s0003-9969(03)00114-6. [DOI] [PubMed] [Google Scholar]
- 40.Lindemeyer RG, Gibson CW, Wright TJ. Amelogenesis imperfecta due to a mutation of the enamelin gene: clinical case with genotype-phenotype correlations. Pediatr Dent. 2010;32:56–60. [PMC free article] [PubMed] [Google Scholar]
- 41.Hu JC, Hu Y, Smith CE, McKee MD, Wright JT, Yamakoshi Y, Papagerakis P, Hunter GK, Feng JQ, Yamakoshi F, Simmer JP. Enamel defects and ameloblast-specific expression in Enam knock-out/lacz knock-in mice. J Biol Chem. 2008;283:10858–10871. doi: 10.1074/jbc.M710565200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawada T, Sekiguchi H, Uchida T, Yamashita H, Shintani S, Yanagisawa T. Histological and immunohistochemical analyses of molar tooth germ in enamelin-deficient mouse. Acta Histochem. 2010;113:542–546. doi: 10.1016/j.acthis.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Lee MJ, Lee SK, Lee KE, Kang HY, Jung HS, Kim JW. Expression patterns of the Fam83h gene during murine tooth development. Arch Oral Biol. 2009;54:846–850. doi: 10.1016/j.archoralbio.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 44.Lee SK, Lee KE, Jeong TS, Hwang YH, Kim S, Hu JC, Simmer JP, Kim JW. FAM83H mutations cause ADHCAI and alter intracellular protein localization. J Dent Res. 2011;89:1378–1382. doi: 10.1177/0022034510389177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wright JT. Oral manifestations in the epidermolysis bullosa spectrum. Dermatol Clin. 2010;28:159–164. doi: 10.1016/j.det.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGrath JA, Gatalica B, Li K, Dunnill MG, McMillan JR, Christiano AM, Eady RA, Uitto J. Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol. 1996;148:1787–1796. [PMC free article] [PubMed] [Google Scholar]
- 47.Murrell DF, Pasmooij AM, Pas HH, Marr P, Klingberg S, Pfendner E, Uitto J, Sadowski S, Collins F, Widmer R, Jonkman MF. Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J Invest Dermatol. 2007;127:1772–1775. doi: 10.1038/sj.jid.5700766. [DOI] [PubMed] [Google Scholar]
- 48.Almaani N, Liu L, Dopping-Hepenstal PJ, Lovell PA, Lai-Cheong JE, Graham RM, Mellerio JE, McGrath JA. Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol. 2009;160:1094–1097. doi: 10.1111/j.1365-2133.2008.08977.x. [DOI] [PubMed] [Google Scholar]
- 49.Kirzioglu Z, Ulu KG, Sezer MT, Yuksel S. The relationship of amelogenesis imperfecta and nephrocalcinosis syndrome. Med Oral Patol Oral Cir Bucal. 2009;14:e579–e582. doi: 10.4317/medoral.14.e579. [DOI] [PubMed] [Google Scholar]
- 50.Martelli-Junior H, Santos Neto PE, Aquino SN, Santos CC, Borges SP, Oliveira EA, Lopes MA, Coletta RD. Amelogenesis imperfecta and nephrocalcinosis Syndrome: a case report and review of the literature. Nephron Physiol. 2011;118:p62–p65. doi: 10.1159/000322828. [DOI] [PubMed] [Google Scholar]
- 51.Nusier M, Yassin O, Hart TC, Samimi A, Wright JT. Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:220–230. doi: 10.1016/j.tripleo.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 52.Lu Y, Papagerakis P, Yamakoshi Y, Hu JC, Bartlett JD, Simmer JP. Functions of KLK4 and MMP-20 in dental enamel formation. Biol Chem. 2008;389:695–700. doi: 10.1515/BC.2008.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Z, Fan D, Fan Y, Du C, Moradian-Oldak J. Enamel proteases reduce amelogenin-apatite binding. J Dent Res. 2008;87:1133–1137. doi: 10.1177/154405910808701212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith CE, Richardson AS, Hu Y, Bartlett JD, Hu JC, Simmer JP. Effect of kallikrein 4 loss on enamel mineralization: comparison with mice lacking matrix metalloproteinase 20. J Biol Chem. 2011;286:18149–18160. doi: 10.1074/jbc.M110.194258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caterina JJ, Skobe Z, Shi J, Ding Y, Simmer JP, Birkedal-Hansen H, Bartlett JD. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002;277:49598–49604. doi: 10.1074/jbc.M209100200. [DOI] [PubMed] [Google Scholar]
- 56.Bartlett JD, Beniash E, Lee DH, Smith CE. Decreased mineral content in MMP-20 null mouse enamel is prominent during the maturation stage. J Dent Res. 2004;83:909–913. doi: 10.1177/154405910408301204. [DOI] [PubMed] [Google Scholar]
- 57.Simmer JP, Hu Y, Lertlam R, Yamakoshi Y, Hu JC. Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. J Biol Chem. 2009;284:19110–19121. doi: 10.1074/jbc.M109.013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simmer J, Hu Y, Richardson A, Bartlett J, Hu JC-C. Why does enamel in Klk4 null mice break above the dentino–enamel junction? Cells Tissues Organs. 2011;194:211–215. doi: 10.1159/000324260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, Al-Bahlani S, Al Harasi S, Kirkham J, Inglehearn CF, Mighell AJ. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 2009;85:699–705. doi: 10.1016/j.ajhg.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krebsbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada K, Yamada Y. Full-length sequence, localization, and chromosomal mapping of ameloblastin: a novel tooth-specific gene. J Biol Chem. 1996;271:4431–4435. doi: 10.1074/jbc.271.8.4431. [DOI] [PubMed] [Google Scholar]
- 61.Iwasaki K, Bajenova E, Somogyi-Ganss E, Miller M, Nguyen V, Nourkeyhani H, Gao Y, Wendel M, Ganss B. Amelotin – a Novel Secreted, Ameloblast-specific protein. J Dent Res. 2005;84:1127–1132. doi: 10.1177/154405910508401207. [DOI] [PubMed] [Google Scholar]
- 62.Moffatt P, Smith CE, Sooknanan R, St-Arnaud R, Nanci A. Identification of secreted and membrane proteins in the rat incisor enamel organ using a signal-trap screening approach. Eur J Oral Sci. 2006;114 Suppl 1:139–146. doi: 10.1111/j.1600-0722.2006.00318.x. [DOI] [PubMed] [Google Scholar]
- 63.O’Sullivan J, Bitu CC, Daly SB, Urquhart JE, Barron MJ, Bhaskar SS, Martelli-Junior H, Dos Santos Neto PE, Mansilla MA, Murray JC, Coletta RD, Black GC, Dixon MJ. Whole-exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet. 2011;88:616–620. doi: 10.1016/j.ajhg.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee SK, Hu JC-C, Bartlett JD, Lee KE, Lin BP-J, Simmer JP, Kim JW. Mutational spectrum of FAM83H: the C-terminal portion is required for tooth enamel calcification. Hum Mutat. 2008;29:E95–E99. doi: 10.1002/humu.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sekiguchi H, Kiyoshi M, Yakushiji M. DNA diagnosis of X-linked amelogenesis imperfecta using PCR detection method of the human amelogenin gene. Dent Japan. 2001;37:109–112. [Google Scholar]
- 66.Lagerstrom-Fermer M, Nilsson M, Backman B, Salido E, Shapiro L, Pettersson U, Landegren U. Amelogenin signal peptide mutation: correlation between mutations in the amelogenin gene (AMGX) and manifestations of X-linked amelogenesis imperfecta. Genomics. 1995;26:159–162. doi: 10.1016/0888-7543(95)80097-6. [DOI] [PubMed] [Google Scholar]
- 67.Lench NJ, Winter GB. Characterisation of molecular defects in X-linked amelogenesis imperfecta (AIH1) Hum Mutat. 1995;5:251–259. doi: 10.1002/humu.1380050310. [DOI] [PubMed] [Google Scholar]
- 68.Aldred MJ, Crawford PJ, Roberts E, Thomas NS. Identification of a nonsense mutation in the amelogenin gene (AMELX) in a family with X-linked amelogenesis imperfecta (AIH1) Hum Genet. 1992;90:413–416. doi: 10.1007/BF00220469. [DOI] [PubMed] [Google Scholar]
- 69.Lench NJ, Brook AH, Winter GB. SSCP detection of a nonsense mutation in exon 5 of the amelogenin gene (AMGX) causing X-linked amelogenesis imperfecta (AIH1) Hum Mol Genet. 1994;3:827–828. doi: 10.1093/hmg/3.5.827. [DOI] [PubMed] [Google Scholar]
- 70.Kida M, Sakiyama Y, Matsuda A, Takabayashi S, Ochi H, Sekiguchi H, Minamitake S, Ariga T. A novel missense mutation (p.P52R) in amelogenin gene causing X-linked amelogenesis imperfecta. J Dent Res. 2007;86:69–72. doi: 10.1177/154405910708600111. [DOI] [PubMed] [Google Scholar]
- 71.Hart PS, Aldred MJ, Crawford PJ, Wright NJ, Hart TC, Wright JT. Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol. 2002;47:261–265. doi: 10.1016/s0003-9969(02)00003-1. [DOI] [PubMed] [Google Scholar]
- 72.Sekiguchi H, Alaluusua S, Minaguchi K, Yakushiji M. A new mutation in the amelogenin gene causes X-linked amelogenesis imperfecta. J Dent Res. 2001;80:617. [Google Scholar]
- 73.Greene SR, Yuan ZA, Wright JT, Amjad H, Abrams WR, Buchanan JA, Trachtenberg DI, Gibson CW. A new frameshift mutation encoding a truncated amelogenin leads to X-linked amelogenesis imperfecta. Arch Oral Biol. 2002;47:211–217. doi: 10.1016/s0003-9969(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 74.Lee K-E, Lee S-K, Jung S-E, Song SJ, Cho SH, Lee ZH, Kim J-W. A novel mutation in the AMELX gene and multiple crown resorptions. Eur J Oral Sci. 2011;119 Suppl. 1:324–328. doi: 10.1111/j.1600-0722.2011.00858.x. [DOI] [PubMed] [Google Scholar]
- 75.Kindelan SA, Brook AH, Gangemi L, Lench N, Wong FS, Fearne J, Jackson Z, Foster G, Stringer BM. Detection of a novel mutation in X-linked amelogenesis imperfecta. J Dent Res. 2000;79:1978–1982. doi: 10.1177/00220345000790120901. [DOI] [PubMed] [Google Scholar]
- 76.Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 2001;10:1673–1677. doi: 10.1093/hmg/10.16.1673. [DOI] [PubMed] [Google Scholar]
- 77.Urzua OB, Ortega PA, Rodriguez ML, Morales BI. Genetic, clinical and molecular analysis of a family affected by amelogenesis imperfecta. Rev Med Chil. 2005;133:1331–1340. doi: 10.4067/s0034-98872005001100009. [DOI] [PubMed] [Google Scholar]
- 78.Gutierrez SJ, Chaves M, Torres DM, Briceno I. Identification of a novel mutation in the enamalin gene in a family with autosomal-dominant amelogenesis imperfecta. Arch Oral Biol. 2007;52:503–506. doi: 10.1016/j.archoralbio.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 79.Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, Ozdemir D, Firatli S, Aren G, Firatli E. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003;40:900–906. doi: 10.1136/jmg.40.12.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wright JT, Frazier-Bowers S, Simmons D, Alexander K, Crawford P, Han ST, Hart PS, Hart TC. Phenotypic variation in FAM83H-associated amelogenesis imperfecta. J Dent Res. 2009;88:356–360. doi: 10.1177/0022034509333822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hart PS, Becerik S, Cogulu D, Emingil G, Ozdemir-Ozenen D, Han ST, Sulima PP, Firatli E, Hart TC. Novel FAM83H mutations in Turkish families with autosomal dominant hypocalcified amelogenesis imperfecta. Clin Genet. 2009;75:401–404. doi: 10.1111/j.1399-0004.2008.01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hyun HK, Lee SK, Lee KE, Kang HY, Kim EJ, Choung PH, Kim JW. Identification of a novel FAM83H mutation and microhardness of an affected molar in autosomal dominant hypocalcified amelogenesis imperfecta. Int Endod J. 2009;42:1039–1043. doi: 10.1111/j.1365-2591.2009.01617.x. [DOI] [PubMed] [Google Scholar]
- 83.El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. Ultrastructural analyses of deciduous teeth affected by hypocalcified amelogenesis imperfecta from a family with a novel Y458X FAM83H nonsense mutation. Cells Tissues Organs. 2010;191:235–239. doi: 10.1159/000252801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ozdemir D, Hart PS, Ryu OH, Choi SJ, Ozdemir-Karatas M, Firatli E, Piesco N, Hart TC. MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J Dent Res. 2005;84:1031–1035. doi: 10.1177/154405910508401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee SK, Seymen F, Kang HY, Lee KE, Gencay K, Tuna B, Kim JW. MMP20 hemopexin domain mutation in amelogenesis imperfecta. J Dent Res. 2010;89:46–50. doi: 10.1177/0022034509352844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. Hypomaturation amelogenesis imperfecta due to WDR72 mutations: a novel mutation and ultrastructural analyses of deciduous teeth. Cells Tissues Organs. 2011;194:60–66. doi: 10.1159/000322036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.