

Abstract
The γ134.5 protein of herpes simplex viruses (HSV) is essential for viral pathogenesis, where it precludes translational arrest mediated by double-stranded-RNA-dependent protein kinase (PKR). Paradoxically, inhibition of PKR alone is not sufficient for HSV to exhibit viral virulence. Here we report that γ134.5 inhibits TANK binding kinase 1 (TBK1) through its amino-terminal sequences, which facilitates viral replication and neuroinvasion. Compared to wild-type virus, the γ134.5 mutant lacking the amino terminus induces stronger antiviral immunity. This parallels a defect of γ134.5 for interacting with TBK1 and reducing phosphorylation of interferon (IFN) regulatory factor 3. This activity is independent of PKR. Although resistant to IFN treatment, the γ134.5 amino-terminal deletion mutant replicates at an intermediate level between replication of wild-type virus and that of the γ134.5 null mutant in TBK1+/+ cells. However, such impaired viral growth is not observed in TBK1−/− cells, indicating that the interaction of γ134.5 with TBK1 dictates HSV infection. Upon corneal infection, this mutant replicates transiently but barely invades the trigeminal ganglia or brain, which is a difference from wild-type virus and the γ134.5 null mutant. Therefore, in addition to PKR, γ134.5 negatively regulates TBK1, which contributes viral replication and spread in vivo.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infects epithelial cells of mucosal tissues and establishes latency in the trigeminal ganglia (TG). Upon primary infection or reactivation, viral gene expression, DNA replication, and maturation ensue, which can lead to pathological conditions, including ocular lesions and encephalitis (48). Although multiple factors are involved, the γ134.5 protein encoded by HSV-1 plays a key role in this complex process (9, 28). HSV-1 γ134.5 facilitates viral replication in the central nervous system and causes encephalitis in experimental animal models (2, 9, 28). It also dictates viral replication in the peripheral tissues and subsequent spread to the central nervous system (29, 34, 47). Similarly, the γ134.5 protein from HSV-2 serves as an essential factor in viral pathogenesis (27). It is thought that these phenotypes are related to the capacity of γ134.5 to cope with type I interferon (IFN) responses (5, 17, 18, 25).
Type I IFN is a family of cytokines which are produced in response to viral infection. In HSV-infected cells, several mechanisms operate to induce antiviral immunity in a cell-type- and time-dependent manner (37). For example, Toll-like receptor 3 (TLR3) senses HSV-1 and stimulates IFN expression (50) (Fig. 1). In the cytoplasm, RNA helicases, retinoid acid-inducible gene I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5) recognize viral 5′-triphosphate RNA or double-stranded RNA (dsRNA) (30, 36). Additionally, intracellular DNA sensors, such as the DNA-dependent activator of interferon regulatory factor (DAI), RNA polymerase III, and interferon-inducible protein 16 (IFI16), detect HSV-1 in infected cells (7, 42, 44). Small interfering RNA (siRNA) knockdown of DAI as well as IFI16 or treatment with an RNA polymerase III inhibitor reduces cytokine induction by HSV-1 but not RNA viruses. Consistently, HSV oligonucleotide-induced IFN expression is diminished when introduced into cells devoid of IFI16. Although recognizing different pathogen-associated molecular patterns, TLRs and cytosolic receptors relay signals to TANK-binding kinase 1 (TBK1), which phosphorylates interferon regulatory factor 3/7 (IRF3/7) and induces interferon-stimulated genes (ISGs), chemokines, and alpha/beta IFN (IFN-α/β) (43). Of note, IFN-α/β upregulates a spectrum of antiviral molecules through the JAK/STAT pathway (39, 41). Among them is the dsRNA-dependent protein kinase (PKR), which is normally present at basal levels in normal cells. Upon activation by viral dsRNA, PKR phosphorylates the α subunit of translation initiation factor eIF-2 (eIF-2α) and thereby inhibits viral replication (Fig. 1).
Fig 1.

An outline of type I IFN responses. Upon virus infection, TLR3 on the endosomal membrane, RIG-I, MDA5, and DNA sensors in the cytoplasm are available to detect viral nucleic acids. These sensors transmit signals to TBK1 via multiple adaptor proteins; TBK1 subsequently phosphorylates transcription factor IRF-3/7, leading to production of IFN-α/β, RANTES, and ISGs. Secreted IFN-α/β acts in an autocrine or paracrine manner, where it induces the expression of a wide array of ISGs. Collectively, these gene products promote the establishment of an antiviral state. PKR, which is constitutively expressed in normal cells, is also upregulated by IFN-α/β. Once bound to viral dsRNA, PKR is activated to phosphorylate eIF2α and shuts off protein synthesis.
The γ134.5 gene is located in the inverted repeats of the HSV genome flanking the unique long sequence and is present in two copies per genome (11). In HSV-infected cells, onset of viral DNA replication activates PKR, which induces the translational arrest (8, 10). As a countermeasure, γ134.5 recruits protein phosphatase 1 (PP1) via its carboxyl terminus to dephosphorylate eIF-2α. This activity is linked to viral resistance to interferon-α/β and HSV pathogenesis (17, 18, 45). It has been reported that the γ134.5 null mutant is virulent in PKR knockout mice but not in wild-type mice (9, 26). However, a γ134.5 null mutant with a secondary mutation in the Us11 promoter inhibits PKR but remains avirulent (31, 32). Similarly, a γ134.5 null mutant with a secondary mutation elsewhere partially restores virulence (3). Thus, HSV γ134.5 may have an additional function(s) required for viral infection or virulence, which cannot be compensated simply by inhibition of PKR. In a search for HSV functions, we recently noted that γ134.5 targets TBK1 (46). When ectopically expressed, the amino terminus of γ134.5 associates with and inhibits TBK1. Nevertheless, its physiological role relevant to HSV infection remains to be established. We hypothesize that the amino terminus of γ134.5 promotes viral virulence by interfering with TBK1. Here we report that γ134.5 blocks the induction of antiviral genes independently of PKR in HSV-infected cells. Notably, γ134.5 overcomes TBK1-mediated restriction of HSV replication via its amino-terminal domain. Furthermore, we demonstrate that this domain is crucial to facilitate viral replication and spread to the central nervous system in vivo. Thus, regulation of TBK1 by γ134.5 represents a key step that determines the outcome of HSV infection.
MATERIALS AND METHODS
Mice, cells, and viruses.
BALB/c mice were purchased from Harlan Sprague Dawley Inc. and housed under specific-pathogen-free conditions in biosafety level 2 containment. Groups of 6-week-old mice were selected for this study. Experiments were performed in accordance with the guideline of the University of Illinois at Chicago. Vero and 293T cells were obtained from the American Type Culture Collection and propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). TBK1+/+ and TBK1−/− mouse embryonic fibroblasts (MEFs) were gifts from Wen-Chen Yeh. PKR−/− MEFs were gifts from Bryan Williams. All cells were propagated in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HSV-1(F) is a prototype HSV-1 strain used in this study (13). In recombinant virus R3616, a 1-kb fragment from the coding region of the γ134.5 gene was deleted (9). In recombinant virus H1001, the region encoding amino acids 1 to 146 was deleted. This was constructed by homologous recombination as described previously (17, 23). In recombinant virus H1002, the deleted coding region was repaired with wild-type γ134.5. Preparation of viral stock and titration of infectivity were carried out with Vero cells.
Plasmids and reporter assays.
The FLAG-γ134.5 and FLAG-ΔN146 plasmids were constructed by inserting PCR-amplified fragments into the BamHI and XhoI sites of pcDNA3. To construct glutathione S-transferase (GST)-IRF3, a DNA fragment encoding amino acids 380 to 427 from IRF3 was ligated into the BamHI and EcoRI sites of pGEX4-T1. To construct HA-TBK1, the TBK1 insert was PCR amplified and cloned into the BamHI and XhoI sites of pcDNA3. Plasmids HA-γ134.5, pTK-Luc, IFNB, and pISG56-Luc were described elsewhere (46). Reporter assays were carried out as described previously (46).
Viral infections.
Cells were infected with viruses at appropriate multiplicities of infection (MOI). At various time points, virus yields were determined on Vero cells (5). For interferon assays, cells were untreated or treated with mouse alpha interferon (250 units/ml; Sigma) for 20 h. Cells were then infected with viruses at a MOI of 0.05. Samples were harvested at 48 h postinfection, and viruses were released by three cycles of freezing and thawing and then titrated on Vero cells. For in vivo analysis, groups of mice were inoculated with 4 × 105 PFU of each virus by corneal scarification as described previously (45). At different time points after infection, tissues were collected from sacrificed mice and subjected to titration on Vero cells or immunohistochemistry analysis (45).
Quantitative real-time PCR assay.
Cells were mock infected or infected with viruses at 5 PFU per cell in serum-free DMEM. At 1 h after infection, cells were grown in DMEM with 1% fetal bovine serum. At the indicated time points, total RNA was harvested from cells using an RNeasy kit (Qiagen) and subjected to DNase I digestion (New England BioLabs). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using an Applied Biosystems ABI Prism 7900HT instrument with ABI SYBR green master mix (Applied Biosystems), and results were normalized to endogenous control 18S rRNA as follows: ΔCT(normalized gene expression) = CT(target gene) − CT(18S rRNA). Relative expression was calculated using the formula 2−ΔΔCT, where ΔΔCT = ΔCT (normalized gene expression at different time points) − ΔCT (normalized gene expression at 0 h), expressed as arbitrary units. Primers for each gene were chosen according to the recommendation of the qPrimerDepot database (12). Primer sequences were as follows: mouse IFN-β, AATTTCTCCAGCACTGGGTG and AGTTGAGGACATCTCCCACG; mouse ISG54, GCAAGATGCACCAAGATGAG and CACTCTCCAGGCAACCTCTT; mouse ISG56, CAAGGCAGGTTTCTGAGGAG and zAAGCAGATTCTCCATGACCTG; mouse RANTES, CTGCTGCTTTGCCTACCTCT and CACTTCTTCTCTGGGTTGGC; 18s rRNA, CCTGCGGCTTAATTTGACTC and AACCAGACAAATCGCTCCAC.
Western blot and immunoprecipitation analyses.
To analyze protein expression, cells were harvested and resuspended in disruption buffer containing 50 mM Tris-HCl (pH 7.0), 5% 2-mercaptoethanol, 2% SDS, and 2.75% sucrose. Samples were subjected to electrophoresis and reacted with antibodies against β-actin (Sigma), FLAG (Sigma), HA (Santa Cruz Biotechnology), IRF3 (Santa Cruz Biotechnology), phosphorylated IRF3 (Ser396) (Cell Signaling Technology, Inc.), ICP27 (Virusys Inc.), and γ134.5, respectively. The membranes were rinsed in phosphate-buffered saline and reacted with the corresponding secondary antibody conjugated to horseradish peroxidase. Protein bands were detected by enhanced chemiluminescence (Amersham Biosciences). To examine protein interactions, 293T cells were transfected with the indicated amounts of pcDNA3, HA-TBK1, FLAG-γ134.5, and FLAG-ΔN146. At 40 h after transfection, cells were harvested and lysed in 50 mM Tris-HCl (pH 7.4) buffer containing 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride, 1 μg/ml aprotinin-leupeptin-pepstatin, 1 mM Na3VO4, and 1 mM NaF. Lysates were incubated overnight at 4°C with anti-HA antibody (Applied Biological Materials Inc.) plus protein A/G-agarose beads (Santa Cruz Biotechnology). Immunocomplexes were subjected to electrophoresis and immunoblotting analysis.
Kinase assays.
293T cells were transfected with pcDNA3, FLAG-TBK1, FLAG-ΔN146, and HA-γ134.5. At 40 h after transfection, cell lysates were prepared in 20 mM Tris-HCl (pH 7.4) containing 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 50 mM sodium glycerophosphate, 20 mM sodium pyrophosphate, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM Na3VO4, and 5 mM benzamidine. TBK1 was immunoprecipitated with anti-HA antibody (Applied Biological Materials Inc.) plus protein A/G agarose beads (Santa Cruz Biotechnology). Immunocomplexes captured on the beads were incubated with recombinant GST-IRF3(380–427) for 20 min at 30°C in 25 mM HEPES buffer (pH 7.5) containing 10 mM MgCl2, 25 mM sodium–β-glycerophosphate, 5 mM benzamidine, 1 mM Na3VO4, 0.5 mM dithiothreitol, and 100 μM ATP (46). Samples were subjected to electrophoresis and immunoblotting analysis with rabbit anti-phospho-IRF3 (Ser396).
Immunohistochemistry analysis.
Tissue sections were deparaffinized with xylene and rehydrated through a series of graded ethanol. Endogenous peroxidase activity was quenched using a 0.3% H2O2-methanol bath followed by several washes with phosphate-buffered saline. HSV-1 antigens were detected with antibody against HSV-1 (Dako) as described previously (45). Tissue sections were incubated with primary antibody prior to the addition of biotinylated anti-rabbit immunoglobulin secondary antibody, avidin-horseradish peroxidase, and 3,3′-diaminobenzidine tetrahydrochloride (0.04%) in 0.05 M Tris-HCl (pH 7.4) and 0.025% H2O2 as a chromogen (Ventana Medical Systems, Tucson, AZ).
RESULTS
Removal of the γ134.5 amino-terminal sequences stimulates the expression of antiviral genes.
To study γ134.5 in the context of HSV infection, we constructed a virus, H1001, in which the amino terminus of γ134.5, spanning amino acids 1 to 146, was removed. As a control, we constructed a repair virus, H1002, where wild-type γ134.5 was restored (Fig. 2A). The virus constructs were verified by restriction digestion and PCR analysis (data not shown). Expression of γ134.5 was detected by Western blot analysis with anti-γ134.5 antibody. As shown in Fig. 2B, wild-type HSV-1(F), as well as the repair virus, H1002, expressed full-length γ134.5, whereas the γ134.5 null mutant, R3616, did not. The N-terminal deletion mutant, H1001, expressed a truncated form of γ134.5, with a molecular mass of 21 kDa.
Fig 2.

(A) Schematic representation of the genome structure and sequence arrangements of HSV-1(F) and related mutants. Boxes on the top line denote the inverted repeats flanking the unique long and unique short sequences, represented by thin lines. The expanded sections below show the γ134.5 loci, where thin lines represent DNA sequences retained in each virus. HSV-1(F) is wild-type virus, whereas R3616 lacks the entire γ134.5 coding region. H1001 has a deletion of the amino terminus of γ134.5, and H1002 is a repair virus that harbors the wild-type γ134.5 gene. (B) Expression of different γ134.5 variants. Vero cells were infected with the indicated viruses at 0.05 PFU per cell. At 24 h after infection, lysates of cells were subjected to immunoblotting with polyclonal antibody against γ134.5. Size markers are listed on the left.
We next assessed the induction of antiviral immunity in HSV-infected cells by quantitative real-time PCR. As indicated in Fig. 3A, HSV-1(F) or H1002 triggered a low level of IFN-β mRNA expression in mouse embryonic fibroblasts (MEFs). In contrast, R3616 stimulated robust IFN-β expression, with a 180-fold increase of IFN-β mRNA at 3 h postinfection. This effect persisted as infection progressed to the 6-h time point. Likewise, H1001 triggered drastic IFN-β expression, with a 400-fold increase in the IFN-β mRNA level at 3 h postinfection. This stimulation decreased at 6 h postinfection but remained significant compared to that with HSV-1(F) or H1002. A similar phenotype was observed for ISG56, although the magnitude of gene induction varied (Fig. 3B). However, the viral effects on ISG54 were indistinguishable until 6 h postinfection (Fig. 3C). Similarly, virus infection did not trigger RANTES induction until 6 h postinfection (Fig. 3D). These results suggest that the amino terminus of γ134.5 is crucial for impeding the induction of antiviral immunity early in HSV infection.
Fig 3.

The γ134.5 protein inhibits the induction of antiviral genes via the amino terminus. Mouse embryonic fibroblasts (MEFs) were either mock infected or infected with HSV-1(F), R3616, H1001, or H1002 (5 PFU/cell). At indicated time points, total RNA extracted from cells was subjected to quantitative real-time PCR amplification for expression of IFN-β (A), ISG56 (B), ISG54 (C), or RANTES (D). The data were normalized to that for 18S rRNA, and fold induction was calculated as described in Materials and Methods. Results are expressed as fold activation with standard deviations among triplicate samples.
The γ134.5 protein suppresses IRF3 phosphorylation by TBK1 through the amino-terminal domain.
Based on the above analysis, we evaluated the impact of γ134.5 on IRF3 phosphorylation. As illustrated in Fig. 4A, IRF3 was constitutively expressed in MEF cells, where it remained unphosphorylated in mock-infected cells (lanes 1 and 6). At 3 h postinfection, R3616 as well as H1001 stimulated IRF3 phosphorylation at serine 396 (lanes 3 and 4), which is a hallmark of IRF3 activation. As infection progressed, IRF3 phosphorylation remained evident for R3616 but was attenuated for H1001 (lanes 8 and 9). As expected, HSV-1(F) and H1002 did not induce IRF3 phosphorylation throughout infection (lanes 2, 5, 7, and 10). Although reduced at 6 h postinfection (lanes 8 and 9), H1001-induced IRF3 phosphorylation was notable compared to that seen for HSV-1(F) and H1002 (Fig. 4B). All viruses were able to infect efficiently, as indicated by a similar level of ICP27 at 3 h postinfection (Fig. 4A). However, at 6 h postinfection, higher levels of ICP27 were observed for HSV-1(F) and H1002 than for R3616 and H1001, suggesting that a defect in γ134.5 already impeded infection by R3616 or H1001 as early as 6 h postinfection. These results suggest that the amino terminus of γ134.5 is required to suppress IRF3 activation during HSV infection.
Fig 4.

The amino terminus of γ134.5 is required to inhibit IRF3 phosphorylation. (A) MEFs were infected with indicated viruses at 5 PFU/cell, and cell lysates were subjected to immunoblotting analysis with antibodies against IRF3, phosphorylated IRF3 (Ser396), and ICP27, respectively, at 3 h and 6 h postinfection. (B) Quantification of IRF3 phosphorylation. The protein bands shown in panel A were quantified using NIH ImageJ software. The data are presented as the relative amount of phosphorylated IRF3 normalized to the total level of IRF3 in each sample, with mock infection arbitrarily set at 1.0.
Since TBK1 phosphorylates IRF3 (40), we examined how γ134.5 exerted its effect in this process by immunoprecipitation coupled with kinase assays. As shown in Fig. 5A, proteins were expressed in 293T cells transfected with HA-TBK1, FLAG-ΔN146 (the N-terminal-deletion γ134.5 mutant), and FLAG-γ134.5 (full-length) (upper panel). Expression of TBK1 resulted in IRF3 phosphorylation (lane 1, bottom panel). Addition of wild-type γ134.5 reduced IRF3 phosphorylation (lane 3). Nevertheless, expression of ΔN146 failed to inhibit IRF3 phosphorylation by TBK1 (lane 5). Among all samples tested, TBK1 was present at a comparable level in the immunoprecipitates (middle panel). Quantification from multiple experiments confirmed that the ΔN146 mutant was unable to suppress TBK1 kinase activity compared with full-length γ134.5 (Fig. 5B). In correlation, wild-type γ134.5 associated with TBK1, whereas the ΔN146 mutant did not (Fig. 5C, lanes 3 and 4). Reporter assays revealed that unlike wild-type γ134.5, the ΔN146 mutant was unable to inhibit the activation of IFN-β and ISG56 promoters by TBK1 (Fig. 5D and E). Collectively, these data suggest that the amino terminus of γ134.5 works as a module to inhibit IRF3 activation by TBK1.
Fig 5.

(A) Effect of γ134.5 variants on IRF3 phosphorylation by TBK1. 293T cells were transfected with HA-TBK1 along with empty vector, FLAG-γ134.5, or FLAG-ΔN146. At 40 h after transfection, aliquots of cell lysates were processed for protein expression with antibodies against FLAG and HA. In parallel, lysates were immunoprecipitated with anti-HA antibody. The immunoprecipitates were incubated with GST-IRF3 (amino acids [aa] 380 to 427) and ATP for kinase assays. Samples were subjected to electrophoresis and probed with antibodies against phosphorylated IRF3, FLAG, and HA, respectively. (B) Quantitation of in vitro kinase assays. The relative kinase activity was expressed as a ratio of phosphorylated IRF3 to TBK1 in the immunoprecipitate, with the control TBK1 arbitrarily set to 100 (NIH ImageJ software). The data represent four independent experiments with standard deviations (*, P < 0.05). (C) Interactions of γ134.5 variants with TBK1. 293T cells were cotransfected with FLAG-γ134.5 or FLAG-ΔN146 along with an empty vector, FLAG-TBK1. At 36 h after transfection, lysates of cells were immunoprecipitated with anti-HA antibody. Samples were processed for immunoblotting analysis with antibodies against FLAG and HA, respectively. (D and E) Effects of γ134.5 variants on promoter activation by TBK1. 293T cells were transfected with an empty vector or a plasmid expressing HA-γ134.5, FLAG-TBK1, FLAG-ΔN146, or FLAG-γ134.5, along with an IFN-β or ISG56 reporter. At 36 h posttransfection, cells were harvested for luciferase assays. Results are expressed as fold activation with standard deviations among triplicate samples.
The γ134.5 protein prevents the induction of IFN-β independently of PKR in HSV-infected cells.
In addition to translational arrest, PKR is involved in the induction of type I IFN (15, 49). We asked whether γ134.5 suppressed type I IFN induction through PKR. Accordingly, PKR−/− MEFs were infected with viruses, and IFN-β expression was then determined by real-time PCR. As seen in Fig. 6A, infection by HSV-1(F) or H1002 initially triggered a low level of IFN-β mRNA, which diminished at 6 h postinfection. However, R3616 induced dramatic IFN-β mRNA expression, which increased about 100-fold by 6 h postinfection. Interestingly, H1001 exhibited different induction kinetics. At 3 h postinfection, this mutant stimulated 160-fold IFN-β expression, which then attenuated to 40-fold at 6 h postinfection. Thus, HSV γ134.5 variants induced or inhibited IFN-β expression independently of PKR in HSV-infected cells.
Fig 6.

(A) HSV γ134.5 inhibits IFN-β expression independently of PKR. PKR−/− MEFs were either mock infected or infected with indicated viruses (5 PFU/cell). At 3 and 6 h after infection, total RNA extracted from cells was subjected to quantitative real-time PCR amplification. The level of IFN-β mRNA was normalized to the level of 18S rRNA, and fold induction was calculated as described in Materials and Methods. Results are representative of three independent experiments with standard deviations among triplicate samples. (B) Deletion of the γ134.5 amino terminus has no effect on HSV resistance to IFN-α. Vero cells were untreated or pretreated with IFN-α (Sigma) for 20 h. Cells were then infected with indicated viruses at 0.05 PFU per cell, and viral yields were determined at 48 h postinfection.
Because inhibition of PKR-mediated translational arrest is coupled to HSV resistance to IFN (5, 17), we determined the role of the γ134.5 amino terminus in this process. As indicated in Fig. 6B, all viruses replicated efficiently in untreated Vero cells, which are devoid of IFN-α/β genes. Upon treatment with IFN-α, replication of HSV-1(F) and H1002 was virtually unchanged. Replication of R3616 was inhibited by approximately 1,000-fold. Intriguingly, replication of H1001 was marginally reduced (6-fold) by IFN-α, a result which was close to those seen for HSV-1(F) and H1002. Herein, deletion of the amino terminus of γ134.5 does not affect HSV resistance to type I IFN treatment.
HSV replication is functionally linked to the amino terminus of γ134.5 and TBK1.
Since γ134.5 inhibits TBK1 via its amino terminus, we examined its role in viral replication. As shown in Fig. 7, HSV-1(F) replicated efficiently in both TBK1+/+ and TBK1−/− cells, reaching titers of 2.5 × 105 and 4.4 × 105 PFU/ml, respectively. As expected, R3616 barely replicated in TBK1+/+ cells, and its replication was partially restored to a titer of 3.6 × 102 PFU/ml in TBK1−/− cells, which was still 1,000-fold lower than that for wild-type virus. Interestingly, H1001 displayed a different phenotype. In TBK1+/+ cells, this amino terminus deletion mutant replicated to a titer of 6.4 × 102 PFU/ml, an intermediate level between titers of wild-type virus and R3616. However, H1001 replicated efficiently in TBK1−/− cells, reaching a titer of 6.7 × 104 PFU/ml, which is close to that of wild-type virus. H1002 behaved like wild-type virus. We conclude that the amino terminus of γ134.5 acts to overcome TBK1-mediated restriction on HSV replication.
Fig 7.
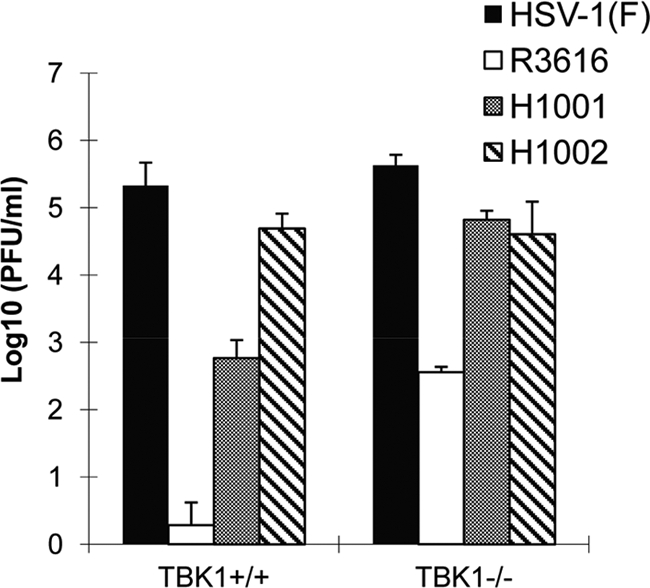
Viral replication in TBK1+/+ or TBK1−/− cells. Cells were infected with indicated viruses (0.05 PFU/cell). At 24 h after infection, virus yields were titrated on Vero cells. Data are averages for three independent experiments with standard deviations.
The γ134.5 protein facilitates viral replication as well as spread via amino-terminal sequences in vivo.
To evaluate γ134.5 in vivo, we investigated viral replication in a mouse ocular infection model. Mice were infected with viruses, and viral yields were determined on day 3. As shown in Fig. 8A, wild-type HSV-1 replicated efficiently in the eye (3.6 × 102 PFU/eye), whereas R3616 failed to replicate. Under this condition, H1001 barely replicated, with no infectious virus being detectable. Replication of H1002 resembled that of wild-type virus (4.6 × 102 PFU/eye). Similar phenotypes were seen in the trigeminal ganglia and brain. To examine histopathology, thin sections of the eye were stained with anti-HSV-1 antibody. As shown Fig. 8B, viral antigens were detected in the corneal epithelium infected with HSV-1(F) and H1002 (panels b and e). This was accompanied by corneal thickening with ulceration, reflecting inflammation in the epithelium and stroma. No viral antigen or inflammation was detected in the eyes mock infected or infected with R3616 or H1001 (panels a, c, and d).
Fig 8.

(A) The amino terminus of γ134.5 is required to facilitate neuroinvasion. Mice were mock infected or infected with HSV-1(F), R3616, H1001, or H1002 at 4 × 105 PFU through corneal scarification. At 3 days postinfection, eyes, trigeminal ganglia, and brain tissues were collected and virus yields were determined. Data are expressed as means ± standard deviations for six mice for each group. (B) Immunohistochemistry analysis. The sections from eye tissues described above were reacted with anti-HSV-1 antibody, and immunohistochemistry was performed. Specific HSV-1 staining is shown in brown. Representative images from each group were chosen for the panels.
We further examined the kinetics of viral replication and spread in vivo. As illustrated in Fig. 9A, HSV-1(F) replicated efficiently in the eye on day 1 (1.4 × 104 PFU/eye). Similarly, H1001 replicated to a titer of 1.8 × 103 PFU/eye. Removal of the amino terminus marginally reduced viral replication in the eye on day 1 (the first 24 h). This is presumably due to the ability of H1001 to overcome the PKR-mediated antiviral effect. As infection continued, HSV-1(F) maintained viral yields at 4.5 × 103 PFU/eye and 1.4 × 103 PFU/eye in the eye on days 3 and 5, respectively. However, replication of H1001 was reduced drastically to an undetectable level in the eye on day 3 and 5. This reduction may have resulted from a defect(s) in blocking TBK1-mediated responses. In the trigeminal ganglia (Fig. 9B), HSV-1(F) appeared after day 1 and replicated to a titer of 1.7 × 103 PFU/TG on day 3 and 4.4 × 103 PFU/TG on day 5, respectively. However, neither R3616 nor H1001 replicated to an appreciable level. In the brain (Fig. 9C), only HSV-1(F) began to appear on day 3 and replicated to 5.0 × 102 PFU/BS by day 5, whereas H1001 and R3616 were undetectable. These results suggest that the amino terminus of γ134.5 promotes viral replication and neuroinvasion.
Fig 9.

Kinetics of viral replication in vivo. Mice were infected with HSV-1(F), R3616, H1001, or H1002 at 4 × 105 PFU via corneal scarification. Mice were sacrificed on days 1, 3, and 5. Tissue samples from the eye (A), trigeminal ganglia (B), or brain stem (C) were analyzed for viral yields on Vero cells. Data are expressed as means ± standard deviations for data from four mice for each group.
DISCUSSION
The γ134.5 protein of HSV-1 consists of 263 amino acids with an amino-terminal domain, a linker region with triplet repeats (ATP), and a carboxyl-terminal domain (11). It has been demonstrated that γ134.5 prevents the PKR response via its carboxyl terminus and thereby confers viral virulence (18, 26). Recently, we identified TBK1 as a novel cellular target of HSV γ134.5 (46). This led us to reason that regulation of TBK1 by γ134.5 may contribute to viral virulence. In this study, we provide evidence that γ134.5 inhibits TBK1-mediated antiviral immunity independently of PKR. This activity requires its amino-terminal domain, which facilitates viral replication in the peripheral tissue and spread to the central nervous system. Therefore, in addition to PKR, γ134.5 negatively modulates TBK1 and promotes viral replication.
Our work suggests that γ134.5 inhibits the induction of antiviral immunity via its amino-terminal sequences. Notably, HSV γ134.5 associated with TBK1, which inhibited IRF3 phosphorylation and antiviral gene expression. Deletion of the amino-terminal domain disrupted its activity. Wild-type virus induced a low level of IFN-β or ISG expression, which could be triggered by viral entry before γ134.5 was expressed. In contrast, the virus lacking the amino-terminal domain of γ134.5 stimulated robust expression of antiviral molecules compared to results with wild-type virus. Additionally, replication of the amino-terminus deletion mutant was impaired in TBK+/+ cells but not in TBK1−/− cells. These results establish a functional link between γ134.5 and TBK1. In mammalian cells, TBK1 forms a complex with a number of proteins, such as TRAF family member-associated NF-κB activator (TANK), NAK-associated protein 1 (NAP1), similar to NAP1TBK1 adaptor (SINTBAD), TRAF3, and Sec5 (6, 14, 16, 33, 35, 38). These interactions probably occur in a signal-specific manner (4, 20–22, 51). Once activated, the TBK1 ubiquitin-like domain makes contacts with its kinase domain, leading to IRF-3/IRF-7 phosphorylation and cytokine expression (19). It is conceivable that the amino terminus of γ134.5 may prevent the recruitment of TBK1 by one or more adaptor proteins. Alternatively, it may block the contact between the ubiquitin-like domain and the kinase domain to prevent kinase activation. While additional work is required, these results highlight the importance of γ134.5 and TBK1 in HSV infection.
PKR mediates the translational arrest as well as cytokine expression (15, 49). In HSV-infected cells, γ134.5 recruits PP1 via its carboxyl-terminal domain and precludes translation arrest. This confers viral resistance to IFN-α/β (5, 17, 18). Importantly, site-specific mutations in the PP1 binding site cripple viral replication (45). We observed that the γ134.5 mutant lacking the amino-terminal sequences remained resistant to IFN-α/β. Thus, the amino-terminal sequences are dispensable in counteracting the PKR response. On the other hand, the deletion of the amino terminus impaired HSV replication in TBK1+/+ cells. Such a defect was fully repaired in TBK1−/− cells. The amino-terminal sequences have likely evolved to control TBK1. We postulate that γ134.5 may facilitate viral replication via distinct domains, which coordinately regulate PKR and TBK1 responses. In this context, it is notable that PKR is implicated in type I IFN expression (15). This raises the possibility that PKR may be a key factor in this process in HSV-infected cells. The data in the present study do not favor this idea, because PKR deficiency did not disrupt IFN-β induction or inhibition by HSV γ134.5 variants. Apparently, TBK1 and PKR act differentially in type I IFN responses upon HSV infection.
Although essential in viral virulence, exactly how γ134.5 exerts its activity in vivo has remained incompletely defined. Previous studies established that γ134.5 prevents protein synthesis shutoff mediated by PKR and facilitates viral replication in experimental models (9, 10, 18, 26). Furthermore, inhibition of autophagy by γ134.5 is required for HSV virulence (24). This occurs relatively late in HSV infection (around day 7), where it inhibits CD4+ T cell responses independently of IRF3 in vivo (1, 24). Since γ134.5 inhibits TBK1, we evaluated viral replication in a mouse ocular infection model. Intriguingly, an HSV mutant lacking the amino terminus of γ134.5 initially (day 1) replicated in the eye but disappeared shortly thereafter (day 3). This is a difference from wild-type virus, which persisted throughout infection, or the γ134.5 null mutant, which was unable to replicate. A simple explanation is that persistent viral replication in vivo relies on the capacity of γ134.5 to overcome innate immunity mediated by both PKR and TBK1. We suspect that initial replication of the amino-terminal deletion mutant is presumably due to the ability of γ134.5 to counteract PKR. On the other hand, its relative early clearance from the eye may have resulted from a defect of γ134.5 in inhibiting TBK1.
It is notable that unlike the γ134.5 null mutant, the wild type spread efficiently to the trigeminal ganglia and brain. Although it had traveled to the trigeminal ganglia by day 3, the amino-terminal deletion mutant barely replicated. No infectious virus was detectable in the brain. One possibility is that transient viral replication in the eye may have accumulated beyond a threshold where a small fraction escaped to the trigeminal ganglia. Alternatively, the majority of the amino-terminal deletion mutant may have spread to the trigeminal ganglia, where it went into latency. Due to restriction by TBK1, the amino-terminal deletion mutant was crippled in initiating the lytic cycle in the trigeminal ganglia and failed to travel to the brain. This raises the question of whether γ134.5 interacts with TBK1 functionally in a tissue-specific manner. It is likely that control of TBK1 by γ134.5 may contribute to efficient viral replication in vivo. Our future studies will focus on the precise mechanism by which γ134.5 promotes HSV virulence.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (AI092230; to B.H.) and the National Natural Science Foundation of China (30870128; to Y.C.).
We thank Bernard Roizman, Wen-Chen Yeh, Rongtuan Lin, Bryan Williams, and Ulrich Siebenlist for providing valuable reagents.
Footnotes
Published ahead of print 14 December 2011
REFERENCES
- 1. Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. 2007. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81:12128–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bolovan CA, Sawtell NM, Thompson RL. 1994. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J. Virol. 68:48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cassady KA, Gross M, Gillespie GY, Roizman B. 2002. Second-site mutation outside of the U(S)10–12 domain of Δγ134.5 herpes simplex virus 1 recombinant blocks the shutoff of protein synthesis induced by activated protein kinase R and partially restores neurovirulence. J. Virol. 76:942–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chau TL, et al. 2008. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 33:171–180 [DOI] [PubMed] [Google Scholar]
- 5. Cheng G, Brett ME, He B. 2001. Val193 and Phe195 of the γ134.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-α/β. Virology 290:115–120 [DOI] [PubMed] [Google Scholar]
- 6. Chien Y, et al. 2006. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127:157–170 [DOI] [PubMed] [Google Scholar]
- 7. Chiu YH, Macmillan JB, Chen ZJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2α and premature shutoff of protein synthesis after infection with γ134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 92:10516–10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science 250:1262–1266 [DOI] [PubMed] [Google Scholar]
- 10. Chou J, Roizman B. 1992. The γ134.5 gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell death in neuronal cells. Proc. Natl. Acad. Sci. U. S. A. 89:3266–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chou J, Roizman B. 1990. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn+. J. Virol. 64:1014–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui W, Taub DD, Gardner K. 2007. qPrimerDepot: a primer database for quantitative real time PCR. Nucleic Acids Res. 35:D805–D809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 14. Fujita F, et al. 2003. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 23:7780–7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia MA, et al. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70:1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo B, Cheng G. 2007. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J. Biol. Chem. 282:11817–11826 [DOI] [PubMed] [Google Scholar]
- 17. He B, Gross M, Roizman B. 1998. The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273:20737–20743 [DOI] [PubMed] [Google Scholar]
- 18. He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikeda F, et al. 2007. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J. 26:3451–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii KJ, et al. 2008. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451:725–729 [DOI] [PubMed] [Google Scholar]
- 21. Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 455:674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jing X, Cerveny M, Yang K, He B. 2004. Replication of herpes simplex virus 1 depends on the γ134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J. Virol. 78:7653–7666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. 2009. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 83:12164–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leib DA, et al. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. U. S. A. 97:6097–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. MacLean A, Robertson L, McKay E, Brown SM. 1991. The RL neurovirulence locus in herpes simplex virus type 2 strain HG52 plays no role in latency. J. Gen. Virol. 72:2305–2310 [DOI] [PubMed] [Google Scholar]
- 28. MacLean AR, Ul-Fareed M, Robertson L, Harland J, Brown SM. 1991. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 72:631–639 [DOI] [PubMed] [Google Scholar]
- 29. Mao H, Rosenthal KS. 2003. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J. Virol. 77:3409–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melchjorsen J, et al. 2010. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 84:11350–11358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mohr I, Gluzman Y. 1996. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 15:4759–4766 [PMC free article] [PubMed] [Google Scholar]
- 32. Mohr I, et al. 2001. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J. Virol. 75:5189–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oganesyan G, et al. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211 [DOI] [PubMed] [Google Scholar]
- 34. Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J. Virol. 70:2883–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rasmussen SB, et al. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rasmussen SB, et al. 2007. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 81:13315–13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryzhakov G, Randow F. 2007. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 26:3180–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sen GC. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255–281 [DOI] [PubMed] [Google Scholar]
- 40. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151 [DOI] [PubMed] [Google Scholar]
- 41. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264 [DOI] [PubMed] [Google Scholar]
- 42. Takaoka A, et al. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505 [DOI] [PubMed] [Google Scholar]
- 43. Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820 [DOI] [PubMed] [Google Scholar]
- 44. Unterholzner L, et al. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11:997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verpooten D, et al. 2009. Dephosphorylation of eIF2α mediated by the γ134.5 protein of herpes simplex virus 1 facilitates viral neuroinvasion. J. Virol. 83:12626–12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Whitley RJ, Kern ER, Chatterjee S, Chou J, Roizman B. 1993. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 91:2837–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518 [DOI] [PubMed] [Google Scholar]
- 49. Zhang P, Samuel CE. 2008. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283:34580–34587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang SY, et al. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527 [DOI] [PubMed] [Google Scholar]
- 51. Zhong B, et al. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550 [DOI] [PubMed] [Google Scholar]