

Gelsolin is a dynamic actin-filament-severing and -capping protein (1) capable of the demolition of the actin cytoskeleton. Gelsolin undergoes large conformational changes on binding calcium and actin, transformations that can be reversed on interaction with phosphatidylinositol 4,5-bisphosphate (PIP2) and the removal of calcium. This structural plasticity goes askew in familial amyloidosis (Finnish type) (FAF), where a single amino acid mutation (2), at residue 187, produces an aberrant, protease-sensitive molecule. A series of protease cleavages follow that result in a fragment of gelsolin domain 2 (residues 173–243; Fig. 1) that spontaneously associates into amyloid fibrils. These fibrils are deposited mainly in the facial regions of FAF sufferers, producing physical and cosmetic dysfunction. Ratnaswamy et al. (3) provide insight into the mechanism of FAF-gelsolin degeneration and, hence, the progression of this disease, by assessing the conformational stability of the FAF mutant domain 2 in comparison with that of the wild type.
Figure 1.
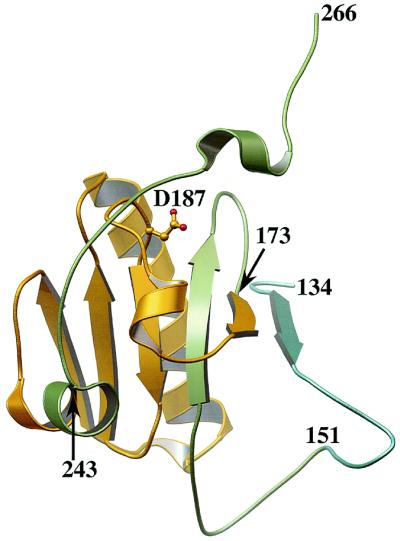
The structure of the second domain of gelsolin (G2) in the absence of calcium. Residue 187 is the mutation site in FAF, and residues 173–243 (gold) comprise the amyloid fragment. Fersht and coworkers studied a fragment comprising residues 151–266 (green and gold) and Ratnaswamy et al. examined the entire G2, residues 134–266 (blue, green and gold) (3, 5).
Theories of the structural disintegration of FAF-gelsolin abound. On elucidating the structure of nonactivated gelsolin (4), we proposed that the network of interactions formed by residue D187 (the mutation site in FAF) stabilizes the central β-sheet of gelsolin domain 2 (G2). Hence, in FAF G2, this β-sheet is relatively destabilized, allowing the structure to open up and allow protease cleavage. This hypothesis, whereby the FAF G2 unfolds to some degree before cleavage can take place, will be referred to as the disintegration mechanism. Fersht and coworkers (5) investigated this hypothesis by studying the stability of both FAF and wild-type fragments of G2 (residues 151–266; Fig. 1). They found that the two possible FAF mutations (D187N and D187Y) destabilized the G2 fragment by 1.22 and 2.16 Kcal⋅mol−1, respectively, and concluded that, at body temperature, a significant fraction of FAF gelsolin would be at least partially unfolded. So, point proven? Far from it. Ratnaswamy et al. (3) have now extended this study by examining the entire G2 domain (residues 134–266; Fig. 1). They report that the FAF G2 shares a similar stability to the wild type. This all appears to leave us at an impasse, where we know that this single mutation has devastating consequences for gelsolin's structural integrity yet the stability of G2 appears to remain unimpaired.
Clues to this enigmatic behavior lie in considering G2's role in the actin-severing process. Gelsolin comprises six homologous domains (G1–G6) that in the absence of calcium adopt a conformation in which the actin-binding sites are obscured (ref. 4; Fig. 2). On binding calcium, a series of conformational changes ensue that expose the three actin-binding sites. Unfortunately, no high resolution structural data are yet available on this activated form. G2 then binds to the side of an actin filament, orientating the other two actin-binding domains (G1 and G4) in a fashion where they can cooperatively disrupt the filament, resulting in a gelsolin-capped filament. We have an excellent idea of what at least part of this capped filament looks like (Fig. 2). Holmes et al. (6) provide a good model of the actin filament, McLaughlin et al. (7) have described G1 binding to actin, we have shown G4–G6 bound to actin (8), and, at lower resolution, McGough et al. (9) have described G2 and the possible G3 interactions with actin.
Figure 2.

A schematic diagram highlighting the structural changes in gelsolin that occur on binding calcium and actin. (Right) The inactive, calcium-free form of gelsolin and (Left) a gelsolin-capped filament. The arrangements of calcium-free gelsolin, G1 bound to actin, and G4–G6 bound to actin are based on x-ray structures whereas the model for the actin filament and the placing of G2–G3 on the filament are derived from electron microscopy data. G2 and G3 are drawn as unique shapes, when bound to the actin filament, because of the uncertainty of their structures in this state (4, 6–9).
Combining all these structural data confirms a prediction of McGough and Way (10) that G1 and G2 bind to two adjacent actins along the length of the actin filament. This binding requires a linker between G1 and G2 of around 30 Å, yet, in the non-calcium-activated conformation, these two domains abut each other. To bridge this distance, some unfolding of either the C terminus of G1 or the N terminus of G2 is required. From the 10 high resolution structures of gelsolin domains elucidated thus far, no dramatic variation has been observed in the C-terminal regions; however, the N-termini are seen to adopt wildly different conformations. Hence, a reasonable model would be that the first β-strand of G2 (residues 137–141; Fig. 1), which is not present in four of the six gelsolin domains anyway, becomes dislodged from the β-sheet and forms this linker (approximately residues 134–160). This conformation has recently been modeled by Puius et al. (11). Some additional evidence of conformational changes in this region is provided by Xian et al. (12). These researchers were concerned with how PIP2 removes the gelsolin cap from a filament. In studying the structures of a peptide consisting of G2 residues 150–169, they observed conformational changes on binding PIP2, verifying the structural mutability of this region.
Where does all this hypothesizing about G2 conformational change lead us in trying to understand the progression of FAF? And what does it say about the somewhat conflicting observations of isolated G2 stability? At the very least, we can say that the structure of G2 in calcium-activated gelsolin is very different from that of calcium-free gelsolin. Hence, drawing conclusions from the structure of the nonactivated form is shortsighted, when it is entirely possible that the FAF protease cascade may act on the activated form. If we are prepared to be a little bolder, and consider the model of G2 conformational changes that we outlined in the preceding paragraph, we come to a much more far-reaching conclusion. The construct studied by Ratnaswamy et al. (3) is clearly that of the calcium-free form of gelsolin. This FAF-mutant shows no structural destabilization relative to wild type. However, the smaller construct studied by Isaacson et al. (5) would represent the hypothetical structure of G2 in the activated molecule. This situation leads to the conclusion that protease must act on the activated form of FAF gelsolin and that the G2 disintegration mechanism may still be viable.
In another recent paper, Fersht and coworkers (13) investigated structural and dynamic aspects of their short G2 fragment (residues 151–266; Fig. 1) by using NMR techniques. They observed that, at 25°C, the FAF mutation of D147N results in small but widespread perturbations involving approximately one third of the residues. Of particular note is that residues 254–258 become unstructured in FAF G2. Kazmirski et al. (13) propose that conformational changes in these residues (254) reveal the nearby protease cleavage site, allowing the FAF protease cleavage cascade to proceed. We shall refer to this as the exposure mechanism. They also observe the loss of a mainchain–mainchain hydrogen bond (D187-G202) in FAF G2 that may be suspected to destabilize the central β-sheet and perhaps support the disintegration mechanism. Clearly, it would be valuable to have access to these types of data at the physiological temperature, but such experiments may be challenging because of the instability of FAF G2 at these temperatures (5). Whether the exposure theory is sufficient to produce the full effects of FAF gelsolin cleavage, or whether the exposure mechanism is simply a smaller part of the disintegration mechanism, has yet to be established.
As with the majority of good science, an advance in understanding leaves us with more questions than answers, as we greater appreciate the complexity of system with which we are dealing. This principle is very true in understanding the dynamics of gelsolin and, in particular, the aberrant dynamics of FAF-gelsolin. What exactly is the conformation of FAF-G2 when the protease strikes? Is it bound to calcium, actin, or PIP2 at this time? Is it partially unfolded or is there a much smaller conformational change that exposes the cleavage site? These are just a few of the many questions that must be addressed before arriving at a convincing explanation of FAF-gelsolin disintegration. Both Kelly and Fersht and their coworkers have taken the first steps to disseminate the mechanism of the progression of FAF. We look forward to learning of their future results as they continue on their quest.
Footnotes
See companion article on page 2334.
References
- 1.Yin H. BioEssays. 1987;7:176–179. doi: 10.1002/bies.950070409. [DOI] [PubMed] [Google Scholar]
- 2.Maury C P, Alli K, Baumann M. FEBS Lett. 1990;260:85–87. doi: 10.1016/0014-5793(90)80072-q. [DOI] [PubMed] [Google Scholar]
- 3.Ratnaswamy G, Huff M E, Su A I, Rion S, Kelly J W. Proc Natl Acad Sci USA. 2001;98:2334–2339. doi: 10.1073/pnas.041452598. . (First Published February 20, 2001; 10.1073/pnas.041452598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burtnick L D, Koepf E K, Grimes J, Jones E Y, Stuart D I, McLaughlin P J, Robinson R C. Cell. 1997;90:661–670. doi: 10.1016/s0092-8674(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 5.Issacson R L, Weeds A G, Fersht A R. Proc Natl Acad Sci USA. 1999;96:11247–11252. doi: 10.1073/pnas.96.20.11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmes K C, Popp D, Gebhard W, Kabsch W. Nature (London) 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin P J, Gooch J T, Mannhertz H G, Weeds A G. Nature (London) 1993;364:685–692. doi: 10.1038/364685a0. [DOI] [PubMed] [Google Scholar]
- 8.Robinson R C, Mejillano M, Le V P, Burtnick L D, Yin H L, Choe S. Science. 1999;286:1939–1942. doi: 10.1126/science.286.5446.1939. [DOI] [PubMed] [Google Scholar]
- 9.McGough A, Chiu W, Way M. Biophys J. 1998;74:764–772. doi: 10.1016/S0006-3495(98)74001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGough A, Way M. J Struct Biol. 1995;115:144–150. doi: 10.1006/jsbi.1995.1038. [DOI] [PubMed] [Google Scholar]
- 11.Puius Y A, Fedorov E V, Eichinger L, Schleicher M, Almo S C. Biochemistry. 2000;39:5322–5331. doi: 10.1021/bi992364d. [DOI] [PubMed] [Google Scholar]
- 12.Xian W, Vegners R, Jamney P A, Braunlin W H. Biophys J. 1995;69:2695–2702. doi: 10.1016/S0006-3495(95)80140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazmirski S L, Howard M J, Isaacson R L, Fersht A R. Proc Natl Acad Sci USA. 2000;97:10706–10711. doi: 10.1073/pnas.180310097. . (First Published September 19, 2000; 10.1073/pnas.180310097) [DOI] [PMC free article] [PubMed] [Google Scholar]