

Abstract
The deleterious mutation rate plays a key role in a number of important topics in biology, from mating system evolution to human health. Despite this broad significance, the nature and causes of variation in mutation rate are poorly understood, especially in multicellular organisms. We test whether genetic quality, the presence or absence of deleterious alleles, affects the mutation rate in Drosophila melanogaster by using a modified mutation accumulation approach. We find evidence that genotypes constructed to carry deleterious “treatment” alleles on one chromosome during mutation accumulation experience an elevated mutation rate on a different chromosome. Further, this elevation is correlated with the effect of the treatment alleles on phenotypic condition, measured as body mass. Treatment alleles that reduce mass by 10% cause a doubling in the rate of mutational decline. Our results show that mutation rates are sensitive to genetic stress, such that individuals with low-quality genotypes will produce offspring of even lower genetic quality, in a mutational positive feedback loop. This type of variation in mutation rate is expected to alter a variety of predictions based on mutation load theory and accelerate adaptation to new environments. Positive mutational feedback could affect human health by increasing the rate of germline mutation, and possibly somatic mutation, in individuals of poor health because of genetic or environmental stress.
Keywords: genetic load, mutational meltdown
Mutation is a constant source of harmful genetic variation that can dramatically reduce population fitness (1), and mutation rates are sufficiently high to generate substantial mutation loads in multicellular organisms, including humans (2, 3). Consequently, the deleterious mutation rate plays a key role in a number of important topics in biology, including the magnitude of inbreeding depression, the maintenance of genetic variation, mating system evolution, senescence, the persistence of endangered populations, and human health (4, 5).
There is considerable variation in mutation rate among species, strains, and environmental contexts (2, 6–8). The sources and mechanisms underlying this variation have been best studied in microbes (8–11), but the sources of variation in microbes may differ from those in multicellular eukaryotes for several reasons. First, most microbes usually replicate asexually, and theory predicts that selective forces on mutation rates differ dramatically depending on the degree of linkage between a mutator allele and the new mutations it creates (8, 12). Second, microbial genomes are more directly exposed to environmental influences than the germline DNA of animals. Moreover, it is particularly important to understand variation in mutation rate in multicellular organisms because it is in these taxa that genomewide per-generation rates of deleterious mutation tend to be sufficiently high for mutation loads to act as a major evolutionary force (2–5).
In animals, mutation rate varies among genotypes (6, 13, 14), although the functional sources of this variation are unknown. Some variation is likely caused by genes known to affect DNA replication and repair (e.g., DNA polymerases, endonucleases; refs. 2, 8). However, it is plausible that the number of mutations transmitted to the next generation could also depend on individual quality, i.e., an axis of individual heterogeneity closely related to fitness (15). Individuals with suboptimal genotypes because of the presence of deleterious alleles (i.e., low genetic quality) will tend to be in poor phenotypic condition (16). This effect observed at the organismal level must be mediated by the direct or indirect effects of deleterious alleles on cell state. In turn, various aspects of the cellular environment (e.g., cell metabolism, cell cycle dynamics) are known or speculated to affect the risk of DNA damage, the functioning of replication and repair processes, and rates of molecular evolution (17–20). Thus, it is probable that genetic quality influences mutation rate, provided that genetic quality affects the cellular environment of germ cells rather than just somatic cells. Indeed, there is evidence that genetic quality and related aspects of condition affect traits likely to influence mutational mechanisms, including resistance to oxidative damage (21) and the regulation of DNA replication and repair processes that differ in fidelity (22–24). Here we use a modification of the classical mutation accumulation (MA) approach to study the impact of genetic quality on the spontaneous mutation rate in Drosophila melanogaster.
In a traditional MA experiment, many initially identical lines are repeatedly bottlenecked, preventing selection against most new mutations, which fix at random. After many generations, the mean fitness of such lines decreases, and the variance in fitness among lines increases, as deleterious mutations accumulate. In our experiment, we assessed the accumulation of mutations on the second chromosome while manipulating the genetic quality of the third chromosome. All MA lines (and outbred controls) shared an initially identical second chromosome copy; MA line third chromosomes were drawn at random from an outbred population, except that one or two deleterious marker alleles (henceforth “treatment alleles”) were introgressed into a subset of third chromosomes to manipulate genetic quality. We conducted MA in 11 groups of lines: one group with no treatment alleles (unloaded treatment) and 10 groups with one or two treatment alleles (loaded treatments). The loaded treatments consisted of six groups with one treatment allele each (AntpNs, Bsb1, Dr1, Gl1, Ki1, sensLy-1) and four groups with two treatment alleles each (Dr1 KgV, Bsb1 sensLy-1, Gl1 H2, Ki1 kD). None of the genes in question have been annotated with Gene Ontology terms relating to DNA repair or DNA replication (25). Rather, these genes are known to be involved in various other functions including pattern formation, cell fate determination, regulation of cell death, and transcription regulation. More information regarding these genes and their alleles is available on FlyBase (25).
We assessed fitness, measured as competitive viability, on three occasions over 46 generations of MA (∼21 mo). For each assay, the focal MA chromosome (chromosome 2) was “extracted” from each MA line and placed on a standardized genetic background by using standard crossing techniques involving markers and balancer chromosomes (Fig. S1). This means that, in each line, new mutations accumulated on chromosome 2 in the presence of treatment third chromosomes but fitness assays were conducted in the absence of these treatment third chromosomes; thus, fitness differences among groups following MA cannot be attributed to the direct effects of the treatment third chromosomes, but rather to differential effects of treatment alleles on either the number or average effect of mutations accumulating on chromosome 2. At each assay point, we also measured the fitness of “control” second chromosomes (Fig. S2). Control chromosomes came from three populations each maintained at moderate size (N ∼ 450), at which selection can prevent the accumulation of mutations with detectable effects.
Results
Across all assays, we obtained more than 3,800 measures of relative fitness for more than 380 MA lines. After 46 generations of MA, fitness was significantly lower in the loaded treatments than in the unloaded treatment (permutation test, P < 0.02). To assess the rate of fitness decline, we performed a regression of fitness on generation number for each treatment, using only lines present in all three assays. The regression slope, ΔM, represents the change in mean fitness per generation of MA. On average, ΔM for the loaded treatments was approximately 2.9 times that of the unloaded treatment, and was significantly more negative in the loaded treatments relative to the unloaded treatment (permutation test, P < 0.03; Fig. 1 A and B). We obtained similar results for the per-generation change in among-line variance in fitness, ΔV, which was approximately 3.6 times more rapid in the loaded treatments than in the unloaded treatment (P < 0.0002 based on simulation results; Fig. 1 C and D; Materials and Methods provides further details).
Fig. 1.

Change in the mean and variance in fitness for 11 treatment groups over 46 generations of MA. (A) Mean fitness relative to controls over time for second chromosomes that accumulated mutations in different genetic backgrounds with respect to the third chromosome (solid line, unloaded third chromosome; dashed lines, one deleterious treatment allele; dotted lines, two deleterious treatment alleles). (B) The rate of decline in mean fitness, ΔM, was significantly greater in the loaded treatment (L) than in the unloaded treatment (UL; randomization test, P < 0.03). Error bars represent ± 1 SE among different backgrounds within the loaded treatment. Compared with the unloaded treatment, ΔM was approximately 2.8 times greater in the loaded treatment (∼2.7 times for those carrying one treatment allele, L1, and ∼3.2 times for those carrying two treatment alleles, L2). (C) Variance in fitness among lines within each genetic background over time. (D) The rate of increase in variance, ΔV, was significantly greater in the loaded treatments than in the unloaded treatment (P < 0.0002, based on simulation results; Materials and Methods provides further details). Compared with the unloaded treatment ΔV was approximately 3.6 times greater for loaded treatments (∼3.3 times for those carrying one treatment allele and ∼4 times for those carrying two treatment alleles).
Changes in the mean and variance in fitness, ΔM and ΔV, are expected to depend on the rate at which new deleterious mutations arise, U, and their average effect on fitness, 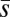 . In principle, the observed differences between our loaded and unloaded treatments with respect to ΔM and ΔV could result from differences in U,
. In principle, the observed differences between our loaded and unloaded treatments with respect to ΔM and ΔV could result from differences in U, 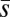 , or both. To attempt to distinguish between these possibilities, we calculated the Bateman–Mukai estimators Umin, the minimum mutation rate, and
, or both. To attempt to distinguish between these possibilities, we calculated the Bateman–Mukai estimators Umin, the minimum mutation rate, and 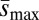 , the maximum average mutational effect, for each group, following Lynch and Walsh (ref. 26, pp. 342–343). Bateman–Mukai estimates should be regarded with caution because the method relies on a number of assumptions and the values obtained can depend strongly on the assay conditions used. However, our use of them here is primarily for comparison between treatments rather than to obtain realistic point estimates of U and
, the maximum average mutational effect, for each group, following Lynch and Walsh (ref. 26, pp. 342–343). Bateman–Mukai estimates should be regarded with caution because the method relies on a number of assumptions and the values obtained can depend strongly on the assay conditions used. However, our use of them here is primarily for comparison between treatments rather than to obtain realistic point estimates of U and 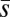 .
.
On average, Umin per haploid second chromosome was approximately 2.8 times greater in the loaded treatments (Fig. 2A), and 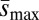 was only approximately 1.3 times greater in the loaded treatments (Fig. 2B), suggesting that the differences in ΔM and ΔV we observed were more likely a result of differences in the mutation rate than differences in mutational effects. Although we do not test for differences in mutational parameters between loaded treatments with one vs. two treatment alleles, we note that, on average, ΔM, ΔV, and Umin, but not
was only approximately 1.3 times greater in the loaded treatments (Fig. 2B), suggesting that the differences in ΔM and ΔV we observed were more likely a result of differences in the mutation rate than differences in mutational effects. Although we do not test for differences in mutational parameters between loaded treatments with one vs. two treatment alleles, we note that, on average, ΔM, ΔV, and Umin, but not 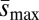 , were greater in loaded treatments that carried two treatment alleles than in those that carried one (Figs. 1 B and D and 2). A summary of sample sizes and results for each genetic background is given in Table S1.
, were greater in loaded treatments that carried two treatment alleles than in those that carried one (Figs. 1 B and D and 2). A summary of sample sizes and results for each genetic background is given in Table S1.
Fig. 2.

Estimates of minimum haploid second chromosome mutation rate, Umin, and maximum average effect of new mutations, 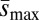 . (A) Compared with the unloaded treatment, UL, average Umin was approximately 2.8 times greater for the loaded treatment, L (∼2.7 times for those carrying one treatment allele, L1, and ∼2.8 times for those carrying two alleles, L2). (B) Compared with the unloaded treatment, average
. (A) Compared with the unloaded treatment, UL, average Umin was approximately 2.8 times greater for the loaded treatment, L (∼2.7 times for those carrying one treatment allele, L1, and ∼2.8 times for those carrying two alleles, L2). (B) Compared with the unloaded treatment, average 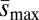 was only approximately 1.3 times greater for loaded treatment (∼1.4 times for those carrying one treatment allele and ∼1.3 times for those carrying two treatment alleles). Error bars represent ± 1 SE among different backgrounds within the loaded treatments.
was only approximately 1.3 times greater for loaded treatment (∼1.4 times for those carrying one treatment allele and ∼1.3 times for those carrying two treatment alleles). Error bars represent ± 1 SE among different backgrounds within the loaded treatments.
If mutation rates depend on individual quality, we expect the effects of treatment alleles on the mutation rate to be related to their effects on quality, i.e., those genotypes of the lowest quality should have the highest mutation rate. Near the beginning of the experiment, we measured the effects of the treatment third chromosomes on body mass as an index of their effect on individual quality. We found that this measure of their effect on mass was strongly and significantly correlated with their effect on ΔM (Spearman ρ = 0.82, P < 0.005; Fig. 3A) and with Umin (Spearman ρ = −0.8, P < 0.01; Fig. 3B). In contrast, there was no significant correlation with 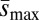 (Spearman ρ = 0.51, P > 0.1; Fig. 3C), providing further support for the interpretation that U rather than
(Spearman ρ = 0.51, P > 0.1; Fig. 3C), providing further support for the interpretation that U rather than 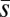 differs among treatments.
differs among treatments.
Fig. 3.

Correlations between the effects of treatment mutations on mass and their effects on ΔM, Umin, and 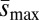 . Mass (measured relative to standard competitor genotypes reared in the same vials) was significantly correlated with ΔM (A) (ρ ∼ 0.82, P < 0.005) and with Umin (B) (ρ = −0.8, P < 0.01), but not with
. Mass (measured relative to standard competitor genotypes reared in the same vials) was significantly correlated with ΔM (A) (ρ ∼ 0.82, P < 0.005) and with Umin (B) (ρ = −0.8, P < 0.01), but not with 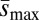 (C) (ρ ∼ 0.51, P > 0.1). Linear regression lines are shown for the two mutational properties ΔM and U that were significantly correlated with mass.
(C) (ρ ∼ 0.51, P > 0.1). Linear regression lines are shown for the two mutational properties ΔM and U that were significantly correlated with mass.
Discussion
The spontaneous deleterious mutation rate is critical to a number of topics in biology, because of the potentially large cumulative effects of deleterious alleles on fitness (1, 4, 5). In the past decade, it has become clear that mutation rates in many multicellular species, including humans, are sufficiently high to cause large mutation loads (2, 3). However, many aspects of this important trait remain unexplored. In particular, few studies have addressed mutation rate heterogeneity within populations. Because variation in phenotypic condition must reflect variation in cellular processes, it is reasonable to expect variation in the mutation rate among individuals when there is variation in condition. There is suggestive evidence that mutation rates in eukaryotes can vary in response to environmental and genetic stress (7, 13, 22, 27). Our experimental design allowed us to compare mutation rates among 11 different genotypes of varying quality. We found that poor-quality genotypes bearing deleterious alleles caused a large and significant increase in the spontaneous mutation rate (Figs. 1 and 2). The striking relationship between each genotype's effect on mass and its effect on mutation rate (Fig. 3) suggests that deleterious alleles indirectly affect the mutation rate through their effects on individual quality (i.e., deleterious alleles reduce quality, causing a decrease in mass and an increase in the mutation rate). As noted earlier, the values of ΔM and Umin presented here depend on the assumptions underlying the estimation procedure and so may differ from the true values. However, because the same statistical procedure was applied to all treatments, the estimation method alone should not cause either differences between loaded and unloaded treatments (Figs. 1 and 2) or correlations between mutation estimates and mass (Fig. 3). These patterns should only occur if genetic quality affects mutation.
Our experiment was not designed to determine the underlying mechanism behind this relationship. Reductions in quality could result in metabolic changes that affect the potential for oxidative DNA damage (but see ref. 28); alternatively, individuals of low quality may invest less in DNA repair or high-fidelity replication. Determining the molecular mechanisms that govern the association between individual quality and mutation rate should be a goal of future studies. In addition, we cannot exclude the possibility that some of the genes we used to manipulate genetic quality have an intimate involvement with the molecular mechanisms of DNA replication fidelity that has yet to be identified.
Based on Fig. 3 A and B, deleterious alleles causing a decrease in mass of 10% relative to WT are expected to cause a 104% increase in the rate of mutational decline (ΔM) and a 77% increase in the deleterious mutation rate (Umin). This high degree of sensitivity of mutation rate to individual quality variation could have a substantial impact on mutation rates and mean fitness in natural populations (29) and could contribute to the variation among empirical mutation rate estimates from different strains within species (6, 14). When mutation rates are related to fitness, as implied by the relationship with genetic quality we observe, theory predicts an increased genetic load and reduced mean fitness of sexual populations relative to asexuals (29). In small populations, deleterious mutations may drift to fixation, leading to negative population growth and further fixations in a process known as mutational meltdown (30, 31). Our results suggest that mutations will occur at a higher rate as fitness declines, thereby accelerating this process, especially in finite asexual populations (32). The positive mutational feedback loop implied by our data, whereby poor-quality genotypes produce offspring of even poorer quality, is also likely to alter classic models for the evolution of selfing (29). Not only would inbred genotypes have a direct reduction in fitness as a result of inbreeding depression, but the genetic stress of inbreeding depression would lead to the production of additional mutations in their offspring. The pattern we observe could also have positive effects on populations by increasing the rate of beneficial mutation in new environments. If novel environmental conditions increase the mutation rate by reducing individual quality through a mismatch between genotype and environment, the resulting increase in genetic variance could accelerate adaptation (27).
Finally, our results complement studies that link environmental stress and toxicity with oxidative DNA damage and other mutational mechanisms in humans and other mammals (33–35). In the context of elevated mutation under environmental and genetic stress, the high existing germline and somatic mutation load of modern human populations (i.e., low genetic quality), coupled with reduced environmental quality from anthropogenic mutagens, may present serious immediate and long-term risks to human health in the form of cancer and heritable genetic disorders (5, 36).
Materials and Methods
Summary.
To examine the effect of genetic quality on the mutation rate, we conducted MA in multiple sets of lines. During MA, many initially identical replicate chromosomes are propagated independently through a series of population bottlenecks, preventing selection against new mutations. We modified this traditional approach by also allowing mutations to accumulate in lines that carried known deleterious treatment alleles. All MA lines shared the same initial copy of chromosome 2, but differed in the genetic quality of chromosome 3, because of the presence or absence of treatment alleles. After many generations, we assessed the fitness of the lines after replacing chromosome 3 in each line with a standard WT copy. This allowed us to examine the effect that genetic quality variation on the third chromosome had on the rate of mutational degradation of the second chromosome, while excluding the confounding fitness effects of the treatment alleles themselves.
Stocks and MA Line Maintenance.
Experiments were conducted by using standard Drosophila protocols. Experimental flies were derived from an outbred population originally collected in Dahomey (now Benin; in West Africa) in 1970 and maintained in the current laboratory for more than 3 y (>75 generations) before this experiment at a population size of several thousand adults. Phenotypic marker mutations were obtained from the Bloomington Drosophila Stock Center or from U. Tepass (University of Toronto, Toronto, ON, Canada) and introgressed into the Dahomey background through serial backcrossing, with the phenotypically dominant mutations used to manipulate genetic quality (i.e., treatment mutations) introgressed for at least 10 generations. All MA lines shared an initially identical copy of chromosome 2, which carried the recessive mutation bw. This was the focal chromosome where the accumulation of new mutations was assessed; we denote this focal chromosome as bw*. Each MA line carried a treatment third chromosome from the unloaded treatment (no mutations) or one of 11 loaded treatments: AntpNs, Bsb1, Dr1, Gl1, Ki1, sensLy-1 (one mutation), Gl1 H2, Ki1 kD, Dr1 KgV, and Bsb1 sensLy-1 (two mutations). Treatment mutations were selected for their dominant visible phenotype and known or suspected effects on fitness; none have been annotated with Gene Ontology terms relating to DNA repair or DNA replication (25).
Third chromosome copies were drawn at random from an outbred population, with one or two treatment alleles introgressed into a subset of these through 10 generations of serial backcrossing to ensure that each third chromosome was independently derived, and that MA lines containing a given treatment allele were not more closely related to one another than to lines from a different treatment, except with respect to the region of the genome closely linked to the treatment allele itself. For each MA line, the third chromosome was independently derived except in a subset of Bsb1 sensLy-1 lines, which were derived from existing lines of the same treatment at MA generation two as a result of unexpected line losses. All lines were treated as independent for the purposes of analysis.
Three control populations were generated at the beginning of the experiment by using flies homozygous for the same focal second chromosome as in the MA lines (Fig. S2). Each control population was maintained in 15 vials using 450 adults, with complete admixture each generation.
The recessive phenotypic marker mutations vg1 and se1 on homologous chromosomes were used to track the focal second chromosome, bw*, and the treatment third chromosome, T, across generations, respectively (Fig. S1). Each generation a single male of the appropriate genotype was placed in a new vial with four virgin females from a large outbred stock homozygous for vg1 and se1, i.e., one bw*/vg;T/se male × four vg/vg;se/se females. The appropriate genotype could then be identified in offspring by the absence of the vg and se phenotypes. In this crossing design chromosomes 2 and 3, as well as the Y chromosome, are transmitted intact to the subsequent generation.
Each generation, each MA line consisted of a primary mating vial, as described earlier, plus one to three backup vials generated in the same fashion by using different flies. More backups were established for some treatments because of higher observed rates of vial failure early in the experiment. During line maintenance, a backup vial was used in place of the primary vial if no flies of the appropriate type were available, generally as a result of a complete absence of offspring, suggesting male death or sterility. All use of backup vials, as well as line extinctions, was recorded. MA lines and control populations were maintained every 14 ± 1 d, with their incubator locations randomized, and flies were allowed to produce offspring for 6 to 7 d and then discarded. Adult flies from the control populations were discarded after 1 d of oviposition.
Fitness Assays.
Following 16, 30, and 46 generations of MA, four males carrying the focal second chromosome but not the treatment third chromosome (bw*/vg;se/se) were collected from each MA line and crossed to a standard marker stock (Fig. S1). Flies from each control population were subjected to the same crossing procedure, but were also bottlenecked to a single focal chromosome during the crossing procedure (Fig. S2). Flies for fitness assays were obtained after several additional crosses to obtain the desired genotypes, each cross consisting of multiple focal males and multiple females from standard marker or balancer stocks (Figs. S1 and S2). The final assay cross was one or two bw*/vg L;+/+ males with two bw*/CyO;+/+ virgin females [where L is a dominant marker (L2), CyO is a balancer chromosome to suppress recombination, and + is an outbred WT chromosome]. Several such vials were generated, and flies from each vial were transferred into new vials after 3 d and discarded after a further 3 d. Each assay vial was treated as an independent subreplicate for the purposes of analysis. Assay vials were coded using random numbers, their incubator locations were randomized, and they were scored “blind” 12 d after mating and again after a further 3 d (a small percentage of replicates were scored on day 12 only); the day-12 and day-15 scores for each vial were summed. Competitive offspring viability, Pfocal, was assessed as the number of bw*/bw* offspring relative to the total number of bw*/bw* and L/CyO offspring (Figs. S1 and S2). Replicates containing no L/CyO offspring or any offspring with an unexpected phenotype were excluded from all analyses. For control lines, crosses were performed to produce heterozygous (bw*A/bw*B) focal offspring, generated by using a “round-robin” approach within each control population (Fig. S2). Line mean Pfocal values were averaged within each control population, and then these values from the three control populations were averaged  . The relative fitness of each MA line i was calculated as follows:
. The relative fitness of each MA line i was calculated as follows:
where Pfocal[MA,i] represents the mean value across subreplicates within line i. A line was classified as lethal when Pfocal[MA,i] was less than 2.5% (i.e., ∼1 bw*/bw* fly per vial). In practice, the value for lethal lines was almost always 0%, indicating complete recessive lethality. Lethal lines were excluded from analyses to estimate mutational parameters. Rates of lethal mutation were calculated as described previously (37), with confidence limits based on the Agresti–Coull method for confidence limits of proportions. Lethal rates were consistent with published estimates for the second chromosome (38, 39), with no obvious differences among treatments (Table S2).
Mass Measurements.
To estimate the effects of treatment mutations on individual quality, approximately four focal males were obtained from each MA vial at generations seven and eight of MA, and their mass was assessed relative to approximately four standard vg/vg;se/se males from the same vial to control for density effects. Flies were dried at 70 °C for approximately 21 h and weighed on a microbalance.
Data Analysis.
Analyses were performed in the R statistical environment (40); all reported P values are from two-sided tests. Fitness data from generation 46 were analyzed by finding the difference between mean fitness in the unloaded treatment and mean fitness averaged over loaded treatments. A permutation test was performed by randomly assigning lines to treatments and calculating the difference in fitness between loaded and unloaded treatments from this permutated data set; 10,000 permutations were performed to generate a null distribution for this statistic. Recall that the lines were derived such that each line represents a random sample of the outbred laboratory stock population; only the treatment mutations and closely linked regions are shared between lines within a given background treatment. To calculate per-generation rates of change in mean and variance in fitness (ΔM and ΔV), only MA lines present in all three fitness assays were considered. ΔM was calculated for each third chromosome type as the slope from a linear regression of fitness on generations of MA, with a fixed intercept of 1. The SE of ΔM was calculated from these values. A null distribution for the difference in ΔM between loaded and unloaded treatments was determined by using randomization with 10,000 replicates.
Among-line variance at each assay time point and the sampling variance of this parameter were determined by using ANOVA to partition variance in fitness, following Lynch and Walsh (ref. 26, p. 556–561). ΔV was then estimated as the slope from a linear regression of among-line (“sire”) variance on generations of MA, with a fixed intercept of 0. To determine the SE of ΔV for each treatment, we used an ad hoc “simulated resampling” procedure. For each genetic background, a variance value for each assay time point was sampled from a normal distribution based on the observed sire variance and its sampling variance. A slope, ΔV , was calculated from these sampled values. This procedure was repeated 10,000 times, and the SD of the resulting distribution of slopes was used as the SE of ΔV in calculating the Bateman–Mukai estimators. (This procedure was tested by using simulated data sets within known ΔV values and performed as expected.) For each resampling, the difference in ΔV between loaded and unloaded treatments was also calculated. All 10,000 values were positive, indicating that ΔV is significantly greater in the loaded than the unloaded treatment. In contrast, when we performed this procedure on 1,000 artificial data sets in which there was no true difference between treatments, we found that almost all (995 of 1,000) had resampling distributions that spanned zero, as expected. Repeating this procedure with artificial data sets drawn from γ-distributions yielded similar results. Umin and 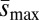 were calculated by using the estimates and sampling errors of ΔM and ΔV, following Lynch and Walsh (ref. 26, p. 342–343). Spearman rank correlation was used to assess the relationship between mass and other variables.
were calculated by using the estimates and sampling errors of ΔM and ΔV, following Lynch and Walsh (ref. 26, p. 342–343). Spearman rank correlation was used to assess the relationship between mass and other variables.
Supplementary Material
Acknowledgments
We thank C. Baer and P. Keightley for helpful discussion and a number of laboratory assistants, especially K. Chu, R. Fan, K.H.E. Ho, M. Kimura, and N. Vasileva. This research was funded by the Natural Sciences and Engineering Research Council (Canada) to N.P.S (Vanier Graduate Scholarship) and A.F.A (Discovery Grant).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118918109/-/DCSupplemental.
References
- 1.Haldane JBS. The effect of variation on fitness. Am Nat. 1937;71:337–349. [Google Scholar]
- 2.Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8:619–631. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- 3.Eöry L, Halligan DL, Keightley PD. Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol Biol Evol. 2010;27:177–192. doi: 10.1093/molbev/msp219. [DOI] [PubMed] [Google Scholar]
- 4.Lynch M, et al. Perspective: Spontaneous deleterious mutation. Evolution. 1999;53:645–663. doi: 10.1111/j.1558-5646.1999.tb05361.x. [DOI] [PubMed] [Google Scholar]
- 5.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baer CF, et al. Comparative evolutionary genetics of spontaneous mutations affecting fitness in rhabditid nematodes. Proc Natl Acad Sci USA. 2005;102:5785–5790. doi: 10.1073/pnas.0406056102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamb BC, Mandaokar S, Bahsoun B, Grishkan I, Nevo E. Differences in spontaneous mutation frequencies as a function of environmental stress in soil fungi at “Evolution Canyon,” Israel. Proc Natl Acad Sci USA. 2008;105:5792–5796. doi: 10.1073/pnas.0801995105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. The evolution of mutation rates: Separating causes from consequences. Bioessays. 2000;22:1057–1066. doi: 10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 9.Foster PL. Adaptive mutation: Implications for evolution. Bioessays. 2000;22:1067–1074. doi: 10.1002/1521-1878(200012)22:12<1067::AID-BIES4>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol Microbiol. 2006;60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- 11.Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci USA. 2011;108:13659–13664. doi: 10.1073/pnas.1104681108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leigh EG., Jr Natural selection and mutability. Am Nat. 1970;104:301–305. [Google Scholar]
- 13.Ávila V, et al. Increase of the spontaneous mutation rate in a long-term experiment with Drosophila melanogaster. Genetics. 2006;173:267–277. doi: 10.1534/genetics.106.056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haag-Liautard C, et al. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature. 2007;445:82–85. doi: 10.1038/nature05388. [DOI] [PubMed] [Google Scholar]
- 15.Wilson AJ, Nussey DH. What is individual quality? An evolutionary perspective. Trends Ecol Evol. 2010;25:207–214. doi: 10.1016/j.tree.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Rowe L, Houle D. The lek paradox and the capture of genetic variance by condition dependent traits. Proc Biol Sci. 1996;263:1415–1421. [Google Scholar]
- 17.Ames BN, Shigenaga MK, Gold LS. DNA lesions, inducible DNA repair, and cell division: Three key factors in mutagenesis and carcinogenesis. Environ Health Perspect. 1993;101(suppl 5):35–44. doi: 10.1289/ehp.93101s535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin AP, Palumbi SR. Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci USA. 1993;90:4087–4091. doi: 10.1073/pnas.90.9.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanaar R, Hoeijmakers JHJ, van Gent DC. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8:483–489. doi: 10.1016/s0962-8924(98)01383-x. [DOI] [PubMed] [Google Scholar]
- 20.Gillooly JF, Allen AP, West GB, Brown JH. The rate of DNA evolution: Effects of body size and temperature on the molecular clock. Proc Natl Acad Sci USA. 2005;102:140–145. doi: 10.1073/pnas.0407735101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada K, Blount JD, Sharma MD, Snook RR, Hosken DJ. Male attractiveness, fertility and susceptibility to oxidative stress are influenced by inbreeding in Drosophila simulans. J Evol Biol. 2011;24:363–371. doi: 10.1111/j.1420-9101.2010.02170.x. [DOI] [PubMed] [Google Scholar]
- 22.Agrawal AF, Wang AD. Increased transmission of mutations by low-condition females: Evidence for condition-dependent DNA repair. PLoS Biol. 2008;6:e30. doi: 10.1371/journal.pbio.0060030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjedov I, et al. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 24.Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19:791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 25.Tweedie S, et al. FlyBase Consortium FlyBase: Enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37(database issue):D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sunderland, MA: Sinauer; 1998. [Google Scholar]
- 27.Goho S, Bell G. Mild environmental stress elicits mutations affecting fitness in Chlamydomonas. Proc Biol Sci. 2000;267:123–129. doi: 10.1098/rspb.2000.0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joyner-Matos J, Bean LC, Richardson HL, Sammeli T, Baer CF. No evidence of elevated germline mutation accumulation under oxidative stress in Caenorhabditis elegans. Genetics. 2011;189:1439–1447. doi: 10.1534/genetics.111.133660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal AF. Genetic loads under fitness-dependent mutation rates. J Evol Biol. 2002;15:1004–1010. [Google Scholar]
- 30.Lynch M, Conery J, Bürger R. Mutation accumulation and the extinction of small populations. Am Nat. 1995;146:489–518. [Google Scholar]
- 31.Lynch M, Conery J, Bürger R. Mutational meltdowns in sexual populations. Evolution. 1995;49:1067–1080. doi: 10.1111/j.1558-5646.1995.tb04434.x. [DOI] [PubMed] [Google Scholar]
- 32.Shaw FH, Baer CF. Fitness-dependent mutation rates in finite populations. J Evol Biol. 2011;24:1677–1684. doi: 10.1111/j.1420-9101.2011.02320.x. [DOI] [PubMed] [Google Scholar]
- 33.Epel ES, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci USA. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yauk C, et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci USA. 2008;105:605–610. doi: 10.1073/pnas.0705896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Somers CM, Cooper DN. Air pollution and mutations in the germline: Are humans at risk? Hum Genet. 2009;125:119–130. doi: 10.1007/s00439-008-0613-6. [DOI] [PubMed] [Google Scholar]
- 36.Baer CF. Does mutation rate depend on itself. PLoS Biol. 2008;6:e52. doi: 10.1371/journal.pbio.0060052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukai T, Chigusa SI, Mettler LE, Crow JF. Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics. 1972;72:335–355. doi: 10.1093/genetics/72.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fry JD, Keightley PD, Heinsohn SL, Nuzhdin SV. New estimates of the rates and effects of mildly deleterious mutation in Drosophila melanogaster. Proc Natl Acad Sci USA. 1999;96:574–579. doi: 10.1073/pnas.96.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashburner M, Golic KG, Hawley RS. Drosophila: A Laboratory Handbook. 2nd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2005. [Google Scholar]
- 40.R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.