

Abstract
The MYC oncogene contributes to the genesis of many human cancers. Recent insights into its expression and function have led to new cancer therapeutic opportunities. MYC’s activation by bromodomain proteins could be inhibited by drug-like molecules, resulting in tumor inhibition in vivo. Tumor growth can also be curbed by pharmacologically uncoupling bioenergetic pathways involving glucose or glutamine metabolism from Myc-induced cellular biomass accumulation. Other approaches to halt Myc on the path to cancer involve targeting Myc-Max dimerization or Myc-induced microRNA expression. Here the richness of our understanding of MYC is reviewed, highlighting new biological insights and opportunities for cancer therapies.
Introduction
MYC belongs to a family that includes MYCL (L-Myc) and MYCN (N-Myc) (Brodeur et al., 1984; Kohl et al., 1984; Maris, 2010; Nau et al., 1985). While the role of L-Myc is less well understood, N-Myc expression is tissue-restricted, and N-Myc could substitute for c-Myc in murine development (Malynn et al., 2000). The proto-oncogene, MYC, lies at the crossroads of many growth promoting signal transduction pathways and is an immediate early response gene downstream of many ligand-membrane receptor complexes (Armelin et al., 1984; Kelly et al., 1983) (Figure 1A). MYC expression is highly regulated, such that its level of expression is tightly control by a number of mechanisms involving many transcriptional regulatory motifs found within its proximal promoter region (Brooks and Hurley, 2010; Hurley et al., 2006; Levens, 2010).
Figure 1.


A. The MYC protooncogene is depicted downstream of receptor signal transduction pathways, which elicit positive or negative regulation of the MYC gene. MYC produces the transcription factor Myc, which dimerizes with Max and bind target DNA sequences or E-boxes (with the sequence 5′-CANNTG-3′) to regulate transcription of genes involved in cell growth and proliferation. The WNT pathway is depicted with APC negatively regulating β-catenin, which upon nuclear translocation participates in the transactivation of MYC, such that loss of APC results in constitutive oncogenic MYC expression. B. When MYC is deregulated, by gene amplication, chromosomal translocation or loss of upstream regulators, such as APC, acute sustained oncogenic MYC expression results in checkpoint activation p53 or Arf. Loss of checkpoint regulation through mutations of p53 or Arf, for example, uncloaks MYC’s full tumorigenic potential.
The road to MYC’s discovery was paved by early studies of fulminant chicken tumors caused by oncogenic retroviruses, leading to the identification of the v-myc oncogene that causes myelocytomatosis (leukemia and sarcoma) (Duesberg and Vogt, 1979; Hu et al., 1979; Sheiness and Bishop, 1979). The v-myc oncogene was co-opted from the host cellular genome containing the proto-oncogenic version or c-myc (Vennstrom et al., 1982). Although the search for comparable human retroviruses failed to recapitulate the retroviral oncogene paradigm in human cancers, the discovery that human MYC is consistently altered by balanced chromosomal translocation in Burkitt lymphoma marked it as a bona fide human oncogene (Dalla-Favera et al., 1982; Taub et al., 1982). MYC is frequently translocated in multiple myeloma (Shou et al., 2000) and is one of the most highly amplified oncogene among many different human cancers (Beroukhim et al., 2010). Defects in the Wnt-APC pathway found in human colon carcinoma results in enhanced TCF transcriptional activation of MYC (He et al., 1998). MYC is downstream of deregulated Notch signaling pathways found in T-cell leukemia (Palomero et al., 2006; Sharma et al., 2006; Weng et al., 2006). Hence, alterations of MYC are commonly found on the path to cancer.
In addition to its role in tumorigenesis, MYC was also identified as one of four genes, including Sox2, Oct4, and KLF4, that could collectively reprogram fibroblasts to a pluripotent stem cell state (Laurenti et al., 2009; Singh and Dalton, 2009; Takahashi and Yamanaka, 2006). Given its pivotal role in cell growth, proliferation, tumorigenesis and stem cells, it appears timely to review what is currently understood about MYC by addressing the following key questions. What are the molecular functions of its protein product, Myc? How does MYC contribute to tumorigenesis? What are the differences between the MYC proto-oncogene and its deregulated form found in a variety of human cancers? Could MYC or Myc’s target genes be targeted for cancer therapy?
Myc, checkpoints and neoplastic transformation
Early in vitro studies of MYC revealed its potential to transform normal embryonic fibroblasts in cooperation with other oncogenes (Land et al., 1983). These studies set the stage for transgenic mouse studies that provided the evidence that deregulated expression of MYC is sufficient to drive tumorigenesis in a number of transgenic mouse tissues (Adams et al., 1985; Chesi et al., 2008; Leder et al., 1986). Retroviral insertional mutagenesis further identified c-Myc as a major murine oncogene (Akagi et al., 2004). In each of these models, however, additional mutagenic events are necessary for tumor formation as evidenced by a predictable time delay before the onset of tumors (Beer et al., 2004; Ellwood-Yen et al., 2003; Felsher and Bishop, 1999a; Pelengaris et al., 1999). Hence, MYC requires other genetic alterations in vivo to enable its tumorigenic potential. Mammary carcinoma triggered by transgenic Myc expression acquired K-ras mutations that rendered tumors aggressive (D’Cruz et al., 2001). Acute overexpression of MYC in normal cells triggers checkpoints including ARF or p53 (Figure 1B), such that many MYC-induced transgenic lymphomas lacks functional Arf or p53 (Eischen et al., 1999; Zindy et al., 1998). The findings from transgenic mouse studies underscore a causal role for MYC in murine cancers and support its tumorigenic role in human cancers.
MYC is documented to play a role in tumor initiation; however, whether MYC participates in tumor maintenance was previously unclear. Knock-down of MYC in established cancer cell lines in vitro appears to uniformly reduce cell proliferation and in some instances induce apoptosis (Cappellen et al., 2007; Koh et al., 2011a; Wang et al., 2008). In transgenic mouse models with inducible MYC, established tumors regress upon withdrawal of MYC ectopic expression, indicating that MYC plays a role in tumor maintenance, and once established these tumors are addicted to MYC (Arvanitis and Felsher, 2006). In fact, expression of a dominant negative inhibitor of Myc heterodimerization in vivo has resulted in tumor regression, suggesting that inhibiting Myc function could be a feasible therapeutic strategy (Soucek et al., 2008).
Molecular functions of Myc
The MYC mRNA generates Myc polypeptides including one that initiates at a CUG upstream of the canonical AUG start codon, and another that starts at an internal AUG (Blackwood et al., 1994). The Myc protein translated from the canonical AUG contains an N-terminal transcriptional regulatory domain followed by a nuclear localization signal and a C-terminal region with a basic DNA binding domain tethered to a helix-loop-helix-leucine zipper (HLH-Zip) dimerization motif. Myc dimerizes with Max to bind DNA and mediates many of its functions (Amati et al., 1993; Amati et al., 1992; Blackwood and Eisenman, 1991; Grinberg et al., 2004; Kato et al., 1992; Kretzner et al., 1992). A distinct function for the longer Myc polypeptide initiated at the upstream CUG is not known (Blackwood et al., 1994; Hann et al., 1992), but the shorter one initiated from an internal AUG appears to play a role in stress response and perhaps serves as a dominant negative Myc protein (Spotts et al., 1997; Xiao et al., 1998). Myc biology is further complicated by the finding that a cytoplasmic cleavage product of Myc (Myc-nick), which lacks the nuclear localization signal and DNA binding domain, can promote alpha-tubulin acetylation by recruiting GCN5 and promote cell differentiation in a non-transcriptional manner (Conacci-Sorrell et al., 2010).
Myc also appears to recruit DNA replication licensing factors to catalyze DNA replication, although whether its transcriptional function at replication origins is part-and-parcel of its DNA replication activity is not yet clear (Dominguez-Sola et al., 2007). Myc also plays an important non-transcriptional role in stimulating cap-dependent translation (Cole and Cowling, 2008; Cowling and Cole, 2007). Lastly, Myc appears to function even in the absence of functional Max protein as documented in PC12 cells and more recently in Drosophila (Hopewell and Ziff, 1995; Steiger et al., 2008). Whether Myc could homo-oligomerize or hetero-oligomerize with other helix-loop-helix proteins to regulate transcription in the absence of Max in cells remains unknown (Nair and Burley, 2003).
The Myc protein contains an unstructured N-terminal transcriptional regulatory domain, which contains conserved Myc Boxes I and II, followed by Myc Box lll and IV and a nuclear targeting sequence (Cowling et al., 2006; Dang and Lee, 1988; Kato et al., 1990; Pineda-Lucena and Arrowsmith, 2001). The C-terminal domain comprises a basic HLH-Zip domain, which is largely unstructured until it dimerizes with Max (Follis et al., 2009; Hu et al., 2005; Mustata et al., 2009; Sauve et al., 2007). The monomers assemble on DNA, and the heterodimer locks onto and bend DNA through binding motifs (5′-CACGTG-3′) termed E-boxes (Park et al., 2004). The N-terminal domain has been documented to form complexes with many factors including TRRAP, GCN5, and TBP, which are likely to induce more structured folding of the N-terminal Myc transcriptional regulatory domain (Fladvad et al., 2005; Liu et al., 2003; McEwan et al., 1996; McMahon et al., 2000; Nikiforov et al., 2002). Hence, it is envisioned that when bound to DNA, the Myc-Max heterodimer would recruit complexes that modify chromatin (Figure 2).
Figure 2.
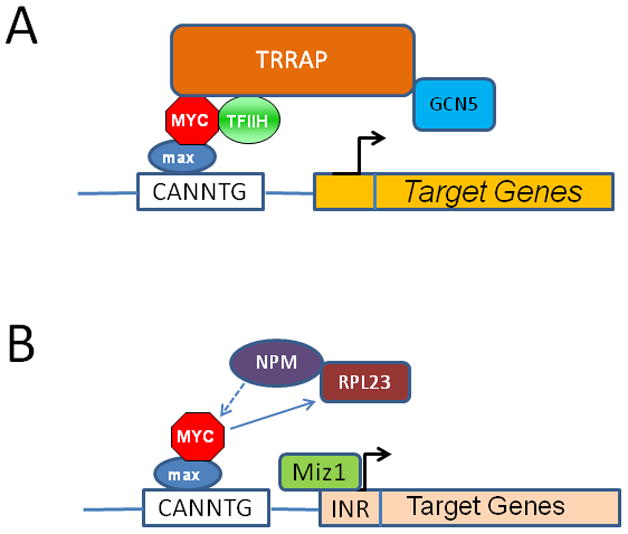
A. The Myc-Max heterodimer is shown to interact with key co-factors such as TFIIH that triggers transcriptional elongation or TRRAP that recruits the GCN5, which acetylates histone, permitting transcription of target genes. B. Myc-Max also mediates gene repression. Miz-1 is shown tethered to the INR element to regulate transcription of target genes, which could be silenced by Myc displacement of NPM, a Miz-1 cofactor, or by Myc induction of the ribosomal protein RPL23, which retains NPM in the nucleolus, keeping it away from Miz-1.
Although the mechanisms by which Myc activates transcription are emerging, including the recruitment of histone acetylase, the mode by which Myc represses gene expression is less well understood. Among the vast numbers of targets that are repressed by Myc, a fraction is linked to Miz-1, which activates these genes (Figure 2) (Schneider et al., 1997). TGFβ signaling best illustrates a role for Myc-Miz-1 interaction. In the absence of TGFβ, Myc represses CDKN2B (p15INK4b) by binding Miz-1 and displacing Miz-1 cofactors to silence CDKN2B (Seoane et al., 2001). With TGFβ, MYC expression is suppressed, and the Smad transcription factor translocates and cooperates with Miz-1 to recruit NPM1 as a Miz-1 cofactor to stimulate CDKN2B transcription and induce cell cycle arrest (Wanzel et al., 2008). Myc, on the other hand, activates many ribosomal protein genes including Rpl23, which binds to and retains NPM1 in the nucleolus, thereby inhibiting Miz-1 activity. Myc itself is modulated by NPM1, which acts as a positive Myc coactivator (Li et al., 2008).
Another critical mode for Myc-mediated gene repression is through its ability to activate microRNAs (Figure 3) (Chang et al., 2008; O’Donnell et al., 2005). Specifically, the activation of the miR-17-92 cluster of microRNAs mediates a number of biological activities of Myc, including the attenuation of E2F1 activity. Intriguingly, the miR-17-92 cluster also targets many components of the TGFβ signaling pathway (Aguda et al., 2008; Dews et al., 2010; Mestdagh et al., 2010; O’Donnell et al., 2005). These observations indicate that Myc repression of gene expression occurs through different modalities which are linked to regulatory loops. Myc also represses many more microRNAs resulting in increased gene expression at the protein level (Figure 3). It is almost certain that Myc would also directly activate long non-coding RNAs (lncRNAs) to mediate gene repression as implicated in studies of embryonic stem cells.
Figure 3.

Myc regulates a network of microRNAs through activation of the miR-17-92 cluster and repression of dozens of miRs including Let-7, which was recently shown to affect insulin signaling, miR-23a/b, which regulates glutaminase expression, and miR-34a, which was shown to regulate lactate dehydrogenase (LDHA) expression. The miR-17 cluster contains microRNAs that targets PTEN, thereby activating AKT, and those that targets the proapoptotic BimL or the transcription factor E2F1 expression. MicroRNAs downstream of Myc have also been implicated in epithelial-mesenchymal transition and angiogenesis.
Transcription: upstream and downstream of Myc
The MYC proto-oncogene itself is under tight transcriptional control as are the mRNA and Myc protein. In fact, MYC is not only regulated by a whole host of transcription factors, such as CNBP, FBP, and TCF that is downstream of the Wnt pathway, but it is also regulated by non-B DNA structures including single-stranded bubbles, G-quadruplexes and Z-DNA (Levens, 2010). The FUSE (Far UpStream Element), melts when bound by FBP (FUSE binding protein), which relieves torsional stresses on DNA from ongoing transcription of MYC (He et al., 2000). TCF is a transcription factor that plays a role in deregulated MYC expression downstream of the WNT pathway, such as with the loss of the tumor suppressor APC that results in constitutive nuclear localization of the TCF co-factor β-catenin. Genome wide association studies further identified common polymorphisms nearby MYC that are associated with multiple cancers (Ahmadiyeh et al., 2010; Tuupanen et al., 2009; Wasserman et al.). Such SNPs lie in enhancers that involve TCF binding and DNA looping, which connects the enhancer to the MYC proximal promoter (Pomerantz et al., 2009; Sotelo et al., 2010; Wright et al., 2010). Recently, the BET domain containing transcriptional regulator, BRD4, was shown to bind to the MYC promoter region and play a critical role in MYC expression in human cancer cells such that a drug-like BET domain chemical inhibitor could inhibit in vivo tumorigenesis (Delmore et al., 2011; Mertz et al., 2011).
The MYC mRNA, which is short-lived, is affected by microRNAs (let-7, miR-34, and miR-145) resulting in translational modulation (Cannell et al., 2010; Christoffersen et al., 2010; Kim et al., 2009; Kress et al., 2011; Sachdeva et al., 2009). The Myc protein itself is post-translationally modified, ubiquitinated and degraded, with a half-life in the order of 15–20 minutes (Gregory and Hann, 2000; Gregory et al., 2003). Myc transcriptional activity is regulated by phosphorylation at Ser-62 followed by Thr-58, and subsequent proteasomal degradation after performing its function (Salghetti et al., 1999; Thomas and Tansey, 2010)(Adhikary et al., 2005; Popov et al., 2010; Popov et al., 2007). Mutations of Myc residues Thr-58 and Ser-62, prevalently found in Burkitt lymphoma, are associated with stabilized mutant proteins that could perturbed transgenic mammary tumorigenesis (Salghetti et al., 1999; Thomas and Tansey, 2010; Wang et al., 2011b). The resulting sustained levels of Myc contributes to tumorigenesis, which in some instances may not require total elevated average levels of Myc but rather depend on deregulated expression of Myc throughout the cell cycle. How then does Myc transcriptional activity contribute to tumorigenesis?
The canonical Myc E-box 5′-CACGTG-3′ is among the most frequently occurring DNA binding motifs in the human genome (Xie et al., 2005). This motif, however, could be bound by different transcription factors such as ChREBP, SREBP, HIF-1, NRF1, USF, TFE3, Clock, and Bmal (Figure 4). It stands to reason that in non-proliferating cells, non-Myc E-box transcription factors regulate basal metabolism to maintain cellular structural and functional integrity. When cells are stimulated to proliferate, Myc levels rise, permitting it to occupy E-box driven genes normally bound by other transcription factors and activate a program of biomass accumulation and enhanced cellular bioenergetics. As such, which of the many E-boxes are occupied by Myc in proliferating cells and does occupancy trigger changes in transcription and mRNA levels of the target genes?
Figure 4.

Myc-Max is shown bound to E-box driven genes, which could also be regulated by other E-box transcription factors, such as the carbohydrate response element binding protein (ChREBP), sterol response element binding protein (SREBP), nuclear respiratory factor 1 (NRF1), circadian transcription factor Clock (and Bmal), and hypoxia inducible factor (HIF). The non-Myc E-box transcription factors regulate genes involved in metabolism, which is maintained for cellular homeostasis when cells are not proliferating. Upon activation of MYC and elevated levels of Myc, mass action favors the binding of Myc-Max to E-box genes to regulate metabolism and genes involved in ribosomal biogenesis and cell mass accumulation. This model suggests that resting cells express metabolic genes through ‘homeostatic’ E-box transcription factors, which regulate a set of genes that overlaps with Myc target genes that are expressed when cells are stimulate to grow and proliferate.
Genome-wide association of Myc protein to chromatin has been documented by a number of approaches. Myc was documented by chromatin immunoprecipitation (ChIP) and quantitative PCR to bind a selected panel of promoters (Fernandez et al., 2003). A genome-wide study in Drosphila documented key dMyc-bound target genes that are involved in many cellular functions, consistent with studies performed in mammalian cells (Orian et al., 2005); most notable among the Myc targets is CDK4 which is also a key target in mammals. Myc binding sites was also mapped using a promoter array containing 4839 promoter sequences (Li et al., 2003). This study suggests that Myc could be considered a general transcriptional factor since promoters (15%) were broadly bound by Myc. This study was corroborated by a subsequent ChIP-chip tiled array study of human chromosomes 21 and 22, which documented widespread association of Myc with chromatin (Cawley et al., 2004). Genome-wide Myc binding sites were mapped in a human B cell line with ChIP followed by paired ends tag sequencing (ChIP-PET) of the immunoprecipitated DNA fragments (Zeller et al., 2006). This study estimated that up to 6000 genes are bound by Myc, and among the 3000 genes deemed high quality Myc bound target genes, only about 700 genes responded to Myc activation with alterations in their mRNA levels. Most of these binding sites are found in the proximal promoter with significant portions found intragenically and about 10% are located in intergenic (>100kb from promoters) regions.
A subsequent study examines the role of Myc in transcription and relief of RNA Pol II paused complexes (Rahl et al., 2010). This study provides evidence suggesting that recruitment of pTEFb by Myc stimulate pause release partly by phosphorylation of RNA Pol II. This view is in contrast to the previous perspective that transcription factors such as Myc assemble co-factors which in turn recruits TBP and RNA Pol II for transcription initiation. In this regard, it is intriguing to note that a significant proportion of Myc binding sites occur in the first introns of genes. Although the position of Myc binding sites and their effects on transcription pause release was not specifically addressed, this work establishes a key role for Myc in transcriptional elongation.
Genome wide mapping of Myc binding sites and associated gene expression profiling established that Myc binding is insufficient to induce changes in mRNA levels of target genes. In fact, many of the Myc bound and induced genes in the human B cell model are associated with E2F DNA binding motifs, suggesting that Myc targets require additional factors for their expression (Li et al., 2003; Zeller et al., 2006). An instructive study of Myc among 6 other factors, including stem cell factors Sox2, Oct4, and KLF4, and their binding sites in murine embryonic stem cells revealed that genes whose expression were changed with stem cell differentiation tended to be those that were bound by multiple transcriptional factors (Kim et al., 2010). Hence, binding by a single transcription factor is generally insufficient to activate a target gene, unless it is bound by multiple transcriptional factors. What then are the Myc target genes and how do they contribute to the biology of the cell?
Myc target genes, stem cells and the path to cancer
Although low throughput approaches to identify and characterize Myc target genes were largely fruitful, a global perpective of Myc function has only recently emerged from unbiased studies of Myc targets. It is notable that early studies rely on changes in mRNA levels in response to manipulation of Myc levels to identify Myc target genes. Recently, the combination of global immunoprecipitation and gene expression profiling provides a better approach to defining Myc targets, particularly if coupled with a genome-wide nuclear run-on assay (Ji et al., 2011).
Through the use of an inducible Myc system coupled with ChIP-seq and gene expression changes, a set of 300 Myc-dependent serum response (MDSR) genes was identified in fibroblasts (Perna et al., 2011). This set comprises about 6% of Myc bound targets, which encompass 22% of all promoters. Intriguingly, these MDSR genes are largely involved in nucleotide metabolism, ribosome biogenesis, RNA processing and DNA replication. In this regard, Myc also regulates RNA polymerases I and III mediated transcription in addition to its role in regulating RNA Pol II genes (Felton-Edkins et al., 2003; Gomez-Roman et al., 2003; Grandori et al., 2005; Kenneth et al., 2007). Thus, the protein biosynthetic machinery is inherent linked to Myc transcriptional activity and the balance between rRNA synthesis, ribosomal protein production, and the availability of adequate bioenergetics is essential for normal cell growth. Many direct Myc target genes encode ribosomal proteins, which are directly involved in the ARF and p53 checkpoint, particularly when ribosome biogenesis is perturbed.
A common 50-gene Myc core signature (MCS) was identified among Myc target genes in four tumorigenic human cell lines and in embryonic stem cells, and expression of the MCS correlates with MYC gene expression among 8,129 microarray samples that include 312 cell and tissue types, attesting to its cell-type independence (Ji et al., 2011). Functional annotation of the MCS reveals enrichment of genes involved in ribosome biogenesis, underscoring Myc’s primordial function in biomass accumulation. This notion is consistent with the observation that diminished Drosophila dMyc function results in small cells and body size, which phenocopies mutations affect ribosomal protein genes found in a large set of mutants termed Minutes (Johnston et al., 1999). Furthermore, the link between Myc and ribosome biogenesis has been documented in specific cell types (Challagundla et al., 2011; Chan et al., 2011; Greasley et al., 2000; Kim et al., 2000; Schlosser et al., 2005; Schlosser et al., 2003) and in Myc-driven tumorigenesis in vivo (Barna et al., 2008; Stumpf and Ruggero, 2011). It is long known that cancer cells are characterized by intense nucleolar hypertrophy; hence, a signature of cancer cells could now be linked mechanistically to Myc function (van Riggelen et al., 2010b).
The role of Myc in reprogramming fibroblasts to pluripotency and in stem cells emerges from a number of studies. One study suggests that Myc drives an embryonic stem cell-like program, without defining the functional significance of this program (Wong et al., 2008). The study by Kim et al (2010), however, dissected the roles of stem cell factors (Sox2, Oct4) versus Myc through ChIP analysis (Kim et al., 2010). In this case, stem cell factors regulate a distinct core stem cell module of target genes, while Myc drives a program common to ES cells and cancer cells but distinct from the core stem cell targets. A third genetic module comprises polycomb-related genes, which are involved in regulation of cell differentiation. The Myc module is enriched in genes involved in ribosome biogenesis, suggesting that the MCS does mark a core function of Myc (Kim et al., 2010). It is notable, however, that other studies implicate Myc in the regulation of chromatin structure which is reprogrammed in stem cells. Myc induces Bmi-1 and EZH2, which are polycomb proteins, and perhaps modulates the expression of long non-coding RNAs (lncRNAs) that are involved in polycomb-mediated gene silencing (Guney et al., 2006; Koh et al., 2011b; Sander et al., 2008). Perhaps, these Myc functions link Myc to the regulation of the polycomb module, the suppression of senescence, terminal cell differentiation, and the maintenance of pluripotency.
The role of Myc in tissue stem cells appears to depend on the tissue type, and multiple studies suggest that MYC is required for the commitment to terminal differentiation, which contrasts with the role of MYC in pluripotency. MYC’s roles in tissue versus pluripotent stem cells are expected to be different; however, additional studies are required to provide a richer mechanistic understanding. MYC is required for production of committed hematopoietic progenitors such that loss of MYC in mice result in the expansion of hematopoietic stem cells (HSC) and diminished progenitors with pancytopenia (Laurenti et al., 2008). On the other hand, overexpression of MYC caused a decrease in the HSC pool. Similarly, skin stem cells and pro-B cells require Myc for differentiation to mature keratinocytes or B cells, respectively (Frye et al., 2003; Gandarillas and Watt, 1997; Habib et al., 2007; Iritani et al., 2002; Watt et al., 2008). Hence, transient Myc-induced cell proliferation is coupled with differentiation in multiple tissues in vivo. While MYC is required for colonic epithelial turnover, deletion of MYC in mouse hepatocytes did not inhibit liver regeneration (Baena et al., 2005; Li et al., 2006; Sansom et al., 2007; Wilkins and Sansom, 2008). It remains unclear, however, whether N-Myc could play a role in the absence of c-Myc in the liver. These studies collectively indicate that Myc plays a critical role in tissue stem cell maturation toward committed progenitors.
The study of the 50-gene Myc core signature (MCS) also allows for an analysis of genes that are cell type specific Myc target genes (Ji et al., 2011). In this regard, B cell restricted genes such as BLMH behave as a direct Myc target and LIN28 behaves as a human embryonic stem cell restricted Myc target, whereas FBL is an MCS gene common to many cell types (Chang et al., 2009; Ji et al., 2011; Koh et al., 2011a). These studies collectively suggest that a key function of Myc is to drive biomass accumulation. Biomass accumulation requires a commensurate level of bioenergetics and building blocks, but genes involved in energy metabolism are not found in the MCS. This observation suggests that the bioenergetics of cells may depend on cell types. Among normal cells there is a remarkable array of different metabolic profiles, from highly aerobic (cardiac, brain) to adaptive anaerobic (skeletal muscle) cells. Hence, it could be the variable uses of different energetic pathways among different cell types that eliminate Myc target genes involved in metabolism from the MCS.
Although the MCS defines a stringent cell-type independent set of Myc targets, other bona fide Myc targets have been firmly identified in a number of systems. In fact, Myc directly regulates genes involved in cell cycle regulation such as CDK4, which is documented in mammalian and Drosophila cells as a Myc target (Hermeking et al., 2000; Orian et al., 2003). In addition, Myc regulates energy metabolism through its direct activation of genes involved in glycolysis, glutamine metabolism and mitochondrial biogenesis (Gao et al., 2009; Li et al., 2005; Osthus et al., 2000; Shim et al., 1997; Wise et al., 2008). In this regard, it seems that Myc as a master regulator affects a broad spectrum of genes to coordinate energy metabolism with biomass accumulation in preparation for DNA replication and cell division. The cooperation of Myc with other transcription factors such as E2F is necessary for the sequential expression of target genes as the cell traverses through the cell cycle (Li et al., 2003; Pickering et al., 2009; Zeller et al., 2006). Under hypoxia, normal Myc can be suppressed by the hypoxia inducible factor, HIF-1 (Gordan et al., 2007; Gordan et al., 2008; Zhang et al., 2007); however, deregulated MYC expression appears to collaborate with HIF-1 to drive glycolytic gene expression for cancer cell proliferation (Dang, 2007; Kim et al., 2007; Qing et al., 2010). The ability of Myc or N-Myc to cooperate with HIF-1 could be highly relevant to the ability of cancer cells to survive and proliferate under moderately hypoxic conditions commonly found in the tumor microenvironment. This perspective suggests that normal cell proliferation is controlled by many of the same genes that are coordinated by Myc in cancer cells. The question then is whether there are distinctive features of normal MYC regulation of cell proliferation versus those related to oncogenic (deregulated) MYC.
Metabolic and MYC oncogene addiction
In contrast to normal cells, in which the MYC proto-oncogene is under stringent regulation downstream of many receptor signaling pathways including WNT, Hedgehog, Notch, TGFβ, as well as many receptor tyrosine kinases, MYC activation in cancer cells can result from constitutive activation of a pathway such as WNT in tumors with loss of APC or through direct alterations of the MYC gene such as amplification or chromosomal translocation. Furthermore, MYC amplification is documented as a means to resist therapeutic inhibition of PI3K, indicating that MYC is downstream of PI3K for tumorigenesis (Ilic et al., 2011; Muellner et al., 2011). The deregulated expression of MYC in cancers presumably causes a sustained increased Myc protein expression, perhaps throughout the cell cycle rather than in a restricted manner. A threshold level for the pathological expression of Myc has been documented in several systems, supporting the concept that constitutive, elevated expression of Myc contributes to tumorigenesis (Murphy et al., 2008; Shachaf et al., 2008; Yustein et al., 2011). It is also reasonable to hypothesize that constitutive Myc expression could cause Myc to promiscuously activate E-box driven genes that would be regulated by other E-box transcription factors in the normal non-proliferative cells.
It could be envisioned that non-proliferating cells express certain E-box genes for homeostatic purposes. For example, lipogenesis in non-proliferating liver or fat cells is mediated by SREBP (Krycer et al., 2010). Mitochondrial biogenesis could be stimulated in normal cells by NRF1, which binds E-boxes and is a target of Myc (Scarpulla, 2008). Certain E-box driven metabolic genes are regulated in a circadian rhythm by Clock/Bmal, while glycolytic and angiogenic genes can be activated by HIF-1 in response to hypoxia (Asher and Schibler, 2011; Semenza, 2007; Shchors et al., 2006). When Myc is elevated, it could bind to the same targets that would be bound by other E-box transcription factors (Dang et al., 2008; Kim et al., 2007). Hence, it is plausible that there is a switch in the regulation of E-box genes from homeostatic transcription to Myc transactivation, which orchestrates cell growth and proliferation. In this regard, the ability of Myc to co-opt the functions of other E-box transcription factors would enable Myc to coordinate a growth program along with angiogenesis (Baudino et al., 2002; Dews et al., 2010; Knies-Bamforth et al., 2004). In the case of lipogenic genes, their induction by Myc would be for lipid membrane synthesis for a growing cell rather than for fat storage. Glycolytic and mitochondrial genes would be induced by Myc to drive the biosynthetic needs of proliferating cells. The metabolic gene, NAMPT which is involved in NAD+ synthesis, is a circadian gene responsive to Clock/Bmal in the liver (Ramsey et al., 2009). The NAMPT gene is prominently bound by Myc and is a direct Myc target gene (Menssen et al., 2012). Collectively, these findings suggest that deregulated Myc commandeers many of the E-box genes to enable the cell to grow and then divide, and in the case of neoplasia Myc also drives angiogenesis (Figure 4).
Normal cells have feedback loops that negatively regulate growth when deprived of nutrients or growth factors. Deprivation of glucose from normal fibroblasts causes them to withdraw from the cell cycle into the G1 phase of the cell cycle (Holley and Kiernan, 1974). Perhaps, this is due partly to the diminished levels of Myc under hypoxia or low glucose conditions (Okuyama et al., 2010). Because glucose is involved in multiple metabolic pathways including glycolysis (providing carbon skeletons for lipid synthesis), the pentose phosphate pathway (ribose synthesis) and glucosamine synthesis, its deprivation is expected to profoundly affect normal cell proliferation. In contrast, glucose withdrawal of Myc-overexpressing cells triggers apoptosis (Shim et al., 1998). Likewise, glutamine withdrawal triggers apoptosis of Myc-overexpressing cells (Yuneva et al., 2007). In the recent study of normal primary activated murine T lymphocytes bearing floxed Myc alleles, it was found that the initial phase of cell growth (cell size increase) was profoundly dependent on Myc expression that drives target genes involved in both glucose and glutamine metabolism. Loss of Myc was associated with profoundly diminished expression of genes involved in metabolism and the inability for normal primary T cells to grow and proliferate (Wang et al., 2011a). It was also documented that HIF-1, which regulates anaerobic glycolysis, was not required for cell growth, but required for T cell fate determination (Dang et al., 2011; Shi et al., 2011). Given that Myc drives biomass accumulation, the sensitivity of Myc-overexpressing cells to nutrient deprivation could reflect their deregulated growth which renders them dependent on and addicted to continual bioenergetic sources, such as glucose and glutamine.
While nutrient dependency characterizes a feature of Myc-transformed cells, it has been recognized in transgenic models that Myc-induced tumors are also addicted to Myc, such that conditional silencing of ectopic Myc expression causes tumor regression in a number of different tumor models. The collapse of the tumor microenvironment and neo-vasculature could account for the oncogene addiction (Giuriato et al., 2006; Sodir et al., 2011). In these cases, the time-dependent changes in the Myc-induced genetic program when Myc is turned off may cause an asynchrony between bioenergetics demands and fuel sources. Inactivation of Myc can also restore the normal TGFβ regulatory circuitry, such that loss of Myc would reactivate SMAD-Miz-1 mediated activation of p21 downstream of TGFβ, culminating in cell cycle arrest and senescence (van Riggelen et al., 2010a). Finally, immune cells are critical components of the tumor microenvironment, and in this regard they affect Myc’s ability to regulate angiogenesis and senescence (Rakhra et al., 2010).
Conceptually, it appears that regulation of Myc expression in normal cells is orchestrated in a fashion that withdrawal of growth signals is followed by an orderly recession of gene expression programs such that there is a balance between fuel needs and fuel supply and utilization by the normal cell. In Myc-overexpressing transformed cells, deregulated Myc presumably alters expression and the balance among the target genes such that Myc withdrawal is followed by an asynchronous recession of the target gene network, resulting in an imbalance between nutrient supply and demand. In some case, a brief suppression of MYC is sufficient to reverse in vivo tumorigenesis (Jain et al., 2002), but not in other cases (Boxer et al., 2004; Jonkers and Berns, 2004; Shachaf et al., 2004), suggesting that tissue specificity and either other mutagenic events or epigenetic alterations affect the reversibility of tumorigenesis upon Myc suppression. Suppression of MYC could trigger senescence or cause an imbalance between apoptotic and anti-apoptotic gene expression could also tip the scale toward cell death with Myc withdrawal (Felsher, 2010; Wu et al., 2007; Zhuang et al., 2008). It is reasonable to hypothesize that the imbalance between energy demand and supply upon Myc withdrawal could also play a role in Myc oncogene addiction.
Metastasis and Genome Instability induced by Myc
Cancer cells are not just characterized by the propensity to proliferate with disregard to extracellular cues but also underscored by increased genomic instability, changes in morphology and function such as epithelial-mesenchymal transition (EMT) and an increased ability to metastasize. Given that Myc can repress genes involved in cell-cell and cell-substratum contact and that these processes would be diminished as normal cells detach from neighboring cells to undergo mitosis, it should not be surprising that overexpressed Myc would also elicit these phenotypes (Dang et al., 2006). Myc has been linked to EMT and metastasis via its regulation of a microRNA miR-9, which targets E-cadherin, as well as its ability to transactivate Bmi-1 which is linked to EMT (Ma et al., 2010; Song et al., 2009). Whether Myc plays a similar parallel role in non-pathologic conditions is not clear.
Overexpression of Myc in a number of cell systems in vitro has been linked to increased genomic instability (Felsher and Bishop, 1999b; Karlsson et al., 2003; Neiman et al., 2006; Prochownik, 2008; Ray et al., 2006). Myc induction of ROS, presumably through its induction of mitochondrial biogenesis and increased metabolism, has been implicated in genomic instability (Egler et al., 2005; Gao et al., 2007; Vafa et al., 2002; Zhang et al., 2007). Here it could be envisioned that normal cells with normal regulated Myc would have the appropriate compensatory mechanisms to detoxify free oxygen radicals, whereas highly active Myc would induce a sustained ROS insult on the genomic, causing instability (Egler et al., 2005; Graves et al., 2009; Wonsey et al., 2002). Whether ROS is central to Myc-induced genomic instability is unclear, particularly since Myc could directly affect telomere function and increase genome instability (Louis et al., 2005). Intriguingly, in a transgenic lymphoma model with inducible Myc, many of the tumors display remarkable chromosomal rearrangements suggesting that Myc affects chromosomal stability but the molecular details remained undefined (Karlsson et al., 2003). Myc is, however, known to regulate a number of components of the mitotic checkpoint; whether deregulated expression of these components contributes to chromosomal instability remains to be studied (Li and Dang, 1999; Menssen et al., 2007).
Therapeutic opportunities
MYC certainly seems to be at the crossroads of many important biological pathways and processes involved in neoplastic cell growth and proliferation. MYC is documented to be involved broadly in many cancers, in which its expression is estimated to be elevated or deregulated in up to 70% of human cancers. High levels of MYC expression have been linked to aggressive human prostate cancer and triple negative breast cancer (Gurel et al., 2008; Palaskas et al., 2011). Experimental models of Myc-mediated tumorigenesis suggest that established tumors are addicted to Myc and that deregulated expression of Myc result in an addiction not only to Myc but also to nutrients. These Myc-induced changes provide a unique opportunity for new therapeutic strategies. Notwithstanding the fact that normal proliferating cells (stem cell compartments and immune cells) also use MYC for renewal, many studies have focused on targeting Myc for cancer therapeutics. Strategies have emerged to inhibit MYC expression, to interrupt Myc-Max dimerization, to inhibit Myc-Max DNA binding, and to interfere with key Myc target genes.
Although there has been significant attention to the G-quadruplex regulatory sequence in the MYC promoter as a therapeutic target, current available compounds that lock the G-quadruplex into an ‘off’ mode are not yet shown to be effective in the preclinical setting (Brown et al., 2011; Dai et al., 2011). Intriguingly, the BET bromain domain regulatory proteins recently emerged as potent regulators of MYC expression in different tumor types. In particular, inhibition of BET with a drug-like molecule in multiple myeloma, a malignant plasma cell tumor, resulted in a remarkable diminution of MYC expression and associated cell death. In this regard, it is also notable that a number of Burkitt lymphoma cell line with MYC translocations were also susceptible to growth inhibition by BET inhibitors. Inhibition of the BET BRD4 protein, thus, proved to be effective in an in vivo preclinical mode, suggesting that targeting MYC expression is feasible in selected cancers (Delmore et al., 2011; Mertz et al., 2011).
The strategy directed toward interrupting Myc-Max dimerization has been forged by several groups (Clausen et al., 2010; Follis et al., 2009; Follis et al., 2008; Hammoudeh et al., 2009; Huang et al., 2006; Mustata et al., 2009; Park et al., 2004; Prochownik and Vogt, 2010). While proof-of-concept has been documented with inhibitors effective in the micromolar concentration range in vitro, evidence for in vivo effectiveness is still lacking. Nothwithstanding this limitation, the proof-of-concept provided to date suggests that this avenue could be fruitful particularly when new chemistry is applied such as click-chemistry that bridges two moderate inhibitors against neighboring protein domains of the target to form a more effective inhibitor. Another approach is to consider the unstructured transcriptional regulatory domain and the DNA binding domain (until it makes contact with DNA) as potential targets for small molecules that would nucleate polypeptide folding and lock the domain in a non-functional conformation.
Other strategies have focused on targeting Myc target genes. For example, Myc repression of miR-26a in a Myc-induced liver model of liver cancer was exploited by treating tumor bearing animals with adenovirus associated viral expression of miR-26a (Kota et al., 2009). This strategy resulted in a remarkable response in this liver cancer model, suggesting that interfering with Myc regulated microRNAs could be therapeutically feasible (Frenzel et al., 2010; Loven et al., 2010). Myc target genes such as ornithine decarboxylase (ODC), lactate dehydrogenase A (LDHA), and glutaminase (GLS) have also been targeted by shRNAs or drug-like small molecules in vivo (Figure 3) (Fantin et al., 2006; Le et al., 2010; Le et al., 2012; Seltzer et al., 2010; Seth et al., 2011; Wang et al., 2010; Xie et al., 2009). These scenarios rely on the necessity of these target genes for the full transforming ability of Myc. It is expected, even with promising preclinical responses to targeting Myc or its target genes, that tumor types and context will add to the complexity and heterogeneity of response to any one strategy. In fact, the avidity of the triple-negative breast cancer subtype for glucose as determined by PET scanning using radiolabeled 18F-deoxyglucose has been linked to a MYC gene expression signature, which contains components of glucose fermentation or glycolysis (Palaskas et al., 2011). Thus, targeting metabolism could be strategically aimed at triple negative breast cancers rather than the estrogen receptor positive tumors.
Recent screens for synthetic lethality in Myc-transformed cells will also likely provide new therapeutic opportunities such as targeting the death receptor pathway or the use of inhibitors against the aurora or cyclin dependent kinases (den Hollander et al., 2010; Yang et al., 2010). A recent synthetic dosage lethality screen uncovered sumoylation of Myc as an essential element for tumor cell growth (Kessler et al., 2012). Further, Myc-induced replicative stress renders transgenic murine lymphomas sensitive to Chk1 inhibitors (Murga et al., 2011). Although synthetic lethality screens are unbiased with respect to the targets, the screens are inherently limited by the choice of tumor cell type. Here, cellular and tissue type context may also affect responses and the spectrum of synthetically lethal targets. In this regard, novel therapies targeting Myc, its target genes or synthetically lethal targets may best be applied in the future together with molecular profiling of cancers for clinical stratification and selection of combination therapies.
Acknowledgments
I thank the reviewers for comments. Our primary work is supported in part by the Leukemia and Lymphoma Foundation, National Institutes of Health, National Cancer Institute, AACR Stand-Up-to-Cancer translational grant, and the Abramson Family Cancer Research Institute. I apologize for omissions, which necessarily happen because of space limitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123:409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc Natl Acad Sci U S A. 2008;105:19678–19683. doi: 10.1073/pnas.0811166106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72:233–245. doi: 10.1016/0092-8674(93)90663-b. [DOI] [PubMed] [Google Scholar]
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature. 1992;359:423–426. doi: 10.1038/359423a0. [DOI] [PubMed] [Google Scholar]
- Armelin HA, Armelin MC, Kelly K, Stewart T, Leder P, Cochran BH, Stiles CD. Functional role for c-myc in mitogenic response to platelet-derived growth factor. Nature. 1984;310:655–660. doi: 10.1038/310655a0. [DOI] [PubMed] [Google Scholar]
- Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313–317. doi: 10.1016/j.semcancer.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinos M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez AC, de Alboran IM. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci U S A. 2005;102:7286–7291. doi: 10.1073/pnas.0409260102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530–2543. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, Arvanitis C, Attardi LD, Feng S, Ruebner B, et al. Developmental Context Determines Latency of MYC-Induced Tumorigenesis. PLoS Biol. 2004;2:E332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Lugo TG, Kretzner L, King MW, Street AJ, Witte ON, Eisenman RN. Functional analysis of the AUG- and CUG-initiated forms of the c-Myc protein. Mol Biol Cell. 1994;5:597–609. doi: 10.1091/mbc.5.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–586. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Brooks TA, Hurley LH. Targeting MYC Expression through G-Quadruplexes. Genes Cancer. 2010;1:641–649. doi: 10.1177/1947601910377493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RV, Danford FL, Gokhale V, Hurley LH, Brooks TA. Demonstration that drug-targeted downregulation of MYC in non-hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J Biol Chem. 2011 doi: 10.1074/jbc.M111.274720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, Elia A, Kress TR, Dickens M, Clemens MJ, et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci U S A. 2010;107:5375–5380. doi: 10.1073/pnas.0910015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, Schlange T, Bauer M, Maurer F, Hynes NE. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 2007;8:70–76. doi: 10.1038/sj.embor.7400849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- Challagundla KB, Sun XX, Zhang X, DeVine T, Zhang Q, Sears RC, Dai MS. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol Cell Biol. 2011;31:4007–4021. doi: 10.1128/MCB.05810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JC, Hannan KM, Riddell K, Ng PY, Peck A, Lee RS, Hung S, Astle MV, Bywater M, Wall M, et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal. 2011;4:ra56. doi: 10.1126/scisignal.2001754. [DOI] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–180. doi: 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther. 2010;335:715–727. doi: 10.1124/jpet.110.170555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD, Cowling VH. Transcription-independent functions of MYC: regulation of translation and DNA replication. Nat Rev Mol Cell Biol. 2008;9:810–815. doi: 10.1038/nrm2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Ngouenet C, Eisenman RN. Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell. 2010;142:480–493. doi: 10.1016/j.cell.2010.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Chandriani S, Whitfield ML, Cole MD. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol Cell Biol. 2006;26:4226–4239. doi: 10.1128/MCB.01959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Cole MD. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol Cell Biol. 2007;27:2059–2073. doi: 10.1128/MCB.01828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, Cox JD, Ha SI, Belka GK, Golant A, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- Dai J, Carver M, Hurley LH, Yang D. Solution Structure of a 2:1 Quindoline-c-MYC G-Quadruplex: Insights into G-Quadruplex-Interactive Small Molecule Drug Design. J Am Chem Soc. 2011;133:17673–17680. doi: 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Found Symp Proc. 2007:35–53. doi: 10.1007/2789_2008_088. [DOI] [PubMed] [Google Scholar]
- Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- Dang CV, Lee WM. Identification of the human c-myc protein nuclear translocation signal. Mol Cell Biol. 1988;8:4048–4054. doi: 10.1128/mcb.8.10.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, Furth E, Enders GH, El-Deiry W, Schelter JM, et al. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010;70:8233–8246. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- Duesberg PH, Vogt PK. Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: evidence for a second class of transforming genes. Proc Natl Acad Sci U S A. 1979;76:1633–1637. doi: 10.1073/pnas.76.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer. 2010;1:597–604. doi: 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999a;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999b;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton-Edkins ZA, Kenneth NS, Brown TR, Daly NL, Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle. 2003;2:181–184. [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fladvad M, Zhou K, Moshref A, Pursglove S, Safsten P, Sunnerhagen M. N and C-terminal sub-regions in the c-Myc transactivation region and their joint role in creating versatility in folding and binding. J Mol Biol. 2005;346:175–189. doi: 10.1016/j.jmb.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Follis AV, Hammoudeh DI, Daab AT, Metallo SJ. Small-molecule perturbation of competing interactions between c-Myc and Max. Bioorg Med Chem Lett. 2009;19:807–810. doi: 10.1016/j.bmcl.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Frenzel A, Loven J, Henriksson MA. Targeting MYC-Regulated miRNAs to Combat Cancer. Genes Cancer. 2010;1:660–667. doi: 10.1177/1947601910377488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development. 2003;130:2793–2808. doi: 10.1242/dev.00462. [DOI] [PubMed] [Google Scholar]
- Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997;11:2869–2882. doi: 10.1101/gad.11.21.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, van Riggelen J, Kopelman AM, Passegue E, Tang F, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A. 2006;103:16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- Graves JA, Metukuri M, Scott D, Rothermund K, Prochownik EV. Regulation of reactive oxygen species homeostasis by peroxiredoxins and c-Myc. J Biol Chem. 2009;284:6520–6529. doi: 10.1074/jbc.M807564200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greasley PJ, Bonnard C, Amati B. Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res. 2000;28:446–453. doi: 10.1093/nar/28.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol. 2000;20:2423–2435. doi: 10.1128/mcb.20.7.2423-2435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- Grinberg AV, Hu CD, Kerppola TK. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Mol Cell Biol. 2004;24:4294–4308. doi: 10.1128/MCB.24.10.4294-4308.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci U S A. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib T, Park H, Tsang M, de Alboran IM, Nicks A, Wilson L, Knoepfler PS, Andrews S, Rawlings DJ, Eisenman RN, Iritani BM. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J Cell Biol. 2007;179:717–731. doi: 10.1083/jcb.200704173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- Hann SR, Sloan-Brown K, Spotts GD. Translational activation of the non-AUG-initiated c-myc 1 protein at high cell densities due to methionine deprivation. Genes Dev. 1992;6:1229–1240. doi: 10.1101/gad.6.7.1229. [DOI] [PubMed] [Google Scholar]
- He L, Liu J, Collins I, Sanford S, O’Connell B, Benham CJ, Levens D. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. Embo J. 2000;19:1034–1044. doi: 10.1093/emboj/19.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway [see comments] Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley RW, Kiernan JA. Control of the initiation of DNA synthesis in 3T3 cells: low-molecular weight nutrients. Proc Natl Acad Sci U S A. 1974;71:2942–2945. doi: 10.1073/pnas.71.8.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopewell R, Ziff EB. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol Cell Biol. 1995;15:3470–3478. doi: 10.1128/mcb.15.7.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Banerjee A, Goss DJ. Assembly of b/HLH/z proteins c-Myc, Max, and Mad1 with cognate DNA: importance of protein-protein and protein-DNA interactions. Biochemistry. 2005;44:11855–11863. doi: 10.1021/bi050206i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SS, Lai MM, Vogt PK. Genome of avian myelocytomatosis virus MC29: analysis by heteroduplex mapping. Proc Natl Acad Sci U S A. 1979;76:1265–1268. doi: 10.1073/pnas.76.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34:1480–1489. doi: 10.1016/j.exphem.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Drug targeting of the c-MYC promoter to repress gene expression via a G-quadruplex silencer element. Semin Oncol. 2006;33:498–512. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani BM, Delrow J, Grandori C, Gomez I, Klacking M, Carlos LS, Eisenman RN. Modulation of T-lymphocyte development, growth and cell size by the Myc antagonist and transcriptional repressor Mad1. EMBO J. 2002;21:4820–4830. doi: 10.1093/emboj/cdf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–104. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al. Cell-Type Independent MYC Target Genes Reveal a Primordial Signature Involved in Biomass Accumulation. PLoS One. 2011;6:e26057. doi: 10.1371/journal.pone.0026057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Berns A. Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004;6:535–538. doi: 10.1016/j.ccr.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, Felsher DW. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc Natl Acad Sci U S A. 2003;100:9974–9979. doi: 10.1073/pnas.1732638100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato GJ, Barrett J, Villa-Garcia M, Dang CV. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol Cell Biol. 1990;10:5914–5920. doi: 10.1128/mcb.10.11.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato GJ, Lee WM, Chen LL, Dang CV. Max: functional domains and interaction with c-Myc. Genes Dev. 1992;6:81–92. doi: 10.1101/gad.6.1.81. [DOI] [PubMed] [Google Scholar]
- Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35:603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci U S A. 2007;104:14917–14922. doi: 10.1073/pnas.0702909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23:1743–1748. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci U S A. 2000;97:11198–11202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knies-Bamforth UE, Fox SB, Poulsom R, Evan GI, Harris AL. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004;64:6563–6570. doi: 10.1158/0008-5472.CAN-03-3176. [DOI] [PubMed] [Google Scholar]
- Koh CM, Gurel B, Sutcliffe S, Aryee MJ, Schultz D, Iwata T, Uemura M, Zeller KI, Anele U, Zheng Q, et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am J Pathol. 2011a;178:1824–1834. doi: 10.1016/j.ajpath.2010.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc Enforces Overexpression of EZH2 in Early Prostatic Neoplasia via Transcriptional and Post-transcriptional Mechanisms. Oncotarget. 2011b;2:669–683. doi: 10.18632/oncotarget.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl NE, Gee CE, Alt FW. Activated expression of the N-myc gene in human neuroblastomas and related tumors. Science. 1984;226:1335–1337. doi: 10.1126/science.6505694. [DOI] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Cannell IG, Brenkman AB, Samans B, Gaestel M, Roepman P, Burgering BM, Bushell M, Rosenwald A, Eilers M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol Cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, Eisenman RN. Myc and Max proteins possess distinct transcriptional activities. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268–276. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- Laurenti E, Varnum-Finney B, Wilson A, Ferrero I, Blanco-Bose WE, Ehninger A, Knoepfler PS, Cheng PF, MacDonald HR, Eisenman RN, et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell. 2008;3:611–624. doi: 10.1016/j.stem.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E, Wilson A, Trumpp A. Myc’s other life: stem cells and beyond. Curr Opin Cell Biol. 2009;21:844–854. doi: 10.1016/j.ceb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder A, Pattengale PK, Kuo A, Stewart TA, Leder P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell. 1986;45:485–495. doi: 10.1016/0092-8674(86)90280-1. [DOI] [PubMed] [Google Scholar]
- Levens D. You Don’t Muck with MYC. Genes Cancer. 2010;1:547–554. doi: 10.1177/1947601910377492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xiang Y, Potter J, Dinavahi R, Dang CV, Lee LA. Conditional deletion of c-myc does not impair liver regeneration. Cancer Res. 2006;66:5608–5612. doi: 10.1158/0008-5472.CAN-05-4242. [DOI] [PubMed] [Google Scholar]
- Li Q, Dang CV. c-Myc overexpression uncouples DNA replication from mitosis. Mol Cell Biol. 1999;19:5339–5351. doi: 10.1128/mcb.19.8.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc Natl Acad Sci U S A. 2008;105:18794–18799. doi: 10.1073/pnas.0806879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem. 2003;278:20405–20412. doi: 10.1074/jbc.M211795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis SF, Vermolen BJ, Garini Y, Young IT, Guffei A, Lichtensztejn Z, Kuttler F, Chuang TC, Moshir S, Mougey V, et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc Natl Acad Sci U S A. 2005;102:9613–9618. doi: 10.1073/pnas.0407512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Zinin N, Wahlstrom T, Muller I, Brodin P, Fredlund E, Ribacke U, Pivarcsi A, Pahlman S, Henriksson M. MYCN-regulated microRNAs repress estrogen receptor-alpha (ESR1) expression and neuronal differentiation in human neuroblastoma. Proc Natl Acad Sci U S A. 2010;107:1553–1558. doi: 10.1073/pnas.0913517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–1399. [PMC free article] [PubMed] [Google Scholar]
- Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan IJ, Dahlman-Wright K, Ford J, Wright AP. Functional interaction of the c-Myc transactivation domain with the TATA binding protein: evidence for an induced fit model of transactivation domain folding. Biochemistry. 1996;35:9584–9593. doi: 10.1021/bi960793v. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, Hermeking H. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle. 2007;6:339–352. doi: 10.4161/cc.6.3.3808. [DOI] [PubMed] [Google Scholar]
- Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, Larsson LG, Hermeking H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, von Stedingk K, Ghesquiere B, Schulte S, Dews M, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010;40:762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, D’Artista L, Schleker T, Guerra C, Garcia E, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–1335. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang H, Prochownik EV, Lazo JS, Bahar I. Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem. 2009;52:1247–1250. doi: 10.1021/jm801278g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, Minna JD. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature. 1985;318:69–73. doi: 10.1038/318069a0. [DOI] [PubMed] [Google Scholar]
- Neiman PE, Kimmel R, Icreverzi A, Elsaesser K, Bowers SJ, Burnside J, Delrow J. Genomic instability during Myc-induced lymphomagenesis in the bursa of Fabricius. Oncogene. 2006;25:6325–6335. doi: 10.1038/sj.onc.1209646. [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Chandriani S, Park J, Kotenko I, Matheos D, Johnsson A, McMahon SB, Cole MD. TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol Cell Biol. 2002;22:5054–5063. doi: 10.1128/MCB.22.14.5054-5063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Okuyama H, Endo H, Akashika T, Kato K, Inoue M. Downregulation of c-MYC protein levels contributes to cancer cell survival under dual deficiency of oxygen and glucose. Cancer Res. 2010;70:10213–10223. doi: 10.1158/0008-5472.CAN-10-2720. [DOI] [PubMed] [Google Scholar]
- Orian A, Grewal SS, Knoepfler PS, Edgar BA, Parkhurst SM, Eisenman RN. Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harb Symp Quant Biol. 2005;70:299–307. doi: 10.1101/sqb.2005.70.019. [DOI] [PubMed] [Google Scholar]
- Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Palaskas N, Larson SM, Schultz N, Komisopoulou E, Wong J, Rohle D, Campos C, Yannuzzi N, Osborne JR, Linkov I, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011;71:5164–5174. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Chung S, Kim KM, Jung KC, Park C, Hahm ER, Yang CH. Determination of binding constant of transcription factor myc-max/max-max and E-box DNA: the effect of inhibitors on the binding. Biochim Biophys Acta. 2004;1670:217–228. doi: 10.1016/j.bbagen.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell. 1999;3:565–577. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R, et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene. 2011 doi: 10.1038/onc.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering MT, Stadler BM, Kowalik TF. miR-17 and miR-20a temper an E2F1-induced G1 checkpoint to regulate cell cycle progression. Oncogene. 2009;28:140–145. doi: 10.1038/onc.2008.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Lucena A, Arrowsmith CH. 1H, 13C and 15N resonance assignments and secondary structure of the c-Myc binding domain (MBD) and the SH3 domain of the tumor suppressor Bin1. J Biomol NMR. 2001;19:191–192. doi: 10.1023/a:1008387817677. [DOI] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov N, Schulein C, Jaenicke LA, Eilers M. Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat Cell Biol. 2010;12:973–981. doi: 10.1038/ncb2104. [DOI] [PubMed] [Google Scholar]
- Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. 2007;9:765–774. doi: 10.1038/ncb1601. [DOI] [PubMed] [Google Scholar]
- Prochownik EV. c-Myc: linking transformation and genomic instability. Curr Mol Med. 2008;8:446–458. doi: 10.2174/156652408785747988. [DOI] [PubMed] [Google Scholar]
- Prochownik EV, Vogt PK. Therapeutic Targeting of Myc. Genes Cancer. 2010;1:650–659. doi: 10.1177/1947601910377494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Skuli N, Mayes PA, Pawel B, Martinez D, Maris JM, Simon MC. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 2010;70:10351–10361. doi: 10.1158/0008-5472.CAN-10-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, Fan AC, Yang Q, Braunstein L, Crosby E, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Atkuri KR, Deb-Basu D, Adler AS, Chang HY, Herzenberg LA, Felsher DW. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006;66:6598–6605. doi: 10.1158/0008-5472.CAN-05-3115. [DOI] [PubMed] [Google Scholar]
- Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. Embo J. 1999;18:717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Moller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Sauve S, Naud JF, Lavigne P. The mechanism of discrimination between cognate and non-specific DNA by dimeric b/HLH/LZ transcription factors. J Mol Biol. 2007;365:1163–1175. doi: 10.1016/j.jmb.2006.10.044. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Schlosser I, Holzel M, Hoffmann R, Burtscher H, Kohlhuber F, Schuhmacher M, Chapman R, Weidle UH, Eick D. Dissection of transcriptional programmes in response to serum and c-Myc in a human B-cell line. Oncogene. 2005;24:520–524. doi: 10.1038/sj.onc.1208198. [DOI] [PubMed] [Google Scholar]
- Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31:6148–6156. doi: 10.1093/nar/gkg794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Peukert K, Eilers M, Hanel F. Association of Myc with the zinc-finger protein Miz-1 defines a novel pathway for gene regulation by Myc. Curr Top Microbiol Immunol. 1997;224:137–146. doi: 10.1007/978-3-642-60801-8_14. [DOI] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–408. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- Seth P, Grant A, Tang J, Vinogradov E, Wang X, Lenkinski R, Sukhatme VP. On-target inhibition of tumor fermentative glycolysis as visualized by hyperpolarized pyruvate. Neoplasia. 2011;13:60–71. doi: 10.1593/neo.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O, Perez OD, Chang M, Mitchel D, Robinson WH, et al. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 2008;68:5132–5142. doi: 10.1158/0008-5472.CAN-07-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, Krishnamoorthy V, Bhasin M, Capobianco AJ, Kelliher MA. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26:8022–8031. doi: 10.1128/MCB.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527–2538. doi: 10.1101/gad.1455706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheiness D, Bishop JM. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J Virol. 1979;31:514–521. doi: 10.1128/jvi.31.2.514-521.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Chun YS, Lewis BC, Dang CV. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc Natl Acad Sci U S A. 1998;95:1511–1516. doi: 10.1073/pnas.95.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI, Soucek L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011;25:907–916. doi: 10.1101/gad.2038411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, Kong QL, Xu LH, Zhang X, Liu WL, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009;119:3626–3636. doi: 10.1172/JCI39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spotts GD, Patel SV, Xiao Q, Hann SR. Identification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by full-length c-Myc proteins. Mol Cell Biol. 1997;17:1459–1468. doi: 10.1128/mcb.17.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger D, Furrer M, Schwinkendorf D, Gallant P. Max-independent functions of Myc in Drosophila melanogaster. Nat Genet. 2008;40:1084–1091. doi: 10.1038/ng.178. [DOI] [PubMed] [Google Scholar]
- Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev. 2011;21:474–483. doi: 10.1016/j.gde.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2010;110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9. [DOI] [PubMed] [Google Scholar]
- Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Bjorklund M, Wei G, Yan J, Niittymaki I, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41:885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- van Riggelen J, Muller J, Otto T, Beuger V, Yetil A, Choi PS, Kosan C, Moroy T, Felsher DW, Eilers M. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010a;24:1281–1294. doi: 10.1101/gad.585710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010b;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- Vennstrom B, Sheiness D, Zabielski J, Bishop JM. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol. 1982;42:773–779. doi: 10.1128/jvi.42.3.773-779.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Mannava S, Grachtchouk V, Zhuang D, Soengas MS, Gudkov AV, Prochownik EV, Nikiforov MA. c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene. 2008;27:1905–1915. doi: 10.1038/sj.onc.1210823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011a doi: 10.1016/j.immuni.2011.09.021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011b;71:925–936. doi: 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanzel M, Russ AC, Kleine-Kohlbrecher D, Colombo E, Pelicci PG, Eilers M. A ribosomal protein L23-nucleophosmin circuit coordinates Miz1 function with cell growth. Nat Cell Biol. 2008 doi: 10.1038/ncb1764. [DOI] [PubMed] [Google Scholar]
- Wasserman NF, Aneas I, Nobrega MA. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 20:1191–1197. doi: 10.1101/gr.105361.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM, Frye M, Benitah SA. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat Rev Cancer. 2008;8:234–242. doi: 10.1038/nrc2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins JA, Sansom OJ. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res. 2008;68:4963–4966. doi: 10.1158/0008-5472.CAN-07-5558. [DOI] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonsey DR, Zeller KI, Dang CV. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci U S A. 2002;99:6649–6654. doi: 10.1073/pnas.102523299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JB, Brown SJ, Cole MD. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol Cell Biol. 2010;30:1411–1420. doi: 10.1128/MCB.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Claassen G, Shi J, Adachi S, Sedivy J, Hann SR. Transactivation-defective c-MycS retains the ability to regulate proliferation and apoptosis. Genes Dev. 1998;12:3803–3808. doi: 10.1101/gad.12.24.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009;8:626–635. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–13841. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yustein JT, Liu YC, Gao P, Jie C, Le A, Vuica-Ross M, Chng WJ, Eberhart CG, Bergsagel PL, Dang CV. Induction of ectopic Myc target gene JAG2 augments hypoxic growth and tumorigenesis in a human B-cell model. Proc Natl Acad Sci U S A. 2011;107:3534–3539. doi: 10.1073/pnas.0901230107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, Berman AE, Giordano TJ, Prochownik EV, Soengas MS, Nikiforov MA. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008;27:6623–6634. doi: 10.1038/onc.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]