

Abstract
A betaproteobacterium within the family Rhodocyclaceae previously identified as a pyrene degrader via stable-isotope probing (SIP) of contaminated soil (designated pyrene group 1 or PG1) was cultivated as the dominant member of a mixed bacterial culture. A metagenomic library was constructed, and the largest contigs were analyzed for genes associated with polycyclic aromatic hydrocarbon (PAH) metabolism. Eight pairs of genes with similarity to the α- and β-subunits of ring-hydroxylating dioxygenases (RHDs) associated with aerobic bacterial PAH degradation were identified and linked to PG1 through PCR analyses of a simplified enrichment culture. In tandem with a ferredoxin and reductase found in close proximity to one pair of RHD genes, six of the RHDs were cloned and expressed in Escherichia coli. Each cloned RHD was tested for activity against nine PAHs ranging in size from two to five rings. Despite differences in their predicted protein sequences, each of the six RHDs was capable of transforming phenanthrene and pyrene. Three RHDs could additionally transform naphthalene and fluorene, and these genotypes were also associated with the ability of the E. coli constructs to convert indole to indigo. Only one of the six cloned RHDs was capable of transforming anthracene and benz[a]anthracene. None of the tested RHDs were capable of significantly transforming fluoranthene, chrysene, or benzo[a]pyrene.
INTRODUCTION
Awide variety of bacteria are capable of transforming polycyclic aromatic hydrocarbons (PAHs), ubiquitous environmental chemicals that at high concentrations can be considered major pollutants, particularly at sites such as former manufactured gas plants (MGPs) (17, 56). A number of PAHs, particularly those with 4 or more rings, are considered potential carcinogens (39). The reasons for their generally incomplete biological removal from contaminated sites (35, 42) have not yet been fully elucidated.
While the genes and pathways of aerobic bacterial PAH metabolism are generally well understood in representative strains from some bacterial genera such as Pseudomonas (4, 5, 10), Mycobacterium (28, 32), and Sphingomonas (57), the variability in genes responsible for the transformation of these compounds in complex environments has only begun to be revealed. Surveys of contaminated sites using degenerate primers for genes associated with PAH degradation have revealed gene clusters with low similarity to homologous genes from described organisms (34, 36) that were presumably derived from organisms that have yet to be cultivated or closely examined in the laboratory.
As with other areas of microbial ecology, the emergent technology of high-throughput pyrosequencing has the potential to dramatically impact the study of PAH biodegradation. With the current relative ease of genomic sequencing, the number of genomes of PAH-degrading organisms is rapidly increasing (3, 6, 24, 54), and more are likely to be obtained in the future. Metagenomic studies also hold the promise of recovering large numbers of genes related to biodegradation even in the absence of an isolated organism. For example, a recent survey of a tidal mudflat for genes associated with biphenyl metabolism coupled with stable-isotope probing (SIP) revealed a high diversity of those genes, many of which were novel (22).
One common target for studies relevant to PAH degradation is the gene for the α-subunit of the ring-hydroxylating dioxygenase (RHD), for which a large number of PCR primer sets have already been developed (23). RHDs are multicomponent enzymes in the family of Rieske nonheme iron oxygenases that catalyze the initial step in aerobic bacterial PAH degradation through the addition of oxygen to an aromatic ring (14, 40). The well-studied naphthalene dioxygenase associated with the nah gene cluster (of Pseudomonas spp.) contains three components: a reductase, ferredoxin, and dioxygenase, with the dioxygenase itself consisting of large (α) and small (β) subunits (40). A functional naphthalene dioxygenase therefore requires the expression of four genes (18). The genes encoding all three of these activities are located in close proximity in the nah gene cluster (10, 58) and in gene clusters associated with PAH metabolism in other organisms (8, 13, 16, 25, 26, 33). However, in some organisms, these genes can be scattered among different operons (2, 7, 30, 43). Understanding these genes, the enzymes they code for, and their substrate specificities may be key to understanding some limitations of PAH biodegradation.
In prior work, we used SIP to identify the bacteria capable of growth on several PAHs, including naphthalene, phenanthrene, and pyrene in soil from a former MGP site in Charlotte, NC, that had been treated in a lab-scale, aerobic slurry bioreactor (49, 51). Of particular interest were several groups of uncultivated proteobacteria capable of growing on pyrene, a four-ring PAH that is commonly used as a model compound for studies of high-molecular-weight PAHs. One of these groups, designated pyrene group 1 (or PG1) dominated the clone library constructed of 13C-enriched DNA and comprised very closely related sequences of Betaproteobacteria within the family Rhodocyclaceae. Organisms associated with these sequences were later shown to be capable of phenanthrene degradation in a separate SIP experiment (48), and sequences related to PG1 sequences were among the most abundant bacterial sequences that were recovered in a laboratory soil column following persulfate oxidation of a PAH-contaminated soil (41). The 16S rRNA gene of the SIP-derived representative clonal sequence possessed low similarity to isolated and characterized organisms (<92% to a Denitratisoma strain at the time of publication and currently <95% to Sulfuritalea hydrogenivorans). However, this sequence was closely related to a variety of other environmental clones from a range of geographically diverse habitats, suggesting a possible widespread distribution of the group.
In this work, we describe the enrichment of a member of pyrene group 1 as a member of a mixed bacterial community, metagenomic sequencing of a portion of its genome, and the description and expression of multiple RHD genes associated with PAH metabolism. This work adds to our incomplete understanding of the diversity of PAH-degrading bacteria and the genetic determinants of PAH metabolism, and it highlights the need for further studies that focus on those organisms that may have environmental relevance but have not yet been isolated in the laboratory.
MATERIALS AND METHODS
Chemicals.
The chemicals used in this study, their purity, and vendors were as follows: naphthalene (99+%, scintillation grade; Aldrich, Milwaukee, WI), phenanthrene (>96%, high-performance liquid chromatography [HPLC] grade; Sigma, St. Louis, MO), anthracene (scintillation grade; Kodak, Rochester, NY), fluorene (98%; Aldrich), pyrene (product P-2146; Sigma), benz[a]anthracene (i.e., 1,2-benzanthracene, 99%; Aldrich), fluoranthene (98%; Aldrich), chrysene (98%; Aldrich), benzo[a]pyrene (97%; Aldrich), and indole (99+%; Aldrich).
Cultivation.
Growth of pyrene-degrading bacteria from PAH-contaminated soil from Charlotte, NC, that had been treated in a lab-scale, aerobic bioreactor (49) was initially performed on solid medium consisting of 5% (vol/vol) autoclaved bioreactor-treated slurry in a 10 mM phosphate buffer amended with 2.3 mM NH4NO3 (pH 7.0) (reactor buffer) and 1.5% agar. Serial dilutions of bioreactor slurry were spread onto the plates before a layer of pyrene was deposited on the agar surface using a thin-layer chromatography plate sprayer with a solution of 2% (wt/wt) pyrene in acetone. The plates were incubated at room temperature in the dark for several weeks. Clear zones that developed in the pyrene overspray were tested for the presence of SIP-identified PAH degraders using previously developed PCR primers (see below). Subsequent maintenance of pyrene-degrading mixed cultures was in SSEM, an autoclave-sterilized liquid medium consisting of 1% of a soil slurry extract in reactor buffer. Soil slurry extract was prepared by adding 7 g (wet weight) of uncontaminated garden soil from Carrboro, NC, to 35 ml reactor buffer, shaking vigorously for at least 1 h, centrifuging to remove solids, and filtering (0.45 μm) the liquid phase. As a carbon source, pyrene or phenanthrene was added in an acetone solution after autoclaving to a final concentration of 0.02% (wt/vol). Serial dilutions of the mixed culture were performed with phosphate-buffered saline (pH 7.4) and occasionally incorporated a 10-s incubation in an ultrasonic bath to disrupt aggregates.
Two cultures were used during this experiment. The first, called the metagenomic enrichment culture, contained at least five bacterial species but appeared dominated by members of pyrene group 1 (PG1). The second culture, derived from the metagenomic enrichment culture and designated the simplified enrichment culture appeared to contain fewer organisms while retaining PG1. Isolates from the simplified enrichment culture capable of growth on nutrient agar (Difco) and nutrient broth were grown on those media. The cultures were stored at −80°C in 15% glycerol.
Screening and sequencing.
Screening for the presence of uncharacterized pyrene-degrading bacteria determined by SIP was performed with conventional PCR using primers specific for those organisms as previously described (51). Cells sampled from clear zones on pyrene-sprayed plates were suspended in water and used as the template for PCR screening. Amplicons were examined on 2% agarose gels. DNA from liquid cultures was obtained using the Wizard Genomic DNA purification kit (Promega, Madison, WI). Quantification of 16S rRNA genes associated with members of the uncharacterized PG1 was performed by quantitative real-time PCR as previously described (51).
Metagenomic sequencing was performed on a DNA sample extracted from the metagenomic enrichment culture in which quantitative real-time PCR (qPCR) indicated that the majority (>90%) of 16S rRNA genes were associated with the organism of interest (PG1). DNA was isolated from 40 ml of SSEM culture as described above and purified with a DNA Clean and Concentrator kit (Zymo Research, Irvine, CA). A total of 6.4 μg of DNA was submitted for pyrosequencing at the University of North Carolina High Throughput Sequencing Facility using a Roche/454 Life Sciences GS-FLX platform, and the resultant reads were assembled using gsAssembler (Roche). The assembled contigs were submitted to the U.S. Department of Energy Joint Genome Institute (DOE JGI) Integrated Microbial Genomes with Microbiome Samples Expert Review system (IMG/MER) for annotation (37). Analyses of the metagenome and searches for 16S rRNA genes and functional genes potentially associated with PAH degradation were carried out with tools within the IMG/MER system. BLAST searches of 16S rRNA genes and predicted protein sequences of functional genes were carried out on the National Center for Biotechnology Information website (1).
DNA sequencing of 16S rRNA genes of isolates and end sequencing of plasmid constructs (see below) was performed by either the University of North Carolina Genome Analysis Facility or Eton Bioscience (Durham, NC). Isolate genes were amplified by whole-cell PCR from single colonies using general 16S rRNA gene primers 8F and 1492R, the amplicons purified using the QIAquick PCR purification kit (Qiagen), and the partial gene sequences obtained using primers 8F, 939R, and 1492R. To compare nucleotide sequences of the RHD genes that have been determined to those of existing PCR primer pairs, DNA sequences were aligned with representative RHD genes using ClustalX 2.0 (55), the target positions were identified in the representative sequences, and the aligned bases were manually compared to the primers. DNA and predicted protein sequences of determined genes were aligned, and trees were generated using a neighbor-joining algorithm and a bootstrap value of 1,000 using ClustalX.
DGGE.
Denaturing gradient gel electrophoresis (DGGE) was performed to examine the complexity of the pyrene-degrading mixed culture. DGGE-PCR was performed with general bacterial primers 341FGC and 517R for 25 cycles of touchdown PCR. The initial annealing temperature for the PCR was 65°C with a decrease of 1°C during each of the first 10 cycles to a final annealing temperature of 55°C for the final 15 cycles. The gel contained 10% acrylamide with 30 to 60% denaturant and was run at 60 V for approximately 16 h.
Construction of plasmids for ring-hydroxylating dioxygenase gene expression.
Genes identified from the metagenomic analysis as possible subunits of a PAH-related RHD or members of an electron transport system involved with PAH metabolism were PCR amplified using template DNA extracted from the simplified enrichment culture. PCR primers were designed to include restriction sites for cloning as well as ribosome binding sites (RBS) for expression in Escherichia coli (see Table S1 in the supplemental material). Forward primers for the first cloned gene additionally incorporated a stop codon prior to the RBS to prevent the addition of a polyhistidine tag to the protein. Putative RHD α- and β-subunit pairs were amplified together as a single product and therefore possessed only a single added RBS upstream of the α-subunit. Cloning was performed by standard methods (44), and vectors and host strains used are described in Table 1. The genes were first PCR amplified and cloned into pCR4-TOPO (Invitrogen), and the sequences were checked against the metagenome-derived sequences by 5′- and 3′-end sequencing using primers targeting M13 sites in the vector. The genes were then excised from the cloning vector using restriction endonucleases (New England BioLabs) targeting sites incorporated into the initial PCR primers and cloned into vector pRSET-A (Invitrogen). Restriction sites internal to the cloned genes of some dioxygenase pairs required partial digestion and gel extraction using a QIAquick gel extraction kit (Qiagen) prior to cloning into the expression vector. The presence of either two or four cloned genes in the final constructs in the correct orientation and size was confirmed by PCR using primers at the 5′ end of the ferredoxin gene and the 3′ end of either the reductase or RHD β-subunit genes prior to transformation into the host strain for expression.
Table 1.
Vectors and strains used in this study
| Plasmid or strain | Relevant phenotype, genotype, or description | Source |
|---|---|---|
| Plasmids | ||
| pCR4-TOPO | Ampr Kanr; cloning vector for sequencing | Invitrogen |
| pRSET-A | Ampr; vector for gene expression | Invitrogen |
| pFR.1 | pRSET-A vector with ferredoxin and reductase genes | This study |
| pFRR.1 | pFR.1 containing α/β subunit genes of putative RHD-1 | This study |
| pFRR.2 | pFR.1 containing α/β subunit genes of putative RHD-2 | This study |
| pFRR.5 | pFR.1 containing α/β subunit genes of putative RHD-5 | This study |
| pFRR.6 | pFR.1 containing α/β subunit genes of putative RHD-6 | This study |
| pFRR.7 | pFR.1 containing α/β subunit genes of putative RHD-7 | This study |
| pFRR.8 | pFR.1 containing α/β subunit genes of putative RHD-8 | This study |
| Strains | ||
| Mach1-T1 | F− ϕ80lacZΔM15 ΔlacX74 hsdR (rK− mK+) ΔrecA1398 endA1 tonA | Invitrogen |
| BL21(DE3)pLysS | F−ompT hsdSB(rB− mB−) gal dcm (DE3) pLysS (Camr) | Invitrogen |
| Variovorax sp. strain LY1 | Isolated from simplified enrichment culture | This study |
| Pseudomonas sp. strain BW1 | Isolated from simplified enrichment culture | This study |
Expression of RHDs in Escherichia coli.
Putative RHDs and associated electron transport genes cloned into the vector pRSET-A were transformed into E. coli host BL21(DE3)pLysS for expression using a method similar to that employed by Kim et al. (27). Single colonies grown overnight on LB plates containing ampicillin (50 μg/ml) and chloramphenicol (35 μg/ml) were inoculated into 5 ml of LB broth (with antibiotics) and incubated overnight at 30°C and 225 rpm. One milliliter of the overnight culture was used to inoculate 200 ml of LB broth containing antibiotics which was shaken at 30°C and 225 rpm until the optical density at 600 nm (OD600) of the culture reached 0.5. At that point, isopropyl-β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce expression, and the flask was returned to the shaker for 2 h. Cells from 90 ml of culture were pelleted by centrifugation and washed with M9 minimal medium containing glucose (M9-glucose) and antibiotics before being resuspended to the initial volume in fresh M9-glucose medium containing antibiotics. Three milliliters each of this culture were aliquoted into 27 screw cap glass tubes to test activity against 9 PAHs in triplicate. PAHs were added individually to the cell suspensions in dimethyl sulfoxide (DMSO) to a target final concentration of 1 μg/ml. The tubes were incubated in the dark for 40 to 42 h at 30°C and shaken at 225 rpm, except for tubes containing no cells, which were incubated for 15 h. PAHs were extracted by the addition of 7 ml of ethyl acetate to the tubes, followed by vortexing for 10 s, and quantified by HPLC as previously described (50). Reductions in the PAH concentrations during incubation of strains containing all four genes of an RHD were compared to a strain containing only the ferredoxin and reductase genes. Abiotic controls of the media and added PAH but without cells were also performed in triplicate.
Test for indigo formation.
E. coli BL21(DE3)pLysS host cells containing a plasmid with the electron transport genes and both subunits of a candidate RHD were streaked on LB plates containing ampicillin and chloramphenicol. A crystal of indole was placed in the center of the lid, and the plates were incubated inverted at 30°C overnight and at room temperature thereafter for several weeks. Formation of a purple product was indicative of indigo formation (11, 45).
Nucleotide sequence accession numbers.
Sequences from this study were deposited into GenBank under accession numbers JQ360171 to JQ360191. The metagenome is publicly available on the IMG/MER website under the name “bacterial pyrene-degrading mixed culture.”
RESULTS
Cultivation.
PCR screening of a clear zone on a pyrene-oversprayed agar plate derived from lab-scale bioreactor-treated soil led to a mixed culture containing a betaproteobacterium with high 16S rRNA gene sequence similarity to a number of uncharacterized environmental sequences, including a group of pyrene-degrading bacteria recovered from a prior SIP experiment on that same soil designated pyrene group 1 or PG1 (51). Quantitative real-time PCR (qPCR) of a liquid culture that had been inoculated from the clear zone on the plate and successively transferred for over 2 years indicated that 16S rRNA genes associated with PG1 comprised >90% of the total bacterial 16S rRNA genes in that culture. We refer to this as the metagenomic enrichment culture, as DNA extracted from it was used for metagenomic sequencing. A second culture called the simplified enrichment culture was derived from the metagenomic enrichment culture and was the product of successive transfers and serial dilutions for another 2 years. DNA from the simplified enrichment culture was used to PCR amplify genes of interest for expression.
DGGE analyses of the simplified enrichment culture (Fig. 1) indicated three bands, each of which could be matched to either an isolate cultivated from the culture on nutrient agar medium (identified through 16S rRNA gene sequencing as members of the Pseudomonas or Variovorax genus and designated strains BW1 and LY1, respectively) or a 16S rRNA gene associated with PG1 (Fig. 1). The 16S rRNA gene of Variovorax sp. strain LY1 possessed 99.6% similarity to a partial Variovorax 16S rRNA gene in the metagenomic library (see below) and was identical to Variovorax paradoxus strain IHB B. The isolated Pseudomonas strain possessed 99.9% 16S rRNA gene similarity to a number of sequences recovered by SIP with [U-13C]naphthalene (49) of the same bioreactor-treated soil from which PG1 was cultivated and 99.8% similarity to Pseudomonas fluorescens strain A1XB1-4. The relative abundance of PG1 16S rRNA genes in this simplified culture was 37% as determined by qPCR. This mixed culture required approximately 10 days to clear crystalline pyrene from a liquid medium. Both strains BW1 and LY1 independently grew (increased turbidity) in SSEM liquid medium containing pyrene as the dominant carbon source. The PG1 organism was not successfully cultivated in isolation.
Fig 1.
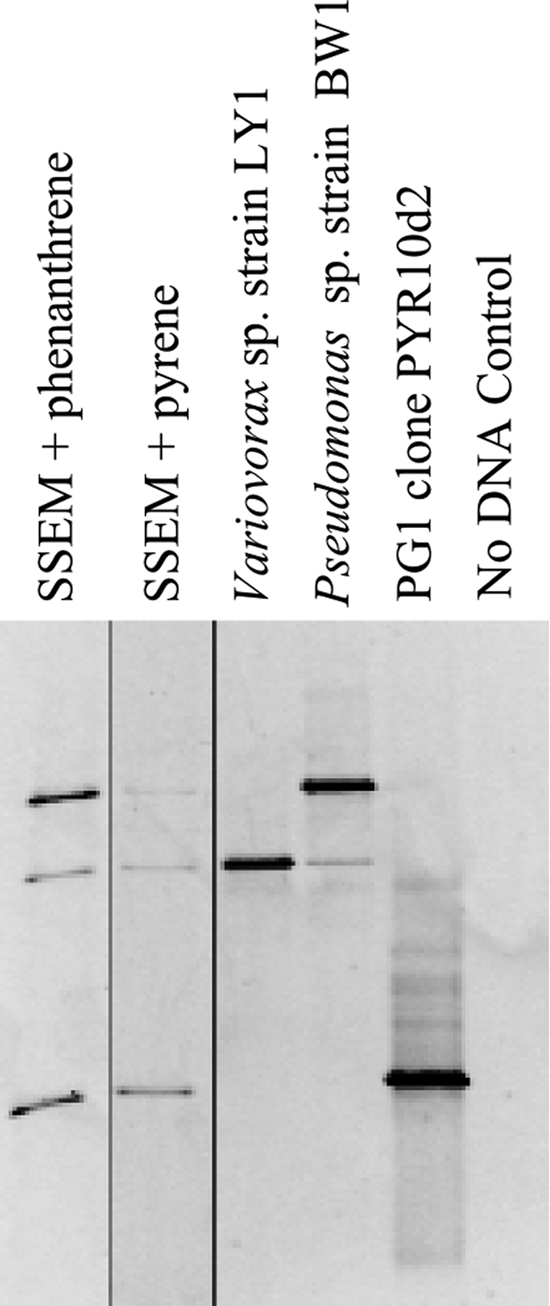
DGGE showing the diversity of bacterial 16S rRNA genes in the simplified enrichment culture from which RHD genes were amplified for expression studies. All lanes were run on a single gel, but the image was cropped to remove intervening lanes and areas without obvious bands. The vertical thin black lines indicate where lanes were digitally removed. In the two leftmost lanes, the simplified enrichment culture was grown with either amended phenanthrene or pyrene. In the middle two lanes, genomic DNA from isolated bacteria is shown. In lane PG1 clone PYR10d2, a PG1 clonal sequence from the earlier SIP experiment is shown. A no-DNA control is shown in the rightmost lane.
Metagenomic analyses.
A metagenomic library was constructed from DNA extracted from the culture in which 16S rRNA genes associated with PG1 were >90% of the total 16S rRNA genes. A total of 288,933 reads were obtained, resulting in 115,626,839 total base pairs of raw data. After elimination of poor-quality reads and assembly, these data were condensed to 5,512,543 bases on 5,446 contigs (maximum contig size, 356,464 bp). The largest contigs were presumed to have been derived from the most abundant DNA in the culture (PG1), so further analyses concentrated only on DNA fragments of ≥10 kbp. These 33 contigs totaled 2,848,788 bp (51.7% of the assembled fragments) and contained 2,751 predicted protein-encoding genes. As evidence for common derivation from a single species, the largest contigs had similar G+C contents (54% ± 2%; maximum 59%, minimum 48%). The smaller contigs had a wider range of G+C content, with the majority (82%) possessing at least 60% G+C (see Fig. S1 in the supplemental material).
Only one complete 16S rRNA gene (assembled from multiple fragments; total size, 1,562 bp) was recovered from the metagenomic library. This gene possessed 99.8% sequence similarity to the representative sequence of PG1 from the prior SIP experiment (see Fig. S2 in the supplemental material). Additional partial 16S rRNA gene fragments in the metagenomic library were associated with organisms related to the Stenotrophomonas or Burkholderia (100% identity over 348 bp), Methylobacterium (100%; 494 bp), Variovorax (99%; 688 bp), and Cupriavidus or Ralstonia (100%; 153 bp) genera.
Using the Integrated Microbial Genomics/Microbiome Expert Review (IMG/MER) system, the largest contigs were examined for genes associated with ring-hydroxylating dioxygenases (RHDs). Eight RHD gene pairs (α- and β-subunits) on the 33 largest contigs possessed significant similarity to and clustered with known PAH-associated RHD genes (Fig. 2). All of these putative RHD α-subunit genes possessed the expected conserved residues for a Rieske center, iron binding, and electron transfer. The predicted amino acid sequences of the α-subunit of these genes possessed 42 to 79% identity (57 to 86% similarity) to established RHD protein sequences. Genes putatively encoding the β-subunit of an RHD were found adjacent to and downstream of the α-subunit genes in each of these eight instances. The predicted amino acid sequences in the α-subunits of two pairs of RHDs possessed significant amino acid similarity to one another (PahAc1 and PahAc3, 75% similarity; PahAc2 and PahAc4, 90%), so only one member of each pair was used for expression experiments.
Fig 2.

Neighbor-joining tree of predicted RHD α-subunit protein sequences. Sequences from this experiment are shown in boldface type. The open and closed circles at the nodes indicate ≥95% and ≥50% bootstrap support, respectively. GenBank accession numbers are indicated in parentheses. A biphenyl dioxygenase was used as an outgroup.
Recommended PCR primer sets developed for the amplification of PAH-associated RHDs and Rieske centers (23) were tested in silico against the eight RHD α-subunit sequences (see Table S2 in the supplemental material). Primers targeting the RHD Rieske center generally possessed few mismatches to the RHD gene sequences and would likely amplify those regions. In contrast, primers recommended for amplification of RHD genes from both Gram-negative and Gram-positive bacteria possessed a greater number of mismatches and, depending on the specific PCR conditions, would be unlikely to amplify the genes described in this study.
Genes potentially associated with PAH degradation in PG1 generally did not cluster in complete and clearly defined operons (Fig. 3). However, one of the more significant clusters of putative PAH metabolism genes appeared on contig_05431, which contained genes for all four subunits of an RHD (Fig. 3). This cluster also contained a putative hydratase/aldolase (open reading frame [ORF] F) and isomerase (ORF G), which were homologous to the nahE and nahD genes of Pseudomonas putida G7, respectively (52). The remaining two ORFs in this cluster (ORFs C and D) had strong predicted amino acid similarity (93% and 82%, respectively) to genes similarly located downstream of a ferredoxin-encoding gene in the betaproteobacteria Burkholderia sp. strain DBT1 (in a gene cluster responsible for dibenzothiophene transformation), but those genes did not have an obvious relationship to PAH degradation (9). This entire cluster of PAH-related genes on contig_05431 may be controlled by a divergently transcribed lysR-type transcriptional regulator (ORF A).
Fig 3.

Gene neighborhoods of RHD genes on contigs putatively associated with the PG1 organism. Diagrams are derived from information provided by the IMG/MER system. Predicted RHD α- and β-subunits are shown with a black background. Ferredoxin and reductase genes are shown on a gray background. The highest BLASTP hits to the predicted amino acid sequence of each open reading frame (ORF) can be found in supplemental information (see Table S3 in the supplemental material).
A second, incomplete cluster of putative genes for PAH metabolism appeared on contig_05432, with a putative dihydrodiol dehydrogenase (ORF B), catechol 2,3-dioxygenase (ORF D), and two pairs of RHD genes. The gene arrangement of an aromatic RHD β-subunit (ORF F, unrelated to a PAH-associated RHD), succinate dehydrogenase/fumarate reductase (ORF G), and ferredoxin (ORF H) also appeared in the genome of Mycobacterium sp. strain KMS, and several other ORFs in this region bore high similarity to genes in Mycobacterium spp. (see Table S3 in the supplemental material).
The other five identified RHD gene pairs did not appear in regions with significant numbers of genes potentially associated with PAH metabolism. Additional genes that did appear to encode several “lower pathway” enzymes, including a tautomerase, decarboxylase, aldolase, hydratase, and semialdehyde dehydrogenase (with similarity to nahJ, nahK, nahM, nahL, and nahI of P. putida G7, respectively) appeared in a cluster on the largest contig (data not shown) but were not located close to any identified RHD gene. The full description of genes in close proximity to the identified RHDs presented in Fig. 3 can be found in Table S3 in the supplemental material.
Substrate specificity of cloned RHDs.
Pairs of RHD genes (α- and β-subunits) were cloned into an expression vector behind the ferredoxin and reductase genes identified on contig_05431 (Fig. 3). The template DNA for cloning reactions was extracted from the simplified enrichment culture. PCR primers specific for each of the RHDs were additionally tested against isolated strains from this culture, but no PCR primer set produced a product (data not shown). This strongly suggests that the cloned RHD genes were derived from the unisolated PG1 organism or some other, undetected, minor member of that community.
In order to evaluate whether a particular RHD possessed activity against an added substrate, the amount of PAH remaining after induction and incubation with an E. coli host expressing a putative PG1 RHD was compared to an equivalent incubation with an E. coli host strain containing only the cloned electron transport genes (the ferredoxin and reductase) and no RHD (i.e., a no-RHD control, plasmid pFR.1). Given the variability that could arise during the addition of the substrates, incubations, and PAH extractions, we conservatively considered only results with at least a 25% decrease in PAH concentration and statistically significant difference to the no-RHD control (Student's t test; P < 0.05) as evidence of enzymatic activity against a given substrate (determined in part from analyses of abiotic controls; data not shown).
Six RHDs were evaluated for their ability to transform nine PAHs ranging in size from 2 to 5 rings: naphthalene, phenanthrene, anthracene, fluorene, pyrene, fluoranthene, benz[a]anthracene, chrysene, and benzo[a]pyrene. Substrates for which at least one RHD was capable of transformation under the criteria specified above are shown in Fig. 4. All six RHDs significantly transformed both phenanthrene and pyrene. No significant removal was observed for chrysene, benzo[a]pyrene, or fluoranthene from any cloned RHD.
Fig 4.

Decrease in PAH parent compound concentration by E. coli cells expressing all four PG1 RHD genes relative to E. coli cells expressing only the RHD ferredoxin and reductase genes. Values are averages plus standard deviations (error bars) of triplicate incubations. Experiments for which an expressed RHD displayed at least 25% removal of the PAH and significantly lower PAH concentration than the control without expressed RHD α- and β-subunits are indicated by asterisks. Abbreviations: NAP, naphthalene; FLU, fluorene; ANT, anthracene; BAA, benz[a]anthracene; PHN, phenanthrene; PYR, pyrene.
Three RHDs (RHD-1, RHD-2, and RHD-5) were additionally capable of transforming the low-molecular-weight PAHs naphthalene and fluorene (Fig. 4). These RHDs also conferred upon the E. coli host the ability to convert indole to indigo, additional evidence of a functional RHD. The gene products of RHD-5 displayed a particularly strong reaction when exposed to indole, with rapid and strong production of indigo even in the absence of gene induction. PG1 RHD-6, RHD-7, and RHD-8 did not appear capable of transforming indole to indigo. RHD-1 was the only dioxygenase tested capable of transforming anthracene and benz[a]anthracene. It is possible that a negative result for a given PAH may have been due to undetermined factors (e.g., poor expression, enzyme instability) other than substrate specificity.
DISCUSSION
Previously, SIP of PAH-contaminated soil treated in a bioreactor identified three groups of uncultivated proteobacteria capable of incorporating carbon from pyrene, none of which were closely related to described genera (51). One of the groups, which we named pyrene group 1, did not appear to be an abundant member of the contaminated soil prior to bioreactor treatment. We hypothesized that conditions utilized during biological treatment of the soil in a lab-scale, aerobic bioreactor provided a more favorable environment for the growth of these organisms. With this in mind, we set out to isolate members of the uncultivated pyrene-degrading groups by creating a medium that mimicked the conditions of the bioreactor environment, with the bioreactor slurry itself as the major component.
We were successful in obtaining a culture containing PG1 as a predominant member of a mixed culture. After construction of the metagenomic library, successive dilution series and transfers in liquid media produced a mixed culture appearing to comprise three organisms: the unclassified PG1 organism (or organisms), and Pseudomonas and Variovorax strains. Interestingly, a prior study of bacteria from an Arctic soil also identified Pseudomonas and Variovorax strains as part of a biofilm consortium growing on solid pyrene (12). The surface environment of solid pyrene may be analogous to the conditions on our initial isolation plate, with crystalline pyrene deposited on the agar surface. It is likely that both of these organisms were carryovers from the initial cultivation efforts and subsisted and thrived due to their possible pyrene-degrading capabilities. As 16S rRNA gene sequences highly similar to the PG1 organism have appeared in clone libraries from environmental sources around the world (15, 19–21) but no isolates have been recovered, a reliance on other bacteria for a required nutrient or activity may be one explanation for this lack of cultivation.
The metagenomic library provided several new gene sequences putatively associated with PAH metabolism. There are several lines of evidence that many, if not all, of these genes are associated with PG1. First, the culture from which the metagenomic library was created was dominated by members of PG1. As such, we expected that the greatest number of sequences and therefore the largest assembled contigs would be derived from that organism. As support, the only complete 16S rRNA gene recovered from the library was associated with PG1. Second, the G+C percentages of each of the 33 largest contigs were similar to one another, suggesting a common source organism. The majority of the smaller fragments, presumably derived primarily from the minor members of the community, possessed significantly higher G+C percentages. Third, most of the genes studied were not highly similar to homologous PAH degradation genes from described organisms, further suggesting derivation from a previously unstudied organism or genus. Finally, in a simplified enrichment culture, no PCR product was obtained in screens for the PAH degradation genes from isolated members of the consortium, suggesting that it was an unisolated member of the culture from which the genes were amplified. These data strongly suggest that the PG1 organism was the source of the genes characterized in this study, although we cannot entirely exclude the possibility of an additional, undetected organism in the culture which may have been the source of one or more of the RHD genes.
While it was unlikely that the entirety of the PG1 organism's genome was represented in the library, a large number of genes putatively associated with PAH degradation were acquired, including eight RHD gene pairs. There is precedent for such a large number of RHD genes in a single organism—for example, Mycobacterium vanbaalenii PYR-1 expresses 10 RHDs, with six of them induced in the presence of pyrene (31). An additional possibility is that these genes are associated with several strains of PG1 organisms in the culture, which might explain the high diversity and number of RHDs. However, whether this might be the case cannot be determined from the available data. Also similar to M. vanbaalenii, we found significantly fewer genes for enzymes putatively catalyzing the remaining two steps of the “upper pathway” of PAH degradation (a dihydrodiol dehydrogenase and ring cleavage dioxygenase, respectively) among the contigs presumed to be derived from PG1. The induction of genes associated with PAH degradation and the characterization of the remaining genes in the upper pathway in PG1 will be the subjects of future research.
Based on genomic proximity, only one set of electron carrier genes appeared to be associated with PAH-RHDs in the available genomic data for PG1. The cloned and expressed ferredoxin and reductase genes were located in close proximity to RHD-1 (genes pahAc1 and pahAd1) and were likely controlled by the same transcriptional regulator. RHD-1 was also the enzyme with the broadest substrate range of the six enzymes tested in this study. It is possible that this expanded substrate range for RHD-1 was a result of a complete dioxygenase enzyme consisting of the four subunits that would typically comprise the holoenzyme in the PG1 organism and that the cloned and expressed ferredoxin and reductase were not typically associated with the other five RHDs. The role of electron carrier subunits in RHDs associated with PAH metabolism is not always clear. Some dioxygenases have been successfully expressed heterologously with electron carrier genes from the target organism (25, 45, 53), while others functioned only with replacement genes from a different organism (27, 29, 46). Still other RHDs have been expressed using electron transfer proteins native to an E. coli host (7, 26, 33, 38, 47). In some cases, the addition of specific electron carrier genes significantly increased activity of an expressed dioxygenase, suggesting that the specific electron carrier proteins present can be important for activity of the enzyme (7, 29). We did not explicitly test whether the cloned ferredoxin and reductase genes from PG1 were required for RHD activity, but the successful transformation of at least two PAHs by each of the six RHDs suggests that either the cloned genes for electron transfer or native E. coli proteins were sufficient to provide some level of activity. It is noteworthy that all six RHDs transformed phenanthrene and pyrene, two growth substrates for members of PG1 as previously revealed by SIP (48, 51).
Supplementary Material
ACKNOWLEDGMENT
We thank our funding source, the U.S. National Institute of Environmental Health Sciences (NIEHS) Superfund Research Program, grant number 5 P42 ES005948.
Footnotes
Published ahead of print 16 March 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Armengaud J, Happe B, Timmis KN. 1998. Genetic analysis of dioxin dioxygenase of Sphingomonas sp. strain RW1: catabolic genes dispersed on the genome. J. Bacteriol. 180:3954–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bergmann F, et al. 2011. Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulfate-reducing Deltaproteobacterium N47. Environ. Microbiol. 13:1125–1137 [DOI] [PubMed] [Google Scholar]
- 4. Bosch R, Garcia-Valdes E, Moore ERB. 1999. Genetic characterization and evolutionary implications of a chromosomally encoded naphthalene-degradation upper pathway from Pseudomonas stutzeri AN10. Gene 236:149–157 [DOI] [PubMed] [Google Scholar]
- 5. Bosch R, García-Valdés E, Moore ERB. 2000. Complete nucleotide sequence and evolutionary significance of a chromosomally encoded naphthalene-degradation lower pathway from Pseudomonas stutzeri AN10. Gene 245:65–74 [DOI] [PubMed] [Google Scholar]
- 6. Chauhan A, et al. 2011. Draft genome sequence of the polycyclic aromatic hydrocarbon-degrading, genetically engineered bioluminescent bioreporter Pseudomonas fluorescens HK44. J. Bacteriol. 193:5009–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Demaneche S, et al. 2004. Identification and functional analysis of two aromatic-ring-hydroxylating dioxygenases from a Sphingomonas strain that degrades various polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 70:6714–6725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denome SA, Stanley DC, Olson ES, Young KD. 1993. Metabolism of dibenzothiophene and naphthalene in Pseudomonas strains: complete DNA sequence of an upper naphthalene catabolic pathway. J. Bacteriol. 175:6890–6901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Gregorio S, et al. 2004. Identification of two new sets of genes for dibenzothiophene transformation in Burkholderia sp. DBT1. Biodegradation 15:111–123 [DOI] [PubMed] [Google Scholar]
- 10. Eaton RW, Chapman PJ. 1992. Bacterial metabolism of naphthalene: construction and use of recombinant bacteria to study ring cleavage of 1,2-dihydroxynaphthalene and subsequent reactions. J. Bacteriol. 174:7542–7554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ensley BD, et al. 1983. Expression of naphthalene oxidation genes in Escherichia coli results in the biosynthesis of indigo. Science 222:167–169 [DOI] [PubMed] [Google Scholar]
- 12. Eriksson M, Dalhammar G, Mohn WW. 2002. Bacterial growth and biofilm production on pyrene. FEMS Microbiol. Ecol. 40:21–27 [DOI] [PubMed] [Google Scholar]
- 13. Fuenmayor SL, Wild M, Boyes AL, Williams PA. 1998. A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J. Bacteriol. 180:2522–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibson DT, Parales RE. 2000. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 11:236–243 [DOI] [PubMed] [Google Scholar]
- 15. Gihring TM, et al. 2006. The distribution of microbial taxa in the subsurface water of the Kalahari Shield, South Africa. Geomicrobiol. J. 23:415–430 [Google Scholar]
- 16. Goyal AK, Zylstra GJ. 1996. Molecular cloning of novel genes for polycyclic aromatic hydrocarbon degradation from Comamonas testosteroni GZ39. Appl. Environ. Microbiol. 62:230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haeseler F, Blanchet D, Durelle V, Werner P, Vandecasteele J-P. 1999. Analytical characterization of contaminated soils from former manufactured gas plants. Environ. Sci. Technol. 33:825–830 [Google Scholar]
- 18. Harayama S, Rekik M. 1989. Bacterial aromatic ring-cleavage enzymes are classified into two different gene families. J. Biol. Chem. 264:15328–15333 [PubMed] [Google Scholar]
- 19. He Z, Xiao S, Xie X, Hu Y. 2008. Microbial diversity in acid mineral bioleaching systems of Dongxiang copper mine and Yinshan lead-zinc mine. Extremophiles 12:225–234 [DOI] [PubMed] [Google Scholar]
- 20. He Z, Xie X, Xiao S, Liu J, Qiu G. 2007. Microbial diversity of mine water at Zhong Tiaoshan copper mine, China. J. Basic Microbiol. 47:485–495 [DOI] [PubMed] [Google Scholar]
- 21. Humphries JA, Ashe AM, Smiley JA, Johnston CG. 2005. Microbial community structure and trichloroethylene degradation in groundwater. Can. J. Microbiol. 51:433–439 [DOI] [PubMed] [Google Scholar]
- 22. Iwai S, et al. 2010. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 4:279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwai S, Johnson TA, Chai B, Hashsham SA, Tiedje JM. 2011. Comparison of the specificities and efficacies of primers for aromatic dioxygenase gene analysis of environmental samples. Appl. Environ. Microbiol. 77:3551–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin HM, et al. 2011. Complete genome sequence of the polycyclic aromatic hydrocarbon-degrading bacterium Alteromonas sp. strain SN2. J. Bacteriol. 193:4292–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasai Y, Shindo K, Harayama S, Misawa N. 2003. Molecular characterization and substrate preference of a polycyclic aromatic hydrocarbon dioxygenase from Cycloclasticus sp. strain A5. Appl. Environ. Microbiol. 69:6688–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khan AA, et al. 2001. Molecular cloning, nucleotide sequence, and expression of genes encoding a polycyclic aromatic ring dioxygenase from Mycobacterium sp. strain PYR-1. Appl. Environ. Microbiol. 67:3577–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim SJ, et al. 2006. Molecular cloning and expression of genes encoding a novel dioxygenase involved in low- and high-molecular-weight polycyclic aromatic hydrocarbon degradation in Mycobacterium vanbaalenii PYR-1. Appl. Environ. Microbiol. 72:1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim SJ, et al. 2007. Complete and integrated pyrene degradation pathway in Mycobacterium vanbaalenii PYR-1 based on systems biology. J. Bacteriol. 189:464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krivobok S, et al. 2003. Identification of pyrene-induced proteins in Mycobacterium sp. strain 6PY1: evidence for two ring-hydroxylating dioxygenases. J. Bacteriol. 185:3828–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kulakov LA, Chen S, Allen CCR, Larkin MJ. 2005. Web-type evolution of Rhodococcus gene clusters associated with utilization of naphthalene. Appl. Environ. Microbiol. 71:1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kweon O, et al. 2011. Polycyclic aromatic hydrocarbon metabolic network in Mycobacterium vanbaalenii PYR-1. J. Bacteriol. 193:4326–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kweon O, et al. 2007. A polyomic approach to elucidate the fluoranthene degradative pathway in Mycobacterium vanbaalenii PYR-1. J. Bacteriol. 189:4635–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laurie AD, Lloyd-Jones G. 1999. The phn genes of Burkholderia sp. strain RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J. Bacteriol. 181:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lozada M, et al. 2008. Novel aromatic ring-hydroxylating dioxygenase genes from coastal marine sediments of Patagonia. BMC Microbiol. 8:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lundstedt S, Haglund P, Oberg L. 2003. Degradation and formation of polycyclic aromatic compounds during bioslurry treatment of an aged gasworks soil. Environ. Toxicol. Chem. 22:1413–1420 [PubMed] [Google Scholar]
- 36. Marcos MS, Lozada M, Dionisi HM. 2009. Aromatic hydrocarbon degradation genes from chronically polluted subantarctic marine sediments. Lett. Appl. Microbiol. 49:602–608 [DOI] [PubMed] [Google Scholar]
- 37. Markowitz VM, et al. 2008. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 36:D534–D538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moser R, Stahl U. 2001. Insights into the genetic diversity of initial dioxygenases from PAH-degrading bacteria. Appl. Microbiol. Biotechnol. 55:609–618 [DOI] [PubMed] [Google Scholar]
- 39. National Toxicology Program 2011. Report on carcinogens, 12th ed National Toxicology Program, National Institute of Environmental Health Services, National Institutes of Health, Public Health Service, US Department of Health and Human Services, Bethesda, MD [Google Scholar]
- 40. Parales R, Resnick SM. 2004. Aromatic hydrocarbon dioxygenases, p 175–196 In Singh A, Ward OP. (ed), Soil biology, vol 2. Biodegradation and bioremediation. Springer-Verlag, Heidelberg, Germany [Google Scholar]
- 41. Richardson SD, Lebron B, Miller CT, Aitken MD. 2011. Recovery of phenanthrene-degrading bacteria after simulated in situ persulfate oxidation in contaminated soil. Environ. Sci. Technol. 45:719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ringelberg DB, et al. 2001. Succession of phenotypic, genotypic, and metabolic community characteristics during in vitro bioslurry treatment of polycyclic aromatic hydrocarbon-contaminated sediments. Appl. Environ. Microbiol. 67:1542–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romine MF, et al. 1999. Complete sequence of a 184-kilobase catabolic plasmid from Sphingomonas aromaticivorans F199. J. Bacteriol. 181:1585–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 45. Schell MA. 1983. Cloning and expression in Escherichia coli of the naphthalene degradation genes from plasmid NAH7. J. Bacteriol. 153:822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schuler L, et al. 2009. Characterization of a ring-hydroxylating dioxygenase from phenanthrene-degrading Sphingomonas sp. strain LH128 able to oxidize benz[a]anthracene. Appl. Microbiol. Biotechnol. 83:465–475 [DOI] [PubMed] [Google Scholar]
- 47. Simon M, et al. 1993. Sequences of genes encoding naphthalene dioxygenase in Pseudomonas putida strains G7 and NCIB 9816-4. Gene 127:31–37 [DOI] [PubMed] [Google Scholar]
- 48. Singleton DR, Hunt M, Powell SN, Frontera-Suau R, Aitken MD. 2007. Stable-isotope probing with multiple growth substrates to determine substrate specificity of uncultivated bacteria. J. Microbiol. Methods 69:180–187 [DOI] [PubMed] [Google Scholar]
- 49. Singleton DR, et al. 2005. Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl. Environ. Microbiol. 71:1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singleton DR, Richardson SD, Aitken MD. 2008. Effects of enrichment with phthalate on polycyclic aromatic hydrocarbon biodegradation in contaminated soil. Biodegradation 19:577–587 [DOI] [PubMed] [Google Scholar]
- 51. Singleton DR, Sangaiah R, Gold A, Ball LM, Aitken MD. 2006. Identification and quantification of uncultivated Proteobacteria associated with pyrene degradation in a bioreactor treating PAH-contaminated soil. Environ. Microbiol. 10:1736–1745 [DOI] [PubMed] [Google Scholar]
- 52. Sota M, et al. 2006. Genomic and functional analysis of the IncP-9 naphthalene-catabolic plasmid NAH7 and its transposon Tn4655 suggests catabolic gene spread by a tyrosine recombinase. J. Bacteriol. 188:4057–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stingley RL, Khan AA, Cerniglia CE. 2004. Molecular characterization of a phenanthrene degradation pathway in Mycobacterium vanbaalenii PYR-1. Biochem. Biophys. Res. Commun. 322:133–146 [DOI] [PubMed] [Google Scholar]
- 54. Tang H, et al. 2011. Genome sequence of Pseudomonas putida strain B6-2, a superdegrader of polycyclic aromatic hydrocarbons and dioxin-like compounds. J. Bacteriol. 193:6789–6790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thompson JD, Gibson TJ, Higgins DG. 2002. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics. 2002: 2.3.1–2.3.22 [DOI] [PubMed] [Google Scholar]
- 56. US Environmental Protection Agency 2000. A resource for MGP site characterization and remediation. Expedited site characterization and source remediation at former manufactured gas plant sites. US Environmental Protection Agency report EPA-542-R-00-005 US Environmental Protection Agency, Washington, DC [Google Scholar]
- 57. van Herwijnen R, et al. 2003. Elucidation of the metabolic pathway of fluorene and cometabolic pathways of phenanthrene, fluoranthene, anthracene and dibenzothiophene by Sphingomonas sp. LB126. Res. Microbiol. 154:199–206 [DOI] [PubMed] [Google Scholar]
- 58. Yen KM, Gunsalus IC. 1982. Plasmid gene organization: naphthalene/salicylate oxidation. Proc. Natl. Acad. Sci. U. S. A. 79:874–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.