

Abstract
Predicting organismal phenotypes from genotype data is important for plant and animal breeding, medicine, and evolutionary biology. Genomic-based phenotype prediction has been applied for single-nucleotide polymorphism (SNP) genotyping platforms, but not using complete genome sequences. Here, we report genomic prediction for starvation stress resistance and startle response in Drosophila melanogaster, using ∼2.5 million SNPs determined by sequencing the Drosophila Genetic Reference Panel population of inbred lines. We constructed a genomic relationship matrix from the SNP data and used it in a genomic best linear unbiased prediction (GBLUP) model. We assessed predictive ability as the correlation between predicted genetic values and observed phenotypes by cross-validation, and found a predictive ability of 0.239±0.008 (0.230±0.012) for starvation resistance (startle response). The predictive ability of BayesB, a Bayesian method with internal SNP selection, was not greater than GBLUP. Selection of the 5% SNPs with either the highest absolute effect or variance explained did not improve predictive ability. Predictive ability decreased only when fewer than 150,000 SNPs were used to construct the genomic relationship matrix. We hypothesize that predictive power in this population stems from the SNP–based modeling of the subtle relationship structure caused by long-range linkage disequilibrium and not from population structure or SNPs in linkage disequilibrium with causal variants. We discuss the implications of these results for genomic prediction in other organisms.
Author Summary
The ability to accurately predict values of complex phenotypes from genotype data will revolutionize plant and animal breeding, personalized medicine, and evolutionary biology. To date, genomic prediction has utilized high-density single-nucleotide polymorphism (SNP) genotyping arrays, but the availability of sequence data opens new frontiers for genomic prediction methods. This article is the first application of genomic phenotype prediction using whole-genome sequence data in a substantial sample of a higher eukaryote. We use ∼2.5 million SNPs with minor allele frequency greater than 2.5% derived from genomic sequences of the “Drosophila Genetic Reference Panel” to predict phenotypes for two traits, starvation resistance and startle-induced locomotor behavior. We systematically address prediction within versus across sexes, genomic best linear unbiased prediction (GBLUP) versus a Bayesian approach, and the effect of SNP density. We find that (i) genomic prediction can be efficiently implemented using sequence data via GBLUP, (ii) there is little gain in predictive ability if the number of SNPs is increased above 150,000, and (iii) neither implicit nor explicit marker selection substantially improves the predictive ability. Although the findings must be seen against the background of small sample sizes, the results illustrate both the potential of the approach and the challenges ahead.
Introduction
Most efforts to understand the genetic architecture of quantitative traits have focused on mapping the variants causing phenotypic variation in quantitative trait locus (QTL) mapping populations derived from crosses between lines genetically divergent for the trait, or in association mapping populations, with the goal of understanding the biological underpinnings of trait variation [1]. However, the ability to accurately predict quantitative trait phenotypes from information on genotypic variation in the absence of knowledge of causal variants will revolutionize evolutionary biology, medicine and human biology, and breeding of agriculturally important plant and animal species. The premise of personalized medicine is based on prediction of individual genetic risk to disease from genome-wide association studies [2], [3], and the ability to select individuals or lines in animal and plant breeding programs based on genotypic information circumvents the costly process of progeny testing and reduces the generation interval in applied breeding programs, leading to greater efficiency [4], [5].
In classical animal and plant breeding, the genetic quality of individuals or lines is predicted from phenotypic values of selection candidates and their relatives. The widely used Best Linear Unbiased Prediction (BLUP, [6]) method models the covariance structures between individuals via the numerator relationship matrix, which is constructed from known pedigree information and thus reflects expected relationships between individuals (i.e. the proportion of shared alleles of identical ancestral origin) given the pedigree. The advent of high-throughput genotyping platforms for many agronomic species [7] enabled genotyping large numbers of individuals for dense panels of single nucleotide polymorphisms (SNPs) spanning the genome. The expected, pedigree-based numerator relationship matrix can then be replaced by a realized, genome-based relationship matrix (often called the “genomic” relationship matrix, [8]). This approach is equivalent to a random regression approach in which all SNP genotypes are simultaneously accounted for as explanatory variables in a multiple regression model [9]. In animal and plant breeding, selection based on genome-based predictions of genetic values is expected to massively increase genetic progress [4], [10] and has quickly found its way into widespread practical application (see [4], [5] for reviews).
Genome based-prediction follows a different paradigm than genome wide association studies (GWAS). GWAS identify single molecular variants associated with phenotypic variability using individual statistical tests for significance of each variant. Genome-based prediction uses the entire genomic variability captured by the available marker set to explain the observed phenotypic variation, and does not rely on selection of single loci based on significance tests. Standard prediction methods are thought to work for traits with a highly polygenic or even infinitesimal [11] genetic architecture, where the effect of a single variant is too small to be captured by a statistical test in a GWAS. There is strong empirical evidence that many quantitative traits have such a highly polygenic genetic architecture in farm animals [12], agriculturally used plants [13], model organisms and humans [14], [15].
With the advent of next generation sequencing technologies, it is now feasible to implement genomic prediction based on complete genome sequences of higher organisms. While these techniques have only been applied to individuals or cohorts of limited size [16] to date, initiatives to sequence larger panels are under way [17], [18], and genotyping by whole genome resequencing will become a standard technology in the foreseeable future.
The accuracy of prediction methods based on marker data depends on the heritability of the trait, its genetic architecture (number of loci affecting trait variation, mode of inheritance, and distribution of allelic effects, [19]), the LD reflecting effective population size, the size of the genome, the marker density and the sample size used in the statistical analysis [20]. Various methods of prediction incorporating genomic information have been studied on real and simulated data, including Genomic Best Linear Unbiased Prediction (GBLUP) approaches with genomic relationship matrices [8], Random Regression BLUP (RRBLUP), Bayesian linear regression methods [10], [21] or fully non-parametric approaches [22]–[25].
GBLUP approaches are based on a linear model for the phenotypic values, which encompasses a vector of random genetic values of individuals whose covariance structure is inferred from genomic data. The linear model underlying the RRBLUP approach includes a vector of random marker effects (instead of a vector of genetic values) which are assumed to be drawn from the same normal distribution and uncorrelated. The model primarily provides estimates of SNP effects, but estimated genetic values of individuals can be derived as linear combinations of the estimated SNP effects, yielding the same predictions of individual genotypic or phenotypic values as GBLUP. The BayesB method [10], on the other hand, fits only a small fraction of the available markers to conform with the assumption that most loci are expected to have zero effect on the phenotype, and the remaining non-zero marker effects are drawn from normal distributions with random variances.
It has been suggested [26] that differences between prediction methods will become more pronounced with the availability of full genome sequence data. According to a study with simulated data [26], RRBLUP and equivalent GBLUP procedures do not take full advantage of high-density marker data if the number of causal SNPs is small, while approaches with an implicit feature selection such as BayesB might be more accurate. If, on the other hand, the number of causal loci is large, RRBLUP or GBLUP methods may yield accurate predictions because the assumption that every SNP has an effect is closer to reality.
Implementing genomic prediction with full genome sequence data raises a number of questions. What is the most efficient way to incorporate the complete genomic information in prediction? How much predictive ability is gained by using whole genome sequence data compared to high density SNP panels? Is it possible to increase predictive ability by a pre-selection of SNPs or models with an internal feature selection? How comparable are the results of genomic prediction and genome wide association? Here, we address these questions empirically based on full genomic sequences of a population of Drosophila melanogaster inbred lines. The inbred lines have been sequenced, and constitute the Drosophila Genetics Reference Panel (DGRP), a new community resource for genetic studies of complex traits [27].
We report the results of a full sequence based genomic prediction for two quantitative traits, starvation stress resistance and locomotor startle response, both of which display considerable genetic variation in natural populations and respond rapidly to artificial selection [28]–[30]. We used whole-genome sequences determined on the Illumina platform for  DGRP-lines for starvation resistance (startle response) [27]. Our reference method is a GBLUP approach in which ∼2.5 million polymorphic SNPs are used to derive a genomic relationship matrix [8]. We evaluated predictive ability via cross-validation (CV), and compared prediction within vs. across sexes, various SNP densities, and training set sizes. We assessed whether BayesB is superior over GBLUP given full genome sequence data [26], and compared our genomic prediction results with those of GWAS conducted on the same DGRP lines [27].
DGRP-lines for starvation resistance (startle response) [27]. Our reference method is a GBLUP approach in which ∼2.5 million polymorphic SNPs are used to derive a genomic relationship matrix [8]. We evaluated predictive ability via cross-validation (CV), and compared prediction within vs. across sexes, various SNP densities, and training set sizes. We assessed whether BayesB is superior over GBLUP given full genome sequence data [26], and compared our genomic prediction results with those of GWAS conducted on the same DGRP lines [27].
To our knowledge, this is the first application of genomic prediction on empirical whole genome sequence in a substantial sample of a higher organism. However, this study, as well as all previous association studies, only assesses the effects of common SNPs, since the effects of rare alleles cannot be estimated due to the small sample of sequenced lines. The results illustrate both the potential of the approach and challenges to be addressed in the future.
Results
Genomic Best Linear Unbiased Prediction (GBLUP)
We constructed a genomic relationship matrix [8] from ∼2.5 million SNPs for which the minor allele was present in at least four of the DGRP lines [27]. A histogram of the off-diagonal elements of this matrix for  DGRP lines used in the GBLUP analyses (Figure 1) and a corresponding heatmap (Figure 2) show that there were no large blocks of high genomic relationship among the lines. The average genomic relationship is close to zero, as expected, but there is considerable variance around this average (Figure 1), as indicated by two block of lines with average genomic relationships within each block of
DGRP lines used in the GBLUP analyses (Figure 1) and a corresponding heatmap (Figure 2) show that there were no large blocks of high genomic relationship among the lines. The average genomic relationship is close to zero, as expected, but there is considerable variance around this average (Figure 1), as indicated by two block of lines with average genomic relationships within each block of  and
and  (Figure 2). We performed genomic prediction for starvation stress resistance and locomotor startle response. The phenotypes used were the medians of many (
(Figure 2). We performed genomic prediction for starvation stress resistance and locomotor startle response. The phenotypes used were the medians of many ( ) individually tested males and females for each line, or the average of the male and female medians (Table S1). We used several cross-validation (CV) procedures for each trait (Table 1). In the
) individually tested males and females for each line, or the average of the male and female medians (Table S1). We used several cross-validation (CV) procedures for each trait (Table 1). In the  -fold CV, predictive ability was
-fold CV, predictive ability was  for starvation resistance and
for starvation resistance and  for startle response. In human studies the efficiency of a predictor is reported as the squared correlation
for startle response. In human studies the efficiency of a predictor is reported as the squared correlation  rather than
rather than  [31], so that in terms of variance explained the estimates were
[31], so that in terms of variance explained the estimates were  for starvation resistance and
for starvation resistance and  for startle response. The observed accuracy depends on the size of the training set (Figure 3), with decreasing accuracies obtained with smaller training sets. Predictive abilities are roughly halved for both traits when using only
for startle response. The observed accuracy depends on the size of the training set (Figure 3), with decreasing accuracies obtained with smaller training sets. Predictive abilities are roughly halved for both traits when using only  instead of
instead of  of the data to train the model. Maximum likelihood estimates of narrow-sense heritabilities based on the GBLUP model using the genomic relationship matrix were
of the data to train the model. Maximum likelihood estimates of narrow-sense heritabilities based on the GBLUP model using the genomic relationship matrix were  in all analyses (Table S2), reflecting the fact that phenotypes are averages over many replicates and thus residual variance is minimal. Hence, the phenotypes used represent the line genotypes with maximum accuracy, which is the ideal case for training the genomic model.
in all analyses (Table S2), reflecting the fact that phenotypes are averages over many replicates and thus residual variance is minimal. Hence, the phenotypes used represent the line genotypes with maximum accuracy, which is the ideal case for training the genomic model.
Figure 1. Histogram of the offdiagonal elements of the genomic relationship matrix  .
.

The genomic relationship matrix  was calculated according to [8] using 157 lines and 2.5 million SNPs.
was calculated according to [8] using 157 lines and 2.5 million SNPs.
Figure 2. Heatmap of the genomic relationship matrix  .
.

The genomic relationship matrix  was calculated according to [8] using 157 lines and 2.5 million SNPs. The “S” after the line-ID indicates that the line belongs to the set of lines for which phenotypic records for startle response were also available (in addition to the phenotypic records of starvation resistance).
was calculated according to [8] using 157 lines and 2.5 million SNPs. The “S” after the line-ID indicates that the line belongs to the set of lines for which phenotypic records for startle response were also available (in addition to the phenotypic records of starvation resistance).
Table 1. Average correlations between predicted genetic values and observed phenotypes for different CV procedures with GBLUP and different traits.
| type of CV | starvation resistance | startle response |
| (4∶1)-CV1 all2 | 0.2393 (0.008) | 0.230 (0.012) |
| (3∶2)-CV all | 0.213 (0.006) | 0.216 (0.011) |
| (2∶3)-CV all | 0.176 (0.006) | 0.181 (0.010) |
| (1∶4)-CV all | 0.124 (0.006) | 0.128 (0.006) |
| (4∶1)-CV male - female4 | 0.164 (0.007) | 0.217 (0.011) |
| (4∶1)-CV female - male | 0.182 (0.007) | 0.235 (0.012) |
| (4∶1)-CV male - male | 0.203 (0.008) | 0.230 (0.012) |
| (4∶1)-CV female - female | 0.254 (0.009) | 0.216 (0.011) |
“ -CV” means:
-CV” means:  parts are used as training set and
parts are used as training set and  parts are used as validation set.
parts are used as validation set.
The average of the medians of male and female measurements was used to predict line phenotypes. Predicted phenotypes were then correlated with the averages of the medians of male and female measurements.
Average correlation between predicted genetic values and observed phenotypes. Results are averages over 20 replicates. Standard errors of the means in parentheses.
“CV sex sex
sex ” means: Medians of measurements of sex
” means: Medians of measurements of sex were used in the training set, medians of sex
were used in the training set, medians of sex were used in the validation set.
were used in the validation set.
Figure 3. Accuracy of prediction of GBLUP for CVs with different numbers of lines in the training set.

Each boxplot illustrates the average accuracies for 20 replicates of the CV procedure using GBLUP. The left (right) plot shows accuracies for starvation resistance (startle response). The solid line is the curve of [20] fitted to the empirical data, which results in estimates of  and
and  for starvation resistance and startle response. All 2.5 million SNPs were used to construct the genomic relationship matrix in the GBLUP model.
for starvation resistance and startle response. All 2.5 million SNPs were used to construct the genomic relationship matrix in the GBLUP model.
Using male performance data to train the model and using the results to predict the female performance (or vice versa) does not affect the predictive ability for startle response, but substantially reduces the predictive ability for starvation resistance, reflecting a higher degree of genotype by sex interaction in this trait ([27], and see below). Prediction is more accurate in females than in males ( vs.
vs.
 ) for starvation resistance, while there is little difference for startle response.
) for starvation resistance, while there is little difference for startle response.
A series of  -fold CVs for starvation resistance using different SNP densities showed that predictive ability remained almost constant if every
-fold CVs for starvation resistance using different SNP densities showed that predictive ability remained almost constant if every  SNP (∼150,000 SNPs) was used to construct the genomic relationship matrix (Figure 4). The predictive ability began to deteriorate when fewer than
SNP (∼150,000 SNPs) was used to construct the genomic relationship matrix (Figure 4). The predictive ability began to deteriorate when fewer than  SNPs were used, but only vanished completely when as few as ∼2,500 SNPs (every
SNPs were used, but only vanished completely when as few as ∼2,500 SNPs (every  SNP) were used. The corresponding LD distribution for SNP neighbors for different SNP densities is shown in Figure 5, illustrating the extreme short-range extent of LD in the D. melanogaster genome. The average LD between SNPs (after imputation) whose distance lay in the interval
SNP) were used. The corresponding LD distribution for SNP neighbors for different SNP densities is shown in Figure 5, illustrating the extreme short-range extent of LD in the D. melanogaster genome. The average LD between SNPs (after imputation) whose distance lay in the interval  bp was
bp was  for the autosomes and
for the autosomes and  for the X-chromosome. Long-range LD between pairs of loci at the opposite ends of chromosome arms or across different chromosome arms was on average
for the X-chromosome. Long-range LD between pairs of loci at the opposite ends of chromosome arms or across different chromosome arms was on average  both for the autosomes and the X-chromosome.
both for the autosomes and the X-chromosome.
Figure 4. Predictive ability of 5-fold CV with GBLUP for starvation resistance using different numbers of SNPs.

Each boxplot shows the average predictive abilities for 20 replicates of 5-fold CV using GBLUP. For the CVs leading to the  -th boxplot, every 2
-th boxplot, every 2 -th SNP was used to build the genomic relationship matrix
-th SNP was used to build the genomic relationship matrix  according to [8]. This was done for the thinning factors
according to [8]. This was done for the thinning factors  0
0 10. The red dots indicate the average predictive abilities.
10. The red dots indicate the average predictive abilities.
Figure 5. The distribution of  between SNP neighbors for different SNP densities.
between SNP neighbors for different SNP densities.

For the  -th stacked bar, every
-th stacked bar, every  -th SNP was used,
-th SNP was used,  0
0 10. Then, the distribution of
10. Then, the distribution of  for the resulting SNP neighbors was calculated.
for the resulting SNP neighbors was calculated.
For starvation resistance, the influence of the minor allele frequency of the SNPs used on the predictive ability was assessed with a series of 5-fold CVs using SNP sets with different average minor allele frequency. We find that the variability of the predictive ability increases when the average minor allele frequency of the SNPs used to construct the genomic relationship matrix is decreased (Figure S1). In  replicates of an additional 5-fold CV, in which we randomly chose
replicates of an additional 5-fold CV, in which we randomly chose  SNPs to build the genomic relationship matrix, an average predictive ability of
SNPs to build the genomic relationship matrix, an average predictive ability of  was obtained, which is in the range obtained when every
was obtained, which is in the range obtained when every  SNP (∼77,817 SNPs) was used (
SNP (∼77,817 SNPs) was used ( , Figure 4). Running
, Figure 4). Running  replicates of a 5-fold CV using
replicates of a 5-fold CV using  randomly chosen blocks of adjacent SNPs (each block consisting of
randomly chosen blocks of adjacent SNPs (each block consisting of  SNPs) led to an average predictive ability of
SNPs) led to an average predictive ability of  .
.
To analyze whether the predictive ability is due to lines which are more highly related, we ran an additional 5-fold CV with  replicates in which the two groups of higher overall relatedness (Figure 2) were excluded. Here we found an average predictive ability of
replicates in which the two groups of higher overall relatedness (Figure 2) were excluded. Here we found an average predictive ability of  for starvation resistance, which is larger than the average predictive ability we obtained using all lines (
for starvation resistance, which is larger than the average predictive ability we obtained using all lines ( ). For startle response, excluding the two groups led to a decrease in predictive ability (
). For startle response, excluding the two groups led to a decrease in predictive ability ( in comparison to
in comparison to  ).
).
Effective population size derived from empirical accuracies of genomic prediction
The accuracy of genomic prediction is a function of a number of quantities, including the size of the training set and the effective population size  [20].
[20].  has an effect on the number of independently segregating chromosome segments,
has an effect on the number of independently segregating chromosome segments,  , in a population (the larger
, in a population (the larger  , the larger
, the larger  ); and the predictive ability of GBLUP is higher when the number of segments is small. By varying the size of the training set in a series of CVs, we can estimate
); and the predictive ability of GBLUP is higher when the number of segments is small. By varying the size of the training set in a series of CVs, we can estimate  by fitting a curve through the empirical accuracies obtained (Figure 3).
by fitting a curve through the empirical accuracies obtained (Figure 3).
We estimated  for starvation resistance and
for starvation resistance and  for startle response. The coefficient of determination of the fitted curve was
for startle response. The coefficient of determination of the fitted curve was  for starvation resistance (startle response). The bias corrected empirical
for starvation resistance (startle response). The bias corrected empirical  confidence intervals for the
confidence intervals for the  estimates obtained with bootstrapping [32] were
estimates obtained with bootstrapping [32] were  for starvation resistance and
for starvation resistance and  for startle response.
for startle response.
The effective population size in the Raleigh population (from which the DGRP-lines were drawn) was estimated to be ∼19,000 in  , with a massive fluctuation between years [33]. Our estimates of
, with a massive fluctuation between years [33]. Our estimates of  correspond to
correspond to  independently segregating chromosome segments. In this formula
independently segregating chromosome segments. In this formula  is the length of the female genome in Morgans (there is no recombination in male Drosophila). Since the sequenced animals resulted from
is the length of the female genome in Morgans (there is no recombination in male Drosophila). Since the sequenced animals resulted from  generations of full sib mating following the original sampling from the Raleigh population, the DGRP lines are not expected to have the same
generations of full sib mating following the original sampling from the Raleigh population, the DGRP lines are not expected to have the same  as the original population and are consequently expected to have a different
as the original population and are consequently expected to have a different  .
.
We can use the curves fitted through the empirical accuracies (Figure 3), to predict the expected accuracy of prediction for an arbitrarily large size of the training set: If  lines were available in the training set, the curve would predict accuracies of ∼0.58 for starvation resistance and startle response. This value was obtained by using
lines were available in the training set, the curve would predict accuracies of ∼0.58 for starvation resistance and startle response. This value was obtained by using  and
and  as well as
as well as  and
and  in the modified formula of [20].
in the modified formula of [20].
Effective population size derived directly from linkage disequilibrium
We also estimated the effective population size based on LD directly. For a distance bin of  Morgan we obtained average LD-values of
Morgan we obtained average LD-values of  for chromosome 2L (2R, 3L, 3R, X). These values correspond to an estimated effective population size of
for chromosome 2L (2R, 3L, 3R, X). These values correspond to an estimated effective population size of  , approximately
, approximately  generations ago. The average estimated effective population size is
generations ago. The average estimated effective population size is  , which is in the range of the estimates based on the observed accuracies.
, which is in the range of the estimates based on the observed accuracies.
Genomic prediction with SNP selection
Genomic prediction might be improved if we only fit SNPs which are associated with variance in a trait, because we then concentrate on the biologically relevant genomic regions, and excluding SNPs which are not associated with the trait reduces statistical noise. We tested this hypothesis using the starvation resistance data. We identified the  SNPs with the highest absolute estimated effect or the highest estimated genetic variance, respectively, in the training set of the respective
SNPs with the highest absolute estimated effect or the highest estimated genetic variance, respectively, in the training set of the respective  of the folds in a
of the folds in a  -fold CV. We then used these subsets of selected SNPs to predict the phenotype in the remaining
-fold CV. We then used these subsets of selected SNPs to predict the phenotype in the remaining  of the fold. Predictive ability was improved by
of the fold. Predictive ability was improved by  over the reference scenario when using the
over the reference scenario when using the  SNPs with largest effects (average predictive ability of
SNPs with largest effects (average predictive ability of  in comparison to
in comparison to  ). Using the
). Using the  SNPs with greatest variance explained, predictive ability was improved by
SNPs with greatest variance explained, predictive ability was improved by  (average predictive ability of
(average predictive ability of  ). In both cases, the improvement is marginal and provides little support for the idea of SNP pre-selection.
). In both cases, the improvement is marginal and provides little support for the idea of SNP pre-selection.
We also compared our GBLUP results to those from a method which does not assume that all SNP effects are drawn from the same normal distribution and carries out an internal feature selection. We ran  replicates of a
replicates of a  -fold CV for starvation resistance using BayesB [10]. In each round of the Markov Chain Monte Carlo based procedure (see Methods),
-fold CV for starvation resistance using BayesB [10]. In each round of the Markov Chain Monte Carlo based procedure (see Methods),  of the SNPs were assumed to have no effect and the effects of the remaining
of the SNPs were assumed to have no effect and the effects of the remaining  of the SNPs were drawn from normal distribution with random variances. In most folds of each single CV and for all replicates of CV, the observed predictive abilities differed only marginally between BayesB and GBLUP (Figure 6). The average predictive ability obtained with BayesB was
of the SNPs were drawn from normal distribution with random variances. In most folds of each single CV and for all replicates of CV, the observed predictive abilities differed only marginally between BayesB and GBLUP (Figure 6). The average predictive ability obtained with BayesB was  which is not appreciably different from the result obtained with GBLUP (
which is not appreciably different from the result obtained with GBLUP ( ).
).
Figure 6. Predictive ability for GBLUP versus BayesB using phenotypic values of starvation resistance.

Predictive abilities are plotted for 20 replicates of a 5-fold CV, each replicate consisting of 5 corresponding folds of CV.
Genomic prediction versus GWAS
Although genomic prediction follows a different paradigm than genome-wide association studies, it is informative to compare significant SNP positions from the GWAS to areas of large estimated SNP effects resulting from the GBLUP model. Previously [27], a GWAS of  DGRP lines (of which the material used here is a subset) identified
DGRP lines (of which the material used here is a subset) identified  SNPs associated with starvation resistance and
SNPs associated with starvation resistance and  SNPs associated with startle response at a nominal p-value
SNPs associated with startle response at a nominal p-value in the analyses of sex-averaged data. We estimated SNP effects using RRBLUP and compared them to the significant SNPs from the GWAS study (Figure S2, Figure S3). There is excellent concordance of signals from both approaches in some regions (e.g. the genome-wide largest SNP effects on chromosome 3L for starvation resistance and 2L for startle response), while concordance is poor in other regions, especially on the X chromosome.
in the analyses of sex-averaged data. We estimated SNP effects using RRBLUP and compared them to the significant SNPs from the GWAS study (Figure S2, Figure S3). There is excellent concordance of signals from both approaches in some regions (e.g. the genome-wide largest SNP effects on chromosome 3L for starvation resistance and 2L for startle response), while concordance is poor in other regions, especially on the X chromosome.
We further investigated whether the most significant SNPs detected in the GWAS are reflected by large SNP effects in the GBLUP study using a different approach. For each significant SNP position from the GWAS we took the  neighboring SNPs (
neighboring SNPs ( on each side) and calculated the sum of the absolute values of their estimated effects using the GBLUP model. To avoid an effect of different sample size, we used the
on each side) and calculated the sum of the absolute values of their estimated effects using the GBLUP model. To avoid an effect of different sample size, we used the  most significant loci from the GWAS for both traits. We compared these sums to the sums of the absolute values of estimated SNP effects in
most significant loci from the GWAS for both traits. We compared these sums to the sums of the absolute values of estimated SNP effects in  sliding windows spanning the whole genome (with each window containing
sliding windows spanning the whole genome (with each window containing  neighboring SNPs). We observed a clear separation of the density functions of these sums for both startle response and starvation resistance (Figure 7).
neighboring SNPs). We observed a clear separation of the density functions of these sums for both startle response and starvation resistance (Figure 7).
Figure 7. Distribution of absolute SNP effects.

The density of the sum of the absolute SNP effects from GBLUP is plotted for sliding windows of 100 adjacent SNPs covering the whole genome (black) and for windows around the 75 most significant SNPs (red) according to the GWAS of [27]. The left (right) plot shows the densities for starvation resistance (startle response). The blue vertical line indicates the 90% quantile of the black density function.
The density resulting from the sliding window approach reflects the overall distribution of the suggested statistic in the sample. For starvation resistance (startle response) a threshold value of  , cf. Figure 7, cuts off the upper
, cf. Figure 7, cuts off the upper  of the respective distribution. Applying the same threshold with the density function reflecting the statistic for the significant GWAS positions,
of the respective distribution. Applying the same threshold with the density function reflecting the statistic for the significant GWAS positions,  of the distribution exceeds the threshold, indicating that signals found in the GWAS are also associated with large estimates of the SNP effects in the genomic model.
of the distribution exceeds the threshold, indicating that signals found in the GWAS are also associated with large estimates of the SNP effects in the genomic model.
Analyses of individual trait data
In addition to the line means we also analyzed individual records ( individual flies per line tested for starvation resistance and
individual flies per line tested for starvation resistance and  for startle response) to assess whether the variance between lines can be fully explained by additive gene effects or if non-additive mechanisms have an impact. This was done by modeling the covariance structure between lines based on the additive and additive
for startle response) to assess whether the variance between lines can be fully explained by additive gene effects or if non-additive mechanisms have an impact. This was done by modeling the covariance structure between lines based on the additive and additive additive genomic relationship matrix and testing the goodness of fit of the respective models. Most applications of genomic prediction are for outbred populations, for which the additive genetic variance and corresponding narrow-sense heritability determine the extent to which phenotypes in the next generation can be predicted from information obtained on the current generation. However, the variance among DGRP lines is the total genetic variance, and is possibly inflated by additive by additive epistatic variance [34]. Therefore, we performed several analyses on measurements of individual flies to determine the nature of the total genetic variance, especially to what extent the presence of non-additive genetic variance might have affected predictive abilities. We fitted three different models to the individual phenotype data: Model 1 contained a random line effect, and lines were assumed to be unrelated. In Model 2, a random additive line effect
additive genomic relationship matrix and testing the goodness of fit of the respective models. Most applications of genomic prediction are for outbred populations, for which the additive genetic variance and corresponding narrow-sense heritability determine the extent to which phenotypes in the next generation can be predicted from information obtained on the current generation. However, the variance among DGRP lines is the total genetic variance, and is possibly inflated by additive by additive epistatic variance [34]. Therefore, we performed several analyses on measurements of individual flies to determine the nature of the total genetic variance, especially to what extent the presence of non-additive genetic variance might have affected predictive abilities. We fitted three different models to the individual phenotype data: Model 1 contained a random line effect, and lines were assumed to be unrelated. In Model 2, a random additive line effect  was added, whose covariance structure was modeled via the genomic relationship matrix
was added, whose covariance structure was modeled via the genomic relationship matrix  . In Model 3, an additional random additive
. In Model 3, an additional random additive additive epistatic effect
additive epistatic effect  was included, whose covariance structure was modeled via the Hadamard product
was included, whose covariance structure was modeled via the Hadamard product  . Since the between line variance relates to inbred lines, while the additive and additive
. Since the between line variance relates to inbred lines, while the additive and additive additive variance component pertain to the non-inbred base population (or a hypothetical random mating
additive variance component pertain to the non-inbred base population (or a hypothetical random mating  produced from the inbred lines), the variance between inbred lines in Model 1 is expected to be twice the additive genetic variance in Model 2 or 3 under a fully additive model.
produced from the inbred lines), the variance between inbred lines in Model 1 is expected to be twice the additive genetic variance in Model 2 or 3 under a fully additive model.
We estimated variance components for all three models pooled across sexes and separately for males and females (Table S3, Table S4). We find little evidence for non-additive genetic variance for these traits. The estimate of  from Model 2 is
from Model 2 is  from Model 1, and Model 2 gave a significantly better fit than Model 1 when applying the likelihood ratio test, again indicating that the observed between line variance is due to additive gene action. Inclusion of the
from Model 1, and Model 2 gave a significantly better fit than Model 1 when applying the likelihood ratio test, again indicating that the observed between line variance is due to additive gene action. Inclusion of the  component was not significant for either of the traits. We found significant sex by line interaction variance for starvation resistance, but not for startle response (Tables S3, S4), which is in accordance with the findings of the genomic prediction across sexes (Table 1) and previous analyses of these data [27].
component was not significant for either of the traits. We found significant sex by line interaction variance for starvation resistance, but not for startle response (Tables S3, S4), which is in accordance with the findings of the genomic prediction across sexes (Table 1) and previous analyses of these data [27].
Discussion
We report the first (to our knowledge) application of genomic prediction to a real set of full genomic sequencing data in a eukaryotic organism. Although predictive abilities obtained with starvation resistance and startle behavior are only moderate to low, and although we limited our analysis to SNPs that are common due to the small sample size of lines, this study can be seen as a proof of concept for this approach. There are several reasons for the limited predictive ability obtained in this study. First, the training set is small, with a maximum of  observations in the
observations in the  -fold CV, and the accuracy of genomic prediction is a function of the size of the training set [20]. Using the curves fitted through the empirical accuracies (Figure 3), we predict accuracies of
-fold CV, and the accuracy of genomic prediction is a function of the size of the training set [20]. Using the curves fitted through the empirical accuracies (Figure 3), we predict accuracies of  for starvation resistance and startle response, if
for starvation resistance and startle response, if  sequenced lines were available for the training set.
sequenced lines were available for the training set.
The second important factor affecting accuracy of prediction is the number of independently segregating chromosome segments,  [20]. In our study we obtained
[20]. In our study we obtained  . This is larger than usually observed for Holstein cattle (
. This is larger than usually observed for Holstein cattle ( with
with  and genome length
and genome length  Morgans [35]), but is smaller than the corresponding value in the human genome (
Morgans [35]), but is smaller than the corresponding value in the human genome ( with
with  Morgans, [36]). (Note that in mammalian species, there is recombination in both sexes and
Morgans, [36]). (Note that in mammalian species, there is recombination in both sexes and  [9].)
[9].)
Accuracy of genomic prediction is thought to come from two sources: (i) SNPs in useful LD with causal loci; and (ii) SNPs reflecting the relationship structure between the training set and the set to be predicted [37]. Due to the very fast decay of LD in the D. melanogaster genome, few SNPs are in useful LD with any causal polymorphism. Even if we define “useful LD” very conservatively as  , then on average only a region of
, then on average only a region of  bp around a causal polymorphism was in useful LD on an autosome (
bp around a causal polymorphism was in useful LD on an autosome ( bp on the X chromosome). This means that on average
bp on the X chromosome). This means that on average  (
( ) SNPs were in useful LD with a causal autosomal (X-linked) polymorphism, as the average distance between neighboring SNPs was
) SNPs were in useful LD with a causal autosomal (X-linked) polymorphism, as the average distance between neighboring SNPs was  bp (
bp ( bp) on an autosome (X chromosome). If predictive ability was mainly driven by SNPs in LD with causal polymorphisms, reducing the SNP density should lead to a massive decay of predictive ability of the models, which was not observed. Little decrease in accuracy was seen, even if every
bp) on an autosome (X chromosome). If predictive ability was mainly driven by SNPs in LD with causal polymorphisms, reducing the SNP density should lead to a massive decay of predictive ability of the models, which was not observed. Little decrease in accuracy was seen, even if every  SNP was used in the model, in which case hardly any SNP would be in useful LD with causal polymorphisms. The underlying mechanism therefore seems to depend on a sufficient number of SNPs being in low LD with causal polymorphisms, rather than few SNPs in close physical association and high LD. In the DGRP population, LD approaches a small but positive baseline level with increasing physical distance [27], so that even with large physical distances a minimum level of LD is maintained, which was on average
SNP was used in the model, in which case hardly any SNP would be in useful LD with causal polymorphisms. The underlying mechanism therefore seems to depend on a sufficient number of SNPs being in low LD with causal polymorphisms, rather than few SNPs in close physical association and high LD. In the DGRP population, LD approaches a small but positive baseline level with increasing physical distance [27], so that even with large physical distances a minimum level of LD is maintained, which was on average  with
with  being the sample size.
being the sample size.
The number of SNPs for maximal accuracy of genomic prediction with unrelated individuals has been estimated as  [38], corresponding to
[38], corresponding to  SNPs in the present study.
SNPs in the present study.
For starvation resistance, we find that the empirical accuracy levels off when approximately every  SNP is used, which is equivalent to
SNP is used, which is equivalent to  or
or  SNPs. Adding more SNPs beyond this value does not lead to any improvement in the genomic prediction of starvation resistance, but also does not reduce accuracy, which one might expect when using more SNPs than actually needed. While fitting large numbers of “superfluous” SNPs may be considered as noise in the RRBLUP model, these SNPs can also be seen to provide a better basis to estimate the realized relationship matrix in the GBLUP model, which leads to a higher accuracy of the estimated realized relationships. Since both models are fully equivalent [9] no penalty is expected in the prediction of genomic values.
SNPs. Adding more SNPs beyond this value does not lead to any improvement in the genomic prediction of starvation resistance, but also does not reduce accuracy, which one might expect when using more SNPs than actually needed. While fitting large numbers of “superfluous” SNPs may be considered as noise in the RRBLUP model, these SNPs can also be seen to provide a better basis to estimate the realized relationship matrix in the GBLUP model, which leads to a higher accuracy of the estimated realized relationships. Since both models are fully equivalent [9] no penalty is expected in the prediction of genomic values.
Since pedigree information for the founders of the inbred lines was not available, our estimates of heritability and genomic prediction are based on the actual degree of identity-by-descent sharing between relatives [39]. There is little pedigree structure in the DGRP lines, with the exception of two distinct blocks of higher relatedness, comprising  and
and  lines, respectively, with a genomic relationship within blocks of
lines, respectively, with a genomic relationship within blocks of  and
and  . When these blocks were excluded from the data, predictive accuracy in a
. When these blocks were excluded from the data, predictive accuracy in a  -fold CV increased (decreased) for starvation resistance (startle response), suggesting that prediction in the DGRP population does not rely on distinct family structures. Given this together with the short-range extent of LD in the D. melanogaster genome and the robustness of the accuracy of genomic prediction with reduced marker density, we conclude that the observed accuracy of prediction for starvation resistance and startle response is primarily due to the long-range LD in the population, or equivalently, the subtle relationship structure as reflected by the genomic relationship matrix.
-fold CV increased (decreased) for starvation resistance (startle response), suggesting that prediction in the DGRP population does not rely on distinct family structures. Given this together with the short-range extent of LD in the D. melanogaster genome and the robustness of the accuracy of genomic prediction with reduced marker density, we conclude that the observed accuracy of prediction for starvation resistance and startle response is primarily due to the long-range LD in the population, or equivalently, the subtle relationship structure as reflected by the genomic relationship matrix.
We restricted our analyses to SNPs for which the minor allele was present in at least four DGRP lines (a minor allele frequency of  ). We applied this threshold to avoid computational limitations, especially when applying the BayesB method; and for consistency with the GWAS in the DGRP [27], which used the same filtering criterion. Thus, we did not utilize the
). We applied this threshold to avoid computational limitations, especially when applying the BayesB method; and for consistency with the GWAS in the DGRP [27], which used the same filtering criterion. Thus, we did not utilize the  million SNPs with minor allele frequencies less than this, nor did we take other forms of molecular variation into account.
million SNPs with minor allele frequencies less than this, nor did we take other forms of molecular variation into account.
Structural variations such as transposable elements have been repeatedly reported to be associated with phenotypic variation [40], therefore we must consider to what extent not including these variants in the models affected prediction accuracy. Given that we do not observe an increase in accuracy when increasing the number of SNPs from  to
to  million, we do not expect that increasing the marker density by adding more SNPs and other variants will have a significant effect on predictive ability. Additionally, SNPs with low minor allele frequencies were shown to be highly variable in predictive ability, so that the potential amount of information possibly added by the
million, we do not expect that increasing the marker density by adding more SNPs and other variants will have a significant effect on predictive ability. Additionally, SNPs with low minor allele frequencies were shown to be highly variable in predictive ability, so that the potential amount of information possibly added by the  million low frequency SNPs is limited. However, accounting for all polymorphisms in the model means that some fraction of the genetic variants must causally affect the trait. Simulations [26] including the causal polymorphism in the model improves the predictive ability over models based only on neutral SNPs in LD with the causal variants. Further research is needed to understand these mechanisms in the context of genomic prediction based on empirical data.
million low frequency SNPs is limited. However, accounting for all polymorphisms in the model means that some fraction of the genetic variants must causally affect the trait. Simulations [26] including the causal polymorphism in the model improves the predictive ability over models based only on neutral SNPs in LD with the causal variants. Further research is needed to understand these mechanisms in the context of genomic prediction based on empirical data.
The accuracy of BayesB has outperformed that of GBLUP in several simulation studies [10], [37]. Simulation results have suggested that GBLUP did not take full advantage of genome sequence data, suggesting that Bayesian methods are needed to obtain maximum accuracy [26]. The superiority of BayesB over GBLUP is expected to increase with marker density, and decrease when the size of the training data set is increased [38]. However, we did not find that BayesB yielded a significantly higher predictive ability than GBLUP in the  replicates of
replicates of  -fold CV with starvation resistance implemented in the present study. We used a very high marker density and a small training set, and yet GBLUP performed as well as BayesB. These conclusions should be taken with caution, since the available size of the training set was extremely small in our study due to the limited availability of fully sequenced lines. In [20], BayesB yielded a higher accuracy than GBLUP, when the number of simulated QTL was low; but GBLUP slightly outperformed BayesB, when the number of QTL became large, since the GBLUP model is equivalent to RRBLUP, in which all SNPs are assumed to have an effect drawn from the same normal distribution. Although this model may not seem biologically plausible, it performed as well as BayesB in the present study, consistent with several studies on real data from dairy cattle for different traits [4], [41].
-fold CV with starvation resistance implemented in the present study. We used a very high marker density and a small training set, and yet GBLUP performed as well as BayesB. These conclusions should be taken with caution, since the available size of the training set was extremely small in our study due to the limited availability of fully sequenced lines. In [20], BayesB yielded a higher accuracy than GBLUP, when the number of simulated QTL was low; but GBLUP slightly outperformed BayesB, when the number of QTL became large, since the GBLUP model is equivalent to RRBLUP, in which all SNPs are assumed to have an effect drawn from the same normal distribution. Although this model may not seem biologically plausible, it performed as well as BayesB in the present study, consistent with several studies on real data from dairy cattle for different traits [4], [41].
The finding that BayesB did not outperform GBLUP in the present study is consistent with a quasi-infinitesimal genetic architecture; and results indicate that starvation resistance and startle response are complex traits with a highly polygenic genetic architecture rather than being driven by a few major causal genes. This is in agreement with previous studies stating that starvation resistance and startle response can be considered to be model traits with a complex (i.e. quasi-infinitesimal) genetic background [28]–[30]; and it is also in line with the results from the GWAS [27]. One reasonable conclusion might be that there are so many causal polymorphisms, each with a small effect, that the  effective chromosome segments are saturated with causal variants and the effects of segments follow a normal distribution. Under this circumstance, GBLUP is expected to perform as well as BayesB. However, these hypotheses clearly need further investigation. More systematic model comparisons based on the available data were not considered here due to the prohibitive computing time required for BayesB.
effective chromosome segments are saturated with causal variants and the effects of segments follow a normal distribution. Under this circumstance, GBLUP is expected to perform as well as BayesB. However, these hypotheses clearly need further investigation. More systematic model comparisons based on the available data were not considered here due to the prohibitive computing time required for BayesB.
Previously, gene centered multiple regression and partial least square (PLS) regression models were used to predict starvation resistance and startle response phenotypes from genotypic data [27]. In both cases only SNPs that had nominal significance levels of  from the GWAS were used. The gene centered prediction models found that a few SNPs explained a large fraction of the genetic and phenotypic variance of the traits, while the PLS models found that the significant SNPs explained a high fraction of the phenotypic variance. The purpose of these studies was a comparison with human association studies, in which the faction of the variance explained by significant variants in the entire sample is commonly quoted. These approaches are fundamentally different from the BLUP approach used in this study. The BLUP approach includes random components and their covariance structure in the model, whereas regression models do not incorporate random terms except from the residuals; and the BLUP approach does not rely on a pre-selection of SNPs based on a GWAS. Most critically, we evaluated the robustness of the BLUP predictions using
from the GWAS were used. The gene centered prediction models found that a few SNPs explained a large fraction of the genetic and phenotypic variance of the traits, while the PLS models found that the significant SNPs explained a high fraction of the phenotypic variance. The purpose of these studies was a comparison with human association studies, in which the faction of the variance explained by significant variants in the entire sample is commonly quoted. These approaches are fundamentally different from the BLUP approach used in this study. The BLUP approach includes random components and their covariance structure in the model, whereas regression models do not incorporate random terms except from the residuals; and the BLUP approach does not rely on a pre-selection of SNPs based on a GWAS. Most critically, we evaluated the robustness of the BLUP predictions using  -fold cross-validation; whereas the previous analyses only tested the explanatory power of the most significant associated SNPs using the entire sample. Had we done the same analysis using GBLUP, we would be able to predict
-fold cross-validation; whereas the previous analyses only tested the explanatory power of the most significant associated SNPs using the entire sample. Had we done the same analysis using GBLUP, we would be able to predict  of the variance.
of the variance.
The imperfect concordance of the positions of the most significant SNPs from the GWAS and the largest estimates of SNP effects from RRBLUP is a consequence of the different objectives of the two approaches. A sequence-based GWAS is conducted to identify causal polymorphisms and provide estimates of allelic effects and frequencies. Also, the GWAS suffers from estimating one effect at a time and so does not necessarily position the QTL accurately. The goal of RRBLUP is to predict the phenotype using all available SNP information simultaneously. Here, estimated SNP effects are a by-product and mapping causal variants is not the primary objective. Given that the number of SNP effects to estimate is much larger than the number of observations, effects are estimated using penalized multiple regression approaches, shrinking estimated effect sizes towards zero. In addition, the magnitude of estimated SNP effects from RRBLUP is a function of the marker density. The higher the marker density, the more SNPs will be in LD with a causal mutation; therefore, the true allele substitution effect of a causal polymorphism will be split up and assigned in parts to a series of SNPs in the respective haplotype block. This can mask both the effect size, because one large effect may come in many small pieces; and the mapping position, because any SNP in LD with the causal polymorphism may have a substantial estimated effect. Nevertheless, some of the largest SNP effects from RRBLUP are in the proximity of prominent SNPs identified in the GWAS, so that to some extent positional information can still be retrieved from the RRBLUP results.
A methodology combining the strengths of both approaches – unbiased effect estimates and high positional resolution of GWAS with the simultaneous analysis of all SNPs, high predictive power and quality control via CV of genomic approaches – still needs to be developed. Results obtained in our study cannot be directly compared to predictive abilities in human studies due to the extremely small training set size ( in CV), and Drosophila has much larger
in CV), and Drosophila has much larger  and rapid decline of LD compared to humans. When genomic prediction in human studies was based on large training sets (thousands), substantial SNP panels (
and rapid decline of LD compared to humans. When genomic prediction in human studies was based on large training sets (thousands), substantial SNP panels ( k) and a highly heritable trait (
k) and a highly heritable trait ( ), predictive ability of genomic models was found to exceed what has been previously reported using a reduced number of markers pre-selected based on GWAS [31] and genomic prediction based on pre-selected SNPs was found to be of limited use in human studies of height [42].
), predictive ability of genomic models was found to exceed what has been previously reported using a reduced number of markers pre-selected based on GWAS [31] and genomic prediction based on pre-selected SNPs was found to be of limited use in human studies of height [42].
In the near future individual whole genome sequences will become increasingly available for large numbers of individuals in many species [17], [18]. Sequence-based predictions will therefore be relevant for prediction of risk disease and individualized medicine in humans, and for genome-based selection in farm animals and crops. The main findings of our study are: (i) genomic prediction can be efficiently implemented via GBLUP with full genome sequence data; (ii) there is little, if any, gain in predictive ability if the number of SNPs is increased above  (equivalent to
(equivalent to  in Holstein cattle and
in Holstein cattle and  in humans); and (iii) approaches based on external or internal (BayesB) selection of subsets of SNPs were not found to provide a substantial gain in accuracy of prediction compared to GBLUP. All findings must be seen against the background of the small sample size and the specific genetic constellation, with almost unrelated inbred lines and highly accurate phenotypes. Nevertheless, these results provide a realistic assessment of the potential benefits of sequenced-based prediction applied to non-model organisms and indicate avenues for future research.
in humans); and (iii) approaches based on external or internal (BayesB) selection of subsets of SNPs were not found to provide a substantial gain in accuracy of prediction compared to GBLUP. All findings must be seen against the background of the small sample size and the specific genetic constellation, with almost unrelated inbred lines and highly accurate phenotypes. Nevertheless, these results provide a realistic assessment of the potential benefits of sequenced-based prediction applied to non-model organisms and indicate avenues for future research.
Materials and Methods
The Drosophila Genetic Reference Panel (DGRP)
The full Drosophila Genetic Reference Panel (DGRP) [27], a recently developed new community resource for genetic studies of complex traits, consists of  D. melanogaster lines derived by
D. melanogaster lines derived by  generations of full sib mating from wild-caught females from the Raleigh, North Carolina population. Whole genome sequence data of
generations of full sib mating from wild-caught females from the Raleigh, North Carolina population. Whole genome sequence data of  DGRP lines (Freeze 1.0) have been obtained using a combination of Illumina and
DGRP lines (Freeze 1.0) have been obtained using a combination of Illumina and  next generation sequencing technology, which are available from the Baylor College of Medicine, http://www.hgsc.bcm.tmc.edu/project-species-i-DGRP_lines.hgsc. We used the Illumina sequences for
next generation sequencing technology, which are available from the Baylor College of Medicine, http://www.hgsc.bcm.tmc.edu/project-species-i-DGRP_lines.hgsc. We used the Illumina sequences for  DGRP lines in this study.
DGRP lines in this study.
Data preprocessing
SNPs were called from the raw sequence data as described previously [27]. We used SNPs with a coverage greater than 2X but less than 30X, for which the minor allele frequency was present in at least four lines, and for which SNPs were called in at least 60 lines. This series of filters gave a total of  SNPs for this analysis;
SNPs for this analysis;  on 2L,
on 2L,  on 2R,
on 2R,  on 3L,
on 3L,  on 3R and
on 3R and  on the X chromosome. We did not consider the few SNPs on the very short chromosome 4. In total there were
on the X chromosome. We did not consider the few SNPs on the very short chromosome 4. In total there were  missing SNP genotypes (
missing SNP genotypes ( ), which we imputed using Beagle Version 3.3.1 software [43].
), which we imputed using Beagle Version 3.3.1 software [43].
Phenotypic values
Phenotypic measurements for starvation resistance were available for all  DGRP lines, and for startle response on
DGRP lines, and for startle response on  lines [27]. We used the average of the medians of measurements for each trait in males and females as the phenotypic value
lines [27]. We used the average of the medians of measurements for each trait in males and females as the phenotypic value  of the
of the  line, i.e.
line, i.e.
 , where
, where  and
and  are the medians of the measurements for female and male individuals of the
are the medians of the measurements for female and male individuals of the  line. We used medians because of the skewed distribution of traits; however, medians are highly correlated with line means. For starvation resistance (startle response) there were on average
line. We used medians because of the skewed distribution of traits; however, medians are highly correlated with line means. For starvation resistance (startle response) there were on average  measurements for females, and
measurements for females, and  measurements for males (Table S1). Measurements were taken in several replicates for each trait [27].
measurements for males (Table S1). Measurements were taken in several replicates for each trait [27].
Cross-validation
We used different cross-validation (CV) procedures [44]–[46] to assess the predictive ability of different methods. In one replicate of a CV, the lines are randomly divided into a training set, which is used for parameter estimation; and a validation set, for which genetic values are predicted. The CV procedures differ in the ratios of the numbers of lines belonging to the training and validation sets: In a  -CV (with integers
-CV (with integers  and
and  ), the lines are randomly divided into
), the lines are randomly divided into  groups. The
groups. The  groups build the training set, and the remaining
groups build the training set, and the remaining  groups build the validation set. For this classification, there are
groups build the validation set. For this classification, there are  possibilities. For each of these possibilities (“folds”), total genetic values for the lines of the validation set are predicted and the corresponding predictive ability is calculated. The
possibilities. For each of these possibilities (“folds”), total genetic values for the lines of the validation set are predicted and the corresponding predictive ability is calculated. The  predictive abilities are then averaged to obtain one average correlation per CV replicate. For example, one (3∶2)-CV, consists of
predictive abilities are then averaged to obtain one average correlation per CV replicate. For example, one (3∶2)-CV, consists of  CV folds, over which predictive abilities are averaged. A
CV folds, over which predictive abilities are averaged. A  -CV is also called
-CV is also called  -fold CV.
-fold CV.
We used (4∶1)-, (3∶2)-, (2∶3)- and (1∶4)-CVs to analyze the effect of decreasing training set size. The CVs also differed in the constellations of phenotypic records used for the training and validation set. For example, the notation “(4∶1) male – female” indicates that only the medians of male records were used in the training set, and that the predicted genetic values were correlated with the medians of female records of the validation set to obtain the predictive ability in a (4∶1)-CV. CVs were also run for different marker densities, using every  -th SNP (
-th SNP ( ). Additionally, 5-fold CVs using only the
). Additionally, 5-fold CVs using only the  SNPs with the largest absolute values of estimated effects (obtained in the training set), or using only the
SNPs with the largest absolute values of estimated effects (obtained in the training set), or using only the  SNPs with the largest SNP variances (obtained in the training set) were performed. The additive genetic variance marked by the
SNPs with the largest SNP variances (obtained in the training set) were performed. The additive genetic variance marked by the  SNP was calculated as
SNP was calculated as  with allele frequency
with allele frequency  and estimated SNP effect
and estimated SNP effect  . In another series of 5-fold CVs we randomly chose
. In another series of 5-fold CVs we randomly chose  SNPs to build the genomic relationship matrix or we randomly chose
SNPs to build the genomic relationship matrix or we randomly chose  blocks of adjacent SNPs (each block consisting of
blocks of adjacent SNPs (each block consisting of  SNPs). In an additional 5-fold CV we excluded the lines in the two blocks of higher relatedness (Figure 2) from the data. Each type of CV was replicated
SNPs). In an additional 5-fold CV we excluded the lines in the two blocks of higher relatedness (Figure 2) from the data. Each type of CV was replicated  times, resulting in
times, resulting in  average predictive abilities.
average predictive abilities.
We also analyzed the influence of minor allele frequency on the predictive ability by another series of 5-fold CV. For this, we sorted all SNPs by their minor allele frequency and divided the sorted vector into  blocks. For each block we ran
blocks. For each block we ran  replicates of a 5-fold CV using GBLUP and the corresponding
replicates of a 5-fold CV using GBLUP and the corresponding  SNPs.
SNPs.
Predictive ability and accuracy
Predictive ability was measured in terms of correlation between predicted genetic values and observed phenotypic values. The corresponding accuracy  , defined as the correlation between true and predicted genetic value, was obtained by dividing the observed predictive ability by the square root of the observed heritability
, defined as the correlation between true and predicted genetic value, was obtained by dividing the observed predictive ability by the square root of the observed heritability  [47]. The heritability was based on the GBLUP model (see below).
[47]. The heritability was based on the GBLUP model (see below).
Genomic prediction with GBLUP
The underlying statistical model is
| (1) |
In this model, the  component of the
component of the  -vector
-vector  is the phenotypic value of the
is the phenotypic value of the  line that is used for prediction, i.e. the average of the medians of the phenotypic measurements for males and females for this line. Moreover,
line that is used for prediction, i.e. the average of the medians of the phenotypic measurements for males and females for this line. Moreover,  is the overall mean;
is the overall mean;  is assumed to be multivariate normal, with
is assumed to be multivariate normal, with  the genomic relationship matrix of all
the genomic relationship matrix of all  lines [8] and
lines [8] and  the additive genetic variance among lines. The matrix
the additive genetic variance among lines. The matrix  is an
is an  -incidence matrix, whose rows consist of unit vectors with one component being
-incidence matrix, whose rows consist of unit vectors with one component being  and all the others zero, indicating the respective positions of lines used for prediction in the
and all the others zero, indicating the respective positions of lines used for prediction in the  -vector of genetic values of all lines.
-vector of genetic values of all lines.  is the residual term, where
is the residual term, where  is the residual variance. Following the approach of [8],
is the residual variance. Following the approach of [8],  was defined as
was defined as
 |
where  is the
is the  -matrix of SNP genotype vectors for the
-matrix of SNP genotype vectors for the  lines with the
lines with the  SNPs coded as
SNPs coded as  and the
and the  column of
column of  is
is  , where
, where  is the frequency of the second allele at locus
is the frequency of the second allele at locus  .
.
Variance components were estimated via maximum likelihood (ML) using the R-package “RandomFields”, Version 2.0.46 (http://CRAN.R-project.org/package=RandomFields), and its function “fitvario”. The BLUP approach to obtain the vector of genetic values is equivalent to solving the so-called Mixed Model Equations (MME):
 |
A narrow-sense heritability based on the GBLUP model (1) was calculated as
Estimation of SNP effects
The GBLUP model (1) is equivalent to the following linear model in which all SNPs are assumed to have an effect drawn from the same normal distribution [9]:
where  and
and  are as described above and
are as described above and  is the vector of SNP effects with
is the vector of SNP effects with  . Using this equivalence, the SNP effects can be predicted as
. Using this equivalence, the SNP effects can be predicted as
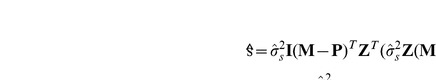 |
To estimate the SNP effects resulting from GBLUP for a single trait, we used all of the available lines, i.e.
 in model (1) contained the phenotypic values of all lines so that
in model (1) contained the phenotypic values of all lines so that  in the corresponding formulas. Note that only the inversion of a matrix of size equal to the number of sequenced lines is required.
in the corresponding formulas. Note that only the inversion of a matrix of size equal to the number of sequenced lines is required.
Distribution of linkage disequilibrium
We used  [48] as a measure of LD between a pair of loci. With two biallelic loci
[48] as a measure of LD between a pair of loci. With two biallelic loci  and
and  with alleles
with alleles  and
and  and frequencies
and frequencies  and
and  , we denote the frequencies of the genotypes
, we denote the frequencies of the genotypes  and
and  as
as  and
and  respectively. Then,
respectively. Then,
We performed the LD analyses using the imputed SNP matrix of  million SNPs for the
million SNPs for the  lines. We calculated the distribution of LD between all pairs of neighboring SNPs for different marker densities, using every
lines. We calculated the distribution of LD between all pairs of neighboring SNPs for different marker densities, using every  -th SNP (
-th SNP ( ). The extent of long-range LD was calculated for
). The extent of long-range LD was calculated for  pairs of SNPs randomly sampled from the first and the last
pairs of SNPs randomly sampled from the first and the last  SNPs per chromosome arm. Moreover, the average LD was calculated between SNPs on different chromosome arms, by sampling
SNPs per chromosome arm. Moreover, the average LD was calculated between SNPs on different chromosome arms, by sampling  pairs of SNPs for each combination of chromosome arms.
pairs of SNPs for each combination of chromosome arms.
Effective population size derived from empirical accuracies of genomic prediction
We modified the formula [20] for the expected accuracy,  of GBLUP given different population parameters (see Text S1 for more details on the derivation in the case of D. melanogaster):
of GBLUP given different population parameters (see Text S1 for more details on the derivation in the case of D. melanogaster):
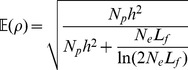 |
(2) |
 is the effective population size,
is the effective population size,  is the size of the training set,
is the size of the training set,  is the length of the female genome in Morgans and
is the length of the female genome in Morgans and  is the narrow-sense heritability of the trait estimated from model (1). The term
is the narrow-sense heritability of the trait estimated from model (1). The term  describes the number of independently segregating genome segments [9].
describes the number of independently segregating genome segments [9].
We ran CVs with different numbers of lines ( for starvation resistance and
for starvation resistance and  for startle response) in the training set (
for startle response) in the training set ( replicates each). Average numbers of lines in the training set are reported, which are non-integer values for starvation resistance because in a
replicates each). Average numbers of lines in the training set are reported, which are non-integer values for starvation resistance because in a  -CV, division of
-CV, division of  lines into
lines into  groups may give unequal numbers of lines in the different partitions. Given the corresponding average accuracies
groups may give unequal numbers of lines in the different partitions. Given the corresponding average accuracies  for the CV replicates, we estimated
for the CV replicates, we estimated  by fitting a curve to the points
by fitting a curve to the points  . To fit the curve, we chose
. To fit the curve, we chose  such that the sum of the squared differences of the observed accuracies and the accuracies obtained by (2) was minimized:
such that the sum of the squared differences of the observed accuracies and the accuracies obtained by (2) was minimized:
 |
using  and
and  Morgan. We calculated the length of the female genome in Morgans by summing the lengths of the chromosomes in base-pairs (
Morgan. We calculated the length of the female genome in Morgans by summing the lengths of the chromosomes in base-pairs ( (
( ,
,  ,
,  ,
,  ) Mbp for chromosome 2L (2R, 3L, 3R, X), [49]) and multiplying by the average recombination rates of females for the different chromosomes in Morgans per base-pair [50].
) Mbp for chromosome 2L (2R, 3L, 3R, X), [49]) and multiplying by the average recombination rates of females for the different chromosomes in Morgans per base-pair [50].
After performing bootstrapping ( replicates), the bias corrected empirical
replicates), the bias corrected empirical  confidence intervals (
confidence intervals ( error in each tail) for the
error in each tail) for the  estimates [32], [51] were calculated as
estimates [32], [51] were calculated as
where  is the
is the  -percentile of the bootstrap cumulative distribution function,
-percentile of the bootstrap cumulative distribution function,  is the
is the  -percentile of the standard normal distribution function
-percentile of the standard normal distribution function  ,
,  and
and  .
.
Effective population size derived directly from linkage disequilibrium
To estimate the effective population size based on LD, the following formula was used [52]:
 |
where  is the number of lines and
is the number of lines and  is the recombination rate in female individuals, cf. Text S1 for more details on this formula.
is the recombination rate in female individuals, cf. Text S1 for more details on this formula.
Genomic prediction with BayesB
The underlying model for the Markov Chain Monte Carlo based BayesB [10] method is
where  and
and  are as defined previously and
are as defined previously and  is the vector of normally distributed and independent SNP effects. The variance of the
is the vector of normally distributed and independent SNP effects. The variance of the  SNP effect,
SNP effect,  , is assigned an informative prior. The prior distribution of the genetic variances aims to resemble a situation where there are many loci with zero variance and only some loci with variance not equal to zero. Therefore, the prior distribution of the variance of a marker effect is a mixture of distributions which is given by
, is assigned an informative prior. The prior distribution of the genetic variances aims to resemble a situation where there are many loci with zero variance and only some loci with variance not equal to zero. Therefore, the prior distribution of the variance of a marker effect is a mixture of distributions which is given by
Note that this implies that the unconditional distribution of each single marker effect is a mixture of a point mass at  (with probability
(with probability  ) and of a t-distribution with zero mean,
) and of a t-distribution with zero mean,  degrees of freedom and scale parameter
degrees of freedom and scale parameter  [21], i.e. BayesB assigns the same unconditional prior distribution to each marker effect.
[21], i.e. BayesB assigns the same unconditional prior distribution to each marker effect.
In our studies, we used  and the scale parameter
and the scale parameter  was calibrated as
was calibrated as
 |
We chose  , such that approximately
, such that approximately  markers were contributing to the additive genetic variance. For the residual variance,
markers were contributing to the additive genetic variance. For the residual variance,  , the prior distribution was
, the prior distribution was  with
with  and
and
Values for  and
and  were chosen in the order of magnitude of the variance components of the GBLUP model (1), which were estimated using all lines and “fitvario”. The BayesB procedure is described in detail in [10]. It consists of running a Gibbs chain, where additionally a Metropolis-Hastings algorithm (
were chosen in the order of magnitude of the variance components of the GBLUP model (1), which were estimated using all lines and “fitvario”. The BayesB procedure is described in detail in [10]. It consists of running a Gibbs chain, where additionally a Metropolis-Hastings algorithm ( iterations) is used to sample from
iterations) is used to sample from  , where
, where  denotes the data
denotes the data  corrected for the mean
corrected for the mean  and all genetic effects other than the marker effect
and all genetic effects other than the marker effect  . Following graphical inspection, we ran BayesB with a chain length of
. Following graphical inspection, we ran BayesB with a chain length of  iterations including a burn in of
iterations including a burn in of  iterations that were discarded. To perform the BayesB approach, we used the software “GenSel”, Version 2.36, by R. Fernando and D. Garrick (cf. http://taurus.ansci.iastate.edu/Site/Welcome.html), which is implemented in C++. BayesB is computationally very intensive. The analyses were run on a Mac Pro 2× 2.93 GHz 6-Core Intel Xeon with 64 GB RAM running Mac OS X Server 10.6.7. One fold of a
iterations that were discarded. To perform the BayesB approach, we used the software “GenSel”, Version 2.36, by R. Fernando and D. Garrick (cf. http://taurus.ansci.iastate.edu/Site/Welcome.html), which is implemented in C++. BayesB is computationally very intensive. The analyses were run on a Mac Pro 2× 2.93 GHz 6-Core Intel Xeon with 64 GB RAM running Mac OS X Server 10.6.7. One fold of a  -fold CV for starvation resistance took approximately
-fold CV for starvation resistance took approximately  hours.
hours.
Comparing areas with large SNP effects with significant SNP positions
A genome-wide association study (GWAS) revealed  significant SNP positions for starvation resistance (startle response) [27], where a SNP position was considered significant if at least one of the three p-values, obtained using only male, only female or sex-pooled phenotypic records, was
significant SNP positions for starvation resistance (startle response) [27], where a SNP position was considered significant if at least one of the three p-values, obtained using only male, only female or sex-pooled phenotypic records, was  . We considered the subset of SNPs for which p-values of SNP effects of pooled data were
. We considered the subset of SNPs for which p-values of SNP effects of pooled data were  , to be more conservative and to be consistent with the previous analyses, leading to
, to be more conservative and to be consistent with the previous analyses, leading to  significant SNPs for starvation resistance (startle response).
significant SNPs for starvation resistance (startle response).
We compared genomic regions for which GBLUP estimated large SNP effects to these significant SNP positions of the GWAS. To avoid an effect of different sample sizes, we chose the  most significant SNPs from the GWAS analysis for each trait. For each of these SNPs, we chose the
most significant SNPs from the GWAS analysis for each trait. For each of these SNPs, we chose the  closest (neighboring) SNPs (
closest (neighboring) SNPs ( on each side) and calculated the sums of absolute values of the corresponding
on each side) and calculated the sums of absolute values of the corresponding  SNP effects (resulting from the GBLUP model). We compared the distribution of these sums to the distribution of the sums of the absolute values of estimated SNP effects in
SNP effects (resulting from the GBLUP model). We compared the distribution of these sums to the distribution of the sums of the absolute values of estimated SNP effects in  windows of
windows of  neighboring SNPs covering the whole genome by plotting the corresponding density functions. To obtain the sums of the absolute values of estimated SNP effects covering the whole genome, the windows were overlapping, displaced by
neighboring SNPs covering the whole genome by plotting the corresponding density functions. To obtain the sums of the absolute values of estimated SNP effects covering the whole genome, the windows were overlapping, displaced by  SNP positions. If the genomic regions for which GBLUP estimated large SNP effects coincide with the significant SNP positions of the GWAS, we expect the density functions to be separated.
SNP positions. If the genomic regions for which GBLUP estimated large SNP effects coincide with the significant SNP positions of the GWAS, we expect the density functions to be separated.
Variance component estimation using ASReml and individual trait records
For each trait, we fitted three different models using individual trait records. The first model included a fixed sex effect, a random line effect, a random line-sex-interaction term and a random term accounting for the different replicates in which measurements of the traits were taken:
In the second model, an additional random genetic effect  was added for each line. The variance-covariance matrix of the vector of these genetic effects was assumed to be given by the genomic relationship matrix
was added for each line. The variance-covariance matrix of the vector of these genetic effects was assumed to be given by the genomic relationship matrix  of [8]:
of [8]:
In the third model, an additional random additive additive epistatic effect
additive epistatic effect  was included for each line. The variance-covariance matrix of the vector of these genetic effects was given by the Hadamard product
was included for each line. The variance-covariance matrix of the vector of these genetic effects was given by the Hadamard product  [53] of the genomic relationship matrix
[53] of the genomic relationship matrix  of [8]:
of [8]:
Other two-way epistatic interactions, like additive dominance or dominance
dominance or dominance dominance, should not exist in inbred lines, provided inbreeding is complete. Variance components and their standard errors were estimated using ASReml 2.0 [54]. The analyses were done pooled across sexes as well as separately for males and females. The analyses of separate sexes did not include the sex term, and the replicate(sex
dominance, should not exist in inbred lines, provided inbreeding is complete. Variance components and their standard errors were estimated using ASReml 2.0 [54]. The analyses were done pooled across sexes as well as separately for males and females. The analyses of separate sexes did not include the sex term, and the replicate(sex line) term was reduced to replicate(line).
line) term was reduced to replicate(line).
Heritabilities
The broad-sense heritability for Model 1 was calculated as
cf. [28]. Narrow sense heritabilities for Models 2 and 3 were calculated as
and
These heritabilities are based on individual trait records.
Unless stated otherwise, all statistical analyses were performed using R software [55]. The R-package “ff”, Version 2.2-1 (http://CRAN.R-project.org/package=ff), was used to handle the large amount of SNP data efficiently in terms of memory capacity.
Supporting Information
Predictive ability of 5-fold CV with GBLUP for starvation resistance using different set of SNPs with different average minor allele frequencies. Each boxplot shows the average predictive abilities for  replicates of 5-fold CV using GBLUP and SNPs with different average minor allele frequencies. The different average minor allele frequencies are plotted as green dots. To choose the SNPs for each bin of minor allele frequency the SNPs were sorted by minor allele frequency and then divided into
replicates of 5-fold CV using GBLUP and SNPs with different average minor allele frequencies. The different average minor allele frequencies are plotted as green dots. To choose the SNPs for each bin of minor allele frequency the SNPs were sorted by minor allele frequency and then divided into  blocks, i.e. each bin contained
blocks, i.e. each bin contained  SNPs. The horizontal green line indicates the average accuracy obtained using every
SNPs. The horizontal green line indicates the average accuracy obtained using every  SNP (resulting in
SNP (resulting in  SNPs as well), which was
SNPs as well), which was  .
.
(PDF)
Manhattan plot of the estimated SNP effects for starvation resistance for different chromosomes. The SNP effects were estimated using the GBLUP approach and sex-averaged phenotypic values of  lines. Vertical lines indicate the
lines. Vertical lines indicate the  significant SNP positions according to the GWAS of [27] using sex-pooled records.
significant SNP positions according to the GWAS of [27] using sex-pooled records.
(PDF)
Manhattan plot of the estimated SNP effects for startle response for different chromosomes. The SNP effects were estimated using the GBLUP approach and sex-averaged phenotypic values of  lines. Vertical lines indicate the
lines. Vertical lines indicate the  significant SNP positions according to the GWAS of [27] using sex-pooled records.
significant SNP positions according to the GWAS of [27] using sex-pooled records.
(PDF)
Mean and standard deviation of phenotypic values and of the number of individual records per line.
(PDF)
Variance components and heritabilities estimated from GBLUP using all lines. Variance components were estimated by maximum likelihood using the R-package “RandomFields” and its function “fitvario.”
(PDF)
Results of variance component estimation using ASReml for starvation resistance. Different linear models for individual trait records were investigated.
(PDF)
Results of variance component estimation using ASReml for startle response. Different linear models for individual trait records were investigated.
(PDF)
We give more details on the formula of [52] for the expected linkage disequilibrium as well as the derivation of the number of independently segregating chromosome segments  [9] and the expected accuracy of prediction
[9] and the expected accuracy of prediction  [20] in the case of D. melanogaster. We also derive the expected value of the genomic relationship matrix
[20] in the case of D. melanogaster. We also derive the expected value of the genomic relationship matrix  of [8] and show that
of [8] and show that  , where
, where  is the numerator relationship matrix.
is the numerator relationship matrix.
(PDF)
Acknowledgments
We thank Stephanie M. Rollman and Michael M. Magwire for collecting the starvation resistance data and Katherine W. Jordan for collecting the startle response data. We thank three anonymous reviewers for their valuable comments on previous versions of the manuscript.
Footnotes
The authors have declared that no competing interests exist.
This research was funded by the German Federal Ministry of Education and Research (BMBF) within the AgroClustEr “Synbreed - Synergistic plant and animal breeding” (FKZ 0315528C) in association with the Deutsche Forschungsgemeinschaft (DFG) research training group “Scaling problems in statistics” (RTG 1644) and by National Institutes of Health grant R01GM 45146 to TFCM and EAS and NHGRI U54 HG003273 to RAG. DG was supported by the Alexander von Humboldt Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Mackay TFC, Stone EA, Ayroles JF. The genetics of quantitative traits: Challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- 2.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–1528. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de los Campos G, Gianola D, Allison DB. Predicting genetic predisposition in humans: The promise of whole-genome markers. Nat Rev Genet. 2010;11:880–886. doi: 10.1038/nrg2898. doi: 10.1038/nrg2898. [DOI] [PubMed] [Google Scholar]
- 4.Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME. Genomic selection in dairy cattle: Progress and challenges. J Dairy Sci. 2009;92:433–443. doi: 10.3168/jds.2008-1646. [DOI] [PubMed] [Google Scholar]
- 5.Lorenz AJ, Chao S, Asoro FG, Heffner EL, Hayashi T, et al. Genomic selection in plant breeding: Knowledge and prospects. Adv Agron. 2011;110:77–123. [Google Scholar]
- 6.Henderson CR. Sire evaluation and genetic trends. J Anim Sci . 1973;1973:10–41. [Google Scholar]
- 7.Ranade K, Chang MS, Ting CT, Pei D, Hsiao CF, et al. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:1262–1268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 9.Goddard M. Genomic selection: Prediction of accuracy and maximisation of long-term response. Genetica. 2009;185:1021–1031. doi: 10.1007/s10709-008-9308-0. [DOI] [PubMed] [Google Scholar]
- 10.Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genomewide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher RA. The correlation between relatives under the supposition of mendelian inheritance. Trans Roy Soc Edinburgh. 1918;52:399–433. [Google Scholar]
- 12.Pimentel E, Erbe M, Koenig S, Simianer H. Genome partitioning of genetic variation for milk production and composition traits in Holstein cattle. Front Gene. 2011;2 doi: 10.3389/fgene.2011.00019. doi: 10.3389/fgene.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schön CC, Utz HF, Groh S, Truberg B, Openshaw S, et al. Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics. 2004;167:485–498. doi: 10.1534/genetics.167.1.485. doi: 10.1534/genetics.167.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackay TFC. The genetic architecture of quantitative traits: Lessons from Drosophila. Curr Opin Genetics Dev. 2004;14:253–257. doi: 10.1016/j.gde.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Flint J, Mackay TFC. Genetic architecture of quantitative traits in mice, ies, and humans. Genome Res. 2009;19:723–733. doi: 10.1101/gr.086660.108. doi: 0.1101/gr.086660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eck SH, Benet-Pagès A, Flisikowski K, Meitinger T, Fries R, et al. Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 2009;10 doi: 10.1186/gb-2009-10-8-r82. doi: 10.1186/gb-2009-10-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The 1000 Genomes Project Consortium. A map of human genome variation from populationscale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e0019379. doi: 10.1371/journal.pone.0019379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME. Genetic architecture of complex traits and accuracy of genomic prediction: Coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010;6:e1001139. doi: 10.1371/journal.pgen.1001139. doi: 10.1371/journal.pgen.1001139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA. The impact of genetic architecture on genome-wide evaluation methods. Genetics. 2010;185:1021–1031. doi: 10.1534/genetics.110.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gianola D, de los Campos G, Hill WG, Manfredi E, Fernando R. Additive genetic variability and the Bayesian alphabet. Genetics. 2009;183:347–363. doi: 10.1534/genetics.109.103952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gianola D, van Kaam JBCHM. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics. 2008;178:2289–2303. doi: 10.1534/genetics.107.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de los Campos G, Gianola D, Rosa GJM. Reproducing kernel Hilbert spaces regression: A general framework for genetic evaluation. J Anim Sci. 2009;87:1883–1887. doi: 10.2527/jas.2008-1259. [DOI] [PubMed] [Google Scholar]
- 24.Long N, Gianola D, Rosa GJM, Weigel KA, Kranis A, et al. Radial basis function regression methods for predicting quantitative traits using SNP markers. Genet Res. 2010;92:209–225. doi: 10.1017/S0016672310000157. [DOI] [PubMed] [Google Scholar]
- 25.Ober U, Erbe E, Long N, Porcu E, Schlather M, et al. Predicting genetic values: A kernelbased best linear unbiased prediction with genomic data. Genetics. 2011;188:695–708. doi: 10.1534/genetics.111.128694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meuwissen T, Goddard M. Accurate prediction of genetic values for complex traits by wholegenome resequencing. Genetics. 2010;185:623–631. doi: 10.1534/genetics.110.116590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, et al. The Drosophila Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, et al. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet. 2009;41:299–307. doi: 10.1038/ng.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harbison ST, Yamamoto AH, Fanara JJ, Norga KK, Mackay TFC. Quantitative trait loci affecting starvation resistance in Drosophila melanogaster. Genetics. 2004;166:1807–1823. doi: 10.1534/genetics.166.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jordan KW, Carbone MA, Yamamoto A, Morgan TJ, Mackay TFC. Quantitative genomics of locomotor behavior in Drosophila melanogaster. Genome Biol. 2007;8 doi: 10.1186/gb-2007-8-8-r172. doi: 10.1186/gb-2007-8-8-r172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makowsky R, Pajewski NM, Klimentidis YC, Vazquez AI, Duarte CW, et al. Beyond missing heritability: Prediction of complex traits. PLoS Genet. 2011;7:e1002051. doi: 10.1371/journal.pgen.1002051. doi: 10.1371/journal.pgen.1002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Statist Sci. 1986;1:54–75. [Google Scholar]
- 33.Kusakabe S, Yamaguchi Y, Baba H, Mukai T. The genetic structure of the Raleigh natural population of Drosophila melanogaster revisited. Genetics. 2000;154:679–685. doi: 10.1093/genetics/154.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falconer DS, Mackay TFC. Introduction to quantitative genetics. Harlow, England: Pearson; 1996. [Google Scholar]
- 35.Qanbari S, Pimentel E, Tetens J, Thaller G, Lichtner P, et al. The pattern of linkage disequilibrium in german Holstein cattle. Anim Genet. 2010;41:346–356. doi: 10.1111/j.1365-2052.2009.02011.x. doi: 10.1111/j.1365-2052.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 36.Tenesa A, Navarro P, Hayes BJ. Recent human effective population size estimated from linkage disequilibrium. Genom Res. 2007;17:520–526. doi: 10.1101/gr.6023607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habier D, Fernando RL, Dekkers JCM. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–2397. doi: 10.1534/genetics.107.081190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meuwissen THE. Accuracy of breeding values of ‘unrelated’ individuals predicted by dense SNP genotyping. Genet Sel Evol. 2009;41 doi: 10.1186/1297-9686-41-35. doi: 10.1186/1297-9686-41-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full sublings. PLoS Genet. 2006;2:e0020041. doi: 10.1371/journal.pgen.0020041. doi: 10.1371/journal.pgen.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.González J, Petrov DA. The adaptive role of transposable elements in the Drosophila genome. Gene. 2009;448:124–133. doi: 10.1016/j.gene.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.VanRaden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, et al. Reliability of genomic predictions for North American Holstein bulls. J Dairy Sci. 2009;92:16–24. doi: 10.3168/jds.2008-1514. [DOI] [PubMed] [Google Scholar]
- 42.Aulchenko YS, Struchalin MV, Belonogova NM, Axenovich TI, Weedon MN, et al. Predicting human height by Victorian and genomic methods. Eur J Human Genet. 2009;17:1070–1075. doi: 10.1038/ejhg.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stone M. Cross-validation choice and assessment of statistical predictions. J Roy Statist Soc B. 1974;36:111–147. [Google Scholar]
- 45.Stone M. An aymptotic equivalence of choice of model by cross-validation and Akaike's criterion. J Roy Statist Soc B. 1977;39:44–47. [Google Scholar]
- 46.Allen D. The relationship between variable selection and data augmentation and a method of prediction. Technometrics. 1977;16:125–127. [Google Scholar]
- 47.Legarra A, Robert-Grani_e C, Manfredi E, Elsen JM. Performance of genomic selection in mice. Genetics. 2008;180:611–618. doi: 10.1534/genetics.108.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill WG, Weir BS. Maximum likelihood estimation of gene location by linkage disequilibrium. Am J Hum Genet. 1995;54:704–714. [PMC free article] [PubMed] [Google Scholar]
- 49.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 50.Fiston-Lavier AS, Singh ND, Lipatov M, Petrov DA. Drosophila melanogaster recombination rate calculator. Gene. 2010;463:18–20. doi: 10.1016/j.gene.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 51.Efron B. Better bootstrap confidence intervals. J Am Stat Assoc. 1987;82:171–185. [Google Scholar]
- 52.Sved JA. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol. 1971;2:125–141. doi: 10.1016/0040-5809(71)90011-6. [DOI] [PubMed] [Google Scholar]
- 53.Henderson CR. Applications of Linear Models in Animal Breeding. Guelph, Canada: University of Guelph; 1984. [Google Scholar]
- 54.Gilmour AR, Gogel BJ, Cullis BR, Thompson R. ASReml user guide release 2.0. Hemel Hempstead, UK: VSN International Ltd.; 2006. [Google Scholar]
- 55.Ihaka R, Gentleman R. R: A language for data analysis and graphics. J Comput Graph Statist. 1996;5:299–314. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Predictive ability of 5-fold CV with GBLUP for starvation resistance using different set of SNPs with different average minor allele frequencies. Each boxplot shows the average predictive abilities for replicates of 5-fold CV using GBLUP and SNPs with different average minor allele frequencies. The different average minor allele frequencies are plotted as green dots. To choose the SNPs for each bin of minor allele frequency the SNPs were sorted by minor allele frequency and then divided into blocks, i.e. each bin contained SNPs. The horizontal green line indicates the average accuracy obtained using every SNP (resulting in SNPs as well), which was .
(PDF)
Manhattan plot of the estimated SNP effects for starvation resistance for different chromosomes. The SNP effects were estimated using the GBLUP approach and sex-averaged phenotypic values of lines. Vertical lines indicate the significant SNP positions according to the GWAS of [27] using sex-pooled records.
(PDF)
Manhattan plot of the estimated SNP effects for startle response for different chromosomes. The SNP effects were estimated using the GBLUP approach and sex-averaged phenotypic values of lines. Vertical lines indicate the significant SNP positions according to the GWAS of [27] using sex-pooled records.
(PDF)
Mean and standard deviation of phenotypic values and of the number of individual records per line.
(PDF)
Variance components and heritabilities estimated from GBLUP using all lines. Variance components were estimated by maximum likelihood using the R-package “RandomFields” and its function “fitvario.”
(PDF)
Results of variance component estimation using ASReml for starvation resistance. Different linear models for individual trait records were investigated.
(PDF)
Results of variance component estimation using ASReml for startle response. Different linear models for individual trait records were investigated.
(PDF)
We give more details on the formula of [52] for the expected linkage disequilibrium as well as the derivation of the number of independently segregating chromosome segments
[9] and the expected accuracy of prediction
[20] in the case of D. melanogaster. We also derive the expected value of the genomic relationship matrix of [8] and show that , where is the numerator relationship matrix.
(PDF)