

Abstract
The canonical Wnt/β-catenin signaling pathway classically functions through the activation of target genes by Tcf/Lef–β-catenin complexes. In contrast to β-catenin-dependent functions described for Tcf1, Tcf4 and Lef1, the known embryonic functions for Tcf3 in mice, frogs and fish are consistent with β-catenin-independent repressor activity. In this study, we genetically define Tcf3–β-catenin functions in mice by generating a Tcf3ΔN knock-in mutation that specifically ablates Tcf3–β-catenin. Mouse embryos homozygous for the knock-in mutation (Tcf3ΔN/ΔN) progress through gastrulation without apparent defects, thus genetically proving that Tcf3 function during gastrulation is independent of β-catenin interaction. Tcf3ΔN/ΔN mice were not viable, and several post-gastrulation defects revealed the first in vivo functions of Tcf3–β-catenin interaction affecting limb development, vascular integrity, neural tube closure and eyelid closure. Interestingly, the etiology of defects indicated an indirect role for Tcf3–β-catenin in the activation of target genes. Tcf3 directly represses transcription of Lef1, which is stimulated by Wnt/β-catenin activity. These genetic data indicate that Tcf3–β-catenin is not necessary to activate target genes directly. Instead, our findings support the existence of a regulatory circuit whereby Wnt/β-catenin counteracts Tcf3 repression of Lef1, which subsequently activates target gene expression via Lef1–β-catenin complexes. We propose that the Tcf/Lef circuit model provides a mechanism downstream of β-catenin stability for controlling the strength of Wnt signaling activity during embryonic development.
Keywords: Tcf3, Wnt signaling, beta-catenin, Embryonic stem cell, Mouse embryogenesis
INTRODUCTION
The canonical Wnt signaling pathway is required for morphogenesis of many organs, and its activity is necessary in adults through the regulation of stem cell properties. Overactivation of Wnt/β-catenin signaling causes cancer (Moon et al., 2002), which demonstrates a need to regulate the pathway through adulthood. Activity of the pathway is controlled through the regulation of β-catenin protein stability (MacDonald et al., 2009). In the absence of a Wnt ligand, β-catenin is targeted for ubiquitin- and proteasome-mediated degradation by phosphorylation at serine and threonine residues near the N-terminus (Aberle et al., 1997; Liu et al., 2002). Phosphorylation of β-catenin occurs within a polyprotein complex that includes adenomatous polyposis coli (APC), Axin, and the kinases casein kinase 1 and glycogen synthase kinase 3 (GSK3) (Behrens et al., 1998; Ikeda et al., 1998; Liu et al., 2002). Wnt ligands bind to Frizzled-Lrp5/6 receptor complexes and initiate a downstream cascade that inhibits GSK3 phosphorylation of β-catenin. This effect stabilizes β-catenin and stimulates its interaction with DNA-binding transcriptional regulators, of which Tcf/Lef proteins are the best characterized (Molenaar et al., 1996; van de Wetering et al., 1997). When bound to Tcf/Lef proteins, β-catenin can function as a transcriptional co-activator by recruiting several nuclear factors to chromatin, such as BCL9, Pygopus, Brg-1 and CBP (Barker et al., 2001; Hecht et al., 2000; Kramps et al., 2002; Sun et al., 2000; Takemaru and Moon, 2000).
Mammals have four Tcf/Lef factors: Tcf1 (Tcf7), Tcf3 (Tcf7L1), Tcf4 (Tcf712) and Lef1. They possess nearly identical DNA-binding domains that allow them to bind the same consensus sequence (A/AT/TCAAAG) and regulate the transcription of target genes (Atcha et al., 2007; van Beest et al., 2000; van de Wetering and Clevers, 1992). All Tcf/Lefs also possess conserved β-catenin interaction domains near the N-terminus, which are necessary for Tcf/Lef proteins to stimulate target gene transcription in response to Wnt-stabilized β-catenin (Behrens et al., 1996; Brannon et al., 1997; Molenaar et al., 1996; van de Wetering et al., 1997). Full-length Tcf/Lef proteins also have β-catenin-independent activities as transcriptional repressors. Although all Tcf/Lef proteins have been reported to interact with co-repressor proteins such as Groucho (Brantjes et al., 2001), the extent to which the embryonic function of Tcf/Lefs depends on β-catenin-independent activities appears to be different for each Tcf/Lef protein (Yi and Merrill, 2007). Lef1 sits at one end of the spectrum, as knockout and transgenic mouse models predominantly show Wnt/β-catenin-dependent functions for Lef1 (van Genderen et al., 1994; Zhou et al., 1995).
At the other end of the spectrum, Tcf3 appears to function primarily as a transcriptional repressor. Transgenic mice showed that full-length wild-type Tcf3 and β-catenin interaction-defective ΔNTcf3 cause essentially identical phenotypes when ectopically expressed in the epidermis (Merrill et al., 2001). Importantly, transgenic mice engineered to overexpress repressor-defective mutant forms of Tcf3 did not exhibit an abnormal phenotype (Merrill et al., 2001). Subsequently, through the use of inducible Tcf3 expression in the skin, the Tcf3 overexpression phenotype was shown to be caused by a reversion of adult cells to an embryonic-like state, and analysis of downstream transcriptional effects showed that Tcf3 did not require Wnt/β-catenin (Nguyen et al., 2006). Genetic ablation of Tcf3 affected gastrulation to produce embryos that display ectopic and partially duplicated axes, which ostensibly phenocopies several embryos engineered for overactivation of Wnt/β-catenin signaling during gastrulation (Ishikawa et al., 2003; Kemler et al., 2004; Popperl et al., 1997; Zeng et al., 1997). In frogs and fish, loss-of-function experiments showed that Tcf3 acts as a transcriptional repressor in its first necessary function in embryos (Dorsky et al., 2003; Houston et al., 2002; Kim et al., 2000).
Despite the lack of direct evidence of a physiological requirement for Tcf3–β-catenin interaction in vivo, all Tcf3 homologs encode a conserved β-catenin-binding domain. Analysis of Tcf3 overexpression has focused on effects of Tcf3 in cells that do not endogenously activate Tcf/Lef–β-catenin target genes (Merrill et al., 2001; Nguyen et al., 2006). Thus, a potential role for the Tcf3–β-catenin interaction in vivo seemed plausible; however, the predominant repressor activity of Tcf3 obscured elucidation of any in vivo processes that require the Tcf3–β-catenin interaction.
We set out to determine genetically whether the Tcf3–β-catenin interaction is necessary for mouse embryogenesis or viability. We generated a Tcf3ΔN knock-in mutation and examined Tcf3ΔN/ΔN mice, which specifically lack the Tcf3–β-catenin interaction. We show that Tcf3ΔN/ΔN embryos progress through gastrulation without the defects associated with Tcf3–/– embryos, demonstrating that the first requirement for Tcf3 in the mouse is independent of Tcf3–β-catenin interaction. After gastrulation, Tcf3ΔN/ΔN embryos exhibited a diverse array of phenotypes. Interestingly, Tcf3–β-catenin is needed for Tcf/Lef–β-catenin activation of target genes; however, the mechanism of target gene activation is indirect and involves Wnt–β-catenin inhibiting Tcf3 repression of Lef1, followed by Lef1–β-catenin activation of transcription in the eyelid.
The findings presented here indicate a novel Tcf/Lef circuit through which Tcf3-expressing cells respond to Wnt/β-catenin signaling. Given the expression of Tcf3 in stem cells (DasGupta and Fuchs, 1999; Ivanova et al., 2002; Pereira et al., 2006) and poorly differentiated tumors (Ben-Porath et al., 2008), this work impacts not only developmental contexts but has the potential to be important for the manipulation of stem cells and treatment of aggressive cancers.
MATERIALS AND METHODS
Generation and genotyping of Tcf3ΔN/ΔN mice
Two independent Tcf3+/ΔN embryonic stem cell (ESC) lines from Yi et al. (Yi et al., 2011) were injected into C57BL/6 blastocysts to produce male chimeras that were selected for mating with wild-type C57BL/6 females. Tcf3+/ΔN heterozygotes were backcrossed ten times to the C57BL/6 background prior to analysis of viability, penetrance of phenotypes and expressivity of phenotypes. Tcf3+/– and Tcf3+/ΔN mice were maintained on a C57BL/6 background throughout the study. Primers used for genotyping are: forward, 5′-AGTCGTCCCTGGTCAACGAAT-3′; reverse, 5′-ACAGAGTAGCTATCTGGAGCTCGG-3′.
Embryo in situ hybridization and immunofluorescence
The age of embryos was determined based on the time of day harvested assuming that noon on the day of plug discovery corresponds to embryonic day (E) 0.5. Whole-mount in situ hybridizations were performed essentially as described previously (Merrill et al., 2004). Probes in this study were specific for Tcf3 (B.J.M laboratory), brachyury (D. Wilkinson, MRC National Institute For Medical Research, London, UK), Shh (A. P. McMahon, Harvard University, MA, USA), patched 1 (M. P. Scott, Stanford University, CA, USA), Fgf8 and gremlin 1 (G. Martin, University of California San Francisco, CA, USA), Msx1 and Msx2 (R. Harland, University of California Berkeley, CA, USA and P. Sharpe, King’s College, London, UK), and Tbx2 (V. Papaionnou, Columbia University, NY, USA).
All immunofluorescent staining was performed on 8-μm frozen sections of embryos embedded in OCT (Tissue-Tek) and fixed in 4% paraformaldehyde (PFA) following sectioning. For immunofluorescent detection of proteins in mouse eyelid using mouse primary antibody, the MOM kit (Vector Laboratories) was used according to the manufacturer’s protocol. The primary antibodies used were: Tcf3 (Merrill et al., 2001; Pereira et al., 2006), Lef1 (Cell Signaling), Tcf1 (Cell Signaling), Tcf4 (Cell Signaling), phospho-histone H3 (Millipore), cleaved caspase 3 (Cell Signaling), β-galactosidase (Developmental Studies Hybridoma Bank), Ki67 (Abcam), phospho-ERK (Cell Signaling), phospho-JNK (Cell Signaling) and phospho-c-Jun (Cell Signaling). Donkey antibodies conjugated with Cy5, FITC or Texas Red (Jackson Labs) were used as secondary antibodies. All fluorescent images were taken with a Zeiss LSM 5 Pascal system. Fluorescein-conjugated phalloidin (Invitrogen) was used to detect filamentous actin.
X-Gal assays
Embryos were dissected in 1× PBS and prefixed for 20 minutes in 4% PFA in PBS at 4°C. The embryos were washed five times in 1× PBS and then stained in X-Gal stain solution [100 mM sodium phosphate pH 7.3, 1.3 mM MgCl2, 3 mM K3Fe(CN)6, 3 mM K4Fe(CN)6, 1 mg/ml X-Gal, 0.1% sodium deoxycholate, 0.2% NP40]. Staining was performed in the dark at 37°C for 2-3 hours. After staining, the embryos were post-fixed in 4% PFA at 4°C overnight. Frozen sections (8 μm) were fixed with 0.5% glutaraldehyde for 2 minutes. After washing seven or eight times in 1× PBS, slides were transferred into X-Gal stain solution without detergent. Staining was performed in the dark at 37°C for 30 minutes and, after mounting in 80% glycerol, images were taken with a Zeiss Axiovert 200M inverted microscope.
Lef1 promoter luciferase assays
For each experiment, 105 ESCs were transfected in 24-well plates using Lipofectamine 2000 following the manufacturer’s protocol (Invitrogen). After 6 hours, Wnt3a-conditioned media or control media were added. After 30 hours, cells were harvested and luciferase activity was measured using a Clarity luminometer (Bio-Tek) and the Dual-Luciferase Reporter Assay System (Promega). Each transfection was carried out in duplicate and repeated at least twice. Relative activity was calculated as the ratio of the firefly reporter to Renilla luciferase (pRL-CMV) control.
Western blot analysis
Primary antibodies and dilutions were Tcf3 [1:3000 (Pereira et al., 2006)], Tcf1 (1:1000; Cell Signaling), Lef1 (1:1000; Cell Signaling), Tcf4 (1:3000; Cell Signaling), β-catenin (1:2000; Sigma), active β-catenin (1:1000; Millipore) and tubulin (1:3000; Developmental Studies Hybridoma Bank). Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies at 1:3000 dilution (Jackson Laboratory). Signals were detected with ECL western blotting detection reagents (Amersham Biosciences).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed essentially as described previously (Yi et al., 2011). Briefly, 2 × 107 ESCs were cultured and processed for each ChIP experiment. Sonication was carried out on ice by using a Branson digital sonifier (Model 450) for 20 × 30-second pulses (60 seconds pause between pulses) at 60% amplitude. The resulting chromatin contained DNA fragments with an average size of 500 bp. Chromatin was immunoprecipitated overnight at 4°C using magnetic beads (Protein G Dynabeads, Invitrogen) that had been pre-incubated with 5 μg of the Tcf3 (B.J.M. lab) or β-catenin (Invitrogen) antibody. Following phenol-chloroform extraction and ethanol precipitation, DNA was dissolved in 60 μl TE (10 mM Tris, 1 mM EDTA). For each qPCR reaction, 1.5 μl of DNA sample was used. DNA oligo sequences used for qPCR reactions are listed in supplementary material Table S1.
RESULTS
Tcf3ΔN knock-in mutation demonstrates that the requirement for Tcf3 during gastrulation is independent of β-catenin interaction
Knockout mouse experiments have shown that Wnt3 and β-catenin are required for induction of the primitive streak and specification of mesoderm (Huelsken et al., 2000; Liu et al., 1999). Subsequently, Wnt3a and the combined effects of Tcf1 and Lef1 specify paraxial mesoderm in a process characterized by activation of Tcf/Lef target genes (Arnold et al., 2000; Galceran et al., 1999; Galceran et al., 2001). Overactivation of Wnt/β-catenin signaling stimulated ectopic mesoderm and partial duplication of the anteroposterior axis (Ishikawa et al., 2003; Kemler et al., 2004; Popperl et al., 1997; Zeng et al., 1997). Ablation of the Tcf3 gene product caused a remarkably similar phenotype to overactivation of Wnt/β-catenin signaling, with partial axis duplication and ectopic mesoderm, but Tcf3–/– mutants also displayed reduced paraxial and lateral mesoderm (Merrill et al., 2004). Thus, it was not clear whether the requirement for Tcf3 during gastrulation reflected a combination of Tcf3 repressor and Tcf3–β-catenin activities.
To determine the function of Tcf3–β-catenin interaction in vivo, we engineered a Tcf3ΔN mutation that replaced ten Tcf3 residues necessary for β-catenin binding with a single alanine (Fig. 1A) (Graham et al., 2000). When wild-type (WT) Tcf3 was transiently expressed in Cos-7 cells, binding to endogenous β-catenin was detected by immunofluorescence (Fig. 1B) and immunoprecipitation (Fig. 1C) assays. By contrast, Tcf3ΔN was unable to relocalize endogenous β-catenin (Fig. 1B), was deficient in co-immunoprecipitation of β-catenin (Fig. 1C), and blocked β-catenin activation of the TOPFlash luciferase reporter (supplementary material Fig. S1A). The Tcf3ΔN mutation and a more extensive Tcf3ΔN71 mutation caused similar defects (Fig. 1C). Previously, homologous recombination in ESCs incorporated the Tcf3ΔN mutation into the genome (Merrill et al., 2004; Yi et al., 2011), and Tcf3+/ΔN ESCs were used to generate new Tcf3+/ΔN lines of mice. The Tcf3ΔN allele generated a stable Tcf3ΔN mRNA (supplementary material Fig. S1B) and Tcf3ΔN protein (supplementary material Fig. S1C) in mice. Tcf3+/ΔN mice did not display any detectable abnormalities during development or as adult mice. Thus, the Tcf3ΔN knock-in mutation provided an excellent means of determining the role of Tcf3–β-catenin interaction in vivo.
Fig. 1.

Tcf3–β-catenin interaction is required after gastrulation. (A) Three-dimensional structure of the Tcf3–β-catenin interaction (Graham et al., 2000). Amino acid sequence of Tcf3 proteins from Xenopus and mouse are aligned above the structure. Red denotes Tcf3 residues of the core β-catenin interaction domain mutated in Tcf3ΔN. (B) Immunofluorescent staining detects nuclear co-localization in Cos-7 cells of endogenous β-catenin (green) and transiently transfected Tcf3 (red, top panels only). Expression of the Tcf3ΔN mutant protein (+Tcf3ΔN, right panels) failed to cause nuclear β-catenin localization. (C) Co-immunoprecipitation (IP) using a Tcf3-specific antibody and total cell lysate (TCL) from Cos-7 cells transiently transfected with the Tcf3 expression plasmid for wild-type (WT) (Tcf3), a mutant lacking the first 71 residues (Tcf3ΔN71), or the Tcf3ΔN knock-in mutation. (D) Whole-mount in situ hybridization with a labeled brachyury cRNA probe on Tcf3+/+ (WT), Tcf3ΔN/ΔN (knock-in, KI) and Tcf3–/– (knockout, KO) embryos. Expression patterns and morphology of KI embryos resembled those of WT embryos, and KI embryos did not display the defects observed in KO embryos. (E) Recovery of embryos of the indicated genotypes obtained from timed pregnancies of Tcf3+/ΔN × Tcf3+/ΔN matings.
Tcf3ΔN/ΔN embryos, hereafter referred to as knock-in (KI) embryos, were first examined between E7.5 and E9.0. All embryos exhibited a normal morphology (Fig. 1D), and genotyping revealed the expected Mendelian ratios of Tcf3+/+, Tcf3+/ΔN and Tcf3ΔN/ΔN embryos (not shown). Whole-mount in situ hybridization using cRNA probes to detect brachyury (Fig. 1D) and Foxa2 (not shown) mRNAs revealed normal patterns of expression in KI mutants through E9.0 (Fig. 1D). By contrast, Tcf3–/– mutants displayed characteristic ectopic and expanded brachyury expression when stained alongside KI and control embryos (Fig. 1D). Normal viability of KI embryos was observed through E11.5 (Fig. 1E), which is well past the point of lethality in Tcf3–/– embryos (Merrill et al., 2004). This result demonstrates genetically that the first requirement for Tcf3 in mouse is independent of Tcf3–β-catenin activity, indicating that Tcf3 is needed to function exclusively as a repressor during gastrulation.
Tcf3–β-catenin interaction is necessary for viability
To determine whether Tcf3–β-catenin interaction is necessary for any stage of development in mice, we examined the frequency and morphology of KI embryos after gastrulation. A drop in the frequency of KI embryos was first observed at E15.5 and was maintained at E18.5 and postnatal day (P) 1 (Fig. 1E). No viable KI mice survived past P1, demonstrating that the Tcf3–β-catenin interaction is necessary for viability.
Mutant KI embryos displayed a variety of phenotypes with variable penetrance (proportion of mutants affected) and expressivity (extent of phenotypic defect). Neural tube defects in the form of exencephaly were observed in KI embryos (Fig. 2A); the low frequency (less than 5% of embryos were affected) indicated that exencephaly was not the primary cause of KI lethality (Fig. 2B). Edema occurred at high frequency between E12.5 and E15.5 (76% of KI embryos), and was not detected at later stages (Fig. 2A,B). Vascular integrity defects and hemorrhages were externally visible in 50% of embryos between E15.5 and E18.5 (Fig. 2A,B). The vascular integrity defects after E15.5 developed multiple presentations as telangiectasia (Fig. 2A) or rupture of major blood vessels including the internal carotid artery (Fig. 2C-F′). Since the frequency of KI inviability (100%) was greater than that of externally visible vascular integrity defects, we subjected E18.5 KI embryos lacking externally visible signs of defects to a comprehensive histopathological examination. Upon internal examination, each KI embryo displayed vascular integrity defects in organs such as the submandibular salivary gland (Fig. 2G) and liver (Fig. 2H). Based on the frequency and severity of the vascular integrity defects in KI embryos, we suggest that they are the primary cause of lethality in the mutants.
Fig. 2.

Diverse requirements for Tcf3–β-catenin interaction during mouse embryogenesis. (A) Representative images showing externally visible abnormal phenotypes of E15.5 to E18.5 KI embryos. (B) The penetrance of abnormal phenotypes observed in KI mutants. NA, not applicable or not determined. (C-D′) Intact Tcf3+/+ (C,D) and KI (C′,D′) E18.5 embryos. Arrow (C′,D′) points to externally visible hemorrhage. Low (C,C′) and high (D,D′) magnification views of the same region are shown. (E-F′) Hematoxylin and Eosin (H&E) staining of tissue sections of the neck from embryos in C-D′. The boxed region in E,E′ is shown at higher magnification in F,F′. Arrows (F,F′) indicate the internal carotid artery. Chevrons (E′) point to abnormal accumulation of blood in the KI embryo at the site of the externally visible hemorrhage. (G,H) H&E-stained tissue sections from submandibular salivary gland (G) and liver (H) of separate KI E18.5 embryos showing abnormal red blood cell accumulation. Sections of WT embryos are shown in supplementary material Fig. S1D,E.
Tcf3–β-catenin interaction in early limb buds is necessary to specify posterior digits
In contrast to the variable expressivity of vascular integrity defects, two phenotypes exhibited consistent expressivity and high penetrance in KI embryos, simplifying elucidation of the etiology of the morphogenetic defects. First, distal limb morphogenesis was defective in 62% of KI embryos, which displayed oligodactyly characterized by the absence of the most posterior digit (digit #5) (Fig. 2A,B; Fig. 3A,A′). Four KI mutants also lacked digit #4 (not shown), and defects always manifested only in the forelimbs. Alizarin Red staining of mineralized tissue revealed the complete absence of digit #5 and the mineralization of bone tissue in other digits in KI limbs (Fig. 3B,B′).
Fig. 3.
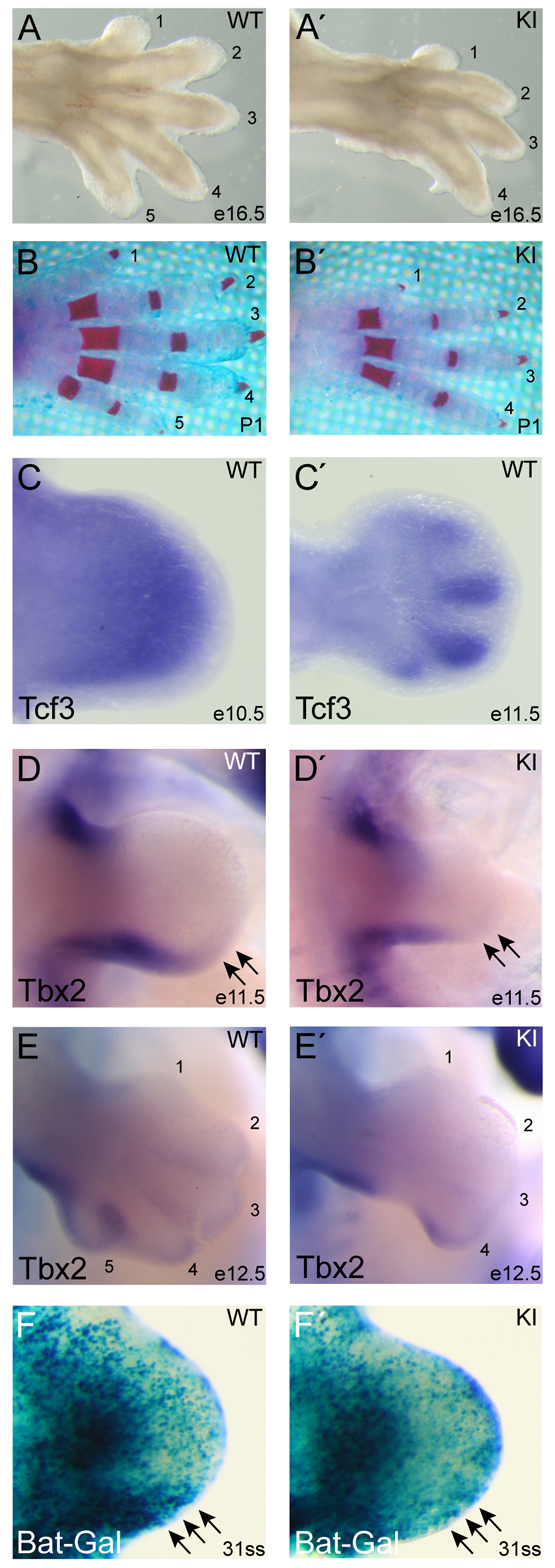
Tcf3–β-catenin interaction is required for post-axial digit formation. (A-B′) Gross morphology of autopod limb segment in WT (A,B) and KI (A′,B′) mouse limbs at E16.5 (A,A′) and P1 (B,B′). Limbs were stained with Alizarin Red (B,B′) and numbers correspond to anterior (#1) to posterior (#5) digits. (C,C′) In situ hybridization of limb buds from E10.5 (C) and E11.5 (C′) WT embryos using a cRNA probe for Tcf3. (D-E′) Whole-mount in situ hybridization using a cRNA probe for Tbx2 mRNA on E11.5 (D,D′) and E12.5 (E,E′) embryos. The posterior edge of Tbx2 expression (arrows) in WT limb buds (D,E) was absent in KI limb buds (D′,E′). (F,F′) Tcf3–β-catenin activity was detected with the BAT-Gal transgene in WT (F) and KI (F′) limb buds at E10.0 [31-somite stage (ss)]. Arrows point to the high BAT-Gal activity in the posterior mesenchyme region of WT limb buds (F), which is absent from KI limb buds (F′).
To determine the etiology of the oligodactyly, we sought to identify the earliest defect affecting morphogenesis of KI limbs. Tcf3 mRNA is expressed in the mesenchyme of the WT limb bud prior to E10.5 (Fig. 3C) and becomes restricted to digit condensates by E11.5 (Fig. 3C′). By E11.5, Tbx2 expression was reduced specifically at the distal posterior of the KI limb bud compared with control limb buds (arrows, Fig. 3D,D′), indicating that the defect occurred prior to E11.5. By E12.5, loss of the posterior limb bud became apparent, the visualization of which being aided by the pattern of Tbx2 expression, which outlines the forming digit #5 (Fig. 3E,E′).
The BAT-Gal transgene, in which Tcf/Lef binding sites drive the expression of lacZ, was used here to determine the pattern of Tcf/Lef–β-catenin-dependent transactivation in the limb bud (Maretto et al., 2003). BAT-Gal was active in several compartments of the forming limb bud, including the apical ectodermal ridge (AER), the proximal mesenchyme and the posterior mesenchyme (Fig. 3F). When BAT-Gal was examined in KI embryos, it was slightly diminished throughout the limb, but showed a substantial loss of activity specifically in the posterior mesenchyme at E10.0 (arrows, Fig. 3F′). Immunofluorescent staining for each Tcf/Lef factor in WT E10.0 limb buds showed Tcf3 throughout the mesenchyme, Lef1 and Tcf1 in anterior and posterior mesenchyme, and Tcf4 in central mesenchyme (supplementary material Fig. S2). In KI limb buds, there was a small but reproducible decrease in Lef1 and Tcf1 expression in the central and distal mesenchyme, and Lef1 was reduced in the distal posterior mesenchyme (supplementary material Fig. S2). These observations indicate that the Tcf3–β-catenin interaction has a direct effect on the posterior mesenchyme of the limb bud during early stages of morphogenesis.
The posterior mesenchyme of the limb bud is the location of an important organizer called the zone of polarizing activity (ZPA). The ZPA is necessary for posterior digit identity through Shh signaling and for maintenance of the AER (Laufer et al., 1994; Niswander et al., 1994; Riddle et al., 1993). In addition to Shh, reciprocal signaling between the AER and ZPA involves the activity of other signaling pathways, including Fgf, Bmp and Wnt. Among the important interactions, Fgf and Wnt induce and maintain the expression of Shh (Laufer et al., 1994; Niswander et al., 1994; Parr and McMahon, 1995), whereas Bmp inhibits Shh signaling (Kengaku et al., 1998; Zuniga et al., 1999). Wnt7a has been shown to maintain Shh expression, but this effect was suggested to be independent of Tcf/Lef–β-catenin because it was not inhibited by ΔNLef1 expression in chicken (Kengaku et al., 1998).
We tested the role of Tcf3–β-catenin interaction in regulating the relationship between the signaling pathways during early limb morphogenesis. Shh expression was induced normally in KI limb buds at E9.5 compared with WT limb buds (Fig. 4A,A′), but it did not increase after E10.0 in KI limb buds in contrast to WT limb buds (Fig. 4B-D′). The reduced Shh expression in KI limb buds reduced the activity of the pathway as detected by expression of the Shh target gene patched 1 (Fig. 4E-F′). Reduction of Shh occurred prior to reduction of Fgf8 (Fig. 4G-H′), indicating that reduced Shh activity led to a reduction of Fgf8 in the AER (Fig. 4H′). Expression of gremlin 1, a Bmp antagonist important for maintenance of Shh in the ZPA (Khokha et al., 2003), was decreased in KI compared with WT limb buds (Fig. 4I-J′). To test whether Shh reduction in KI limb buds could be due to increased Bmp signaling, we examined expression of the Bmp target genes Msx1 and Msx2 (Fig. 4K-N′). The decreased levels of Msx1 and Msx2 in KI limb buds indicated that decreased Shh was not caused by increased Bmp activity. Furthermore, phospho-histone H3 and cleaved caspase 3 staining did not reveal a significant difference in proliferation or cell death, respectively, between WT and KI limb buds (supplementary material Fig. S2). Therefore, we suggest that the Tcf3–β-catenin interaction is necessary for the Wnt/β-catenin-mediated maintenance of high levels of Shh in the ZPA. The partially decreased activity in the ZPA leads to secondary effects reducing signaling throughout the limb bud.
Fig. 4.

Maintenance of Shh expression in the zone of polarizing activity requires Tcf3–β-catenin interaction. (A-B′) Whole-mount in situ hybridization using a cRNA probe specific for Shh mRNA in E9.5 (A,A′) and E10.0 (B,B′) mouse embryos. Higher magnification views of forelimbs are shown in C-D′. (C-N′) Whole-mount in situ hybridization of WT (C-N) and KI (C′-N′) limb buds using labeled cRNA probes for Shh (C-D′), patched 1 (E-F′), Fgf8 (G-H′), gremlin 1 (I-J′), Msx1 (K-L′) and Msx2 (M-N′). Two stages of development observed in E10 embryos are shown that correspond to the 30- to 31-somite (C,C′,E,E′,G,G′,I,I′,K,K′,M,M′) and 33- to 34-somite (D,D′,F,F′,H,H′,J,J′,L,L′,N,N′) stages.
Tcf3–β-catenin interaction in the mucocutaneous junction is necessary for eyelid closure
A second highly penetrant and consistent morphogenetic defect was incomplete eyelid closure, which occurred in 54% of KI mutants (Fig. 2A,B). Mice are normally born with fused eyelids, which stay closed until P12-14 (Findlater et al., 1993). Closure occurs between E15 and E16, stages when periderm cells at the tips of eyelids migrate towards the center of the eye (Fig. 5A) (Findlater et al., 1993). The eyelid primordia follow the periderm projections until the upper and lower eyelids meet and fuse to form a conjunctival sac necessary for corneal development (Fig. 5B) (Findlater et al., 1993). Closure requires actin polymerization and activation of Fgf10, Shh, Tgfα and Mekk1/JNK kinases (supplementary material Fig. S3) (Tao et al., 2005; Vassalli et al., 1994; Zenz et al., 2003; Zhang et al., 2003) and does not occur when Tcf3 is overexpressed in the skin epidermis (Merrill et al., 2001). In KI eyelids, the projection of migrating periderm cells was severely diminished at E15.5 (Fig. 5A′) and remained incomplete at E16.5 (Fig. 5B′). Signaling detected by phospho-ERK, phospho-JNK and phospho-c-Jun antibody staining did not appear to be defective in KI eyelids prior to closure (supplementary material Fig. S3). By contrast, whereas filamentous actin was increased in the migrating cells in the WT, it was not increased in the KI periderm cells (supplementary material Fig. S3), indicating that KI defects occurred during or prior to periderm cell migration.
Fig. 5.

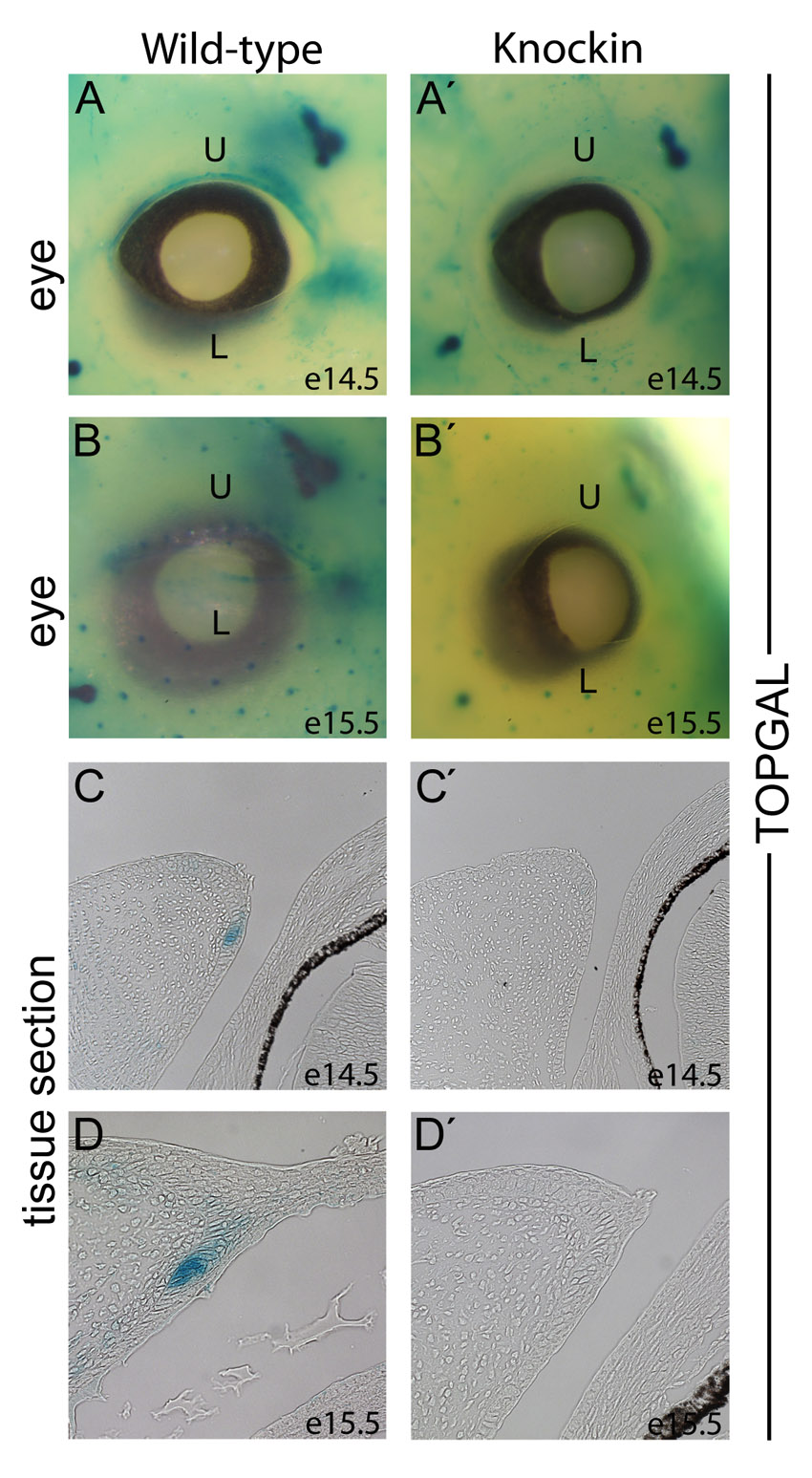
Discrete requirement for Tcf3–β-catenin interaction in the eyelid. (A-B′) H&E staining of the eye and eyelids in WT (A,B) and KI (A′,B′) mouse embryos at E15.5 (A,A′) and E16.5 (B,B′). (C,C′) Tissue sections of E14.5 eyelids from BAT-Gal transgenic mice were stained with X-gal. The mucocutaneous junction (MCJ) stains positively for BAT-Gal activity (dotted line) in WT (C) and is reduced in size in KI (C′) eyelids. Solid lines at the basement membrane denote palpebral epidermis (PE) and palpebral conjunctiva (PC) regions (C). (D,D′) Immunofluorescence staining for β-galactosidase (red) and Ki67 (green) in E14.5 eyelids from BAT-Gal+ WT (D) and BAT-Gal+ KI (D′) embryos. The BAT-Gal activity (bar) is in Ki67-negative cells and its size is reduced in KI eyelids. (E-F″′) Immunofluorescence staining for Lef1 (E,F; red), Tcf3 (E′,F′; green), and β-galactosidase (E″,F″; blue) in E14.5 eyelids from BAT-Gal transgenic WT (E-E″′) and KI (F-F″′) embryos. Arrows point to Lef1-positive nuclei with low levels of Tcf3. Arrowheads point to Tcf3 and Lef1 double-positive nuclei, which were observed in WT (E-E″′) but not KI (F-F″′). The dotted line demarcates the BAT-Gal-positive region of the MCJ.
To identify the cells requiring Tcf3–β-catenin interaction, the activity of Tcf–β-catenin reporter transgenes was examined. In WT eyelids, BAT-Gal and TOPGal activity was present in a band of cells at the forming mucocutaneous junction (MCJ) (Fig. 5C; supplementary material Fig. S4). The MCJ is positioned at the transition between two epithelia: the palpebral conjunctiva on the eyelid surface facing the eye and the palpebral epidermis on the eyelid surface at the exterior of the embryo (Knop et al., 2010; Riau et al., 2008). Intriguingly, based on their slow cycling, cells from the MCJ have been suggested to be the stem cells for conjunctival epithelia in primates and rabbits (Liu et al., 2007; Wirtschafter et al., 1997). The slow cycling of the mouse MCJ cells was made apparent by the lack of immunofluorescent staining for the Ki67 cell proliferation marker in the MCJ patch compared with the highly proliferative cells throughout the growing eyelid (Fig. 5D). Double immunofluorescent staining for β-galactosidase showed that the Ki67-negative MCJ cells were also the BAT-Gal-positive cells in the eyelid epithelium (Fig. 5D). Immunofluorescent staining at E12.5 and E13.5 indicated that the onset of slow cycling coincided with BAT-Gal activation (supplementary material Fig. S5).
In KI eyelids, BAT-Gal activity was detectable in both the mesenchyme and MCJ region; however, the domain of activity in the KI MCJ region was less than half that in the WT (Fig. 5C′). Compared with BAT-Gal, TOPGal displayed weaker activity in WT MCJ, and TOPGal activity was effectively undetectable in KI eyelids at E14.5 and E15.5 (supplementary material Fig. S4). The reduction of Tcf–β-catenin reporter activity in the KI perfectly coincided with the reduced size of the slow-cycling (Ki67-negative) band of cells at the MCJ region (Fig. 5D′). Considering the three-dimensional structure of the band of MCJ cells lining the edge of the eyelid, we suggest that the reduction observed on the KI eyelid is substantial and represents the source of initial defects leading to the open eyelid at birth phenotype observed in KI mice.
The observation that BAT-Gal activity was reduced, but not completely absent, in KI eyelids indicated a potentially interesting effect of Tcf3–β-catenin on target gene activation. Since Tcf3ΔN cannot directly activate Tcf–β-catenin reporters, BAT-Gal activity in KI eyelids indicated the presence of another Tcf/Lef protein. Immunofluorescent staining for each Tcf/Lef factor showed Lef1 in eyelid epithelium from E12.5 to E14.5 (Fig. 5E; supplementary material Fig. S5); Tcf1 was only present in underlying mesenchyme and Tcf4 was not detected (supplementary material Fig. S5). Interestingly, Lef1 (Fig. 5E) and Tcf3 (Fig. 5E′) proteins were expressed in opposing gradients over the typically 10- to 15-cell-wide zone of BAT-Gal activity (Fig. 5E″) at the WT MCJ region (dotted line, Fig. 5E-E″′). Tcf3 was most highly expressed at the epidermal side (Fig. 5E′) and Lef1 was most highly expressed at the conjunctival side of the MCJ (Fig. 5E). BAT-Gal was active in both Tcf3-high (arrowheads, Fig. 5E-E″′) and Tcf3-low (arrows, Fig. 5E-E″′) cells, but only in cells with high levels of Lef1 (Fig. 5E-E″′). Instead of the overlapping, opposing gradients of Tcf3 and Lef1 seen in WT, KI eyelids displayed an abrupt transition from Tcf3-high/Lef1-low (Fig. 5F′) to Tcf3-low/Lef1-high expression (arrows, Fig. 5F). BAT-Gal was active only in Tcf3-low/Lef1-high cells (arrows, Fig. 5F-F″′). Thus, in KI eyelids, the domain of BAT-Gal activity was reduced in size, and this restriction directly corresponded with a restriction of Lef1 expression. We interpret the observation that BAT-Gal was active only in Lef1-expressing cells to suggest that Lef1–β-catenin, and not Tcf3–β-catenin, is necessary for the activation of downstream target genes.
Wnt/β-catenin stimulates Lef1 levels by inhibiting Tcf3 repression
Since Lef1 expression is lost in Tcf3-positive cells in the KI, we examined the possibility that β-catenin inhibits Tcf3 repression of Lef1 transcription. ESCs were used to test this possible relationship because they had been previously found to express high levels of Tcf3 and other Tcf/Lef proteins (Kelly et al., 2011; Pereira et al., 2006). Treating ESCs with recombinant Wnt3a for 24 hours increased Lef1 mRNA levels 2.5-fold (Fig. 6A), demonstrating that they were a good system with which to examine Wnt regulation of Lef1 expression. Chromatin immunoprecipitation followed by quantitative PCR showed that Wnt3a decreased the occupancy of Tcf3 on endogenous target genes in a Tcf3–β-catenin-dependent manner (Fig. 6B,C). As described previously, Tcf3 functioned as a transcriptional repressor in ESCs, whereas Lef1 functioned as a β-catenin-dependent activator of target gene transcription (Fig. 6D) (Yi et al., 2011).
Fig. 6.

Wnt3a stimulates gene expression through regulation of Lef1 levels in ESCs. (A) Quantitative real-time PCR (qPCR) analysis of cDNA made after treating WT ESCs with 50 ng/ml Wnt3a for 12 hours. Values represent fold-changes to the mRNA levels of Tcf3 and Lef1 relative to starting levels. (B,C) Quantitative ChIP-qPCR analysis of Tcf3 binding to target genes Axin2 and Cdx1 in ESC chromatin from WT (B) or KI (C) ESCs treated with control or Wnt3a-conditioned media for 24 hours. Values represent the mean of five biological replicates ± s.d. Neg indicates negative control sites not bound by Tcf3. (D) SuperTOPFlash luciferase reporter assay in ESCs. Tcf3 KO ESCs were transiently transfected with expression plasmids for stable ΔNβ-catenin, Lef1 and/or Tcf3. SuperTOPFlash activity is shown compared with the Renilla luciferase control for each transfection. Values represent the mean of biological triplicates ± s.d. (E) The human LEF1 promoter constructs used for transfections. Numbers refer to base pairs from the LEF1 translation start site. Numbers in parentheses refer to the equivalent sites in the mouse Lef1 gene. (F) Quantitative ChIP-qPCR analysis of Tcf3 binding to different regions of the mouse Lef1 promoter. Chromatin from KO ESCs was used as a negative control. Values represent the mean of three biological replicates ± s.d. (G) Quantitative ChIP-qPCR analysis of β-catenin binding to different regions of the mouse Lef1 promoter. WT ESCs were treated with 50 ng/ml Wnt3a for 24 hours. Values represent the mean of three biological replicates ± s.d. (H,I) Lef1 promoter luciferase reporter plasmids (illustrated in E) were transiently transfected into KO ESCs with a Tcf3 expression plasmid or empty vector control. Cells were treated with Wnt3a-conditioned media or control conditioned media for 24 hours prior to processing for luciferase activity. Values represent the mean of biological triplicates ± s.d. (J,K) Lef1 promoter luciferase reporter constructs were cotransfected with three concentrations of Tcf3 expression constructs and treated for 24 hours with three concentrations of Wnt3a-conditioned media. Values represent the mean of biological triplicates ± s.d. (L) Model illustrating the role of balancing Tcf3 and Lef1 levels in mediating Wnt/β-catenin signaling. Genes are boxed; proteins are in ovals. Wnt/β-catenin-responsive target genes (WRG) are repressed (red line) by Tcf3 and activated (green line) by Lef1–β-catenin. Wnt inhibits Tcf3 repression of WRG and Lef1 gene expression in a β-catenin-dependent manner.
To determine whether Tcf3 directly represses Lef1 transcription, the Lef1 upstream regulatory region was examined. Several potential Tcf/Lef binding sites were noted based on the consensus sequence A/AT/TCAAAG, including a previously identified Wnt responsive element (WRE) (Fig. 6E) (Atcha et al., 2007). Tcf3 occupied positive control sites (Axin2, Cdx1) (Hecht and Stemmler, 2003; Jho et al., 2002) and the –5.3 kb Lef1 site, but not the previously described WRE in ESCs (Fig. 6F).
A DNA fragment consisting of –6713 to –1 bp of human LEF1 was inserted upstream of a luciferase reporter gene to generate a Lef1 reporter plasmid active in ESCs (Fig. 6H) (Rendl et al., 2005). To test the effects of Wnt and Tcf3, the Lef1 reporter was transfected into Tcf3–/– ESCs with and without Wnt3a stimulation and with and without cotransfection with a Tcf3 expression plasmid. Wnt3a stimulated Lef1 reporter activity, whereas Tcf3 repressed activity (Fig. 6H,I). Comparing Lef1 reporter activity in WT and Tcf3 knockout (KO) ESCs indicated that endogenous Tcf3 protein acted only as a repressor and that Tcf3 did not contribute to Wnt3a activation of the Lef1 reporter (supplementary material Fig. S6). Wnt3a stimulated β-catenin occupancy at the –5.3 kb site (Fig. 6G). Removing the Tcf3 binding site at –5.3 kb prevented Wnt3a-mediated activation and Tcf3-mediated repression of the Lef1 reporter (Fig. 6H,I). Further truncation of the Lef1 reporter did not restore significant responsiveness to Wnt3a or Tcf3 (Fig. 6H,I). Tcf3 repression and Wnt3a activation of the Lef1 reporter displayed a competitive concentration dependence (Fig. 6J,K).
Taken together, these data indicate that Tcf3 represses Lef1 and that endogenous Tcf1 and Lef1 mediate Wnt3a stimulation of Lef1 transcription. They support the existence of the circuit illustrated in Fig. 6L.
Transgenic overexpression of Tcf3 represses endogenous Tcf/Lef–β-catenin activity
We hypothesized that Tcf3–β-catenin is not required to activate target genes but instead must alleviate Tcf3 repression of genes, particularly Lef1 (Fig. 6L). Results from previous Tcf3 overexpression experiments in mouse skin ostensibly support this hypothesis (Nguyen et al., 2006); however, it must be noted that the transgenic mouse experiments focused on cells lacking endogenous Tcf/Lef–β-catenin transcriptional activity (Merrill et al., 2001; Nguyen et al., 2006). Thus, it was not known whether Tcf3 would repress or activate transcription when it was transgenically expressed in a cell type that both endogenously expresses Tcf3 and endogenously activates Tcf/Lef–β-catenin targets. Owing to the Tcf3 expression and BAT-Gal activity in the MCJ region, it provided a perfect context in which to test whether Tcf3–β-catenin activates target genes in vivo.
Tcf3 was overexpressed in the MCJ using a bi-transgenic doxycycline-inducible system (Nguyen et al., 2006). The expression of Tcf3 occurred in a mosaic pattern (Fig. 7A), which was useful for distinguishing cell-autonomous from non-cell-autonomous effects. In cells with overexpressed Tcf3, BAT-Gal activity was not detected (Fig. 7A′), indicating that full-length Tcf3 functioned as a transcriptional repressor and not an activator in these cells. Moreover, Tcf3 overexpression in the MCJ repressed Lef1 expression (Fig. 7A″). Both the repression of BAT-Gal and Lef1 occurred in a cell-autonomous manner, as neighboring cells that did not overexpress Tcf3 displayed Lef1 expression and BAT-Gal activity. The mosaic expression of Tcf3 and the mosaic repression of Lef1 in the transgenic mice were sufficient to recapitulate the eyelid closure defect observed in KI mice (Fig. 7C,C′). Given that the MCJ cells activate BAT-Gal in cells without Tcf3 overexpression, these data indicate that Tcf3 only represses target genes. Instead, β-catenin interaction with Tcf3 derepresses Lef1 expression, leading to the formation of Lef1–β-catenin activator complexes (Fig. 6L).
Fig. 7.

Full-length Tcf3 represses Lef1 and BAT-Gal in the MCJ. (A-B″′) Immunofluorescent staining of E14.5 eyelids from Tcf3-overexpressing double-transgenic (A-A″′) and control single-transgenic (B-B″′) mouse embryos. Transgenic Tcf3 (A; green) was expressed in a mosaic pattern in double-transgenic embryos. BAT-Gal activity (A′; blue) and Lef1 expression (A″; red) was low in Tcf3-overexpressing cells (white arrowheads). In neighboring cells that did not overexpress Tcf3 (yellow arrows), BAT-Gal activity and Lef1 expression were similar to levels in the control embryo (B′,B″). (C,C′) The eye region of a control single-transgenic embryo (C) and a Tcf3-overexpressing bi-transgenic littermate (C′) at E16.5.
DISCUSSION
The core components of the canonical Wnt/β-catenin signaling pathway have been evolutionarily conserved from cnidaria to chordates, and the pathway classically functions by β-catenin binding to Tcf/Lef proteins and converting them to transcriptional activators (Brannon et al., 1997; Molenaar et al., 1996; van de Wetering et al., 1997). Although Tcf3–β-catenin interaction has been well characterized (Graham et al., 2001; Graham et al., 2000), fitting Tcf3 into this model has been somewhat problematic. This is primarily because Tcf3 appeared to act as a transcriptional repressor in loss-of-function experiments in diverse organisms (Dorsky et al., 2003; Houston et al., 2002; Kim et al., 2000; Merrill et al., 2004). In addition, overexpression experiments in mice showed that Tcf3 affected cell fates in vivo independently of β-catenin interaction (Merrill et al., 2001; Nguyen et al., 2006). We investigated this paradox by performing a powerful and direct test of genetically ablating β-catenin binding from Tcf3 with a Tcf3ΔN knock-in mutation. The morphogenetic defects in KI embryos and lethality of KI mice provide formal genetic proof that Tcf3–β-catenin interaction is necessary for embryogenesis and viability. To our knowledge, the limb morphogenesis, vascular integrity and eyelid closure defects in the KI mice identify the first locations where Tcf3 functions in animals have been genetically proven to require β-catenin interaction.
Upon examination of these defects in the KI embryos, it became clear that although Tcf3–β-catenin interaction was necessary, the function of Tcf3 did not fit well into the classical model that suggests transcriptional activator activity for Tcf3–β-catenin. One potential explanation for the poor ability of Tcf3 to activate target genes involves a requirement for an unknown cell-specific co-factor that other Tcf/Lef–β-catenin complexes do not require. This possibility is indirectly supported by the differentiation that occurs upon overexpression of Tcf3 in ESCs but not upon overexpression of ΔNLef1, ΔNTcf1 or ΔNTcf4, indicating that Tcf3 possesses a unique biochemical property not shared by other Tcf/Lefs (Kelly et al., 2011; Yi et al., 2011). This would explain why most cells activate Tcf/Lef reporters when co-expressing Lef1 and β-catenin, but very few cell types activate Tcf/Lef reporters when co-expressing Tcf3 and β-catenin (Merrill et al., 2001; Molenaar et al., 1996).
To test whether a putative Tcf3–β-catenin activator activity was required for normal development, we focused on the eyelid MCJ region. We reasoned that the MCJ provided an ideal in vivo test because Tcf3-expressing MCJ cells displayed strong BAT-Gal and TOPGal activity in WT but not KI embryos. As such, MCJ cells should express the complement of factors essential for Tcf3–β-catenin-mediated transactivation. When full-length Tcf3 with a functional β-catenin-binding domain was overexpressed in MCJ cells, it sharply repressed BAT-Gal activity instead of activating it. We interpret this result to suggest that Tcf3 does not directly activate target genes.
As an alternative to the conventional roles ascribed to mammalian Tcf/Lef factors in the canonical Wnt/β-catenin pathway, we present a model in which Tcf3–β-catenin interaction has an important function that is independent of transactivator activity. Wnt3a-stimulated reduction of Tcf3 occupancy on target genes supports this model by providing a mechanism of inhibiting repression. The Tcf/Lef circuit model suggests that Tcf3 represses an overlapping set of Wnt-responsive genes that Lef1–β-catenin activates (Fig. 6L). The DNA-binding HMG domains of Tcf3 and Lef1 are nearly identical and bind the same consensus sequence, supporting their potential to regulate the same genes (Atcha et al., 2007; van Beest et al., 2000; van de Wetering and Clevers, 1992). Moreover, the model includes a switch from Tcf3 repressor to Lef1–β-catenin activator through Wnt/β-catenin release of Tcf3-mediated repression of Lef1 expression. Our data indicate that Wnt3a stimulates Lef1 expression directly through Tcf/Lef binding sites upstream of its transcription start site.
Several observations support a broad usage of the Tcf/Lef circuit model in embryos. Interestingly, the Lef1 WRE (at –5.3 kb) that we identified in ESCs is the third WRE shown to regulate Lef1 expression in cells. Previously, a Lef1 WRE (at –0.9 kb) was shown to be responsive to Wnt/β-catenin signaling in Jurkat T lymphocytes (Hovanes et al., 2001), and another Lef1 WRE (at –0.8 kb) was shown to be responsive in HEK293 cells (Filali et al., 2002). Like Lef1, Tcf1 gene expression is stimulated directly by Wnt/β-catenin activity (Roose et al., 1999); Tcf1 acts similarly to Lef1 as a transactivator in Tcf3-expressing cells (Yi et al., 2011). In colorectal cancer, Tcf/Lef–β-catenin appears to stimulate the expression of endogenous ΔNTcf1 and full-length Lef1 isoforms, which provide feedback on Tcf/Lef–β-catenin activity (Hovanes et al., 2001; Roose et al., 1999; Waterman, 2004). In limb buds, the expression of Tcf1 and Lef1 was affected in KI mutants (supplementary material Fig. S2). In ESCs, endogenous Tcf1 effectively opposed Tcf3 repressor activity in Wnt3a-treated ESCs in terms of self-renewal and target gene expression (Yi et al., 2011). Although Tcf1 is not included in the model shown in Fig. 6L, it is likely to function similarly to Lef1 in most contexts. Thus, we note that previous work supports a high level of regulation of Tcf/Lef gene product expression by multiple WREs, and we suggest that the Tcf3-Lef1 relationship elucidated here provides a model for additional Tcf/Lef circuits robustly integrated into a single network.
It is also notable that the morphogenetic defects in KI mice bore significant similarity to those of other mutations that cause a decrease, but not complete loss, of Wnt/β-catenin signaling activity. In particular, the constellation of defects in KI mice and their variable penetrance are reminiscent of some effects caused by Lrp5 and Lrp6 mutations. By acting redundantly as co-receptors for Wnt ligands, either Lrp5 or Lrp6 is necessary for activation of Wnt/β-catenin signaling, and their effects are sensitive to gene dosage (Tamai et al., 2000). Since Lrp5 and Lrp6 are each ubiquitously expressed, some allelic combinations of Lrp5 and Lrp6 null mutations cause widespread, but partial, reduction of Wnt/β-catenin activity in embryos (Kelly et al., 2004; Pinson et al., 2000). Interestingly, a relatively weak combination of alleles, Lrp5+/+ Lrp6+/–, causes tail kinks that we also observed, but did not count, in KI mutants (C.I.W., unpublished observation) (Pinson et al., 2000). A slightly stronger combination, Lrp5+/– Lrp6+/–, causes an oligodactyly phenotype essentially identical to that observed in KI mutants (Kelly et al., 2004). Finally, Lrp6+/– and Lrp6–/– embryos each display exencephaly similar to that in KI embryos (Gray et al., 2010; Pinson et al., 2000). Only the combinations of Lrp5/6 alleles that cause partial reduction of Wnt/β-catenin activity resemble KI phenotypes, and more substantial loss of Wnt/β-catenin activity causes more severe defects that were not observed in KI embryos. We suggest that this similarity supports a role for Tcf3–β-catenin in attenuating the output of signaling through the Tcf/Lef circuit, as opposed to a role as a direct activator of target gene expression.
Supplementary Material
Acknowledgments
We thank Michael Rendl for the human LEF1 promoter construct; Linda Degenstein for blastocyst injections; Laura Pereira for transient transfection experiments; Susan Ball-Kell for pathology of mutant mice; and Colin Jamora, Bill Lowry and Ramanuj DasGupta for comments and discussions regarding the preparation of the manuscript.
Footnotes
Funding
This work was funded by grants from the American Cancer Society [RSG GGC 112994 to B.J.M.]; the National Institutes of Health [R01-CA128571 to B.J.M., R01-AR059122 to H.N., R01-AR278883 to E.F.]; and the Howard Hughes Medical Institute (E.F.). Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
C.I.W., J.A.H., B.R.S., E.M.F. and B.J.M. performed experiments; C.I.W. and B.J.M. wrote the manuscript; B.J.M. supervised the project; and all authors analyzed experiments and commented on the manuscript.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.076067/-/DC1
References
- Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. (1997). beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S. J., Stappert J., Bauer A., Kispert A., Herrmann B. G., Kemler R. (2000). Brachyury is a target gene of the Wnt/beta-catenin signaling pathway. Mech. Dev. 91, 249–258 [DOI] [PubMed] [Google Scholar]
- Atcha F. A., Syed A., Wu B., Hoverter N. P., Yokoyama N. N., Ting J. H., Munguia J. E., Mangalam H. J., Marsh J. L., Waterman M. L. (2007). A unique DNA binding domain converts T-cell factors into strong Wnt effectors. Mol. Cell. Biol. 27, 8352–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N., Hurlstone A., Musisi H., Miles A., Bienz M., Clevers H. (2001). The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 20, 4935–4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J., von Kries J. P., Kuhl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W. (1996). Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638–642 [DOI] [PubMed] [Google Scholar]
- Behrens J., Jerchow B. A., Wurtele M., Grimm J., Asbrand C., Wirtz R., Kuhl M., Wedlich D., Birchmeier W. (1998). Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 280, 596–599 [DOI] [PubMed] [Google Scholar]
- Ben-Porath I., Thomson M. W., Carey V. J., Ge R., Bell G. W., Regev A., Weinberg R. A. (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannon M., Gomperts M., Sumoy L., Moon R. T., Kimelman D. (1997). A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 11, 2359–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantjes H., Roose J., van De Wetering M., Clevers H. (2001). All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res. 29, 1410–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R., Fuchs E. (1999). Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 126, 4557–4568 [DOI] [PubMed] [Google Scholar]
- Dorsky R. I., Itoh M., Moon R. T., Chitnis A. (2003). Two tcf3 genes cooperate to pattern the zebrafish brain. Development 130, 1937–1947 [DOI] [PubMed] [Google Scholar]
- Filali M., Cheng N., Abbott D., Leontiev V., Engelhardt J. F. (2002). Wnt-3A/beta-catenin signaling induces transcription from the LEF-1 promoter. J. Biol. Chem. 277, 33398–33410 [DOI] [PubMed] [Google Scholar]
- Findlater G. S., McDougall R. D., Kaufman M. H. (1993). Eyelid development, fusion and subsequent reopening in the mouse. J. Anat. 183, 121–129 [PMC free article] [PubMed] [Google Scholar]
- Galceran J., Farinas I., Depew M. J., Clevers H., Grosschedl R. (1999). Wnt3a–/–-like phenotype and limb deficiency in Lef1–/–Tcf1–/– mice. Genes Dev. 13, 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J., Hsu S. C., Grosschedl R. (2001). Rescue of a Wnt mutation by an activated form of LEF-1: regulation of maintenance but not initiation of Brachyury expression. Proc. Natl. Acad. Sci. USA 98, 8668–8673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T. A., Weaver C., Mao F., Kimelman D., Xu W. (2000). Crystal structure of a beta-catenin/Tcf complex. Cell 103, 885–896 [DOI] [PubMed] [Google Scholar]
- Graham T. A., Ferkey D. M., Mao F., Kimelman D., Xu W. (2001). Tcf4 can specifically recognize beta-catenin using alternative conformations. Nat. Struct. Biol. 8, 1048–1052 [DOI] [PubMed] [Google Scholar]
- Gray J. D., Nakouzi G., Slowinska-Castaldo B., Dazard J. E., Rao J. S., Nadeau J. H., Ross M. E. (2010). Functional interactions between the LRP6 WNT co-receptor and folate supplementation. Hum. Mol. Genet. 19, 4560–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A., Stemmler M. P. (2003). Identification of a promoter-specific transcriptional activation domain at the C terminus of the Wnt effector protein T-cell factor 4. J. Biol. Chem. 278, 3776–3785 [DOI] [PubMed] [Google Scholar]
- Hecht A., Vleminckx K., Stemmler M. P., van Roy F., Kemler R. (2000). The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19, 1839–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston D. W., Kofron M., Resnik E., Langland R., Destree O., Wylie C., Heasman J. (2002). Repression of organizer genes in dorsal and ventral Xenopus cells mediated by maternal XTcf3. Development 129, 4015–4025 [DOI] [PubMed] [Google Scholar]
- Hovanes K., Li T. W., Munguia J. E., Truong T., Milovanovic T., Lawrence Marsh J., Holcombe R. F., Waterman M. L. (2001). Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28, 53–57 [DOI] [PubMed] [Google Scholar]
- Huelsken J., Vogel R., Brinkmann V., Erdmann B., Birchmeier C., Birchmeier W. (2000). Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 148, 567–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S., Kishida S., Yamamoto H., Murai H., Koyama S., Kikuchi A. (1998). Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T. O., Tamai Y., Li Q., Oshima M., Taketo M. M. (2003). Requirement for tumor suppressor Apc in the morphogenesis of anterior and ventral mouse embryo. Dev. Biol. 253, 230–246 [DOI] [PubMed] [Google Scholar]
- Ivanova N. B., Dimos J. T., Schaniel C., Hackney J. A., Moore K. A., Lemischka I. R. (2002). A stem cell molecular signature. Science 298, 601–604 [DOI] [PubMed] [Google Scholar]
- Jho E. H., Zhang T., Domon C., Joo C. K., Freund J. N., Costantini F. (2002). Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K. F., Ng D. Y., Jayakumaran G., Wood G. A., Koide H., Doble B. W. (2011). beta-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 8, 214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly O. G., Pinson K. I., Skarnes W. C. (2004). The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development 131, 2803–2815 [DOI] [PubMed] [Google Scholar]
- Kemler R., Hierholzer A., Kanzler B., Kuppig S., Hansen K., Taketo M. M., de Vries W. N., Knowles B. B., Solter D. (2004). Stabilization of beta-catenin in the mouse zygote leads to premature epithelial-mesenchymal transition in the epiblast. Development 131, 5817–5824 [DOI] [PubMed] [Google Scholar]
- Kengaku M., Capdevila J., Rodriguez-Esteban C., De La Pena J., Johnson R. L., Izpisua Belmonte J. C., Tabin C. J. (1998). Distinct WNT pathways regulating AER formation and dorsoventral polarity in the chick limb bud. Science 280, 1274–1277 [DOI] [PubMed] [Google Scholar]
- Khokha M. K., Hsu D., Brunet L. J., Dionne M. S., Harland R. M. (2003). Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat. Genet. 34, 303–307 [DOI] [PubMed] [Google Scholar]
- Kim C. H., Oda T., Itoh M., Jiang D., Artinger K. B., Chandrasekharappa S. C., Driever W., Chitnis A. B. (2000). Repressor activity of Headless/Tcf3 is essential for vertebrate head formation. Nature 407, 913–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop E., Korb D. R., Blackie C. A., Knop N. (2010). The lid margin is an underestimated structure for preservation of ocular surface health and development of dry eye disease. Dev. Ophthalmol. 45, 108–122 [DOI] [PubMed] [Google Scholar]
- Kramps T., Peter O., Brunner E., Nellen D., Froesch B., Chatterjee S., Murone M., Zullig S., Basler K. (2002). Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 109, 47–60 [DOI] [PubMed] [Google Scholar]
- Laufer E., Nelson C. E., Johnson R. L., Morgan B. A., Tabin C. (1994). Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell 79, 993–1003 [DOI] [PubMed] [Google Scholar]
- Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., He X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- Liu P., Wakamiya M., Shea M. J., Albrecht U., Behringer R. R., Bradley A. (1999). Requirement for Wnt3 in vertebrate axis formation. Nat. Genet. 22, 361–365 [DOI] [PubMed] [Google Scholar]
- Liu S., Li J., Tan D. T., Beuerman R. W. (2007). The eyelid margin: a transitional zone for 2 epithelial phenotypes. Arch. Ophthalmol. 125, 523–532 [DOI] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K., He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S., Cordenonsi M., Dupont S., Braghetta P., Broccoli V., Hassan A. B., Volpin D., Bressan G. M., Piccolo S. (2003). Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 100, 3299–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill B. J., Gat U., DasGupta R., Fuchs E. (2001). Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 15, 1688–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill B. J., Pasolli H. A., Polak L., Rendl M., Garcia-Garcia M. J., Anderson K. V., Fuchs E. (2004). Tcf3: a transcriptional regulator of axis induction in the early embryo. Development 131, 263–274 [DOI] [PubMed] [Google Scholar]
- Molenaar M., van de Wetering M., Oosterwegel M., Peterson-Maduro J., Godsave S., Korinek V., Roose J., Destree O., Clevers H. (1996). XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399 [DOI] [PubMed] [Google Scholar]
- Moon R. T., Bowerman B., Boutros M., Perrimon N. (2002). The promise and perils of Wnt signaling through beta-catenin. Science 296, 1644–1646 [DOI] [PubMed] [Google Scholar]
- Nguyen H., Rendl M., Fuchs E. (2006). Tcf3 governs stem cell features and represses cell fate determination in skin. Cell 127, 171–183 [DOI] [PubMed] [Google Scholar]
- Niswander L., Jeffrey S., Martin G. R., Tickle C. (1994). A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature 371, 609–612 [DOI] [PubMed] [Google Scholar]
- Parr B. A., McMahon A. P. (1995). Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature 374, 350–353 [DOI] [PubMed] [Google Scholar]
- Pereira L., Yi F., Merrill B. J. (2006). Repression of nanog gene transcription by tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol. 26, 7479–7491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinson K. I., Brennan J., Monkley S., Avery B. J., Skarnes W. C. (2000). An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 407, 535–538 [DOI] [PubMed] [Google Scholar]
- Popperl H., Schmidt C., Wilson V., Hume C. R., Dodd J., Krumlauf R., Beddington R. S. (1997). Misexpression of Cwnt8C in the mouse induces an ectopic embryonic axis and causes a truncation of the anterior neuroectoderm. Development 124, 2997–3005 [DOI] [PubMed] [Google Scholar]
- Rendl M., Lewis L., Fuchs E. (2005). Molecular dissection of mesenchymal-epithelial interactions in the hair follicle. PLoS Biol. 3, e331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riau A. K., Barathi V. A., Beuerman R. W. (2008). Mucocutaneous junction of eyelid and lip: a study of the transition zone using epithelial cell markers. Curr. Eye Res. 33, 912–922 [DOI] [PubMed] [Google Scholar]
- Riddle R. D., Johnson R. L., Laufer E., Tabin C. (1993). Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416 [DOI] [PubMed] [Google Scholar]
- Roose J., Huls G., van Beest M., Moerer P., van der Horn K., Goldschmeding R., Logtenberg T., Clevers H. (1999). Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 285, 1923–1926 [DOI] [PubMed] [Google Scholar]
- Sun Y., Kolligs F. T., Hottiger M. O., Mosavin R., Fearon E. R., Nabel G. J. (2000). Regulation of beta-catenin transformation by the p300 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 97, 12613–12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru K. I., Moon R. T. (2000). The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 149, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K., Semenov M., Kato Y., Spokony R., Liu C., Katsuyama Y., Hess F., Saint-Jeannet J. P., He X. (2000). LDL-receptor-related proteins in Wnt signal transduction. Nature 407, 530–535 [DOI] [PubMed] [Google Scholar]
- Tao H., Shimizu M., Kusumoto R., Ono K., Noji S., Ohuchi H. (2005). A dual role of FGF10 in proliferation and coordinated migration of epithelial leading edge cells during mouse eyelid development. Development 132, 3217–3230 [DOI] [PubMed] [Google Scholar]
- van Beest M., Dooijes D., van De Wetering M., Kjaerulff S., Bonvin A., Nielsen O., Clevers H. (2000). Sequence-specific high mobility group box factors recognize 10-12-base pair minor groove motifs. J. Biol. Chem. 275, 27266–27273 [DOI] [PubMed] [Google Scholar]
- van de Wetering M., Clevers H. (1992). Sequence-specific interaction of the HMG box proteins TCF-1 and SRY occurs within the minor groove of a Watson-Crick double helix. EMBO J. 11, 3039–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M., Cavallo R., Dooijes D., van Beest M., van Es J., Loureiro J., Ypma A., Hursh D., Jones T., Bejsovec A., et al. (1997). Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88, 789–799 [DOI] [PubMed] [Google Scholar]
- van Genderen C., Okamura R. M., Farinas I., Quo R. G., Parslow T. G., Bruhn L., Grosschedl R. (1994). Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 8, 2691–2703 [DOI] [PubMed] [Google Scholar]
- Vassalli A., Matzuk M. M., Gardner H. A., Lee K. F., Jaenisch R. (1994). Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 8, 414–427 [DOI] [PubMed] [Google Scholar]
- Waterman M. L. (2004). Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis. Rev. 23, 41–52 [DOI] [PubMed] [Google Scholar]
- Wirtschafter J. D., McLoon L. K., Ketcham J. M., Weinstock R. J., Cheung J. C. (1997). Palpebral conjunctival transient amplifying cells originate at the mucocutaneous junction and their progeny migrate toward the fornix. Trans. Am. Ophthalmol. Soc. 95, 417–432 [PMC free article] [PubMed] [Google Scholar]
- Yi F., Merrill B. J. (2007). Stem cells and TCF proteins: a role for beta-catenin-independent functions. Stem Cell Rev. 3, 39–48 [DOI] [PubMed] [Google Scholar]
- Yi F., Pereira L., Hoffman J. A., Shy B. R., Yuen C. M., Liu D. R., Merrill B. J. (2011). Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 13, 762–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L., Fagotto F., Zhang T., Hsu W., Vasicek T. J., Perry W. L., 3rd, Lee J. J., Tilghman S. M., Gumbiner B. M., Costantini F. (1997). The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90, 181–192 [DOI] [PubMed] [Google Scholar]
- Zenz R., Scheuch H., Martin P., Frank C., Eferl R., Kenner L., Sibilia M., Wagner E. F. (2003). c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev. Cell 4, 879–889 [DOI] [PubMed] [Google Scholar]
- Zhang L., Wang W., Hayashi Y., Jester J. V., Birk D. E., Gao M., Liu C. Y., Kao W. W., Karin M., Xia Y. (2003). A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 22, 4443–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Byrne C., Jacobs J., Fuchs E. (1995). Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev. 9, 700–713 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}