

Summary
Pathogens persist in immunocompetent mammalian hosts using various strategies, including evasion of immune effectors by antigenic variation. Among highly antigenically variant bacteria, gene conversion is used to generate novel expressed variants from otherwise silent donor sequences. Recombination using oligonucleotide segments from multiple donors is a combinatorial mechanism that tremendously expands the variant repertoire, allowing thousands of variants to be generated from a relatively small donor pool. Three bacterial pathogens, each encoded by a small genome (<1.2Mb), illustrate this variant generating capacity and its role in persistent infection. Borrelia burgdorferi VlsE diversity is encoded and expressed on a linear plasmid required for persistence and recent experiments have demonstrated that VlsE recombination is necessary for persistence in the immunocompetent host. In contrast, both Treponema pallidum TprK and Anaplasma marginale Msp2 expression sites and donors are chromosomally encoded. Both T. pallidum and A. marginale generate antigenic variants in vivo in individual hosts and studies at the population level reveal marked strain diversity in the variant repertoire that may underlie pathogen strain structure and the capacity for re-infection and heterologous strain superinfection. Here, we review gene conversion in bacterial antigenic variation and discuss the short- and long-term selective pressures that shape the variant repertoire.
Introduction
Propagation of self is the driving force underlying evolution. For obligate parasites, an infectious agent must be transmitted to new susceptible hosts. The mode and efficiency of transmission are dictated by multiple factors, including the specific niche within the host, the growth rate relative to the infectious dose, maintenance of infectivity outside the host, and availability of new susceptible hosts. At one end of the spectrum, pathogens must rapidly replicate to an infectious threshold and be transmitted within a narrow window of time prior to the onset of the adaptive immune response or the pathogen is cleared. Gastrointestinal and respiratory pathogens are highly represented within this transmission phenotype—rapid replication linked to induced host responses, e.g. coughing, sneezing, diarrhea, that provide essentially continuous opportunities for transmission within a brief time span. Among bacterial pathogens, Vibrio cholera is illustrative: the pathogen replicates rapidly within the host and the induction of watery diarrhea facilitates contamination of water sources with spread to new, susceptible hosts prior to immune clearance from the index host. At the opposite end of the spectrum, persistent pathogens have evolved to replicate to and maintain infectiousness in face of an adaptive immune response. Vector-borne and sexually transmitted pathogens disproportionately represent this phenotype, attributable to the need to maintain an infectious dose over a long period of time (defined as beyond development of specific adaptive immunity) as transmission opportunities are episodic rather than continuous. While the seasonality of arthropod presence and abundance obviously underlies vector-borne pathogen persistence, the same principles apply to sexually transmitted pathogens as mating in all mammalian species is periodic and, in many cases (including primates), can be seasonally restricted. Consequently, these pathogens have evolved sophisticated mechanisms to subvert or evade the host’s continually adaptive immune response.
Persistent pathogens use an amazing diversity of immune evasion mechanisms. This micro-review is centered upon antigenic variation in bacterial pathogens and more specifically on variants generated by gene conversion (non-reciprocal homologous recombination). In contrast to RNA viruses in which continual antigenic variation within an individual infected host occurs primarily through mutation, bacteria most commonly use an existing repertoire of donor alleles or cassettes for recombination into an active expression site (Fig. 1). The variant-generating capacity of a specific bacterial pathogen is defined both by the size of the allelic repertoire and the permissiveness for recombination of segments versus complete allelic donors. While a large number of bacterial pathogens utilize gene conversion to continuously generate antigenic variants within an individual infected host, we will focus on three pathogens, two vector-borne and one sexually transmitted, in order to highlight specific recent advances and challenges in understanding both mechanism and biological significance of antigenic variation. The first of these is Borrelia burgdorferi, for which the vls locus has been definitively shown to be a requirement for antigenic variation and persistence in the mammalian host—a rigorous criterion that has been met for very few pathogens. The second is Treponema pallidum for which recent studies have linked specific segmental tprK gene conversion events with antibody binding and pathogen clearance, and which emphasize strain differences in variant generation. The third pathogen is Anaplasma marginale, which illustrates the balance between structural variation in Msp2 that allows antigenic variation but conserves growth fitness and the consequences at the level of both the individual host and the host population.
Figure 1. Overview of gene conversion as the convergent strategy for antigenic variation in B. burgdorferi, T. pallidum, and A. marginale.
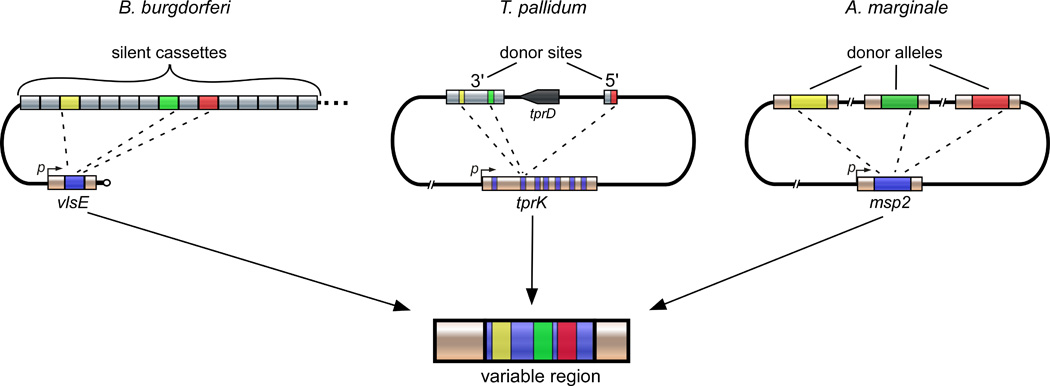
The respective loci of donor and expression regions for each bacterial pathogen are shown. Variant segments act as the donor DNA for nonreciprocal recombination events with the expression locus (vlsE, tprK or msp2). Through this process, segments of the variable regions (blue) are replaced by sections of varied length and location from the donor sequences. In the example shown, three sequential gene conversion events (represented by dashed lines) occur within the expression site through recombination with the colored donor sections (yellow, green or red) to generate a new expression site sequence with a mosaic structure.
Borrelia burgdorferi VlsE
The multisystemic disorder Lyme disease is the most prevalent vector-borne infection affecting humans in North America and Europe (Barbour, 2001; Steere et al., 2004). Causative agents of the disease, the spirochetes Borrelia burgdorferi, B. garinii and B. afzelii, are transmitted by hard-bodied ticks of the genus Ixodes. Infected ticks transmit Borrelia to humans during feeding, which can result in a localized infection (erythema migrans) at the site of the tick bite. Disseminated stages of infection follow that are characterized by neurological, cardiac, and arthritic manifestations. Infection can last from months to years due to avoidance of the host immune response, and key to its successful evasion tactics is recombination at the vls locus located at the telomeric end of a 28-kilobase linear plasmid, lp28-1 (Norris, 2006; Zhang et al., 1997).
The vls locus is comprised of an expression site (vlsE) encoding the 35 kDa lipoprotein VlsE, and a contiguous array of 15 silent (unexpressed) cassettes. These cassettes show strong homology to the central region of VlsE, and contain six variable regions flanked by highly conserved sequences (Zhang et al., 1997). The vlsE locus and the silent cassettes are separated by a short intergenic region that contains an inverted repeat sequence capable of forming a highly stable DNA stem loop (Hudson et al., 2001). The vlsE expression region is comprised of a central variable cassette that is flanked by constant, or invariable, regions. Recombination has been detected as early as four days after infection of mice, and continues throughout infection (Zhang et al., 1998). The sequence changes primarily occur within the six variable regions, and VlsE variation occurs to such a degree that each clone examined from a single mouse skin biopsy can have a different vlsE sequence after only 28 days of infection (Zhang et al., 1998). A recent elaborate study by Norris and colleagues found that the vlsE antigenic variation system promotes both short and long recombination events within each cassette region (Coutte et al., 2009). In addition to gene conversion events, template-independent changes lead to important sequence variation of vlsE. The end result of these accumulated changes is a new vlsE sequence with a mosaic structure, and the total combined potential for sequence variation makes the vlsE switching process one of the most dynamic antigenic variation systems known. A number of studies have provided strong evidence implicating the importance of VlsE antigenic variation for B. burgdorferi persistence. Early work demonstrated that antibodies specific for the variable regions of VlsE were produced during experimental infection of mice (McDowell et al., 2002), and the solved three-dimensional structure of VlsE showed that these variable regions are easily accessible to antibodies, while invariable regions are obscured (Eicken et al., 2002). Despite the apparent inaccessibility to antibodies, a strong humoral response is mounted against the conserved regions of VlsE (most notably invariant region 6). In fact, VlsE-based recombinant proteins or synthetic peptides are used for the immunodiagnosis of Lyme disease (Bacon et al., 2003; Schulte-Spechtel et al., 2003). The function of VlsE independent of its antigenic variation properties is not currently known. There have been indications that VlsE may be involved in tissue tropism, including reports that VlsE production is elevated during mammalian infection (Liang et al., 2004) with higher levels observed in spirochetes recovered from joint and skin tissues than from heart tissue (Crother et al., 2003). However, a recent study has cast doubt on such a role by demonstrating that identical variants can be located throughout these different tissue sites (Coutte et al., 2009). Interestingly, vlsE antigenic switching in B. burgdorferi occurs only during mammalian infections, suggesting that host factor(s) are required to activate the antigenic variation process. More evidence for the role of the vls system in immune evasion came from studies involving the vls-resident plasmid, lp28-1 (Labandeira-Rey et al., 2001; Purser et al., 2000). Clones lacking lp28-1 were shown to exhibit an infectivity phenotype whereby spirochetes are able to disseminate to tissue sites but are unable to persist in the murine host. Notably, these same clones are capable of long-term survival in severe combined immunodeficient (SCID) mice which lack an effective antibody response. Lp28-1 minus clones also grow normally in a dialysis membrane chamber implanted in the peritoneal cavity of rats, where exposure to antibodies and immune cells is restricted (Purser et al., 2003). Together, these studies provided a strong indication that lp28-1, and presumably the vls locus, are required for persistence only in the presence of an effective humoral immune response. Nevertheless, definitive evidence for the role of VlsE antigenic variation in persistence remained elusive, prior to the recent generation of a genetic knockout of the vls locus.
The construction of a vls knockout mutant in B. burgdorferi was made possible by a newly developed technology involving targeted deletion. This method involves integration of a replicated telomere within a linear plasmid (Bankhead et al., 2007; Beaurepaire et al., 2005; Chaconas, 2005), and the end result of the internal placement of this DNA site is deletion of DNA through the action of the Borrelia telomere resolvase, ResT (Chaconas, 2005; Kobryn et al., 2002). The loss of the vls locus due to telomere-mediated removal resulted in complete clearance of spirochetes in immunocompetent C3H mice by 21 days post infection (Bankhead et al., 2007), matching the phenotype observed with the complete loss of lp28-1. Targeted deletion was also carried out to obtain lp28-1 mutants which would contain mainly the vls locus and the necessary genes for autonomous replication of the plasmid. Infectivity experiments showed that these B. burgdorferi clones are fully infectious and persistent in immunocompetent mice, strongly suggesting that the silent vls cassettes and vlsE are the sole lp28-1 components required for spirochete persistence (Bankhead et al., 2007). Moreover, these mutant clones carried out vlsE recombination in immunocompetent C3H mice, indicating that factors required for antigenic switching are not carried on lp28-1, but must be encoded elsewhere. Finally, the vls knockout clones exhibited long-term survival in SCID mice, consistent with the findings that the lp28-1 plasmid is not required for persistent infection in the absence of an adaptive immune response (Labandeira-Rey et al., 2003; Lawrenz et al., 2004; Purser et al., 2003), and confirmed the hypothesis that vls recombination functions to evade the humoral immune response in the mouse host (Norris, 2006; Zhang et al., 1997). Thus, the generation of the vls deletion mutant offered the first definitive evidence that VlsE antigenic variation is absolutely crucial for persistent infection by B. burgdorferi.
Treponema pallidum TprK
Syphilis is a sexually transmitted infection that persists for decades if not treated with antibiotics. During the early infectious stages of disease, very large numbers of T. pallidum are found in the ulcerative chancre (primary stage) and in the disseminated rash (secondary stage). An effective Th1 immune response clears the vast majority of bacteria from skin lesions through macrophage-mediated phagocytosis and killing of opsonized treponemes, resulting in resolution of infectious lesions. However, a few organisms escape clearance (Lukehart et al., 1992) and inapparent infection (“latent” stage) persists for decades. Patients who have been treated with antibiotics for syphilis can be re-infected multiple times, and the contribution of the early acquired immune response to protection from re-infection has not been well-studied. Experimental studies in rabbits have demonstrated that re-infection or symptomatic superinfection (with heterologous strains) can occur, but varies according to the strains of T. pallidum (Turner and Hollander, 1957). These results are compatible with slow development of protective immunity but also with a vigorous mechanism of antigenic variation.
Clearance of T. pallidum from early lesions is mediated by opsonic antibody, making the identity of surface-exposed opsonic targets critical to understanding the host-parasite relationship. TprK (encoded by TP0897) is a member of the T. pallidum repeat (tpr) gene family, comprised of 12 paralogous members (Fraser et al., 1998; Centurion-Lara et al., 1999). TprK is the most highly expressed member of the family, induces early humoral and cellular immune responses, is predicted to be located in the bacterial outer membrane, and is a target of opsonic antibody (Centurion-Lara et al., 1999). Although there is not uniform agreement about the outer membrane location of TprK (Hazlett et al., 2001), one of the most compelling arguments is the impressive variation in the TprK amino acid sequences, differing not only between strains but also within a strain (Centurion-Lara et al., 2000; LaFond et al., 2003). Sequence heterogeneity in the single tprK expression site is restricted to seven discrete variable regions (V1–V7), flanked by highly conserved regions. Each V region is flanked by terminal four-base pair (bp) repeat sequences, each unique to a given V region; additional internal four-bp repeat sequences may be shared among more than one V region (Centurion-Lara et al., 2004). A large collection (>50) of “donor sites”, located in the flanking regions of tprD, also contain four-bp repeats corresponding to those seen in the tprK V regions. In a clonal isolate of T. pallidum Chicago strain (Chicago C), tprK undergoes significant sequence change in the V regions (particularly V6) during infection, and the proportion of variant treponemes increases as the adaptive immune response develops. Sequences found in the new variant V regions can be traced to the “donor sites,” confirming their role as the source. This repertoire can account for ≥105 variant TprK sequences if full donor alleles replace V regions; however, small segments of the V regions, rather than full V regions, are recombined resulting in an exponentially higher number of possible TprK variants containing chimeric or mosaic V region sequences. Similar to B. burgdorferi vlsE and A. marginale msp2, tprK V region heterogeneity occurs by segmental gene conversion (Centurion-Lara et al., 2004).
Variation of TprK strongly suggests a role in immune evasion, supported by the demonstration that the B cell and T cell epitopes of TprK are segregated, with V regions containing antibody targets, while the conserved regions contain T cell epitopes (Morgan et al., 2002). Even a minor change in the amino acid sequence of a V region prevents antibody binding (LaFond et al., 2006a), thus potentially interfering with opsonization. The real test of antigenic variation lies in demonstrating that variants have an advantage in the face of active immunity in vivo. Two recent studies provide direct evidence that TprK variants are selected by acquired immunity (L. Giacani et al, unpublished). In the first study, prior immunization of rabbits with a V6 peptide resulted in more rapid accumulation of TprK V6 variants following infection with the parent strain, compared to unimmunized controls or rabbits immunized with an unrelated V region. In the second study, development of acquired immunity during early infection was inhibited by pharmacological immunosuppression (methylprednisolone), as confirmed by absent or lower titers of antibodies and by vastly diminished interferon-γ expression in the lesions. In immunosuppressed animals, a significantly lower proportion of V6 sequence variants were seen during the 5 weeks of immunosuppression, compared to immunocompetent controls in which variants quickly predominated. These studies clearly demonstrate selection of TprK variants by adaptive immunity in vivo, paving the way for persistent infection.
Interestingly, the published T. pallidum Nichols strain genome, which is virtually the only T. pallidum strain studied for the past 30–40 years, contains only a single tprK sequence (Fraser et al., 1998). The variant nature of tprK was recognized only when its sequence was determined in additional strains of T. pallidum (Centurion-Lara et al., 2000), emphasizing the need to examine multiple strains of any pathogen to gain a full understanding of the host-parasite interaction. Subsequent study of T. pallidum strains has demonstrated that the rate of sequence variation differs significantly among strains. For example, after 22 sequential passes in rabbits over approximately seven months, the Nichols tprK sequence remained unchanged (LaFond et al., 2006b). In fact, the tprK sequence has changed very little among Nichols isolates, all originally obtained from UCLA, that have been passed independently for at least 20 years in different laboratories. However, when the Nichols strain is placed under immune pressure in vivo by passage after ~30–35 days of infection and after induction of acquired immunity, Nichols TprK variants appear and are selected in vivo. Interestingly, these variant Nichols TprK sequences readily revert to the original sequence upon passage in naïve rabbits, suggesting a fitness advantage for the original TprK sequence. In sharp contrast, the Chicago strain contains multiple variant sequences and, when a “clonal” isolate expressing a single TprK allele (e.g. Chicago C) is derived from the parent, new variants appear spontaneously after a single passage, even in the absence of measurable anti-TprK antibody. However, the subsequent development of anti-V region antibodies results in more rapid selection for variants. All non-Nichols strains examined to date have high sequence variability in the tprK expression site, consistent with the finding that samples obtained directly from lesions, cerebrospinal fluid, or blood of syphilis patients also contain heterogeneous tprK sequences (LaFond et al., 2003). Aside from the artificial situation of frequent passage in immunologically naïve rabbits, there appears to be a significant advantage to the maintenance of multiple TprK variants within a population. Whether this advantage is solely attributable to immune evasion or reflects a separate functional role for TprK variants is unknown.
The key to evolutionary success of a pathogen is transmission to a susceptible host. Antigenic variation of TprK permits the persistence of T. pallidum in primary lesions as acquired immunity develops, likely increasing the duration of the highly infectious primary stage. Further, the selection and dissemination of TprK variants allows the development of the secondary rash, prolonging the duration of the infectious period of syphilis to 6 months or more. At the population level, the existence of numerous TprK variants within strains circulating in nature increases the likelihood that a host, even one that has already had syphilis, will lack specific antibodies to TprK variants expressed in the new strain, thus permitting re-infection. The outcome of this strategy is the maintenance of a larger susceptible host population, and maximization of the likelihood that a given strain will be transmitted during episodic sexual activity, contributing to the evolutionary success of T. pallidum.
Anaplasma marginale Msp2
Anaplasma marginale infection persists in its natural hosts, wild and domestic ruminants, to provide a reservoir for transmission to new hosts when competent tick vectors are seasonally abundant. Cyclic waves of bacteremia throughout persistence reflect the sequential emergence and control of Major Surface Protein 2 (Msp2) variants (Palmer et al., 2000). Variant-specific IgG antibodies presumably clear A. marginale bacteria expressing the specific Msp2 variant by opsonization and macrophage phagocytosis during the brief period when the bacteria are extracellular following release from one infected cell and prior to entry into a new target cell (French et al., 1999). Msp2 variants are generated from a small number of donor alleles (<10, the number varies among strains) with recombination into a single expression site (Brayton et al., 2001, 2005). Importantly, the number of distinct variants is exponentially increased by the process of segmental gene conversion in which expression site mosaics are generated by recombination of oligonucleotide segments from multiple donor alleles (Barbet et al., 2000; Brayton et al., 2002). This combinatorial process, similar to that described above for B. burgdorferi and T. pallidum (Fig. 1) has the capacity to generate thousands of distinct Msp2 variants, in agreement with the number of variants required for life-long persistence and observed in vivo within the natural reservoir host.
Although the generation of mosaics greatly expands the number of Msp2 antigenic variants, it does so at a significant fitness cost. Unlike the donor alleles, which are maintained in the genome due to the gene conversion mechanism, the expression site copy is transient. Consequently, only the donor alleles are under long-term evolutionary selection pressure for growth fitness; the expression site copy is under short-term competing pressures to evade existing antibody while maintaining at least minimal growth fitness. The in vivo evidence for this fitness cost includes: (i) high level bacteremia is composed predominately of simple Msp2 variants, derived from recombination of a complete donor allele (Futse et al., 2005); (ii) bacteremia levels decrease by greater than two logs concomitant with the appearance of Msp2 mosaics (Futse et al., 2005; Palmer et al., 2007); and iii) mosaics are maintained only under immune pressure as direct inoculation of bacteria expressing Msp2 mosaics into naive animals or acquisition by a feeding tick rapidly results in predominance by bacteria expressing simple Msp2 variants (Rurangirwa et al., 1999; Palmer et al., 2007).
What are the consequences of this fitness cost? Within an individual infected animal, the consequences in terms of transmission fitness are minimal: ticks efficiently acquire A. marginale even at the lowest bacteremia levels during persistence, and replication within the tick, associated with rapid selection for highly fit simple variants, overcomes the initial low number of bacteria acquired and allows transmission (Rurangirwa et al., 1999; Scoles et al., 2005). In contrast, fitness differences in the Msp2 allelic repertoire among strains appear to drive the dynamic A. marginale strain structure at the population level in natural reservoir hosts. While sequencing of multiple A. marginale strains has revealed an overall closed core genome (Dark et al., 2009), msp2 alleles differ among strains and thus encode strain-specific repertoires of variants (Rodriguez et al., 2005; Futse et al., 2008). That variants encoded by individual alleles differ markedly in fitness in vivo is shown by recent studies showing a non-random, highly biased representation of Msp2 variants derived from specific alleles (Futse et al., 2009). The effect was attributable to the specific alleles rather than the position or structure of the donor locus within the genome, a key difference as compared to other pathogens for which locus structure effects may drive allelic usage. This fitness of specific Msp2 alleles is manifested both in infection of the mammalian host and, as will be discussed below, in the tick vector. The mechanism underlying the growth fitness advantage assignable to specific alleles remains unknown. However studies with the closely related A. phagocytophilum identify Msp2 as a porin (Huang et al., 2007), thus raising the possibility that difference in porin function may confer differential growth. For a given strain, the allelic repertoire would be driven to be sufficiently diverse to encode structurally and antigenically unique variants but balanced by the need to confer at least minimal porin function.
As noted above, there is tremendous strain diversity in A. marginale within the host reservoir population. This diversity appears to be driven by population immunity in the reservoir hosts and results in specific diversification in Msp2 alleles among strains (Futse et al., 2008). The high prevalence of A. marginale in tropical and sub-tropical regions confers a high level of immunity against re-infection with the same (or similar) strain. This paradox, whereby persistently infected animals cannot clear their own infection yet are resistant to re-infection, was resolved by the discovery that ticks transmitted simple variants (Rurangirwa et al., 1999; Palmer et al., 2007). Persistently infected animals develop immunity against simple variants (those generated by recombination from a single donor allele) and, over time, against a diverse array of complex mosaic variants but fail to clear the pathogen due to sequential segmental gene conversion upon the expression site mosaic. In contrast, the A. marginale transmitted by ticks express simple Msp2 variants (due to their fitness advantage in the absence of adaptive immunity) and thus cannot establish infection within a reservoir host population with a high level of immunity to the repertoire of simple variants—unless the strain has diversified to encode a novel set of variants. As a consequence, there is strong selective pressure for Msp2 allelic diversification among strains. This selective pressure for allelic diversification is supported by studies under conditions of natural transmission in which superinfection with strains encoding non-overlapping allelic repertoires is detected and by experimental transmission in which successful strain superinfection was directly linked to expression of Msp2 variants unique to the superinfecting strain (Rodriguez et al., 2005; Futse et al., 2008). Mechanistically, allelic diversification is allowed as the anchoring mechanism of gene conversion in A. marginale limits the requirement for conservation to the 5’ and 3’ regions flanking the hypervariable domains in all alleles and the processes of interallelic gene conversion and mutation are proposed to generate this diversity (Futse et al., 2005; Palmer and Brayton, 2007).
The tremendous advantage of a unique strain, as defined by its Msp2 allelic repertoire and associated capacity to superinfect, in a highly endemic region mimics the advantage of a bacterium expressing a unique variant within the individual. Correspondingly, the same balance between encoding variants that are sufficiently different to evade immune clearance and those that retain growth fitness shapes the strain-specific allelic repertoire. There is no single “optimal” Msp2 allelic repertoire but rather a continual shifting landscape of balancing selection. In infection of the mammalian host, specific alleles and the encoded simple variants have a fitness advantage early in infection, nonetheless there is no compelling evidence that this results in a marked transmission advantage or predominance of the parent strain in the population. Current data do suggest a pronounced effect within the tick vector. As in the mammalian host, there is non-random usage of specific alleles in generating simple variants during rapid replication within the tick, however, there is also evidence that specific strains differ markedly in growth fitness within the tick and that the most highly fit strains outcompete less fit strains and are preferentially transmitted (Ueti et al., 2009; Galletti et al., 2009). In a population of only immunologically naïve hosts, this would be expected to quickly bottleneck to a single optimal strain and the less fit strains would be lost from the population—similar to what has occurred with the rapid passage of the T. pallidum Nichols strain in rabbits. However, the reality of broad population immunity in endemic regions provides the balancing diversification pressure and thus the predicted less fit strains are retained and contribute to the dynamic A. marginale strain structure observed in the mammalian host reservoir population.
Concluding Remarks: Paradoxes and Opportunities
Of the paradoxes common to multiple highly antigenically variant bacterial pathogens is why variation in a single surface protein is sufficient to evade clearance. Notably for both A. marginale and B. burgdorferi, there are multiple well-documented surface proteins that are invariant and immunogenic during infection yet clearance is not effected. Possible explanations are both structural, such as masking of antibody binding in vivo, as well as immunological. Consistent with the latter is the immunodominance of both VlsE and Msp2 in B. burgdorferi and A. marginale infections, respectively. The availability of the vls knockout provides a powerful tool to address this broadly applicable question. Interestingly, B. burgdorferi clones complemented with a non-switching copy of vlsE are cleared faster in immunocompetent mice relative to non-VlsE-complemented clones (Bankhead and Chaconas, 2007), compelling evidence of direct structural and or immunological regulatory effects.
Understanding the pressures at the population level as well as within the individual host represents a second major knowledge gap. While influenza continues to represent a classic example of population level selective pressure for pathogen genetic change, it is also apparent that these pressures apply equally to bacterial pathogen epidemiology. Strain shifts that result in penetration into new host or vector populations underlie emerging diseases and increased knowledge of the drivers of these shifts is essential for improved disease control.
A final note may be the opportunities in vaccine development by understanding the balance between fitness and immune evasion. Highly antigenically variant pathogens cause diseases with high global impact and have proved to be exceedingly difficult to control by vaccines— not surprising given the strong evolutionary adaptation to persistence through immune evasion. However by understanding the fitness cost associated with expression of specific variants, it may be feasible to develop new vaccines that prevent the period of high infectiousness and disease by targeting highly fit variants.
Acknowledgements
We apologize to those authors whose work was not cited due to space limitations. We thank Yvonne Tourand for helpful comments on the manuscript. NIH provided support for research in the Lukehart and Palmer laboratories.
Footnotes
Attributed to Hericlitus, 535-475, B.C., who noted that one can never step in the same river twice.
References
- Bacon RM, Biggerstaff BJ, Schriefer ME, Gilmore RD, Jr, Philipp MT, Steere AC, et al. Serodiagnosis of Lyme disease by kinetic enzyme-linked immunosorbent assay using recombinant VlsE1 or peptide antigens of Borrelia burgdorferi compared with 2-tiered testing using whole-cell lysates. J Infect Dis. 2003;187:1187–1199. doi: 10.1086/374395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead T, Chaconas G. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol. 2007;65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- Barbet AF, Lundgren A, Yi J, Rurangirwa FR, Palmer GH. Antigenic variation of Anaplasma marginale by expression of MSP2 sequence mosaics. Infect Immun. 2000;68:6133–6138. doi: 10.1128/iai.68.11.6133-6138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour AG. Borrelia: a diverse and ubiquitous genus of tick-borne pathogens. In: Scheld MW, Craig WA, Hughes JM, editors. Emerging Infections. Vol. 5. Washington, D.C: American Society for Microbiology; 2001. pp. 153–173. [Google Scholar]
- Beaurepaire C, Chaconas G. Mapping of essential replication functions of the linear plasmid lp17 of B. burgdorferi by targeted deletion walking. Mol Microbiol. 2005;57:132–142. doi: 10.1111/j.1365-2958.2005.04688.x. [DOI] [PubMed] [Google Scholar]
- Brayton KA, Knowles DP, McGuire TC, Palmer GH. Efficient use of a small genome to generate antigenic diversity in tick-borne ehrlichial pathogens. Proc Natl Acad Sci USA. 2001;98:4130–4135. doi: 10.1073/pnas.071056298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayton KA, Palmer GH, Lundgren A, Yi J, Barbet AF. Antigenic variation of Anaplasma marginale msp2 occurs by combinatorial gene conversion. Mol Microbiol. 2002;43:1151–1159. doi: 10.1046/j.1365-2958.2002.02792.x. [DOI] [PubMed] [Google Scholar]
- Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, et al. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci USA. 2005;102:844–849. doi: 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion-Lara A, Castro C, Barrett L, Cameron C, Mostowfi M, Van Voorhis WC, Lukehart SA. Treponema pallidum major sheath protein homologue Tpr K is a target of opsonic antibody and the protective immune response. J Exp Med. 1999;189:647–656. doi: 10.1084/jem.189.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion-Lara A, Godornes C, Castro C, Van Voorhis WC, Lukehart SA. The tprK gene is heterogeneous among Treponema pallidum strains and has multiple alleles. Infect Immun. 2000;68:824–831. doi: 10.1128/iai.68.2.824-831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion-Lara A, LaFond RE, Hevner K, Godornes C, Molini BJ, Van Voorhis WC, Lukehart SA. Gene conversion: a mechanism for generation of heterogeneity in thetprK gene of Treponema pallidum during infection. Mol Microbiol. 2004;52:1579–1596. doi: 10.1111/j.1365-2958.2004.04086.x. [DOI] [PubMed] [Google Scholar]
- Chaconas Hairpin telomere and genome plasticity in Borrelia: all mixed up in the end. Mol Microbiol. 2005;58:625–635. doi: 10.1111/j.1365-2958.2005.04872.x. [DOI] [PubMed] [Google Scholar]
- Coutte L, Botkin DJ, Gao L, Norris SJ. Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog. 2009;5:e1000293. doi: 10.1371/journal.ppat.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crother TR, Champion CI, Wu XY, Blanco DR, Miller JN, Lovett MA. Antigenic composition of Borrelia burgdorferi during infection of SCID mice. Infect Immun. 2003;71:3419–3428. doi: 10.1128/IAI.71.6.3419-3428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dark MJ, Herndon DR, Kappmeyer LS, Gonzales MP, Nordeen E, Palmer GH, et al. Conservation in the face of diversity: multistrain analysis of an intracellular bacterium. BMC Genomics. 2009;10:16–28. doi: 10.1186/1471-2164-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicken C, Sharma V, Klabunde T, Lawrenz MB, Hardham JM, Norris SJ, Sacchettini JC. Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J Biol Chem. 2002;277:21691–21696. doi: 10.1074/jbc.M201547200. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, et al. Complete genome sequence of Treponema pallidum the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- French DM, Brown WC, Palmer GH. Emergence of Anaplasma marginale antigenic variants during persistent rickettsemia. Infect Immun. 1999;67:5834–5840. doi: 10.1128/iai.67.11.5834-5840.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futse JE, Brayton KA, Knowles DP, Palmer GH. Structural basis for segmental gene conversion in generation of Anaplasma marginale outer membrane protein variants. Mol Microbiol. 2005;57:212–221. doi: 10.1111/j.1365-2958.2005.04670.x. [DOI] [PubMed] [Google Scholar]
- Futse JE, Brayton KA, Dark MJ, Knowles DP, Palmer GH. Superinfection as a driver of genomic diversification in antigenically variant pathogens. Proc Natl Acad Sci USA. 2008;105:2123–2127. doi: 10.1073/pnas.0710333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futse JE, Brayton KA, Nydam SD, Palmer GH. Generation of antigenic variants by gene conversion: Evidence for recombination fitness selection at the locus level in Anaplasma marginale. Infect Immun. 2009;77:3181–3187. doi: 10.1128/IAI.00348-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galletti MFBM, Ueti MW, Knowles DP, Brayton KA, Palmer GH. Independence of high and low transmission efficiency Anaplasma marginale strains in the tick vector following simultaneous acquisition by feeding on a superinfected mammalian reservoir host. Infect Immun. 2009;77:1459–1464. doi: 10.1128/IAI.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett KRO, Sellati TJ, Nguyen TT, Cox DL, Clawson ML, Caimano MJ, Radolf JD. The TprK protein of Treponema pallidum is periplasmic and is not a target of opsonic antibody or protective immunity. J Exp Med. 2001;193:1015–1026. doi: 10.1084/jem.193.9.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Wang X, Kikuchi T, Kumagai Y, Rikihisa Y. Porin activity of Anaplasma phagocytophilum outer membrane fraction and purified P44. J Bacteriol. 2007;189:1998–2006. doi: 10.1128/JB.01548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson CR, Frye JG, Quinn FD, Gherardini FC. Increased expression of Borrelia burgdorferi vlsE in response to human endothelial cell membranes. Mol Microbiol. 2001;41:229–239. doi: 10.1046/j.1365-2958.2001.02511.x. [DOI] [PubMed] [Google Scholar]
- Kobryn K, Chaconas G. ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol Cell. 2002;9:195–201. doi: 10.1016/s1097-2765(01)00433-6. [DOI] [PubMed] [Google Scholar]
- Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Seshu J, Skare JT. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun. 2003;71:4608–4613. doi: 10.1128/IAI.71.8.4608-4613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFond RE, Centurion-Lara A, Godornes C, Rompalo AM, Van Voorhis WC, Lukehart SA. Sequence diversity of Treponema pallidum subsp.pallidum tprK in human syphilis lesions and rabbit-propagated isolates. J Bacteriol. 2003;185:6262–6268. doi: 10.1128/JB.185.21.6262-6268.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFond RE, Molini BJ, Van Voorhis WC, Lukehart SA. Antigenic variation of TprK V regions abrogates specific antibody binding in syphilis. Infect Immun. 2006a;74:6244–6251. doi: 10.1128/IAI.00827-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFond RE, Centurion-Lara A, Godornes C, Van Voorhis WC, Lukehart SA. TprK sequence diversity accumulates during infection of rabbits with Treponema pallidum subsp.pallidum Nichols strain. Infect Immun. 2006b;74:1896–1906. doi: 10.1128/IAI.74.3.1896-1906.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenz MB, Wooten RM, Norris SJ. Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect Immun. 2004;72:6577–6585. doi: 10.1128/IAI.72.11.6577-6585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FT, Yan J, Mbow ML, Sviat SL, Gilmore RD, Mamula M, Fikrig E. Borrelia burgdorferichanges its surface antigenic expression in response to host immune responses. Infect Immun. 2004;72:5759–5767. doi: 10.1128/IAI.72.10.5759-5767.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukehart SA, Shaffer JM, Baker-Zander SA. A subpopulation of Treponema pallidum is resistant to phagocytosis: possible mechanism of persistence. J Infect Dis. 1992;166:1449–1453. doi: 10.1093/infdis/166.6.1449. [DOI] [PubMed] [Google Scholar]
- McDowell JV, Sung SY, Hu LT, Marconi RT. Evidence that the variable regions of the central domain of VlsE are antigenic during infection with Lyme disease spirochetes. Infect Immun. 2002;70:4196–4203. doi: 10.1128/IAI.70.8.4196-4203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA, Molini BJ, Lukehart SA, Van Voorhis WC. Segregation of B and T cell epitopes of Treponema pallidum repeat protein K to variable and conserved regions during experimental syphilis infection. J Immunol. 2002;169:952–957. doi: 10.4049/jimmunol.169.2.952. [DOI] [PubMed] [Google Scholar]
- Norris SJ. Antigenic variation with a twist - the Borrelia story. Mol Microbiol. 2006;60:1319–1322. doi: 10.1111/j.1365-2958.2006.05204.x. [DOI] [PubMed] [Google Scholar]
- Palmer GH, Brown WC, Rurangirwa FR. Antigenic variation in the persistence and transmission of the herlichia Anaplasma marginale. Microb Infect. 2000;2:167–176. doi: 10.1016/s1286-4579(00)00271-9. [DOI] [PubMed] [Google Scholar]
- Palmer GH, Futse JE, Leverich CK, Knowles DP, Rurangirwa FR, Brayton KA. Selection for simple Msp2 variants during Anaplasma marginale transmission to immunologically naïve animals. Infect Immun. 2007;75:1502–1506. doi: 10.1128/IAI.01801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GH, Brayton KA. Gene conversion is a convergent strategy for pathogen antigenic variation. Trends Parasitol. 2007;23:408–413. doi: 10.1016/j.pt.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci USA. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez JL, Palmer GH, Knowles DP, Brayton KA. Distinctly different msp2 pseudogene repertoires in Anaplasma marginale strains that are capable of superinfection. Gene. 2005;361:127–132. doi: 10.1016/j.gene.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Rurangirwa FR, Stiller DS, French DM, Palmer GH. Restriction of Major Surface Protein 2 (MSP2) variants during tick transmission of the ehrlichiae Anaplasma marginale. Proc Natl Acad Sci USA. 1999;96:3171–3176. doi: 10.1073/pnas.96.6.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Spechtel U, Lehnert G, Liegl G, Fingerle V, Heimerl C, Johnson BJ, Wilske B. Significant improvement of the recombinant Borrelia-specific immunoglobulin G immunoblot test by addition of VlsE and a DbpA homologue derived from Borrelia garinii for diagnosis of early neuroborreliosis. J Clin Microbiol. 2003;41:1299–1303. doi: 10.1128/JCM.41.3.1299-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoles GA, Broce AB, Lysyk TJ, Palmer GH. Relative efficiency of biological transmission of Anaplasma marginale (Rickettsiales: Anaplasmataceae) by Dermacentor andersoni Stiles (Acari: Ixodidae) compared to mechanical transmission by the stable fly, Stomoxys calcitrans (L.) (Diptera: Muscidae) J Med Entomol. 2005;42:668–675. doi: 10.1603/0022-2585(2005)042[0668:REOBTO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TB, Hollander DH. Biology of the Treponematoses. Geneva: World Health Organization; 1957. [PubMed] [Google Scholar]
- Ueti MW, Knowles DP, Davitt CM, Scoles GA, Baszler TV, Palmer GH. Quantitative differences in salivary pathogen load during tick transmission underlie strain-specific variation in transmission efficiency of Anaplasma marginale. Infect Immun. 2009;77:70–75. doi: 10.1128/IAI.01164-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JR, Hardham JM, Barbour AG, Norris SJ. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–285. doi: 10.1016/s0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- Zhang JR, Norris SJ. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun. 1998;66:3689–3697. doi: 10.1128/iai.66.8.3689-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]