

Abstract
Unraveling how regulatory divergence contributes to species differences and adaptation requires identifying functional variants from among millions of genetic differences. Analysis of allelic imbalance (AI) reveals functional genetic differences in cis regulation and has demonstrated differences in cis regulation within and between species. Regulatory mechanisms are often highly conserved, yet differences between species in gene expression are extensive. What evolutionary forces explain widespread divergence in cis regulation? AI was assessed in Drosophila melanogaster–Drosophila simulans hybrid female heads using RNA-seq technology. Mapping bias was virtually eliminated by using genotype-specific references. Allele representation in DNA sequencing was used as a prior in a novel Bayesian model for the estimation of AI in RNA. Cis regulatory divergence was common in the organs and tissues of the head with 41% of genes analyzed showing significant AI. Using existing population genomic data, the relationship between AI and patterns of sequence evolution was examined. Evidence of positive selection was found in 30% of cis regulatory divergent genes. Genes involved in defense, RNAi/RISC complex genes, and those that are sex regulated are enriched among adaptively evolving cis regulatory divergent genes. For genes in these groups, adaptive evolution may play a role in regulatory divergence between species. However, there is no evidence that adaptive evolution drives most of the cis regulatory divergence that is observed. The majority of genes showed patterns consistent with stabilizing selection and neutral evolutionary processes.
Keywords: Cis regulatory divergence, Drosophila melanogaster, Drosophila simulans, allele-specific expression, adaptive evolution
Introduction
Genetic differences which impact transcript abundance can arise from regulatory sequence variation occurring within regulatory regions of the gene itself (cis effects), in regulatory or coding regions of trans acting factors (trans effects), or through indirect or epistatic effects. Chromosome substitution, eQTL, and allele-specific expression (ASE) studies find abundant regulatory variation both in cis and in trans (Brem et al. 2002; Yan et al. 2002; Lo et al. 2003; Wittkopp et al. 2004; Kirst et al. 2005; Ronald et al. 2005; Hughes et al. 2006; Genissel et al. 2008; Guo et al. 2008; Lemos et al. 2008; Graze et al. 2009; Tirosh et al. 2009; Zhang and Borevitz 2009; McManus et al. 2010). However, there is still debate over the relative contribution of causal genetic differences in cis or trans to variation in gene regulation, with more cis than trans observed in some experiments (Wittkopp et al. 2004) and more trans than cis observed in others (Brem et al. 2002). This is likely fueled both by biological differences between model systems and by differences in analytical or experimental techniques (Genissel et al. 2008; Graze et al. 2009). Researchers have also used different definitions for cis and trans and the biological interpretation of cis and trans can differ between experimental designs (see for review, Rockman and Kruglyak 2006). Regardless, between species studies have revealed widespread functional cis regulatory divergence in gene regulation with hundreds to thousands of cis acting variants identified.
Cis regulatory evolution has been associated with a significant number of trait differences that are hypothesized to be adaptive (see for review, Wray 2007). Patterns of sequence evolution consistent with positive selection are correlated with overall expression divergence (the composite result of cis and trans effects), suggesting a general role for positive selection in regulatory divergence (Nuzhdin et al. 2004; Holloway et al. 2007). Alternatively, regulatory divergence may result primarily from neutral evolutionary processes (Gilad et al. 2006; Whitehead and Crawford 2006a) or as a by-product of adaptive evolution (i.e., through hitchhiking effects Smith and Haigh 1974) or compensatory fixations (True and Haag 2001). Does widespread divergence in expression result from neutral evolutionary processes (Whitehead and Crawford 2006b) or is it a result of positive selection (Wray 2007; Fay and Wittkopp 2008) on regulatory sequence variants? Allelic imbalance (AI) occurs when there are differences in ASE for the two alleles in heterozygous individuals (Yan et al. 2002). In Drosophila, there is no evidence for effects of genetic imprinting in adults (Wittkopp et al. 2006). AI results from genetic differences in regulatory regions, directly identifying causal loci for cis effects (e.g., Yan et al. 2002; Lo et al. 2003; Wittkopp et al. 2004; Guo et al. 2008; Graze et al. 2009). In Drosophila and in yeast, a correspondence between overall nucleotide divergence in 5′ cis regions and 3′ UTRs and AI has been demonstrated (Tirosh et al. 2009; McManus et al. 2010). If signatures of positive selection are identified in loci underlying expression divergence, expression variation resulting from cis regulatory divergence at these loci may be a consequence of adaptive evolution.
Using RNA-seq technology, a comprehensive assessment of cis regulatory divergence in interspecific hybrid female heads was conducted and patterns of sequence evolution (Begun et al. 2007) within causal loci were examined. Genotype-specific references were shown to virtually eliminate the map bias plaguing this technology. A novel Bayesian model, which uses allele representation in F1 hybrid DNA sequence reads as a prior, was used to estimate allele frequencies in RNA sequences. Species differences in cis regulation were identified in 41% of assayed genes. Differences are primarily of small effect, consistent with stabilizing selection on expression combined with neutral evolutionary processes. However, evidence for positive selection is enriched in genes showing cis regulatory divergence. Intriguingly, as AI increases so does the proportion of genes showing positive selection. This suggests that recurrent positive selection may play a role in cis regulatory divergence of these genes.
Materials and Methods
Sample Preparation
Isogenic strains of D. simulans (C167.4; BDSC 4736) and D. melanogaster (Berlin; BDSC 8522) were crossed to produce F1 hybrid progeny. One to one and a half day old adults were flash frozen in synchronized batches. Three independent RNA replicates were formed from progeny collected from distinct sets of bottles. DNA was extracted from bodies of a single pool of 20 individuals. For each RNA extraction, heads were homogenized in 1 ml of Trizol (Invitrogen) and RNA was isolated according to the manufacturer’s protocol. mRNA was purified from total RNA using oligo dT Dynabeads (Invitrogen). DNA was extracted using the MasterPure DNA purification kit (Epicentre). The Genomic DNA Clean and Concentrator kit (Zymo Research) was used to isolate only high molecular weight DNA, following the manufacturer’s protocol. For additional details, see supplementary materials, Supplementary Material online.
Library Construction and Sequencing
For each RNA sample, 100 ng of purified mRNA was fragmented (Ambion Fragmentation buffer) and concentrated to 10 μl in DEPC H2O (Zymo Research RNA concentrator kit). First- and second-strand cDNA synthesis were carried out using Random Hexamer Primers (3 μg/μl, Invitrogen) following standard molecular biology protocols (Chang et al. 2011). Double-stranded cDNA was purified and concentrated to 10 μl (Zymo Research DNA Concentrator kit). Libraries were constructed using reagents from the End-IT Repair kit (Epicentre), Fast-Link DNA Ligation Kit (Epicenter), Illumina Genomic DNA Sample Prep Kit (part # 1000181), addition of an A base (Klenow 3′→5′ exonuclease, NEB) and Zymo Research DNA Concentrator kits. In brief, samples were processed to produce 5′-phosphorylated, blunt-ended DNA followed by 3′-A overhang addition. Illumina genomic DNA adaptors (1 μl adaptor oligo mix 10 mM) were ligated to cDNA fragments and processed cDNA was size selected (200–500 bp) by gel purification. Cleaned, concentrated, size-selected templates were polymerase chain reaction enriched for adaptor-modified cDNA following the Illumina Genomic DNA Sample Prep protocol. For the gDNA sample, approximately 1 μg of gDNA was fragmented (standard probe sonicator, Duty Cycle 80%, Output 2, sets of 20 pulses for five rounds) and concentrated using the Zymo Research DNA Concentrator kit (10 μl). Library construction was completed as described for cDNA libraries starting after cDNA synthesis (2 μl adaptor oligo mix was used for gDNA samples).
RNA and DNA derived libraries were quantified by Qubit (Invitrogen). The three cDNA libraries were sequenced on three lanes (one lane per biological replicate) with 54-bp paired end chemistry using Illumina technology. The gDNA library was sequenced on three lanes (one lane per technical replicate) using 36-bp paired end chemistry. RNA sequencing data are available from the Gene Expression Omnibus database (GEO accession number GSE34591). DNA sequencing data are available from the NCBI Short Read Archive (SRA accession number SRA048616).
Read Mapping and Annotation
Map bias (Degner et al. 2009; Kim and Bartel 2009; Zhang et al. 2009) can lead to false positives (i.e., inferences of cis regulatory differences where none exist). Different analytical methods can vary in the degree and direction of map bias as a result of the alignment algorithm, sequencing error rates and read length (Degner et al. 2009). Genotype-specific references were created for D. melanogaster Berlin and D. simulans C167.4, the two parental genotypes used in this study. Exonic regions (D. melanogaster R5.26 gene models, supplementary data set S1, Supplementary Material online) excised from the D. melanogaster build five syntenic assembly and from the Drosophila Population Genomics Project (DPGP) syntenic assembly (Begun et al. 2007) provided an initial reference set. RNA-seq data from D. melanogaster Berlin (Chang et al. 2011) was aligned to the D. melanogaster reference, whereas RNA-seq data from D. simulans C167.4 (McIntyre et al. 2011) was aligned to the D. simulans reference. Reads were aligned using Bowtie (Langmead et al. 2009) and Last (Frith et al. 2010). Polymorphisms were identified for each genotype separately and the exonic sequence was updated from these alignments. The process was repeated recursively until few additional polymorphisms were identified. The resulting reference set of exonic sequence for D. melanogaster Berlin and D. simulans C167.4 were annotated for TEs and repeats using RepeatMasker (Smit et al. 1996). Details are provided in the supplementary materials, Supplementary Material online.
For ∼10% of exons, genome positions overlapped (e.g., due to alternative transcript initiation sites). The unique genomic positions corresponding to the minimum start and maximum end positions were used in these cases (see supplementary materials, Supplementary Material online) and are referred to as exons throughout the text. Results are reported for 60,028 exons total. Each exon was classified as common or alternative. Exons were classified as common if they were present in all transcript isoforms and alternative if present in some, but not all, isoforms. Genome regions with gene overlaps were annotated and are described and modeled separately (see supplementary materials, Supplementary Material online).
Reads from each sample were mapped to initial species references (D. melanogaster and D. simulans) and to the updated genotype-specific references (Berlin and C167.4) using Bowtie (Langmead et al. 2009) and TopHat (Trapnell et al. 2009). Reads were assigned to an allele based upon the highest quality alignment. Reads mapping equally well to both species references or both genotype-specific references were assigned to a “both” category.
Alignment to genotype-specific references virtually eliminates map bias. The distribution of allele bias (ln[Cm/Cs], see supplementary materials, Supplementary Material online) for each exon was examined for both initial and updated references (supplementary fig. S1, Supplementary Material online). When F1 hybrid reads (for RNA and DNA) were mapped to initial species reference genome sequences the median allelic bias for genomic DNA sequences was 0.22, whereas the median allele bias for genotype-specific (updated) references was 0.0035. The median allele bias for RNA sequences was 0.29 for the initial references, whereas the median allele bias using genotype-specific references was reduced to 0.054. The inclusion of inexact matches did not increase allele bias (see supplementary table S1, Supplementary Material online). Mitochondrial reads were used to measure error in read assignment (following McManus et al. 2010). Mapping to the initial species references produces an incorrect allele assignment 2.1%/3.5% (RNA/DNA) of the time (see supplementary table S2a, Supplementary Material online). When genotype-specific references were used, the percent of RNA (DNA) mitochondrial reads assigned erroneously was 0.09% for RNA and 0.45% for DNA (supplementary table S2b, Supplementary Material online).
Exon Detection
Expression of a given exon was considered detected if the average coverage in RNA samples was greater than 0 for all replicates and greater than five (McIntyre et al. 2011) for at least 2 of the 3 replicates (in alignments to either genotype-specific reference). The power to detect AI is dependent on the number of allele-specific reads (Fontanillas et al. 2010). For analysis of AI, only detected exons with greater than 100 allele-specific reads in both RNA and DNA were retained in the analysis, corresponding to an estimated statistical power of 0.9 when alleles are biased 2-fold (Fontanillas et al. 2010). There was insufficient coverage to analyze junction reads in an allele-specific manner.
Statistical Analysis
Possible sources of bias in the DNA that would be expected to result in bias toward one allele or the other include both sequence bias from the technology and structural variation. DNA from the F1 hybrid genotype was sequenced as a control for these sources of bias in estimates of AI. The Bayesian framework allows the use of these controls as a prior, and the resulting estimates of the AI are adjusted for bias in the DNA allowing a direct interpretation of these estimates. The Bayesian framework has another advantage in this context. Although a generalized linear model based upon a Poisson model or negative binomial model assumes that the number of reads sampled is fixed, the Bayesian model assumes the number of reads sampled is random. Since the number of reads allocated to each exon/allele is not fixed by design, rather sampled from a pool of mRNA, this model more accurately reflects the underlying process. The Bayesian model–based parameter for the allele M (D. melanogaster) in RNA is 1 − θ. Let Xi = number of M reads in the RNA; li = number of S (D. simulans) reads in the RNA; = number of M reads in the DNA, = number of S reads in the DNA. Where, i = 1, 2, … , l and i* = 1, 2, … , l*. Here, l and l* are the number of replicates of the RNA and DNA, respectively.
For the RNA,
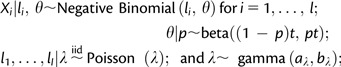 |
and for the DNA,
 |
Here, X (the number of M reads) is the number of “failures” before the first l “successes” and θ is the success rate (the frequency of the allele S). The RNA and DNA models are connected by the parameter p, the probability of sequencing and mapping the allele S in the DNA sample. In the DNA model, p is a parameter to be estimated. In the RNA model, p is a hyperparameter. The expectation in a heterozygous DNA sample is p = 0.5. When p is greater than 0.5 in the DNA there is a bias toward the S allele. The distribution for the RNA S allele frequency (θ) in the model is then centered at 1 − p, to correct for this bias. The estimate for the RNA can be interpreted directly relative to the expected fraction of 0.5. Importantly, the number of counts is random. The final inference for the RNA M allele frequency is based upon the 95% credible interval associated with 1 − θ and is calculated using Markov chain Monte Carlo methods (Robert and Casella 2004). Details of the model specification and the hyperparameter values (i.e., t, v, aλ, bλ, aδ , and bδ) are given in the supplementary materials, Supplementary Material online.
AI in alternative exons was compared with AI in common exons in order to identify differences in isoform usage between the species. There were 623 genes, with common and alternative exons, that were tested for differences in AI using the linear model, Gijkl = μ + Ei + Nj + ak + Ei × Nj + Ei × ak + Nj × ak + Ei × ak × Nj +ϵijkl. Here, Gijkl is the proportion of allele-specific reads. E denotes the exon (i = common, A1, … , An); N denotes the nucleic acid (j = DNA, RNA); and a denotes the allele (k = D. melanogaster, D. simulans). All common exons (C) were combined to estimate the average AI for that gene and compared with each alternative exon (A) using the contrast GC,RNA, mel − GC,RNA, sim − (GC,DNA, mel − GC,DNA, sim) = GA,RNA, mel − GA,RNA, sim − (GA,DNA, mel − GA,DNA, sim). Allele-specific alignments were also examined manually using custom tracks visualized in the UCSC Genome Browser (Kent et al. 2002) and D. melanogaster BDGP R5. Note that the evidence for differential regulation of isoforms is clearest when there are more exons that can be measured.
The relationship between patterns of molecular evolution consistent with positive selection and cis regulatory divergence was examined using polarized (D. simulans lineage) McDonald–Kreitman (MK) tests (McDonald and Kreitman 1991) from Begun et al. (2007) as the criteria for identifying regions with evidence for selection (with cutoff P < 0.05). Begun et al. (2007) constructed MK tests separately for CDS, introns, 5′ UTRs, 3′ UTRs, intergenic regions 5′ of the focal gene, and intergenic regions 3′ of the focal gene. MK tests for noncoding sequences constructed by Begun et al. (2007) are analogous to the traditional MK test. Polymorphism and fixations in the focal region (introns, 5′ UTRs, 3′ UTRs, intergenic regions 5′ of the focal gene, and intergenic regions 3′) were the putatively functional sites (analogous to nonsynonymous sites). Synonymous polymorphism and fixations in the coding regions of the associated gene, for introns and UTRs, or nearest genes, for intergenic regions were used as the putatively neutral sites (for additional details, see Begun et al. 2007). Note that there are a number of limitations of these types of tests which can result in Type I or Type II error and some debate about the conditions under which a significant MK test may be interpreted as evidence for positive selection (Eyre-Walker 2002; Andolfatto 2005, 2008; Begun et al. 2007; Hughes 2007; Haddrill et al. 2008; Parsch et al. 2009). Although Type I or Type II error in inferences of positive selection may inflate or deflate the marginal proportion of genes with significant tests for positive selection, the test of association compares the frequency of positive selection between the two groups (significant/not significant) resulting from the test for AI. There is no reason to suppose that inflation or deflation of the proportion of tests significant for positive selection will affect the association between signatures of selection and AI.
Adaptive evolution was inferred when the proportion of substitutions that were nonsynonymous was greater than the proportion of polymorphisms that were nonsynonymous using the direction of selection (DoS) statistic (Stoletzki and Eyre-Walker 2011; DoS = Dn/(Dn + Ds) − Pn/(Pn + Ps)). Indicator flags were constructed from the tests for each gene region (CDS, introns, 5′ UTRs, 3′ UTRs, intergenic regions 5′ of the focal gene, and intergenic regions 3′ of the focal gene). A single indicator variable (0, no evidence for positive selection; 1, at least one significant MK test with DoS > 0) was also compiled from this data. The 5′ and 3′ intergenic regions were considered with respect to a focal genes position rather than the original organization of the data on the basis of 5′ and 3′ flanking genes. Genes with AI were examined for enrichment of evidence for positive selection for individual MK tests and for the composite indicator (one-tailed Fisher’s exact test).
The enrichment of GO categories was also examined using Fisher’s exact test (Mootha et al. 2003; Rivals et al. 2007). Enrichment was tested for the general test of AI, for bias toward the D. melanogaster allele, bias toward the D. simulans allele and for genes with significant differences in AI between common and alternative exons. Individual GO categories were combined to investigate if broader functional groups were overrepresented among cis regulatory divergent genes with evidence for selection. Five of these categories correspond to those examined previously (see Graze et al. 2009): defense (Lemaitre and Hoffmann 2007; Sackton et al. 2007), sex regulated (Goldman and Arbeitman 2007), vision, olfaction, and nervous system. Two additional categories were tested (see supplementary materials, Supplementary Material online): RNAi/RISC complex and germ line.
Additional documentation, fastq files, reference sequences, tracks, and programs can be found at http://bioinformatics.ufl.edu/McIntyre_Lab/Interspecific_AI.
Results
A large number of the total reads (64%) in RNA samples mapped unambiguously, in either reference, to annotated exons (table 1). There were 47,912 (54,842) exons with at least one read aligned in RNA (DNA) from the Berlin parent and 46,831 (54,250) exons with at least one read from the c167.4 parent. Allele-specific reads accounted for 71% of these (table 2), corresponding to 34,317 exons (8,429 genes; 57% of the transcriptome). Both alleles were detected for 32,889 exons. In the genomic DNA, 37,911 exons (12,655 genes) were detected. There were 16,818 exons (7,823 genes) with sufficient coverage for analysis of allele-specific expression. Exons (1,226) corresponding to regions of complex gene overlap were modeled separately and are reported in the supplementary data set S2, Supplementary Material online. Since reads that map to multiple locations in only one of the species (or reads that do not map due to deletions) could result in mapping bias (Degner et al. 2009), exons with putative deletions or copy number variants were removed from consideration (see supplementary data set S3, Supplementary Material online). There were 14,452 exons (6,369 genes) included in the final analysis of divergence in cis regulation (supplementary data set S2, Supplementary Material online). The distributions of the proportion of reads assigned to the Berlin allele for each exon, for RNA and for DNA, are shown in figure 1.
Table 1.
Summary of Data Acquisition.
| Sample | Totala | Genomeb | Exon (I)c,d | Exon (U)c,e |
| F1 RNA-R1 | 40.95 | 32.30 | 25.92 | 26.41 |
| F1 RNA-R2 | 44.81 | 34.41 | 26.44 | 26.60 |
| F1 RNA-R3 | 42.58 | 32.78 | 28.28 | 29.00 |
| F1 DNA-T1 | 50.47 | 25.07 | 10.51 | 10.74 |
| F1 DNA-T2 | 47.25 | 23.84 | 10.20 | 10.45 |
| F1 DNA-T3 | 44.22 | 22.59 | 9.90 | 10.10 |
The total number of reads (in millions) output by the Illumina GAIIX (including alignments to the mitochondrial genome).
The number of reads that aligned anywhere in the Drosophila melanogaster genome (R5.26) in millions.
The number of reads that aligned unambiguously in exonic regions in millions.
Two initial reference genomes were considered: the melanogaster R 5.26 and a Drosophila simulans reference (initial references, I).
Initial exonic reference sequences were updated based on RNA-seq data from the parental lines and an updated reference (U) was created for both parents.
Table 2.
Allele Assignment.
| Sample | Berlina | C167.4a | Conservedb | Berlin Coveragec,d | C167.4 Coveragec,d |
| F1 RNA-1 | 8.30 | 7.71 | 6.38 | 9.87 (21.40) | 9.77 (21.10) |
| F1 RNA-2 | 8.45 | 7.83 | 7.09 | 10.39 (22.43) | 10.31 (22.06) |
| F1 RNA-3 | 9.64 | 8.99 | 7.26 | 12.00 (25.48) | 11.94 (25.14) |
| F1 DNA-1 | 3.38 | 3.41 | 3.50 | 7.40 (12.84) | 7.32 (13.00) |
| F1 DNA-2 | 3.30 | 3.33 | 3.43 | 6.90 (12.80) | 6.83 (13.03) |
| F1 DNA-3 | 3.19 | 3.24 | 3.32 | 6.45 (12.61) | 6.39 (12.78) |
Reads aligning to exons in nuclear genes are reported for each of the updated reference genomes (in millions).
Reads that align better to one parent are assigned to that parent, whereas reads aligning equally well to both parental references are considered conserved.
The distribution of the coverage (average reads per base pair) across these exons is reported for both Berlin and C167.4.
The median (interquartile range) are reported in columns 5 and 6 for each allele, respectively.
FIG. 1.

Allele frequencies in RNA and DNA. For each exon analyzed (n = 14,452), the distribution of the frequency of the Drosophila melanogaster allele among all allele-specific reads is shown for RNA (dashed) and DNA (dotted) reads and for the Bayesian estimate of the frequency (solid). The median of the RNA frequencies is 0.514 (Q2,DNA = 0.493, Q2,1 − θ = 0.512), the interquartile range is 0.087 (IQRDNA = 0.074, IQR1 − θ = 0.057) and the standard deviation is 0.081 (σDNA = 0.062, σ1 − θ = 0.048).
Allelic Imbalance in Exons and in Genes
The frequency of each allele and the credible interval around that frequency were estimated for each exon using a novel Bayesian model (supplementary data set S2, Supplementary Material online). Of the ∼41% (n = 5,877) of exons that showed significant AI (credible interval excludes 0.5), there were 4,024 exons with AI biased toward the D. melanogaster allele and 1,853 biased toward the D. simulans allele. Significant cis differences are primarily modest in effect with a smaller portion of exons (n = 190) showing prominent or extreme AI (fig. 2). There were 2,866 genes with at least one D. melanogaster biased exon and 1,447 genes with at least one D. simulans biased exon. For the majority of the genes examined, exon level estimates of bias were in the same direction. There were 294 genes (798 exons) in which exon level estimates of AI differed in the direction of bias. Patterns of bias were also examined excluding exons containing TEs or repeats. When exons containing repeats are excluded from the analysis, inferences do not change. Among genes previously identified as differentially expressed between D. melanogaster and D. simulans in adult female heads (Graze et al. 2009), more than 75% showed evidence of AI in this study (e.g., AGO2, Obp49A). This confirms that cis regulatory differences are disproportionately observed among differentially expressed genes. Allele-specific alignments for a selection of genes with significant AI, which also show differential expression between these species, are shown in figure 3A–C.
FIG. 2.

AI. The distribution (black line) of the Bayesian estimates of the frequency of the Berlin allele for all exons with significant AI. Estimates larger than 0.5 are biased toward the Drosophila melanogaster allele and estimates less than 0.5 are biased toward the D. simulans allele. The credible interval (CI) for exons with significant AI does not overlap with 0.5. Exons with CI excluding 0.5, but of magnitude ≥ 0.4 or ≤0.6, were classified as showing modest AI and are shaded in gray. One hundred and seventy-five exons with CI < 0.4 (n = 63) or > 0.6 (n = 112) were classified as showing prominent AI and are depicted in dark gray. Exons with CIs < 0.3 or > 0.7 (n = 15) were classified as showing extreme AI and are depicted in black. Complete information for all estimates and CIs are reported in supplementary data set S2, Supplementary Material online.
FIG. 3.

(A–C). Allele-specific alignments. Allele-specific read alignments showing the mean coverage for Drosophila melanogaster reads (red) and D. simulans reads (blue) per exon in (A) Ugt-35b, (B) Hexo1, (C) Iris. Custom wiggle tracks for reads separated by allele assignment were visualized using the UCSC Genome Browser (Kent et al. 2002). Wiggle tracks for publication including overlays and gene models (Flybase 5.26 annotations) were created using R (R Development Core Team 2011). Gene models are from the FlyBase Protein-Coding Genes track (R5.12). The genome version is D. melanogaster April 2006 BDGP R5/dm3 assembly.
Significant AI is skewed toward increased expression of the D. melanogaster allele. There were 6,369 genes (and 1,146 unique regions corresponding to overlapping gene models) that were assayed. The number of genes with AI biased toward the D. melanogaster allele was twice that of those with AI biased toward D. simulans. This pattern is also observed when only exact matches are considered and is not associated with the update status of reference exons. Technical bias is unlikely as the number of reads assigned to D. melanogaster is approximately equal to the number assigned to D. simulans in DNA alignment. Only in RNA alignments is there an excess of D. melanogaster-specific reads (table 2). It is improbable that the skew results from bias in the alignments because alignment error would impact both RNA and DNA alignments (also see, Fontanillas et al. 2010).
Allelic Imbalance and Alternative Isoforms
Given the paucity of information on expression divergence for multitranscript genes, this study was designed to encompass multiple levels at which evolution of expression can occur, allowing for both gene level and isoform level inferences. Overall, 623 genes with significant AI could be tested for species differences in isoform regulation (having both constitutive and alternative exons), and 232 of these showed significant differences in AI among exons. For the 374 genes with evidence of bias toward the D. melanogaster allele, ∼31% showed evidence for differences in AI among exons (table 3). Of the 143 genes with evidence of bias toward the D. simulans allele, ∼36% showed evidence of significant differences in AI among exons. For 106 of the genes in the isoform analysis, the direction of AI was mixed (table 3). Of these, 62% (n = 65) showed significant differences in AI between exons.
Table 3.
Isoform Differences in AI.
| Direction of AI | Genes with Significant Isoform Variation (# tested)a |
| Drosophila melanogasterb | 7 (28) |
| Drosophila simulansc | 6 (11) |
| Partial D. melanogasterd | 108 (346) |
| Partial D. simulansd | 46 (132) |
| Mixede | 65 (106) |
The number of genes showing significant differences in AI across exons at a false discovery rate level of 0.1 (total number of genes tested in that category).
Genes for which all exons show significant evidence of AI toward the Berlin allele.
Genes for which all exons show significant evidence of AI toward the c167.4 allele.
Genes where some exons are significantly biased, and others are not significantly biased, are classified as partial.
Genes where the direction of AI varies between exons are classified as mixed.
Understanding regulatory divergence at the isoform level is important because these genes are a fundamental component of the biology of sexual dimorphism (Siwicki and Kravitz 2009), immune response (Lemaitre and Hoffmann 2007), and neurological development and function (Li et al. 2007). In these processes, splicing cascades and isoform diversity are important both during development and in the adult fly. Isoform-specific regulatory differences were identified in genes related to these functions (supplementary fig. S2, Supplementary Material online), for example, Jupiter, unc-13, and Doa are sex specifically spliced (McIntyre et al. 2006; Rabinow and Samson 2010) and Doa is itself thought to play a role in the regulation of sexual dimorphism (Rabinow and Samson 2010).
The Association of AI with Selection in the D. simulans Lineage
To test the hypothesis that positive selection drives fixations which result in cis divergence, the correspondence of significant AI and evidence for recurrent positive selection was examined. Positive selection was identified by a significant MK test and positive DoS statistic. Evidence for selection is associated with AI (fig. 4; P = 0.0468). Thirty percent of genes with modest AI (n = 4,415) show evidence of positive selection, whereas 37% of genes with prominent AI (n = 160) and 40% of genes with extreme AI (n = 16) show sequence evolution consistent with positive selection. As the magnitude of AI increases, so does the prevalence of positive selection (P = 0.0348; Cochran-Armitage trend test). Although there are relatively few genes with pronounced or extreme AI, it is intriguing that recurrent adaptive evolution may be associated with larger functional effects of divergence on cis regulation. The amount of sequence divergence in a given gene region is related to AI in these data. Genes with prominent or extreme AI tend to show larger average divergence than genes with moderate or no AI. However, the level of divergence does not affect the association test results (for additional details, see supplementary materials, Supplementary Material online).
FIG. 4.

Patterns of positive selection in allele-imbalanced genes. The percentage of genes with AI that have significant MK tests and positive DoS is shown in black for any significant test, CDS, intron, 5′ and 3′ UTR, and 5′ and 3′ intergenic tests (A–D). The expected percent is shown in gray. Significant enrichment tests (supplementary table S3, Supplementary Material online; table 4) are denoted by an asterix. Percentages are given for all AI genes (All AI), genes with evidence of divergence in isoform regulation (Isoform) and genes with D. simulans biased AI (Sim AI) or Drosophila melanogaster biased AI (Mel AI) (A–D). AI is associated with significant MK tests (positive DoS) overall (P = 0.0468), in the CDS (P = 0.0188) and in the 5′ UTR (P = 0.0133). The percent of genes with significant MK tests (positive DoS) and AI is 30.6% (29.7% expected) overall, 17.4% in the CDS (15.9% exp.), and 14.3% in the 5′ UTR (12.0% exp.). Genes with D. melanogaster-biased AI are enriched in the CDS (P = 0.043) and the 5′ UTR (P = 0.017). The percent of genes with significant MK tests (positive DoS only) and Drosophila melanogaster-biased AI is 17.7% in the CDS (15.8% exp.) and 15.5% in the 5′ UTR (12.4% exp.). The percentage of AI genes with significant MK tests and positive DoS is also shown for Defense (E) and sex-regulated (F) genes. Defense genes are enriched among AI genes with significant MK tests (positive DoS only) overall (P = 0.0039), and for 5′ (P = 0.0077) and 3′ (P = 0.0446) intergenic regions. The percent of adaptively evolving genes with AI that are classified as Defense is 12.4% (9.7% exp.) overall, 14.5% (10.5% exp.) for 5′ and 13.4% (10.3% exp.) for 3′ intergenic regions.
Associations between AI and positive selection were also significant for individual categories of MK tests (fig. 4 and supplementary table S3, Supplementary Material online). Genes with bias toward the D. melanogaster allele and genes with bias toward the D. simulans allele were considered separately. Genes with bias toward the D. melanogaster allele were enriched for selection in coding regions (P = 0.043) and in the 5′ UTR (P = 0.017). Genes with bias toward the D. simulans allele showed a similar trend for association of AI with positive selection in the CDS but were not significantly enriched for any of the individual categories. This is likely due to differences in the number of D. melanogaster and D. simulans biased genes. Genes identified as showing evidence for species differences in isoform regulation were also examined for association with positive selection. The cell numbers in the respective contingency tables were low, and unsurprisingly, no significant associations were observed.
Note that the associations observed may not result from direct selection on regulatory regions. Rather, these associations may be caused by indirect effects of selection due to linkage (e.g., hitchhiking) or result from unknown genomic factors which cross-correlate with both AI and evidence for positive selection.
Functional Enrichments for cis Regulatory Divergent Genes
Gene ontology enrichment analysis (Mootha et al. 2003; Rivals et al. 2007) was conducted between significance of overall AI, bias toward the D. melanogaster allele, and bias toward the D. simulans allele and ontology categories (Fisher’s exact test; supplementary data set S4, Supplementary Material online). Overrepresented categories were predominantly specific to the direction of AI. For example, H3-K9 methyltransferase activity and RNA-induced silencing complex GO terms were enriched among those genes biased toward expression of the D. melanogaster allele. Sensory perception of chemical stimulus and H3-K4 methyltransferase activity GO terms were enriched only among those with bias toward the D. simulans allele.
A common approach to understanding the potential biological implications of expression divergence is to examine enrichments of functional categories among divergent genes. To understand whether these enrichment patterns are influenced by adaptive evolution, gene ontology enrichment was also examined among AI genes with evidence for adaptive evolution (supplementary data set S5, Supplementary Material online). A number of individual GO categories that were overrepresented among these genes belong to functional categories previously identified as enriched among genes with species differences in expression (Graze et al. 2009). Testing for enrichment of these groups (and two additional groups for which multiple individual GO categories were identified) showed that genes with roles in defense/immunity (defense), RNAi/RISC complex, and those regulated downstream of the sex determination hierarchy (sex regulated) are significantly overrepresented among genes showing evidence of both AI and selection (table 4). When possible, enrichments among AI genes with evidence for adaptive evolution were examined for each MK test type individually (CDS, intron, 5′UTR, 3′UTR, 5′ intergenic, and 3′ intergenic regions). AI genes with evidence of adaptive evolution in 5′ intergenic (P = 0.0077) and 3′ intergenic regions (P = 0.0446) were significantly enriched for functions in defense (fig. 4).
Table 4.
Functional Group Enrichments.
| Alla |
Drosophila simulansa |
Drosophila melanogastera |
||||
| Category | P (AI)b | P (AI + S)c | P (AI)b | P (AI + S)c | P (AI)b | P (AI + S)c |
| Defense | 0.0240 | 0.0039 | 0.0182 | 0.1050 | 0.3756 | 0.0493 |
| RNAi/RISC complex | 0.1789 | 0.0408 | 0.3782 | 1.000 | 0.0645 | 0.0054 |
| Sex regulated | 0.2173 | 0.0543 | 0.2200 | 0.0105 | 0.6239 | 0.7945 |
Each test was conducted with AI considered for all significant AI, for AI with greater expression from the D. simulans allele, and for AI with greater expression from the D. melanogaster allele.
P values are for Fisher’s exact one-tailed test for the indicator variable of each specific functional category (Defense, RNAi/RISC complex, or sex hierarchy regulated) by the indicator variable for AI (genes with significant AI/genes with no AI).
P values are for Fisher’s exact one-tailed test for the indicator variable of each specific functional category (Defense, RNAi/RISC complex, or sex hierarchy regulated) by the indicator variable for genes with both AI and significant positive selection (AI + S).
Discussion
Is there a general role for adaptive evolution in cis regulatory divergence? Studies which have examined the evolution of gene expression have concluded that the expression of most genes is subject to stabilizing selection, with fewer genes showing evidence of relaxed selection or positive selection (Rifkin et al. 2003; Lemos et al. 2005). This raises the question of whether cis regulation in female heads is mostly conserved and whether differences that are observed result from neutral evolutionary processes. For ∼70% of genes with AI, no evidence of positive selection was found; suggesting that neutral evolution constrained by modest stabilizing selection (Whitehead and Crawford 2006a) explains most cis regulatory evolution in these tissues. Overall, the results are consistent with the expectation that most cis divergence does not result from positive selection.
Is there any role for positive selection in regulatory evolution? Cis regulatory divergence was observed in multiple genes involved in the basal machinery of regulation, in direct contrast to common molecular models of conservation of regulatory mechanisms. A number of core regulatory genes show quantitative cis regulatory divergence (e.g., AGO1, eiF4G, Cbp20, and Cbp80). In some cases, these genes also show signatures of positive selection. For example, Cbp20 is a component of the RNA cap binding complex and is structurally conserved, showing 75% protein sequence conservation between human and Drosophila (Visa et al. 1996). Together with its cofactor Cbp80, Cbp20 has been implicated as a component of the siRNA and miRNA production pathways (Sabin et al. 2009). Both of these genes show signatures of positive selection within the flanking 5′ intergenic regions. The three elements of global regulation that showed the largest numbers of cis divergent genes (supplementary data sets S4 and S5, Supplementary Material online), RNAi (esiRNAs/siRNA/miRNA)-mediated regulation, chromatin conformation, and splicing can be functionally intertwined (Kavi et al. 2008; Sabin et al. 2009). A subset of these genes, primarily those involved in RISC-based silencing and splicing, are diverging as a result of positive selection (supplementary data set S4, Supplementary Material online). Positive selection may act on core genes as a result of their role in TE suppression or in host defense (e.g., AGO2, Obbard et al. 2011). Adaptive substitutions may have pleiotropic effects on their role as basal components of gene regulation, potentially resulting in compensatory fixations at interacting loci. It may be that the cis regulatory divergence in these regulatory genes is part of a common coevolutionary process (Kopp and McIntyre 2010), which is driven, in part, by adaptive evolution in a subset of these genes.
Coevolution between Drosophila and its pathogens is expected to result in adaptive evolution of defense genes. The protein-coding regions of defense/immunity genes evolve rapidly and show evidence of adaptive evolution (Sackton et al. 2007; Lazzaro 2008; Obbard et al. 2009). Defense genes are enriched among genes with additive variation for expression within species and among genes with overall expression differences between species (Graze et al. 2009; Wayne et al. 2011). In this study, defense genes are enriched among allele imbalanced genes that show evidence of positive selection in 5′ and 3′ intergenic regions. Collectively, these results indicate that cis regulation of defense genes may be evolving in response to coevolutionary processes. In contrast, genes with roles in olfaction were not enriched among genes with AI nor among AI genes with signatures of selection. This suggests that the enrichment for olfaction genes observed for overall expression divergence (Graze et al. 2009) does not arise from positive selection on cis regulation of olfactory genes.
Are there general patterns in AI that may reveal regulatory differences between species? There is a skew toward the D. melanogaster allele that could reflect species differences in regulation. Comparisons of overall expression between D. melanogaster and D. simulans or between D. melanogaster and D. sechellia also show a bias toward increased expression in D. melanogaster (Graze et al. 2009; McManus et al. 2010). A shift in the balance of regulatory mechanisms that favors cis upregulation of the D. melanogaster allele or cis by trans interactions involving common cis regulatory motifs may explain this result. Chromatin conformation or nuclear–cytoplasmic interactions are possible causes of an excess of D. melanogaster allele reads (Fontanillas et al. 2010). Although technical sources of bias cannot be entirely ruled out, there are intriguing clues that could indicate differences in chromosomal biology between these species. For example, chromatin staining patterns and intensities can differ between these species during some stages of the cell cycle, D. simulans has very little inversion polymorphism and a reduced number of induced inversions in artificial mutation experiments, and average recombination rates and suppression of recombination in centromeric regions are different (True et al. 1996; Aulard et al. 2004). In this study, significant AI was observed in H3-K9 and H3-K4 methyltransferase genes, as well as other regulatory genes, that could potentially impact chromatin conformation (supplementary data sets S2, S4, S5, Supplementary Material online).
Transcription factor binding sites and promoter regions have been a natural focus in studies of cis regulatory evolution (Wray et al. 2003; Landry et al. 2007; Wittkopp 2010). Differences between tests for different gene regions may result from differences in power and not differences in the strength or prevalence of positive selection (Begun et al. 2007). However, the association between cis regulatory differences and selection was significant only for genes with evidence of selection in coding regions and in the 5′ UTR (supplementary table S3, Supplementary Material online). Although perhaps unintuitive, the association between divergence in coding regions and divergence for expression is robust and has been observed for different model systems and using different analytical techniques (e.g., Castillo-Davis et al. 2004; Nuzhdin et al. 2004; Holloway et al. 2007; Tirosh et al. 2009). It is possible that the relationship between transcript abundance, protein abundance, and protein activity drives coordinate evolution of regulation and structure.
The MK and related group of tests assess long-term effects of directional selection, by summing multiple recurrent substitutions. The AI assay evaluates extant regulation and misregulation of genes, by comparing closely related species and their hybrids. Here, for the first time, these two sources of evolutionary inference are connected. Intriguingly, as AI increases so does the proportion of genes under selection. It may be that stronger bias requires accumulation of a larger number of regulatory mutations. Furthermore, the effects of these mutations must be codirected, hinting that the direction of the selection on upregulation or downregulation is consistent over long evolutionary time periods. This is akin to Orr's test based on codirection of quantitative trait locus effects between two species, as proving lineage-specific phenotypic evolution under directional selection (Orr 1998). In summary, for both significant CDS MK tests and strong AI, multiple codirected substitutions are required, some of them affecting protein function (MK tests) and others affecting regulation (AI). The concordance between MK tests and AI hints that natural selection may be involved in associating protein and regulatory evolution.
Supplementary Material
Supplementary materials, data sets S1–S5, tables S1–S3, and figures S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank the BDSC for providing fly strains, M.L. Wayne for useful discussions, M.L. McCrory for help with GEO and SRA submissions, M.L. Wayne, C.F. Baer, and H.V. Baker for space for sample collections. This research was supported by the National Institute of Health (R01GM77618, R01GM77618-S1, RGM076643A, 5R01GM081704-03) and by the National Science Foundation (CNS 0821622, DBI 0923513).
References
- Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Controlling type-I error of the McDonald-Kreitman test in genomewide scans for selection on noncoding DNA. Genetics. 2008;180:1767–1771. doi: 10.1534/genetics.108.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulard S, Monti L, Chaminade N, Lemeunier F. Mitotic and polytene chromosomes: comparisons between Drosophila melanogaster and Drosophila simulans. Genetica. 2004;120:137–150. doi: 10.1023/b:gene.0000017637.10230.c4. [DOI] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, et al. (13 co-authors) Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Hartl DL, Achaz G. cis-Regulatory and protein evolution in orthologous and duplicate genes. Genome Res. 2004;14:1530–1536. doi: 10.1101/gr.2662504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Dunham J, Nuzhdin S, Arbeitmans M. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC genomics. 2011;12:364. doi: 10.1186/1471-2164-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. Changing effective population size and the McDonald-Kreitman test. Genetics. 2002;162:2017–2024. doi: 10.1093/genetics/162.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wittkopp PJ. Evaluating the role of natural selection in the evolution of gene regulation. Heredity. 2008;100:191–199. doi: 10.1038/sj.hdy.6801000. [DOI] [PubMed] [Google Scholar]
- Fontanillas P, Landry CR, Wittkopp PJ, Russ C, Gruber JD, Nusbaum C, Hartl DL. Key considerations for measuring allelic expression on a genomic scale using high-throughput sequencing. Mol Ecol. 2010;19(Suppl 1):212–227. doi: 10.1111/j.1365-294X.2010.04472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Wan R, Horton P. Incorporating sequence quality data into alignment improves DNA read mapping. Nucleic Acids Res. 2010;38:e100. doi: 10.1093/nar/gkq010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genissel A, McIntyre LM, Wayne ML, Nuzhdin SV. Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster. Mol Biol Evol. 2008;25:101–110. doi: 10.1093/molbev/msm247. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Rifkin SA. Natural selection on gene expression. Trends Genet. 2006;22:456–461. doi: 10.1016/j.tig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Goldman TD, Arbeitman MN. Genomic and functional studies of Drosophila sex hierarchy regulated gene expression in adult head and nervous system tissues. PLoS Genet. 2007;3:e216. doi: 10.1371/journal.pgen.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, McIntyre LM, Main BJ, Wayne ML, Nuzhdin SV. Regulatory divergence in Drosophila melanogaster and D. simulans, a genomewide analysis of allele-specific expression. Genetics. 2009;183:547–561. doi: 10.1534/genetics.109.105957. , 1SI–21SI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Yang S, Rupe M, Hu B, Bickel DR, Arthur L, Smith O. Genome-wide allele-specific expression analysis using Massively Parallel Signature Sequencing (MPSS) reveals cis- and trans-effects on gene expression in maize hybrid meristem tissue. Plant Mol Biol. 2008;66:551–563. doi: 10.1007/s11103-008-9290-z. [DOI] [PubMed] [Google Scholar]
- Haddrill PR, Bachtrog D, Andolfatto P. Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol. 2008;25:1825–1834. doi: 10.1093/molbev/msn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway AK, Lawniczak MKN, Mezey JG, Begun DJ, Jones CD. Adaptive gene expression divergence inferred from population genomics. PLoS Genet. 2007;3:2007–2013. doi: 10.1371/journal.pgen.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity. 2007;99:364–373. doi: 10.1038/sj.hdy.6801031. [DOI] [PubMed] [Google Scholar]
- Hughes KA, Ayroles JF, Reedy MM, Drnevich JM, Rowe KC, Ruedi EA, Cáceres CE, Paige KN. Segregating variation in the transcriptome: cis regulation and additivity of effects. Genetics. 2006;173:1347–1355. doi: 10.1534/genetics.105.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavi HH, Fernandez H, Xie W, Birchler JA. In: Paddison PJ, Vogt PK, editors. Current topics in microbiology and immunology Vol. 320. Heidelberg (Germany): Springer; 2008. Genetics and Biochemistry of RNAi in Drosophila; pp. 37–75. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Bartel DP. Allelic imbalance sequencing reveals that single-nucleotide polymorphisms frequently alter microRNA-directed repression. Nat Biotechnol. 2009;27:472–477. doi: 10.1038/nbt.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirst M, Basten CJ, Myburg AA, Zeng Z-B, Sederoff RR. Genetic architecture of transcript-level variation in differentiating xylem of a eucalyptus hybrid. Genetics. 2005;169:2295–2303. doi: 10.1534/genetics.104.039198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp A, McIntyre LM. Transcriptional network structure has little effect on the rate of regulatory evolution in yeast. Mol Biol Evol. 2010 doi: 10.1093/molbev/msq283. Advance Access published October 21, 2010, doi:10.1093/molbev/msq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118–121. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro BP. Natural selection on the Drosophila antimicrobial immune system. Curr Opin Microbiol. 2008;11:284–289. doi: 10.1016/j.mib.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Fontanillas P, Hartl DL. Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci U S A. 2008;105:14471–14476. doi: 10.1073/pnas.0805160105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Meiklejohn CD, Cáceres M, Hartl DL. Rates of divergence in gene expression profiles of primates, mice, and flies: stabilizing selection and variability among functional categories. Evolution. 2005;59:126–137. [PubMed] [Google Scholar]
- Li Q, Lee J-A, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, Lee MP. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–1862. doi: 10.1101/gr.1006603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McIntyre LM, Bono LM, Genissel A, Westerman R, Junk D, Telonis-Scott M, Harshman L, Wayne ML, Kopp A, Nuzhdin SV. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol. 2006;7:R79. doi: 10.1186/gb-2006-7-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre LM, Lopiano KK, Morse AM, Amin V, Oberg AL, Young LJ, Nuzhdin SV. RNA-seq?: technical variability and sampling. BMC Genomics. 2011;12:293. doi: 10.1186/1471-2164-12-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res. 2010;20:816–825. doi: 10.1101/gr.102491.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, et al. (21 co-authors) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV, Wayne ML, Harmon KL, McIntyre LM. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol. 2004;21:1308–1317. doi: 10.1093/molbev/msh128. [DOI] [PubMed] [Google Scholar]
- Obbard DJ, Jiggins FM, Bradshaw NJ, Little TJ. Recent and recurrent selective sweeps of the antiviral RNAi gene Argonaute-2 in three species of Drosophila. Mol Biol Evol. 2011;28:1043–56. doi: 10.1093/molbev/msq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Welch JJ, Kim K-W, Jiggins FM. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 2009;5:e1000698. doi: 10.1371/journal.pgen.1000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. Testing natural selection vs. genetic drift in phenotypic evolutionusing quantitative trait locus data. Genetics. 1998;149:2099–2104. doi: 10.1093/genetics/149.4.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J, Zhang Z, Baines JF. The influence of demography and weak selection on the McDonald-Kreitman test: an empirical study in Drosophila. Mol Biol Evol. 2009;26:691–698. doi: 10.1093/molbev/msn297. [DOI] [PubMed] [Google Scholar]
- Rabinow L, Samson M-L. The role of the Drosophila LAMMER protein kinase DOA in somatic sex determination. J Genet. 2010;89:271–277. doi: 10.1007/s12041-010-0038-6. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. [Internet]. Vienna (Austria): R Foundation for Statistical Computing; 2011. [cited 2012 Jan 09]. Available from: http://www.R-project.org. [Google Scholar]
- Rifkin SA, Kim J, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–144. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- Rivals I, Personnaz L, Taing L, Potier M-C. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- Robert CP, Casella G. Monte Carlo statistical methods. New York: Springer; 2004. [Google Scholar]
- Rockman MV, Kruglyak L. Genetics of global gene expression. Nat Rev Genet. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- Ronald J, Akey JM, Whittle J, Smith EN, Yvert G, Kruglyak L. Simultaneous genotyping, gene-expression measurement, and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 2005;15:284–291. doi: 10.1101/gr.2850605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zhou R, Gruber JJ, Lukinova N, Bambina S, Berman A, Lau C-K, Thompson CB, Cherry S. Ars2 regulates both miRNA- and siRNA- dependent silencing and suppresses RNA virus infection in Drosophila. Cell. 2009;138:340–351. doi: 10.1016/j.cell.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–1468. doi: 10.1038/ng.2007.60. [DOI] [PubMed] [Google Scholar]
- Siwicki KK, Kravitz EA. Fruitless, doublesex and the genetics of social behavior in Drosophila melanogaster. Curr Opin Neurobiol. 2009;19:200–206. doi: 10.1016/j.conb.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-2.9. 1996. [Internet]. [cited 2012 Jan 09]. Available from: http://www.repeatmasker.org. [Google Scholar]
- Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Estimation of the neutrality index. Mol Biol Evol. 2011;28:63–70. doi: 10.1093/molbev/msq249. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–662. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JR, Haag ES. Developmental system drift and flexibility in evolutionary trajectories. Evol Dev. 2001;3:109–119. doi: 10.1046/j.1525-142x.2001.003002109.x. [DOI] [PubMed] [Google Scholar]
- True JR, Mercer JM, Laurie CC. Differences in crossover frequency and distribution among three sibling species of Drosophila. Genetics. 1996;142:507–523. doi: 10.1093/genetics/142.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj IW. A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J Cell Biol. 1996;133:5–14. doi: 10.1083/jcb.133.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne ML, Pienaar J, Telonis-Scott M, Sylvestre L-S, Nuzhdin SV, McIntyre LM. Expression of defense genes in Drosophila evolves under a different selective regime from expression of other genes. Evolution. 2011;65:1068–1078. doi: 10.1111/j.1558-5646.2010.01197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A, Crawford DL. Neutral and adaptive variation in gene expression. Proc Natl Acad Sci U S A. 2006a;103:5425–5430. doi: 10.1073/pnas.0507648103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A, Crawford DL. Variation within and among species in gene expression: raw material for evolution. Mol Ecol. 2006b;15:1197–1211. doi: 10.1111/j.1365-294X.2006.02868.x. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ. Variable transcription factor binding: a mechanism of evolutionary change. PLoS Biol. 2010;8:e1000342. doi: 10.1371/journal.pbio.1000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. Evolutionary changes in cis and trans gene regulation. Nature. 2004;430:85–88. doi: 10.1038/nature02698. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. Parent-of-origin effects on mRNA expression in Drosophila melanogaster not caused by genomic imprinting. Genetics. 2006;173:1817–1821. doi: 10.1534/genetics.105.054684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li JB, Gao Y, et al. (12 co-authors) Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods. 2009;6:613–618. doi: 10.1038/nmeth.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Borevitz JO. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics. 2009;182:943–954. doi: 10.1534/genetics.109.103499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.