

Abstract
Recent work in our laboratory has shown that the highly substituted, electronically rich 1,2,3,4–tetrahydroisoquinoline–3–carboxylic acid (THIQ3CA) scaffold is a key building block for a novel class of promising anticoagulants (Al-Horani et al. J. Med. Chem. 2011, 54, 6125–6138). The synthesis of THIQ3CA analogs, especially containing specific, electronically rich substituents, has been a challenge and essentially no efficient methods have been reported in the literature. We describe three complementary, glycine donor-based strategies for high yielding synthesis of highly substituted, electronically rich THIQ3CA esters. Three glycine donors studied herein include hydantoin 1, (±)-Boc-α-phosphonoglycine trimethyl ester 2 and (±)-Z-α-phosphonoglycine trimethyl ester 3. Although the synthesis of THIQ3CA analogs could be achieved using either of the three, an optimal, high yielding approach for the desired THIQ3CA esters was best achieved using 3 in three mild, efficient steps. Using this approach, a focused library of advanced N-arylacyl, N-arylalkyl, and bis-THIQ3CA analogs was synthesized. Variable temperature and solvent-dependent NMR chemical shift studies indicated the presence of two major conformational rotamers in 3:1 proportion for N–arylacyl–THIQ3CA analogs, which were separated by a high kinetic barrier of ~17 kcal/mol. In contrast, N–arylalkyl and bis–THIQ3CA variants displayed no rotamerism, which implicates restricted rotation around the amide bond as the origin for high-barrier conformational interconversion. This phenomenon is of major significance because structure-based drug design typically utilizes only one conformation. Overall, the work presents fundamental studies on the synthesis and conformational properties of highly substituted, electronically rich THIQ3CA analogs.
Introduction
Nitrogen heterocycles are key building blocks for a large number of medicinally-relevant molecules and serve as fundamental scaffolds for discovering drugs. The 1,2,3,4–tetrahydroisoquinoline (THIQ) scaffold is an especially useful nitrogen heterocycle in such efforts for its ability to quickly enhance structural diversity of a potential library. Recent examples highlight the exploitation of several THIQ analogs as direct inhibitors of factor Xa (fXa), a key serine protease of the blood coagulation cascade.1–3 An interesting variant of the THIQ scaffold is the 1,2,3,4–tetrahydroisoquinoline–3–carboxylic acid (THIQ3CA) structure, which has been utilized in the design of several pharmacologically relevant molecules.4–7 Our recent work has exploited the THIQ3CA scaffold in the design of new activators of antithrombin (AT), a plasma glycoprotein and a key regulator of clotting,8 for accelerated inhibition of fXa.9,10
Several methods are available for synthesis of the THIQ3CA analogs.11–19 The Pictet-Spengler11 and Bischler–Napieralski12 reactions, which rely on cyclization of β-arylethylamine derivatives in presence of a protic or Lewis acid,20–31 are among most common protocols for this purpose. Several other methods have also been developed including cycloaddition and enyne metathesis reactions for the synthesis of 1,2–dihydroisoquinoline–3–carboxylic acids13 and 5,6–, 6,7–, or 7,8–functionalized THIQ3CA esters.14,15
An interesting method to synthesize THIQ3CA analogs has been to utilize α,α′–dibromo-O-xylenes as dielectrophiles in reaction with a glycine equivalent. The glycine equivalents studied to date include (2S)–2–isopropyl–3,6–dimethoxy–5–methyl–2,5–dihydropyrazine,32 (R)–di–tert–butyl 2–tert–butyl–4–oxoimidazolidine–1,3–dicarboxylate,33 diethyl acetamidomalonate,34 ethyl acetamidocyano–acetate,35 ethyl 2–(benzylidene amino)acetate,36 (6R)–6–isopropyl–3–methyl–5–phenyl–3,6–dihydro–2H–1,4–oxazin–2–one,37 and 1,4–diacetyl piperazine-2,5-dione.38 An alternative to using dibromo-O-xylenes as electrophiles in the reaction with glycine donors is α-methyl benzyl bromide, which when reacted with (2S,5S)–1–benzoyl–2-tert–butyl-3,5-dimethyl–imidazolidin–4–one afforded α,β–dimethyl THIQ3CA after Pictet-Spengler cyclization.39 At a fundamental level, the glycine equivalents in these reactions undergo base-mediated, simultaneous or sequential C- and N- alkylation to afford racemic or enantiomerically pure products. If alkylation is sequential, then N–alkylation can be achieved by acid–mediated hydrolytic cyclization.32,37
We have been interested in developing a THIQ3CA-based library to gain insight into the mechanism of AT activation by nonsaccharide mimetics of heparin, a highly sulfated polysaccharide.10 The unique highly sulfated nature of the parent biomolecule dictates that the THIQ3CA scaffold contain multiple hydroxyl for sulfation in the last step.40 Unfortunately, appropriate polyphenolic THIQ3CA precursors are either not available commercially or their purported synthesis based on literature reports fails to yield product in high yields and purity. More specifically, the starting materials (dimethoxy–β–arylethylamines) for the synthesis of 5,6–dimethoxy–, 5,8–dimethoxy–, and 6,7–dimethoxy–THIQ3CA esters using Pictet-Spengler and Bischler–Napieralski reactions are not available. The glycine equivalents approach also is not feasible for our targeted THIQ3CA derivatives because of the lack of availability of appropriate α,α′–dibromo-O–xylenes. In fact, a survey of literature shows that each THIQ3CA derivative synthesized to date through the glycine equivalent approach has not carried any functionalization on the aromatic ring of THIQ3CA.32,33,37,39 An added problem has been that these reactions require extended reflux in strong bases35,36,39 and/or concentrated mineral acids.32,33,35,37,39 Such strong conditions severely limit the nature of protecting groups that can be used in the synthesis of polyphenolic THIQ3CAs. Finally although useful, the cyclo-addition approaches have been developed primarily for unsubstituted or minimally substituted THIA3CA esters and are long winding.13–15 Thus, we realized that developing a facile, concise, high yielding synthesis to THIQ3CA analogs containing phenolic groups placed at varying positions was necessary for exploring a library of such molecules as anticoagulants.
In this work, we describe multiple strategies that yield THIQ3CAs containing multiple electron rich methoxy substituents. The work leads to a major conclusion that highly substituted, electron rich THIQ3CA analogs can be synthesized in three, mild, and high yielding steps based on Horner-Wadsworth-Emmons and Pictet-Spengler reactions. Interestingly, NMR studies indicate that the N–arylacyl–THIQ3CA derivatives display two equi-energetic, conformational isomers that are separated by a high kinetic barrier of ~17 kcal/mol. This phenomenon is of major significance for structure-based drug design considering that such efforts typically utilize only one of the rotamers.
Results and Discussion
Glycine Donor based Approach to Synthesize Electronically Rich THIQ3CA Esters
A quick retro-synthetic analysis of the THIQ3CA core suggested that the structure could be built using three building blocks – a benzaldehyde, a formaldehyde, and a glycine equivalent moiety (1–3) (Figure 1). While the benzaldehyde block is available with many different electronically rich substituents, which provides a facile entry to the desired THIQ3CA esters, the glycine block has stringent requirements. One of the requirements is that the glycine unit should possess a more nucleophilic α–carbon than its amine and carboxylic acid groups. This is satisfied by imidazolidine-2,4-dione 1, commonly refered to as hydantoin (Scheme 1). To test its applicability, we utilized 2,3–dimethoxy and 2,5–dimethoxy benzaldehydes (4 and 5) that should yield 5,6–dimethoxy– and 5,8–dimethoxy–THIQ3CA derivatives, which are not readily accessible through other competing approaches and form key scaffold of our novel anticoagulants.10
Figure 1.

Retrosysnthetic analysis for the synthesis of THIQ3CA scaffold using glycine equivalents.
Scheme 1.

Synthesis of 5,6- and 5,8- dimethoxy THIQ3CA esters using 1 and 2 as glycine equivalents.
Reaction of benzaldehyde 4 with 1 at 170 °C gave intermediate 6, which was hydrogenated to imidazolidine-2,4-dione 8 (Scheme 1). The hydantoin moiety of 8 was hydrolyzed and the resulting crude acid was esterified through sequential treatments with SOCl2 and ethanol to afford 2,3-dimethoxy phenylalanine ethyl ester 10. Pictet–Spengler cyclization of 10 under acidic conditions with aqueous formaldehyde resulted in 5,6-dimethoxy-THIQ3CA ethyl ester 12. 5,8-Dimethoxy derivative 13 was also synthesized using the same protocol.
Although useful, the above protocol was not as high yielding and efficient as desired. Two other glycine equivalents, namely (±)-Boc-α-phosphonoglycine trimethyl ester 2 (Scheme 1) and (±)-Z-α-phosphonoglycine trimethyl ester 3 (Scheme 2), were studied to improve yield and enhance feasibility. It is known that the α-proton of trimethylphosphonate is considerably acidic and formation of a strong O-P bond in the by-product dimethyl phosphate drives the addition-elimination reaction.41 Thus, we reasoned that glycine donors 2 and 3 may allow the use of milder conditions to effect similar transformations than those used for hydantion 1. A further advantage is likely to arise from the use of 2 and 3 was easier purification of products because the by-product was known to be water soluble. In contrast, the hydantoin adduct required hot methanol for solubilization. The reaction was also expected to yield a predominantly Z-product42 and thus, these glycine donors offered an excellent opportunity for the synthesis of THIQ3CA esters.43
Scheme 2.

Synthesis of 5,6-, 5,8-, and 6,7- dimethoxy THIQ3CA esters using (±)-Z-α-Phosphonoglycine trimethyl ester 3, as glycine equivalent.
Trimethyl phosphonate 2 was first treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at 0 °C in THF and then with either benzalaldehyde 4 or 5 in a dropwise manner to yield the corresponding benzylidene 14 or 15, respectively, in yields of 77–85%. Catalytic hydrogenation and acidification with equi-volume mixture of TFA and CH2Cl2 gave the α-amino-β-aryl methyl acrylate 18 or 19 in 80–86 % yield. Lastly, Pictet–Spengler cyclization using a milder condition (37% HCHO/TFA in CH2Cl2 at RT) afforded the corresponding THIQ3CA methyl esters 20 and 21 within 5 h and significantly better yields of 70–80%.
Further Improvement in the Glycine Donor Approach
Analysis of the approach described in Scheme 1 led to the realization that a significant improvement in the overall efficiency of the process would be realized if alkene reduction and deprotection steps can be achieved in one step. Thus, benzyloxycarbonyl (Cbz) derivatized α–phosphonoglycine trimethyl ester 3 was treated with either benzaldehyde 4, 5, or 22 to yield benzylidenes 23–25 in 77–85% yield (Scheme 2). The planned simultaneous reduction and deprotection strategy was employed on benzylidene 23 using catalytic hydrogenation (50 psi H2 (g) and 10% Pd/C in ethanol), but conditions gave poor yield of the desired α-amino-β-aryl amino methyl ester 18 (~25%). 1H NMR and mass spectrometry suggested the formation of N–ethoxycarbonyl amino ester as a major side product (see Experimental Section), which appears to have arisen from a benzyloxy–ethoxy exchange.44 To minimize this side reaction, 10% Pd(OH)2 on charcoal in equivolume mixture of CH3OH and t-butanol was studied. This modified condition yielded the desired α-amino-β-aryl amino methyl esters 18, 19, and 26 in good yields (73–76%, Scheme 2). Pictet–Spengler cyclization with formaldehyde under mild conditions afforded the corresponding THIQ3CA methyl esters 20, 21, and 27 in high yields.
Overall, using hydantoin 1 as glycine donor led to two THIQ3CA ethyl esters 12 and 13 in six steps, while using phosphonoglycine trimethyl ester 2 as a glycine donor gave two di-activated THIQ3CA methyl esters 20 and 21 in four steps. An alternative strategy involved glycine donor 3, which utilized three mild and efficient steps for the synthesis of THIQ3CA methyl esters 20, 21, and 27. The hydantoin-based approach afforded THIQ3CA ethyl esters in 21–30% overall yields, while the phosphonoglycine trimethyl esters 2 and 3 led to yields of 43–58% and 40–52%, respectively. Finally, the Horner-Wadsworth-Emmons/Pictet-Spengler approach for synthesis of activated THIQ3CA esters is mild, concise, and high yielding, and also appears to be regioselective.
Synthesis of Advanced N-Arylacyl, N-Arylalkyl, and Bis-THIQ3CA Analogs
The synthetic protocol developed above (Scheme 2) appeared very attractive for preparing a library of electronically rich THIQ3CA derivatives. In particular, (±)-Z-α-phosphonoglycine trimethyl ester 3 was especially useful as several novel THIQ3CA–based compounds could be synthesized in near quantitative yields. These include the N-arylacyl THIQ3CA esters 30–35 and 38–42; the N-arylalkyl THIQ3CA ester 43 and the bis-THIQ3CA esters 44 and 45 (Scheme 3).
Scheme 3.

Synthesis of N-substituted THIQ3CA-based library.
The N–benzoyl and N–acetyl THIQ3CA esters 30–35 were synthesized by direct N-acylation using benzoyl or phenyl acetyl chloride in the presence of triethylamine. The N–propanoyl and N–butanoyl THIQ3CA esters 38–42 were prepared using EDCI–mediated coupling reactions with appropriate propioinc or butanoic acids in presence of DMAP at RT.
The synthesis of N-alkylated derivatives was somewhat more challenging than that described above for N-acylated derivatives. Several bases (K2CO3, Cs2CO3, Et3N, and DIPEA), solvent systems (CH3CN, DMF and their mixtures), and heating methods (conventional refluxing and microwave) were screened to identify high yielding conditions for N-p-methoxybenzyl THIQ3CA methyl ester 43. A 92% yield of 43 was obtained following microwave-assisted heating of 1 equivalent of THIQ3CA methyl ester 27, 2.5 equivalents of DIPEA, 1.1 equivalents of p-methoxy benzyl chloride in CH3CN at 150 °C for 30 min. Combining these optimized conditions with search for best xylene-based electrophile (α,α′-dichloro or dibromo- m- and p-xylenes) led to a high yielding synthesis of bis-THIQ3CA derivatives 44 and 45. The best dimerization yield (62–66%) was obtained by refluxing two equivalents of THIQ3CA 28 with one equivalent of a xylene derivative in presence of 4 equivalents of Cs2CO3 in equivolume mixture of DMF and CH3CN.
Overall, a small library of 14 THIQ3CA esters was synthesized using a combination of Horner-Wadsworth-Emmons/Pictet–Spengler reactions (Schemes 2 and 3) in near quantitative yields. These derivatives vary at different structural levels including the substitution pattern of THIQ3CA (5,6-; 5,8-; or 6,7-disubstituted moiety); the type of ester at position-2 (methyl or ethyl ester); the length of the linker (benzyl, benzoyl, acetyl, propanoyl, or butanoyl); and the type of the linker (amide as in N-arylacyl THIQ3CA or amine as in N-arylalkyl THIQ3CA); and lastly the number and pattern of substituents on the aryl moiety (4-substituted; 2,5-disubstituted; or 3,4,5-trisubstituted moieties). The library of electronically rich THIQ3CA ester analogs, and their chemically modified forms including deprotected poly-phenolic or poly-sulfated derivatives, is likely to be useful in mapping protein binding site(s) so as to deduce optimal binding and/or selectivity properties.
From the perspective of chemical synthesis, several points are noteworthy regarding synthesis using (±)–Z–α–phosphonoglycine trimethyl ester 3. First, this approach can be further exploited in synthesis of many unnatural α-amino acids and their corresponding cyclic rigid forms.45 Second, the Pictet–Spengler reaction is highly amenable to microwave conditions, which greatly expedites the construction of a library. Third, it is possible to control chirality at the 3 position to afford enantiomerically pure THIQ3CA using rhodium (I) or ruthenium (II)–catalyzed asymmetric hydrogenation.46,47 These advantages are expected to further enhance the applicability of the THIQ3CA library for drug discovery purposes.
NMR Studies on N-Arylacyl THIQ3CA esters
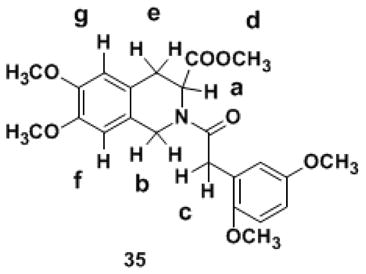
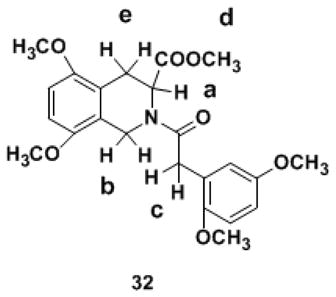
A 1H NMR spectrum is thought of as a classic reflection of the purity of a new compound. The N–arylacyl THIQ3CA esters synthesized in this work were highly pure entities as indicated by RP-HPLC and uPLC-MS analysis, however, each molecule demonstrated complex 1H NMR and 13C NMR spectra (see Experimental Methods), which could not be analyzed on the basis of a single conformational entity composition. We reasoned that the complexity of NMR spectra arises from the –N–C(O)– bond at position–2, which was likely to exhibit partial double-bond character with cis- and trans-isomers. It was likely that the energy barrier for rotation around the amide bond was large resulting in geometrically and magnetically nonequivalent N-substituents. This phenomenon of rotational isomerism has been observed earlier for non-carboxylated THIQ derivatives.48–52 Considering the medicinal importance of the carboxylated THIQ derivatives being studied, we decided to investigate this phenomenon in detail so as to aid development of appropriate computational protocols for the interaction of THIQ3CA with proteins. Derivatives 32 and 35 were chosen as representative THIQ3CA analogs.
For molecule 35, two sets of nine 1H signals were observed at room temperature in CDCl3 (Figure 2, Table 1). The relative proportion of the two pairs of peaks was essentially identical across all seven signals and was approximately 75:25. Likewise, 32 displayed two sets of seven 1H signals (Table 2). In addition, the number of 13C signals for both 32 and 35 were double the number of carbons, although the frequencies for some atoms could not be measured accurately due to lower resolution. These observations suggested the possibility of two conformational isomers.
Figure 2.

1H NMR spectrum of THIQ3CA analog 35 in CDCl3 showing duplicity of signals at ambient temperature. Inset shows 6.0-4.0 δ region, assignable to ‘a’ and ‘b’ protons, for better clarity.
Table 1.
1H NMR-CDCl3 parameters for set of signals that reveal the rotational isomerism in derivative 35.
 | |||
|---|---|---|---|
| Protons | Chemical Shift (δ) | Splitting | Integration |
| Ha, Position-3 | 5.51-5.49 & 4.94-4.92 | dd & dd | 1 |
| Hb, Position-1 | 4.91-4.60 | dd | 1 |
| Hb, Position-1 | 4.52-4.30 | dd | 1 |
| Hc, Methylene linker | 3.76 | broad s | 2 |
| Hd, Methyl ester | 3.67-3.3.66 | d | 3 |
| He, Position-4 | 3.14-3.00 | dt | 1 |
| He, Position-4 | 3.02-2.96 & 2.89-2.84 | dd & dd | 1 |
| Hf, Position-8 | 6.49-6.40 | d | 1 |
| Hg, Position-5 | 6.56-6.53 | d | 1 |
Table 2.
1H NMR-CDCl3 parameters for set of signals that reveal the rotational isomerism in derivative 32.
 | |||
|---|---|---|---|
| Protons | Chemical Shift (d) | Splitting | Integration |
| Ha, Position-3 | 5.64-5.62 & 4.96-4.94 | dd & dd | 1 |
| Hb, Position-1 | 5.02-4.78 | dd | 1 |
| Hb, Position-1 | 4.41-4.14 | dd | 1 |
| Hc, Methylene linker | 3.79-3.73 | d | 2 |
| Hd, Methyl ester | 3.54-3.43 | d | 3 |
| He, Position-4 | 3.42-3.37 | m | 1 |
| He, Position-4 | 2.78-2.72 & 2.55-2.49 | dd & dd | 1 |
To understand whether polarity of solvent affects conformational preferences, the 1H NMR spectrum of 32 was studied in C6D6, CDCl3, CD3CN, and DMSO-d6 at ambient temperature. The effect was studied by monitoring Ha, Hd, and He signals (Figure 3). For proton He the splitting pattern changed dramatically as the solvent polarity increased. The doublet of doublets in C6D6 and CDCl3 merged into an unresolved multiplet in CD3CN, and gave one doublet of doublet in DMSO-d6 (Figure 3A). Protons Ha and Hd belonging to carbon-3 and methyl ester group, respectively, displayed a gradual loss of intensity as the signal spread due to increase in solvent polarity. Whereas the signal spread was 375.7 Hz for Ha in C6D6, it decreased to 62.0 Hz in DMSO-d6 (Figure 3C, see Table 3). Likewise, the spread decreased from 42.6 (C6D6) to 9.7 Hz (DMSO-d6) for Hd (Figure 3B, Table 3) This indicates that the two conformers tend to coalesce with increasing solvent polarity.
Figure 3.

Solvent-dependent changes in the conformational isomerism of 32. A) 3.1-2.4 δ; B) 3.9-2.8 δ; and C) 6.2-3.8 δ
Table 3.
The change in Δδ for protons Ha and Hd of the two detected conformers of THIQ3CA derivative 32 in different solvents.
| Solvents | Ha Δδ (Hz) | Hd Δδ (Hz) |
|---|---|---|
| C6D6 | 374.68 | 42.64 |
| CDCl3 | 269.22 | 46.76 |
| CD3CN | 151.98 | 19.24 |
| DMSO-d6 | 61.98 | 9.68 |
The influence of temperature on the conformational distribution of THIQ3CA analog 32 was studied by measuring spectral changes in DMSO-d6 over the range of 293 – 373 K. Figure 4 shows that the signal spread (Δδ) between the signals of Ha (position-3 proton) and Hb (position-1 protons) gradually decreases as the temperature increases. Both signals retain distinct resonance positions until 353 K and then start to coalesce on the NMR time scale. Measurements at 373 K for both Ha and Hb show that the two conformers have lost their distinction and exhibit a rapidly equilibrating species. Likewise, two singlets at 3.54 and 3.49 δ corresponding to Hd merge as one singlet 3.57 δ and the two doublet of doublet at 2.78-2.72 δ and 2.55-2.49 δ corresponding to He also coalesce as one doublet at 2.82-2.78 δ (not shown). At ambient temperature, the ratio of the major to minor conformer was approximately 3 to 1, which equalized over the range of 353 – 373 K.
Figure 4.

Temperature effect on the conformational isomerism of 32 in DMSO–d6. The entire 1H NMR spectrum of 32 was recorded but only the 5.6-3.8 δ range is shown for enhanced clarity. 1H NMR spectra at 293, 313, 333 and 353 K were recorded on a Varian 300 MHz instrument, while the spectrum at 373 K was recorded on a Bruker 400 MHz.
The variable temperature results can be used to analyze the free energy of activation promotes the dynamic inter-conversion between the conformers. The Eyring equation based on detailed line-shape analysis is the most accurate method, although several other methods have been reported.53–55 Two approximations have been particularly useful include equations I and II that afford reliable estimates of the free energy of inter-conversion (ΔG++) in conjunction with the rate constant (kc) at the coalescence temperature (Tc).54,55 In these equations, a is constant with a value of 0.004575 kcal/mol, T is the temperature in Kelvin (K), and Δν is the maximum peak separation in Hz. This approach has been shown to give reliable estimates for both coalescing singlets and doublets with different intensities as long as Δν > 4 Hz.
| (I) |
| (II) |
Using this approach, evaluation of the two singlets at 3.56 and 3.53 δ, belonging to the methyl ester proton Hd, resulted in values of 16.7 ± 1.0 kcal/mol and 15.5 ± 1.9 kcal/mol for ΔG++ and kc at 353 K, respectively. This value is significantly higher than a value of 7.8 kcal/mol and 15 kcal/mol for N-acetyl-1-benzyl and N-acetyl-1,3-dimethyl THIQ derivatives and comparable to the activation energy range of 17.7 – 20.1 measured for N-acetyl-1-alkylidene THIQ (Figure 5).49–51
Figure 5.

THIQ derivatives display a wide range of energy barriers for conformational transitions. The N–arylacyl THIQ3CA esters studied in this work show a distinct high energy barrier of ~17 kca/mol.
Overall, our work reveals that the duplicity of NMR signals persists for all THIQ3CA derivatives irrespective of groups present on the scaffold as long as the 2-linker is an amide. Conformational isomerism noted for N–arylacyl THIQ3CA analogs arises from rotational restrictions around the amide bond at position 3 with an estimated ΔG++ of ~17 kcal/mol. Lastly, we expect that the two corresponding rotational isomers to be equi-energetic with a calculated energy difference of 0.655 kcal/mol.56
Significance
Despite the fact that THIQ3CA scaffold is widely involved in structures of many medicinal agents, the reported synthetic options to construct this interesting scaffold are limited. This is especially true for electronically activated THIQ3CA derivatives. One of the key reasons is the shortage of availability of appropriate precursors and/or lengthy scheme involved in precursor synthesis. The protocol reported herein utilizes commercially available benzaldehydes and optimized Horner–Wadsworth–Emmons/Pictet–Spengler reactions and glycine donor 3. The approach is mild, expeditious and high yielding. In fact, the approach helped pave the way for the synthesis of a novel library of bicyclic–unicyclic (30–43) and bicyclic–unicyclic–bicyclic (44 and 45) methyl–protected poly–phenolic THIQ3CA derivatives in a straightforward manner. A specific advantage of this protocol is that it is flexible and is likely to be generally applicable.
NMR spectroscopic studies highlighted an important structural property of N-arylacyl THIQ3CA derivatives that is of considerable significance. Two major rotamers – s-trans and s-cis –were found to be present in solution arising from hindered rotation around the amide bond. From the drug design perspective, this observation carries significant implications. Most structure-based drug design studies typically utilize only one conformer. The expectation is that rapid inter-conversion between the different rotamers (or conformers) will eventually induce dynamic equilibrium in favor of the form of that eventually binds to target site.57 This work suggests that such inter-conversion may not occur for N-arylacyl THIQ3CA derivatives because of the high rotational barrier of ~17 kcal/mol. Thus, both conformers will have to be explicitly studied in structure-based drug design.
Experimental Section
Chemicals and Reagents
Anhydrous CH2Cl2, THF, CH3CN, DMF, and toluene were purchased from Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and used as such. All other solvents were of reagent gradient and used as supplied. Other chemical reagents were purchased either from Sigma-Aldrich, Fisher Scientific, or Alfa-Aesar (Ward Hill, MA). Analytical TLC was performed using UNIPLATE™ silica gel GHLF 250 μm pre-coated plates (ANALTECH, Newark, DE). TLC was visualized using light of 254 nm. Chemical reactions sensitive to air or moisture were carried out under nitrogen atmosphere in oven-dried glassware. All reagent solutions unless otherwise noted were handled under an inert nitrogen atmosphere using syringe techniques. Each chemical reaction was optimized to afford a yield of more than 65%.
Purification of Reaction Products
Flash chromatography was performed using Combiflash RF from Teledyne Isco (Lincoln, NE) and disposable normal silica cartridges of 30–70 μ particle size, 230–400 mesh size and 60 Å pore size. The normal phase silica content of cartridges were 4–24 gms (for purification of 250–1000 mg crude samples). Mobile linear gradients utilized either EtOAc or CH3OH in hexanes or CH2Cl2, respectively. The flow rate of the mobile phase was in the range of 18 to 35 ml/min based on the size of the silica cartridge. The purity of all compounds was greater than 95% as determined by uPLC.
NMR Studies
The 1H and 13C NMR spectra were recorded on Bruker 400 MHz spectrometer (unless otherwise noted) in CDCl3, DMSO-d6, CD3OD, C6D6 or CD3COCD3. Signals, in part per million (ppm), are either relative to the internal standard (tetramethyl silane, TMS) or to the residual peak of the solvent. The NMR data are being reported as: chemical shift (ppm), multiplicity of signal (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet, br = broad), coupling constants (Hz), and integration.
1H and 13C NMR spectra of compounds were measured using 5 – 35 mg sample dissolved in 0.6 mL of solvent. For 1H NMR spectrum, 16 scans were recorded with a spectral width of 8223.685 Hz, a FID resolution of 0.125 Hz, relaxation delay of 1 s, and acquisition time of 4 s, while the 13C spectra were recorded using 1024 – 10240 scans with a spectral width of 24038.461 Hz, FID resolution of 0.367 Hz, acquisition time of 1.5 s, and a relaxation delay of 2 s. Variable temperature NMR studies were performed on a Varian 300 MHz in the range of 293–353 K and on a Bruker 400 MHz at temperatures up to 373 K.
Mass Spectrometric Characterization
The ESI MS characterization of all THIQ3CA derivatives was performed using a Waters Acquity TDQ mass spectrometer in positive ion mode. Samples, dissolved in methanol, were infused at a rate of 20 μL/min and mass scans were collected in the range of 200 – 700 amu with a scan time of 1 s. The capillary and cone voltages were varied between 3 – 4 kV and 38 – 103 V, respectively. The extractor voltage was set to 3 V, the Rf lens voltage was 0.1 V, the source block temperature was 150 °C, and desolvation temperature was set to about 250 °C. Ionization conditions were optimized for each compound to maximize ionization of the parent ion.
General Procedure for Preparation of Benzylidene Imidazoline-2,4-dione Derivatives 6 and 7
Hydantoin 1 (5.0 gm, 50.0 mmol), anhydrous sodium acetate (8.2 gm, 100.0 mmol) and aldehyde (4 or 5) (7.5 gm, 45.0 mmol) were successively added in a solution of glacial acetic acid (15 mL) and acetic anhydride (2 mL). The mixture was stirred at 170 °C under nitrogen atmosphere for 3 h. Upon cooling to 110 °C, water (100 mL) was added drop-wise and the mixture left for stirring overnight at RT. The orange precipitate of the desired compound was filtered off and then washed with water to remove acetic acid, the unreacted hydantoin, and sodium acetate. The orange solid was then purified by flash chromatography using mobile phase gradient of 40% to 100% (EtOAc: hexanes) to afford benzylidene imidazoline-2,4-dione derivative 6 or 7 as yellow to white solid in 65–72% yield.
5-(2,3-dimethoxybenzylidene)imidazolidine-2,4-dione (6)
1H NMR (DMSO-d6, 400 MHz): δ = 11.24 (br, 1H, NH), 10.39 (s, br, 1 H, NH), 7.25 (d, 1J = 6.9 Hz, 1 H, Ar), 7.08 (t, 1J = 8.0 Hz, 1H, Ar), 7.03 (d, 1J = 7.2, 1 H, Ar), 6.62 (s, 1 H, CH), 3.81 (s, 3 H, CH3), 3.74 (s, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ = 165.5, 155.5, 152.4, 147.1, 128.7, 126.7, 124.2, 113.3, 102.3, 60.6, 55.7. MS (ESI) calculated for C12H12N2O4, [M+H]+, m/z 249.09, found for [M+Na]+, m/z 271.20.
5-(2,5-dimethoxybenzylidene)imidazolidine-2,4-dione (7)
1H NMR (DMSO-d6, 400 MHz): δ = 7.12 (d, 1J= 2.9 Hz, 1 H, Ar), 6.96 (d, 1J = 9.0 Hz, 1 H, Ar), 6.88-6.85 (dd, 1J= 3.0 Hz, 2J = 9.0 Hz, 1 H, Ar), 6.55 (s, 1H, CH), 3.78 (s, 3H, CH3), 3.77 (s, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ =166.8, 157.1, 153.0, 151.4, 129.4, 122.6, 115.1, 114.1, 112.1, 101.8, 56.0, 55.5. MS (ESI) calculated for C12H12N2O4, [M+H]+, m/z 249.09, found for [M+Na]+, m/z 271.23.
General Procedure for Preparation of Benzyl Imidazoline-2,4-dione Derivatives 8 and 9
Benzylidene imidazolidine-2,4-dione derivative (6 or 7) (7.3 gm, 29.5 mmol) was dissolved in hot ethanol (100 mL) followed by addition of 10% Pd/C (0.73 gm) after cooling. H2 gas was then pumped into the reaction vessel to achieve 50 psi at RT. After stirring the mixture for 5 h, the catalyst was filtered on Celite, which was washed using a 1:1 mixture of EtOAc and CH3OH. The organic filtrate was concentrated in vacuo to afford the corresponding reduced product (8 or 9) in ~100% yield which was used without any further purification.
5-(2,3-Dimethoxybenzyl)imidazolidine-2,4-dione (8)
1H NMR (DMSO-d6, 400 MHz): δ = 10.58 (s, 1 H, NH), 7.76 (s, 1 H, NH), 7.00-6.94 (dd, 1J = 8.0 Hz, 2J = 15.6, 1 H, Ar), 6.94-6.91 (dd, 1J = 1.6 Hz, 2J = 8.2 Hz, 1 H, Ar), 6.81-6.79 (dd, 1J = 1.6 Hz, 2J = 7.4 Hz, 1 H, Ar), 4.26 (t, 1J= 6.5 Hz, 1 H, CH), 3.79 (s, 3H, CH3), 3.73 (s, 3H, CH3), 3.09-3.04 (dd, 1J = 4.9 Hz, 2J = 13.9 Hz, 1 H, CH), 2.81-2.76 (dd, 1J = 7.5 Hz, 2J = 13.9 Hz, 1 H, CH). 13C NMR (DMSO-d6, 100 MHz): δ = 175.4, 157.3, 152.2, 147.1, 129.7, 122.4, 111.6, 60.0, 58.0, 55.5, 31.5. MS (ESI) calculated for C12H14N2O4, [M+H]+, m/z 251.10, found for [M+Na]+, m/z 273.24.
5-(2,5-Dimethoxybenzyl)imidazolidine-2,4-dione (9)
1H NMR (DMSO-d6, 400 MHz): δ = 10.60 (s, 1 H, NH), 7.75 (s, 1 H, NH), 6.87 (d, 1J= 8.6 Hz, 1 H, Ar), 6.78 (d, 1J = 3.0 Hz, 1 H, Ar), 6.76-6.74 (m, 1 H, Ar), 4.26-4.22 (dd, 1J= 5.1 Hz, 2J = 7.2 Hz, 1 H, CH), 3.71 (s, 3H, CH3), 3.68 (s, 3H, CH3), 3.08-3.03 (dd, 1J = 4.8 Hz, 2J = 13.8 Hz, 1 H, CH), 2.72-2.67 (dd, 1J = 7.6 Hz, 2J = 13.8 Hz, 1 H, CH). 13C NMR (DMSO-d6, 100 MHz): δ =175.5, 157.4, 152.7, 151.5, 125.4, 117.1, 112.2, 111.5, 57.4, 55.8, 55.2, 32.2. MS (ESI) calculated for C12H14N2O4, [M+H]+, m/z 251.10, found for [M+Na]+, m/z 273.24.
General Procedure for Preparation of Dimethoxy Phenylalanine Ethyl Esters 10 and 11
A mixture of the benzyl imidazolidine-2,4-dione (8 or 9) (7.0 gm, 28.0 mmol), barium hydroxide (14.4 gm, 84.0 mmol) and water (150 mL) was refluxed for 48 h, then filtered hot and the residue washed with hot water. Following cooling, the filtrate was acidified to pH 6.5 with 50% H2SO4. The resulting BaSO4 precipitate was filtered and the pH of the aqueous filtrate adjusted to 9.0 using NH4OH. The aqueous filtrate was then concentrated, suspended in ethanol (100 mL), treated with thionyl chloride (20 mL of 2.0 M solution in CH2Cl2) at 0 °C, and then mixture refluxed for 4 h. Following neutralization with saturated NaHCO3 solution, the reaction mixture was partitioned between EtOAc and water, and a crude product obtained from the EtOAc layer. Flash chromatography using 70% (EtOAc: hexanes) as the mobile phase was used to yield derivative 10 or 11 as white solid in 43–51% yield.
Ethyl 2,3-dimethoxy phenylalanine (10)
1H NMR (CDCl3, 400 MHz): δ = 6.91 (t, 1J = 7.9 Hz, 1 H, Ar), 6.75-6.73 (dd, 1J = 1.4 Hz, 2J = 8.2 Hz, 1 H, Ar), 6.70-6.68 (dd, 1J = 1.4 Hz, 2J = 7.6 Hz, 1 H, Ar) 4.10-4.04 (q, 1J = 7.2 Hz, 2 H, CH2), 3.78 (s, 3 H, CH3), 3.76 (s, 3 H, CH3), 3.73-3.67 (m, 1 H, CH), 3.03-2.99 (dd, 1J = 5.6 Hz, 2J = 13.3 Hz, 1 H, CH), 2.83-2.78 (dd, 1J = 8.3 Hz, 2J = 13.3 Hz, 1 H, CH), 1.15 (t, 1J= 7.1 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ =175.1, 152.8, 147.7, 131.2, 123.8, 122.8, 111.3, 60.8, 60.5, 55.7, 55.1, 35.8, 14.1. MS (ESI) calculated for C13H19NO4, [M+H]+, m/z 254.14, found for [M+H]+, m/z 254.30.
Ethyl 2,5-dimethoxy phenylalanine (11)
1H NMR (CDCl3, 400 MHz): 6.79-6.76 (m, 1 H, Ar), 6.74 (d, 1J = 3.0 Hz, 1 H, Ar), 6.72 (d, 1J = 1.8 Hz, 1 H, Ar), 4.18-4.11 (q, 1J = 7.2 Hz, 2H, CH2), 3.80-3.78 (m, 1H, CH), 3.77 (s, 3H, CH3), 3.74 (s, 3 H, CH3), 3.09-3.03 (dd, 1J = 6.0 Hz, 2J = 12.2 Hz, 1 H, CH), 2.86-2.79 (dd, 1J = 9.0 Hz, 2J = 12.1 Hz, 1H, CH), 1.15 (t, 1J = 7.2 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ =173.1, 153.5, 151.9, 125.9, 117.4, 112.7, 111.5, 61.2, 56.0, 55.7, 53.8, 33.1, 14.1. MS (ESI) calculated for C13H19NO4, [M+H]+, m/z 254.14, found for [M+H]+, m/z 254.30.
General Procedure for Pictet–Spengler Reaction to Synthesize 12, 13, 20, 21 and 27
A solution of substituted phenylalanine methyl (18, 19, or 26) (0.6 gm, 2.5 mmol) or ethyl ester (10 or 11) (0.5 gm, 2.0 mmol) in CH2Cl2 (10 mL) was treated with formaldehyde (37% aq. solution, 0.2–0.25 ml, 2.5–3.0 mmol) at RT. Trifluoro-acetic acid (TFA, 3.7–4.6 gm, 2.5–3.0 ml, 10.0–12.5 mmol) was then slowly added over 15 min and the reaction mixture was stirred for 5 h following which it was diluted with CH2Cl2 (15 mL) and neutralized with saturated NaHCO3 solution (20 mL). The organic extracts of the aqueous solution were combined, the solvent evaporated under reduced pressure, and the crude mixture was separated by flash chromatography using the mobile phase of 50% (EtOAc: hexanes) to give the desired THIQ3CA ethyl ester (12 or 13) or methyl ester (20, 21 or 27) as yellow oil in 70–82% yield.
Ethyl 5,6-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (12)
1H NMR (CDCl3, 400 MHz): δ = 6.73 (s, 2H, Ar), 4.10-3.80 (m, 5 H, CH, 2 × CH2), 3.80 (s, 3 H, CH3), 3.79 (s, 3 H, CH3), 3.26 (d, 1J = 16.8 Hz, 1 H, CH), 3.04-2.94 (dd, 1J= 6.3 Hz, 2J = 17.1 Hz, 1 H, CH), 1.15 (t, 1J = 7.2 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 173.2, 150.5, 146.5, 127.5, 127.0, 121.7, 109.7, 61.1, 55.7, 55.5, 55.3, 42.8, 26.2, 14.1. MS (ESI) calculated for C14H19NO4, [M+H]+, m/z 266.14, found for [M+H]+, m/z 266.28.
Ethyl 5,8-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (13)
1H NMR (CDCl3, 400 MHz): δ = 6.58 (d, 1J = 8.9 Hz, 1 H, Ar), 6.55 (d, 1J = 8.9 Hz, 1 H, Ar), 4.20-4.18 (dd, 1J = 2.3 Hz, 2J = 7.2 Hz, 1 H, CH), 4.16-4.14 (dd, 1J = 2.2 Hz, 2J = 7.2 Hz, 1 H, CH), 4.13 (d, 1J = 14.2 H, 1 H, CH), 3.80 (d, 1J = 17.0 Hz, 1 H, CH), 3.71 (s, 3 H, CH3), 3.69 (s, 3 H, CH3), 3.59-3.55 (dd, 1J = 4.6 Hz, 2J = 10.4 Hz, 1 H, CH), 3.07-3.01 (ddd, 1J = 1.0 Hz, 2J = 4.5 Hz, 3J =17.1 Hz, 1 H, CH), 2.64-2.57 (dd, 1J = 10.4 Hz, 2J = 17.1 Hz, 1 H, CH), 1.24 (t, 1J = 7.2 Hz, 3 H, CH3). 13C NMR (CDCl3, 400 MHz): δ = 173.1, 151.2, 150.0, 124.9, 123.6, 107.4, 107.1, 61.1, 55.7, 55.5, 55.3, 42.8, 26.2, 14.1. MS (ESI) calculated for C14H19NO4, [M+H]+, m/z 266.14, found for [M+H]+, m/z 266.28.
Methyl 5,6-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (20)
1H NMR (CDCl3, 400 MHz): δ = 6.68 (s, 2H, Ar), 3.97-3.78 (m, 3H, CH, CH2), 3.75 (s, 3H, CH3), 3.73 (s, 3H, CH3), 3.47, (s, 3 H, CH3), 3.22-312 (dd, 1J = 4.3 Hz, 2J = 17.4 Hz, 1H, CH), 2.97-2.91 (dd, 1J = 9.2 Hz, 2J = 16.4 Hz, 1H, CH). 13C NMR (CDCl3, 100 MHz): δ = 173.2, 150.5, 146.5, 127.5, 127.0, 121.7, 110.7, 60.1, 56.9, 55.8, 51.3, 49.3, 25.8. MS (ESI) calculated for C13H17NO4, [M+H]+, m/z 252.12, found for [M+H]+, m/z 252.26.
Methyl 5,8-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylat (21)
1H NMR (CDCl3, 400 MHz): δ = 6.53 (d, 1J = 3.1 Hz, 1 H, Ar), 6.52 (d, 1J = 2.9 Hz, 1 H, Ar), 3.94-3.87 (m, 2H, CH2), 3.86-3.81 (dd, 1J = 7.8 Hz, 2J = 16.4 Hz, 1 H, CH), 3.69 (s, 3H, CH3), 3.67 (s, 3H, CH3), 3.46 (s, 3H, CH3), 3.12-3.13.06 (dd, 1J = 3.9 Hz, 2J = 17.2 Hz,1H, CH), 2.86-2.79 (dd, 1J = 10.2 Hz, 2J = 17.4 Hz, 1H, CH). 13C NMR (CDCl3, 100 MHz): δ = 173.5, 151.2, 150.2, 124.7, 123.2, 107.1, 106.8, 60.1, 56.4, 55.7, 51.2, 45.6, 25.0. MS (ESI) calculated for C13H17NO4, [M+H]+, m/z 252.12, found for [M+H]+ m/z 252.14.
Methyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (27)
1H NMR (CDCl3,400 MHz): δ = 6.52 (s, 1 H, Ar), 6.46 (s, 1 H, Ar), 4.08-3.98 (dd, 1J = 15.8 Hz, 2J= 25.6 Hz, 2 H, CH2), 3.73-3.79 (m, 1 H, CH), 3.78 (s, 3 H, CH3), 3.77 (s, 3 H, CH3), 3.72 (s, 3 H, CH3), 3.00-2.95 (dd, 1J= 4.8 Hz, 2J = 16.1 Hz, 1 H, CH), 2.91-2.84 (dd, 1J = 9.7 Hz, 2J= 16.0 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ = 172.7, 147.8, 125.5, 124.4, 111.7, 108.9, 55.9, 55.6, 52.3, 46.4, 30.6. MS (ESI) calculated for C13H17NO4, [M+H]+, m/z 252.12, found for [M+Na]+, m/z 274.19.
General procedure for Horner–Wadsworth–Emmons Reaction for Synthesis of 14, 15, 23, 24 and 25
To a solution of (±)-Boc-α-phosphono glycine trimethyl ester 2 (2.0 gm, 6.7 mmol) or (±)-Z-α-phosphonoglycine trimethyl ester 3 (2.0 gm, 6.0 mmol) in dry THF (3 mL) was added DBU (0.91–1.0 gm, 6.0–6.7 mmol) and the mixture stirred for 30 min at 0 °C. A solution of either 2,3-dimethoxy benzaldehyde 4, 2,5-dimethoxy benzaldehyde 5, or 3,4-dimethoxybenzaldehyde 22 (0.83–0.93 gm, 5.0–5.6 mmol) in dry THF (3 mL) was then added slowly. The reaction mixture was allowed to warm to RT, stirred for 5 h and the solvent evaporated under reduced pressure. The residue was taken up in EtOAc (50 mL) and following routine organic extraction and evaporation procedures a crude product was obtained. The crude product was then purified by flash chromatography using 30% (EtOAc: hexanes) as eluent to give 14, 15, or 23–25 as white to yellow solid in 77–85% yield.
Methyl-2-(tert-butoxycarbonylamino)-3-(2,3-dimethoxyphenyl)acrylate (14)
1H NMR (CDCl3, 400 MHz): δ = 7.12 (s, 1 H, Ar), 7.06 (d, 1J = 4.3 Hz, 2 H, Ar), 6.91-6.89 (dd, 1J = 0.7 Hz, 2J = 4.8 Hz, 1H, CH), 3.88 (s, 3H, CH3), 3.86 (s, 3H, CH3), 3.80 (s, 3H, CH3), 1.40 (s, 9H, 3 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 166.2, 153.0, 152.9, 147.0, 128.3, 127.6, 124.2, 121.9, 121.7, 112.7, 80.7, 61.4, 55.9, 52.5, 28.1. MS (ESI) calculated for C17H23NO6, [M+H]+, m/z 338.16, found for [M+H]+, m/z 338.32.
Methyl-2-(tert-butoxycarbonylamino)-3-(2,5-dimethoxyphenyl)acrylate (15)
1H NMR (CDCl3, 400 MHz): δ = 7.21 (s, 1H, Ar), 7.01 (s, 1 H, Ar), 6.78 (t, 1J = 0.9 Hz, 1 H, CH), 3.78 (s, 3H, CH3), 3.76 (s, 3H, CH3), 3.68 (s, 3H, CH3), 1.32 (s, 9 H, 3 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 166.1, 153.4, 152.8, 151.5, 126.1, 123.8, 123.5, 116.1, 114.5, 112.6, 80.7, 56.5, 55.7, 52.4, 28.3. MS (ESI) calculated for C17H23NO6, [M+H]+, m/z 338.16, found for [M+Na]+, m/z 360.18.
Methyl-2-(benzyloxycarbonylamino)-3-(2,3-dimethoxyphenyl)acrylate (23)
1H NMR (CDCl3, 400 MHz): δ = 7.32-7.28 (m, 5H, Ar), 7.18 (s, 1 H, Ar), 6.96-6.93 (m, 2 H, Ar), 6.83-6.81 (m, 1 H, CH), 5.00 (s, 2 H, CH2), 3.79 (s, 3H, CH3), 3.73 (s, 3H, CH3) 3.69 (s, 3H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 165.7, 153.7, 153.0, 146.9, 136.0, 128.5, 128.3, 128.2, 128.0, 127.0, 124.3, 123.7, 121.6, 112.9, 67.3, 61.5, 55.8, 52.6. MS (ESI) calculated for C20H21NO6, [M+H]+, m/z 372.14, found for [M+H]+, m/z 372.28.
Methyl-2-(benzyloxycarbonylamino)-3-(2,5-dimethoxyphenyl)acrylate (24)
1H NMR (CDCl3, 400 MHz): δ = 7.34-7.30 (m, 5H, Ar), 6.99 (d, 1J= 2.0 Hz, 1 H, Ar), 6.84 (d, 1J = 2.4 Hz, 2 H, Ar), 6.77-6.73 (m, 1H, CH), 5.08 (s, 2H, CH2), 3.73 (s, 6 H, 2 × CH3), 3.59 (s, 3H, CH3). 13C NMR (CDCl3, 100 MHz): δ =165.7, 153.5, 152.8, 151.4, 136.0, 128.6, 128.4, 128.2, 125.8, 125.0, 123.4, 116.4, 114.4, 112.9, 67.3, 56.6, 55.6, 52.5. MS (ESI) calculated for C20H21NO6, [M+H]+, m/z 372.14, found for [M+H]+, m/z 372.32.
Methyl-2-(benzyloxycarbonylamino)-3-(3,4-dimethoxyphenyl)acrylate (25)
1H NMR (400 MHz, CDCl3): δ = 7.36 (s, 1 H, Ar), 7.35-7.30 (m, 4 H, Ar), 7.13-7.11 (m, 2 H, Ar), 6.90-6.85 (m, 1 H, Ar), 6.82 (d, 1J = 8.9 Hz, 1 H, CH), 6.26 (br, s, 1 H, NH), 5.13 (s, 2 H, CH2), 3.90 (s, 3 H, CH3), 3.81 (s, 3 H, CH3), 3.71 (s, 1 H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 165.9, 150.4, 148.8, 136.0, 133.0, 130.0, 128.5, 128.3, 126.4, 124.2, 124.2, 122.1, 114.2, 112.2, 110.9, 67.5, 55.9, 55.6, 52.6. MS (ESI) calculated for C20H21NO6, [M+H]+, m/z 372.14, found for [M+H]+, m/z 372.28.
General Procedure for Catalytic Hydrogenation of N-Boc and N-Cbz Protected α-Amino-β-aryl methyl acrylates
Method A: Methyl acrylate (14 or 15) (0.5 gm, 1.5 mmol) and 10% Pd/C (0.05 gm) were mixed in ethanol (15 mL) and H2 gas pumped into the reaction mixture at RT. After stirring the solution 5 h, the catalyst was filtered on Celite and the organic filtrate concentrated in vacuo to afford the reduced product (16 or 17) as white solid in ~100% yield. Method B: N-Cbz protected methyl acrylate 23, 24, or 25 (0.5 gm, 1.3 mmol) and 10% Pd(OH)2 (0.05 gm) on activated charcoal were mixed in CH3OH:t-butanol (1:1) mixture (10 mL) and H2 gas pumped into the mixture at RT. After stirring overnight, the catalyst was filtered on Celite, the organic filtrate concentrated in vacuo, and the residue purified by flash chromatography using 70% (EtOAc: hexanes) as mobile phase to afford the corresponding dimethoxy β-aryl-α-amino methyl ester 18 (oil), 19 (oil), or 26 as a yellow solid in 73–76% yield.
Methyl 2-(tert-butoxycarbonylamino)-3-(2,3-dimethoxyphenyl)propanoate (16)
1H NMR (CDCl3, 400 MHz): δ = 7.00 (t, 1J = 1.5 Hz, 1H, Ar), 6.82 (d, 1J= 8.2 Hz, 1 H, Ar), 6.73 (d, 1J= 7.6, 1 H, Ar), 5.32 (d, 1J= 7.0 Hz, 1 H, NH), 4.48-4.43 (dd, 1J= 6.8 Hz, 2J= 13.2 Hz, 1 H, CH), 3.86 (s, 3H, CH3), 3.84 (s, 3H, CH3), 3.71 (s,3H, CH3), 3.06-3.04 (m, 2 H, CH2), 1.39 (s, 9H, 3 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 172.8, 155.4, 152.7, 147.5, 130.1, 124.0, 122.7, 111.6, 79.6, 60.6, 55.7, 54.7, 52.2, 32.4, 28.3. MS (ESI) calculated for C17H25NO6, [M+H]+, m/z 340.18, found for [M+Na]+, m/z 361.82.
Methyl 2-(tert-butoxycarbonylamino)-3-(2,5-dimethoxyphenyl)propanoate (17)
1H NMR (CDCl3, 400 MHz): δ = 6.70-6.68 (m, 2H, Ar), 6.61 (d, 1J= 2.8 Hz, 1 H, Ar), 5.19 (d, 1J= 7.1 Hz, 1 H, NH), 4.45-4.39 (dd, 1J= 13.2 Hz, 2J= 7.0 Hz, 1 H, CH), 3.71 (s, 3H, CH3), 3.67 (s, 3H, CH3), 3.64 (s, 3H, CH3), 2.96 (t, 1J= 4.8 Hz, 2H, CH2), 1.31 (s, 9H, 3 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 172.7, 155.2, 153.5, 151.8, 125.7, 117.2, 112.7, 111.3, 79.5, 55.8, 55.6, 54.1, 52.0, 32.9, 28.2. MS (ESI) calculated for C17H25NO6, [M+H]+, m/z 340.18, found for [M+Na]+, m/z 361.82.
Methyl 3-(2,3-dimethoxyphenyl)-2-(ethoxycarbonylamino)propanoate
1H NMR (CDCl3, 400 MHz): δ = 6.97 (t, 1J = 7.9 Hz, 1 H, Ar), 6.86-6.84 (dd, 1J = 1.3 Hz, 2J = 8.2 Hz, 1H, Ar), 6.80-6.78 (dd, 1J = 1.2 Hz, 2J = 7.6 Hz, 1 H, Ar), 4.12-4.09 (dd, 1J = 5.4 Hz, 2J= 8.5 Hz, 1 H, CH), 3.89 (s, 3H, CH3), 3.86 (s, 3H, CH3), 3.62 (s, 3 H, CH3), 3.54-3.50 (dd, 1J = 5.4 Hz, 2J = 13.4 Hz, 1 H, CH), 3.35-3.30 (dd, 1J = 8.6 Hz, 2J = 13.4 Hz, 1 H, CH), 3.09-3.03 (dd, 1J= 7.2 Hz, 2J = 14.5 Hz, 2 H, CH2), 1.44 (t, 1J = 7.2 Hz, 3 H, CH3). MS (ESI) calculated for C15H21NO6, [M+H]+, m/z 312.34, found for [M+H]+, m/z 312.30.
Methyl 2,3-dimethoxy phenylalanine (18)
1H NMR (CDCl3, 400 MHz): δ = 8.47 (s, br, 2H, NH2), 7.00 (t, 1J= 8.0 Hz, 1 H, Ar), 6.86-6.84 (dd, 1J= 1.2 Hz, 2J= 8.2 Hz, 1 H, Ar), 6.77-6.75 (dd, 1J= 1.1 Hz, 2J= 7.6 Hz, 1 H, Ar), 4.34-4.31 (dd, 1J= 5.4 Hz, 2J= 7.0 Hz, 1 H, CH), 3.82 (s, 3H, CH3), 3.81 (s, 3H, CH3), 3.69 (s, 3H, CH3), 3.36-3.31 (dd, 1J= 5.2 Hz, 1J= 14.2 Hz, 1 H, CH), 3.23-3.18 (dd, 1J= 5.2 Hz, 2J= 14.2 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ = 169.1, 152.7, 147.1, 127.7, 124.7, 123.0, 112.5, 60.7, 55.6, 53.7, 53.0, 31.6. MS (ESI) calculated for C12H17NO4, [M+H]+, m/z 240.12, found for [M+H]+, m/z 240.24.
Methyl 2,5-dimethoxy phenylalanine (19)
1H NMR (CDCl3, 400 MHz): δ = 6.71 (s, 3H, Ar), 4.28-4.25 (dd, 1J= 8.0 Hz, 2J= 5.0 Hz, 1H, CH), 3.70 (s, 3H, CH3), 3.66 (s, 3H, CH3), 3.63 (s, 3H, CH3), 3.33-3.28 (dd, 1J= 5.0 Hz, 2J= 14.1 Hz, 1 H, CH), 3.03-2.97 (dd, 1J= 8.0 Hz, 2J= 14.1 Hz, 1H, CH). 13C NMR (CDCl3, 100 MHz): δ = 169.5, 153.4, 151.8, 123.0, 117.9, 113.9, 111.7, 55.8, 53.0, 52.8, 32.0. MS (ESI) calculated for C12H17NO4, [M+H]+, m/z 240.12, found for [M+Na]+, m/z 262.15.
Methyl 3,4-dimethoxy phenylalanine (26)
1H NMR (CDCl3, 400 MHz): δ = 6.73 (d, 1J= 7.8 Hz,1 H, Ar), 6.66 (d, 1J = 7.4 Hz, 1 H, Ar), 6.65 (s, 1 H, Ar), 3.79-3.65 (m, 1 H, CH), 3.79 (s, 3 H, CH3), 3.78 (s, 3H, CH3), 3.64 (s, 3H, CH3), 2.99-2.95 (dd, 1J= 4.8 Hz, 2J= 13.6 Hz, 1H, CH), 2.79-2.74 (dd, 1J= 7.6 Hz, 2J= 13.6 Hz, 1H, CH), 1.86 (br, s, 2 H, NH2). 13C NMR (CDCl3, 100 MHz): δ = 175.2, 149.0, 148.0, 129.5, 121.4, 112.4, 111.3, 55.9, 55.8, 55.8, 52.0, 40.4. MS (ESI) calculated for C12H17NO4, [M+H]+, m/z 240.12, found for [M+Na]+, m/z 262.22.
General Procedure for N-Boc Deprotection to Synthesize 18 and 19
To a solution of N-Boc α–amino–β-aryl ester (16 or 17) (0.35 gm, 1.0 mmol) in CH2Cl2 (5 mL), trifluoro-acetic acid (TFA, 5 mL) was added drop-wise at 0 °C, and the mixture was warmed to RT. After stirring for 4 h, the reaction mixture was diluted with CH2Cl2 (25 mL) and neutralized by drop-wise addition of saturated aqueous NaHCO3 (20 mL). The organic layer was separated and following routine extraction, evaporation procedure gave a crude product, which was purified by flash chromatography using 70% (EtOAc: hexanes) to give the desired α–amino–β-aryl ester 18 or 19 as yellow oil in 80–86% yield. The NMR spectra for the corresponding product are presented above.
General Procedure for Step–wise Transesterification of THIQ3CA Methyl Esters to Ethyl Esters
To a stirred solution of dimethoxy THIQ3CA methyl ester (20, 21, or 27) (0.15 gm, 0.6 mmol) in methanol (25 mL), lithium hydroxide (0.072 gm, 3.0 mmol) was added. The mixture was stirred at RT for 5 h and then concentrated in vacuo, and washed with ethanol (3 × 10 mL). Ethanol (20 mL) was then added to the reaction solid residue followed by step-wise addition of thionyl chloride (SOCl2, 10 mL of 2 M in CH2Cl2) at 0 °C. The resulting mixture was refluxed for 4 hours, then cooled to RT and neutralized with saturated NaHCO3 solution. The product was extracted with CH2Cl2 (3 × 50 mL) followed by routine extraction and concentration process to give a residue that was purified using 45% (EtOAc: hexanes) as eluent of flash chromatography to give corresponding ethyl ester 12, 13, or 28 as yellow oil in 55–72% yield.
Ethyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (28)
1H NMR (CDCl3, 400 MHz): δ = 6.52 (s, 1 H, Ar), 6.45 (s, 1 H, Ar), 4.18-4.13 (q, 1J = 7.1 Hz, 2 H, CH2), 3.96 (s, 2 H, CH2), 3.80 (s, 3 H, CH3), 3.79 (s, 3 H, CH3), 3.63-3.60 (dd, 1J = 4.7 Hz, 2J = 10.1 Hz, 1 H, CH), 2.94-2.89 (dd, 1J = 4.6 Hz, 2J = 16.0 Hz, 1 H, CH), 2.83-2.77 (dd, 1J = 10.0 Hz, 2J = 15.8 Hz, 1 H, CH), 2.19 (br, s, 1 H, NH), 1.23 (t, 1J = 7.1 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ =173.1, 147.6, 126.7, 124.9, 111.8, 108.9, 61.0, 55.9, 55.9, 55.9, 47.0, 31.1, 14.2. MS (ESI) calculated for C14H19NO4, [M+H]+, m/z 266.14, found for [M+Na]+, m/z 288.21.
General Procedure for N-arylacylation of 1,2,3,4–tetrahydroisoquinoline–3–carboxylic acid Methyl or Ethyl ester (THIQ3CA Ester) and 1,2,3,4–tetrahydroisoquinoline (THIQ)
Method C: To a stirred solution of dimethoxy-1,2,3,4-tetrahydroisoquinoline (12, 13, 20, 21 or 27–29) (0.2 gm, 0.8–1.0 mmol) and triethylamine (0.3 gm, 3.0 mmol) in anhydrous CH2Cl2 (10 mL) at 0 °C was added 3,4,5-trimethoxy benzoyl chloride (0.24 gm, 1.05 mmol) or 2,5-dimethoxylphenyl acetyl chloride (0.24 gm, 1.05 mmol). The reaction mixture was allowed to warm to RT, refluxed for 4 h and then diluted with CH2Cl2 (15 mL). After routine extraction and concentrations steps, the crude product was purified by flash chromatography utilizing 50% (EtOAc: hexanes) to give the desired N-arylacyl THIQ3CA ester (30–35) or N-arylacyl THIQ (36) as colorless or yellow oil in 85–92% yield. Method D: To a stirred solution of either propionic or butanoic acid (0.3 gm, 1.4 mmol) in anhydrous CH2Cl2 (5 mL) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 0.3 gm, 1.55 mmol) and DMAP (0.17 gm, 1.4 mmol) at RT under nitrogen atmosphere. Dimethoxy-1,2,3,4-tetrahydroisoquinoline (12, 21 or 27) (0.4 gm, 1.5–1.6 mmol) in anhydrous CH2Cl2 (5 mL) was then added drop-wise. After stirring overnight, the reaction mixture was partitioned between 2 N HCl solution (20 mL) and CH2Cl2 (30 mL), and the organic layer washed with aqueous solution and concentrated to give a crude product. The crude product was purified using 50% (EtOAC: hexanes) as the mobile phase in flash chromatography to give the desired product (38–42) as colorless or yellow oil in 70–87 % yield.
Ethyl 5,6-dimethoxy-2-(3,4,5-trimethoxybenzoyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (30)
1H NMR (CDCl3, 400 MHz): δ = 6.85-6.68 (m, 2 H, Ar), 6.64-6.60 (m, 2 H, Ar), 5.34-5.32 & 4.78-4.76 (m, 1 H, CH), 5.08-5.04 & 4.50-4.41 (m, 2 H, CH2), 4.13-3.98 (m, 2 H, CH2), 3.80-3.74 (m, 15 H, 5 × CH3), 3.52-3.48 & 3.37-3.33 (d & dd, 1J = 16.3 Hz & 1J = 3.6 Hz, 2J = 16.3 Hz, 1 H, CH), 3.16-3.10 & 2.82-2.77 (dd & dd, 1J = 5.9 Hz, 2J = 16.2 Hz & 1J = 4.8 Hz, 2J = 16.6 Hz, 1 H, CH), 1.18 & 1.07 (t & t, 1J = 6.7 Hz & 1J = 8.8 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 171.5, 170.9, 170.5, 153.5, 153.4, 151.5, 151.0, 146.4, 139.4, 131.1, 126.8, 126.0, 125.7, 125.0, 122.0, 121.1, 111.6, 111.0, 104.4, 103.8, 61.6, 61.4, 60.9, 60.6, 60.4, 56.6, 56.3, 55.9, 52.0, 47.4, 42.9, 25.9, 24.7, 14.1. MS (ESI) calculated for C24H29NO8, [M+H]+, m/z 460.20, found for [M+H]+, m/z 460.40.
Methyl-2-(2-(2,5-dimethoxy phenyl) acetyl)- 5,6-dimethoxy- 1,2,3,4-tetrahydro-isoquinoline-3-carboxylate (31)
1H NMR (CDCl3, 400 MHz): δ = 6.77-6.62 (m, 5 H, Ar), 5.38-5.35 & 4.96-4.94 (dd & dd, 1J = 4.0 Hz, 2J = 6.2 Hz & 1J = 2.5 Hz, 2J = 5.9 Hz, 1 H, CH), 4.85-4.56 (dd, 1J = 17.0 Hz, 2J = 101.0 Hz, 1 H, CH), 4.49-4.34 (dd, 1J = 15.2 Hz, 2J = 43.2 Hz, 1 H, CH), 3.75-3.63 (m, 14 H, CH2, 4 × CH3), 3.47 (d, 1J = 52.2 Hz, 3 H, CH3), 3.48-3.44 & 3.39-3.34 (dd & dd, 1J = 2.5 Hz, 2J = 16.2 Hz & 1J = 4.0 Hz, 2J = 16.3 Hz, 1 H, CH), 2.92-2.86 & 2.67-2.62 (dd & dd, 1J = 6.3 Hz, 2J = 16.3 Hz & 1J = 5.8 Hz, 2J = 16.2 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ = 171.4, 171.3, 171.0, 153.7, 151.4, 151.1, 150.9, 150.5, 146.3, 146.0, 126.7, 125.6, 124.2, 123.8, 122.0, 121.4, 115.8, 113.2, 113.0, 111.5, 111.4, 111.0, 60.5, 60.4, 56.0, 55.8, 55.7, 54.5, 52.3, 52.2, 51.5, 45.3, 43.0, 35.2, 35.0, 25.9, 24.9. MS (ESI) calculated for C23H27NO7, [M+H]+, m/z 430.19, found for [M+H]+, m/z 430.36.
Methyl-2-(2-(2,5-dimethoxy phenyl) acetyl)- 5,8-dimethoxy- 1,2,3,4-tetrahydroisoquinoline-3-carboxylate (32)
1H NMR (CDCl3, 400 MHz): δ = 6.82-6.79 (dd, 1J = 2.9 Hz, 2J = 12.0 Hz, 1 H, Ar), 6.72 (d, 1J = 8.8 Hz, 1 H, Ar), 6.69-6.66 (dd, 1J = 2.8 Hz, 2J = 8.8 Hz, 1 H, Ar), 6.60-6.54 (m, 2 H, Ar), 5.64-5.62 & 4.96-4.94 (dd & dd, 1J = 2.4 Hz, 2J = 6.8 Hz & 1J = 1.6 Hz, 2J = 6.4 Hz, 1 H, CH), 5.02-4.68 (dd, 1J = 18.8 Hz, 2J = 120.8 Hz, 1 H, CH), 4.41-4.14 (dd, 1J = 17.1 Hz, 2J = 88.1 Hz, 1 H, CH), 3.76 (d, 1J = 23.8 Hz, 2 H, CH2), 3.71 (s, 3 H, CH3), 3.70 (s, 3 H, CH3), 3.67 (s, 3 H, CH3), 3.65 (s, 3 H, CH3), 3.48 (d, 1J = 46.7 Hz, 3 H, CH3), 3.42-3.36 (m, 1 H, CH), 2.78-2.72 & 2.55-2.49 (dd & dd, 1J = 6.8 Hz, 2J = 17.0 Hz & 1J = 6.5 Hz, 2J = 16.9 Hz, 1 H, CH). 13C NMR (100 MHz, CDCl3): δ = 171.7, 171.7, 171.5, 171.0, 153.8, 153.6, 151.3, 150.9, 150.6, 150.4, 150.1, 149.6, 124.4, 123.7, 122.3, 121.9, 121.1, 116.0, 115.6, 113.5, 113.0, 111.6, 111.4, 108.1, 107.7, 107.5, 107.4, 56.1, 56.0, 55.8 (× 2), 55.7 (× 3), 55.4, 54.0, 52.4, 52.2, 49.9, 41.2, 39.2, 35.1, 34.9, 25.2, 24.3. MS (ESI) calculated for C23H27NO7, [M+H]+, m/z 430.19, found for [M+Na]+, m/z 452.30.
Ethyl- 2-(2-(2,5-dimethoxy phenyl) acetyl)- 5,8-dimethoxy- 1,2,3,4-tetrahydro-isoquinoline-3-carboxylate (33)
1H NMR (CDCl3, 400 MHz): δ = 6.84-6.80 (dd, 1J = 2.8 Hz, 2J = 14.0 Hz, 1 H, Ar), 6.74-6.72 & 6.60-6.54 (m, 3 H, Ar), 6.69-6.66 (dd, 1J = 2.9 Hz, 2J = 8.9 Hz, 1 H, Ar), 5.61-5.59 & 4.94-4.93 (dd & dd, 1J = 2.1 Hz, 2J = 6.6 Hz & 1J = 1.5 Hz, 2J = 6.4 Hz, 1 H, CH), 5.00-4.68 (dd, 1J = 18.7 Hz, 2J = 110.4 Hz, 1 H, CH), 4.44-4.19 (dd, 1J = 17.1 Hz, 2J = 81.3 Hz, 1 H, CH), 4.06-3.85 (m, 2 H, CH2), 3.80-3.58 (m, 12 H, 3 × CH3), 3.57-3.39 (m, 1 H, CH), 2.77-2.71 & 2.55-2.49 (dd & dd, 1J = 6.7 Hz, 2J = 17.0 Hz & 1J = 6.5 Hz, 2J = 17.2 Hz, 1 H, CH), 1.06 & 0.98 (t & t, 1J = 7.1 Hz & 1J = 7.1 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 171.7, 170.9, 153.8, 153.6, 151.3, 151.0, 150.6, 150.4, 150.2, 149.9, 124.4, 123.7, 123.5, 122.4, 122.0, 121.3,117.2, 116.0, 115.6, 113.5, 113.0, 111.7, 111.4, 108.2, 107.8, 107.6, 61.4, 61.1, 56.2, 56.1, 56.0, 55.9, 55.8, 55.7, 55.7, 55.4, 54.2, 50.1,41.3, 39.3, 35.9, 35.0, 25.3, 24.3, 14.0, 13.9. MS (ESI) calculated for C24H29NO7, [M+H]+, m/z 444.20, found for [M+H]+, m/z 444.34.
Ethyl 6,7-dimethoxy-2-(3,4,5-trimethoxybenzoyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (34)
1H NMR (CDCl3, 400 MHz): δ = 6.85-6.60 (m, 4 H, Ar), 5.35-5.32 & 4.78-4.75 (m, 1 H, CH), 5.10-5.04 & 4.50-4.40 (m, 2 H, CH2), 4.13-3.99 (m, 2 H, CH2), 3.81-3.73 (m, 15 H, 5 × CH3), 3.52-3.48 & 3.37-3.33 (d & dd, 1J = 16.3 Hz & 1J = 3.6 Hz, 2J = 16.3 Hz, 1 H, CH), 3.16-3.10 & 2.82-2.77 (dd & dd, 1J = 5.9 Hz, 2J = 16.2 Hz & 1J = 4.8 Hz, 2J = 16.7 Hz, 1 H, CH), 1.17 & 1.06 (t & t, 1J = 6.7 Hz & 1J = 8.8 Hz, 3 H, CH3). 13C NMR (CDCl3, 400 MHz): δ = 171.5, 170.9, 170.5, 153.5, 153.4, 151.5, 151.0, 146.4, 139.4, 131.1, 126.8, 126.0, 125.7, 125.0, 122.0, 121.1, 111.6, 111.0, 104.4, 103.8, 61.6, 61.4, 60.9, 60.6, 60.4, 56.6, 56.3, 55.9, 52.0, 47.4, 42.9, 25.9, 24.7, 14.1. MS (ESI) calculated for C24H29NO8, [M+H]+, m/z 460.20, found for [M+H]+, m/z 460.34.
Methyl- 2-(2-(2,5-dimethoxy phenyl) acetyl)- 6,7-dimethoxy- 1,2,3,4-tetrahydroisoquinoline-3-carboxylate (35)
1H NMR (CDCl3, 400 MHz): δ = 6.80 (d, 1J = 2.6 Hz, 1 H, Ar), 6.74 (d, 1J = 8.8 Hz, 1 H, Ar), 6.72-6.67 (m, 1 H, Ar), 6.54 (d, 1J = 14.1 Hz, 1 H, Ar), 6.45 (d, 1J = 34.9 Hz, 1 H, Ar), 5.51-5.49 & 4.94-4.92 (dd & dd, 1J = 3.3 Hz, 2J = 6.0 Hz & 1J = 2.3 Hz, 2J = 5.8 Hz, 1 H, CH), 4.91-4.60 (dd, 1J = 17.2 Hz, 2J = 108.8 Hz, 1 H, CH), 4.52-4.30 (dd, 1J = 15.4 Hz, 2J = 71.5 Hz, 1 H, CH), 3.78-3.66 (m, 14 H, CH2, 4 × CH3), 3.50 (d, 1J = 51.5 Hz, 3 H, CH3), 3.14-3.06 (dt, 1J = 3.4 Hz, 2J = 15.8 Hz, 1 H, CH), 3.02-2.97 & 2.89-2.85 (dd & dd, 1J = 6.0 Hz, 2J = 18.8 Hz & 1J = 5.8 Hz, 2J = 15.8 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ =171.5, 171.4 (×2), 171.2, 153.8, 151.2, 150.6, 148.2, 148.1, 148.0, 147.7, 124.3, 124.2, 123.9, 123.8, 122.8, 115.9, 115.8, 113.3, 112.9, 111.6, 111.4, 111.2, 111.0, 109.3, 109.0, 56.1 (×2), 56.0 (×2), 55.9 (×2), 55.7, 55.0, 52.4, 52.3, 51.3, 45.5, 43.3, 35.3, 35.0, 31.2, 30.3. MS (ESI) calculated for C23H27NO7, [M+H]+, m/z 430.19, found for [M+Na]+, m/z 452.36.
1-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)-2-(2,5-dimethoxyphenyl) ethanone (36)
1H NMR (CDCl3, 400 MHz): δ = 6.78-6.64 (m, 5H, Ar), 4.54 (d, 1J= 48.8 Hz, 2H, CH2), 3.76 (s, 3H, CH3), 3.71(s, 3H, CH3), 3.69 (s, 3H, CH3), 3.67(s, 3H, CH3), 3.63-3.60 (m, 3H, CH, CH2), 3.58-3.54 (m, 1H, CH), 2.78 (t, 1J = 6.0 Hz, 1 H, CH), 2.64 (t, 1J = 5.9 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ = 170.4, 170.2, 153.7, 151.2, 151.0, 150.9, 150.8, 146.6, 146.2, 129.4, 128.5, 126.7, 126.2, 124.7, 122.0, 121.2, 117.1, 115.8, 112.9 (×2), 111.6 (×2), 111.5, 110.9, 110.7, 60.3, 56.1 (×2), 56.0, 55.9, 55.7, 55.6, 47.2, 43.9, 43.2, 39.7, 35.9, 35.1, 34.8, 23.6, 22.9. MS (ESI) calculated for C21H25NO5, [M+H]+, m/z 372.18, found for [M+Na]+, m/z 394.33.
Ethyl- 2-(2-(2,5-dimethoxy phenyl) butanoyl)- 5,6-dimethoxy- 1,2,3,4-tetrahydroisoquinoline-3-carboxylate (38)
1H NMR (CDCl3, 400 MHz): δ = 6.79-5.59 (m, 5 H, Ar), 5.40-5.37 & 4.67-4.65 (dd & dd, 1J = 3.6 Hz, 2J = 6.2 Hz & 1J = 2.8 Hz, 2J = 5.8 Hz, 1 H, CH), 4.79-4.74 & 4.52-4.48 (dd, 1J = 16.8 Hz, 2J = 107.7 Hz, 1 H, CH),4.47-4.35 (dd, 1J = 15.1 Hz, 2J = 29.7 Hz, 1 H, CH), 4.05-3.90 (m, 2 H, CH2), 3.80-3.60 (m, 12 H, 4 × CH3), 3.54-3.50 & 3.42-3.37 (dd & dd, 1J = 2.6 Hz, 2J = 16.1 Hz & 1J = 3.6 Hz, 2J = 16.3 Hz, 1 H, CH), 2.86-2.80 (m, 1 H, CH), 2.63-2.55 (m, 2 H, CH2), 2.43-2.26 (m, 2 H, CH2), 1.98-1.81 (m, 2 H, CH2), 1.08 & 1.00 (t & t, 1J = 7.1 Hz, 1J = 7.1 Hz, 3 H, CH3). 13C NMR (CDCl3, 100 MHz): δ = 177.9, 173.1, 171.1, 170.6, 153.5, 153.3, 151.9, 151.6, 151.4, 151.0, 146.4, 146.1, 131.3, 131.0, 129.8, 126.8, 126.3, 126.0, 125.5, 122.0, 121.4, 116.4, 116.3, 111.5, 111.2, 111.0, 61.6, 61.1, 60.6, 60.5, 55.9, 55.8, 55.7 (×2), 54.6, 51.1, 45.0, 42.9, 33.4, 33.1, 29.8, 29.8, 29.5, 26.1, 25.0, 24.9, 24.8, 14.1, 14.0. MS (ESI) calculated for C26H33NO7, [M+H]+, m/z 472.23, found for [M+H]+, m/z 472.40.
Methyl 2-(3-(2,5-dimethoxyphenyl)propanoyl)-5,8-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (39)
1H NMR (CDCl3, 400 MHz): δ = 6.73-6.67 (m, 2 H, Ar), 6.63-6.54 (m, 3 H, Ar), 5.65-5.63 & 4.96-4.94 (dd & dd, 1J = 2.2 Hz, 2J = 6.8 Hz & 1J = 1.6 Hz, 2J = 6.4 Hz, 1 H, CH), 5.03-4.98 & 4.61-4.56 (dd, 1J = 18.7 Hz, 2J = 168.5 Hz, 1 H, CH), 4.43-4.14 (dd, 1J = 17.1 Hz, 2J = 98.6 Hz, 1 H, CH), 3.73 (s, 3 H, CH3), 3.70 (s, 3 H, CH3), 3.69 (s, 3 H, CH3), 3.67 (s, 3 H, CH3), 3.55 (s, 3 H, CH3), 3.52-3.47 & 3.43-3.38 (dd & dd, 1J = 1.4 Hz, 2J = 17.7 Hz & 1J = 2.0 Hz, 2J = 17.1 Hz, 1 H, CH), 2.94-2.85 (m, 2 H, CH2), 2.78-2.53 (m, 3 H, CH, CH2). 13C NMR (CDCl3, 100 MHz): δ = 173.2, 172.9, 171.6, 171.1, 153.6, 153.5, 151.8, 151.6, 150.9, 150.6, 150.1, 149.6, 130.7, 130.3, 122.4, 122.3, 121.7, 121.1, 116.5, 116.4, 111.8, 111.5, 111.2, 111.1, 108.1, 107.8, 107.6, 107.3, 55.8, 55.8, 55.7 (×2), 55.4, 55.3, 54.0, 52.6, 52.3, 49.6, 41.0, 38.9, 34.2, 34.1, 27.2, 26.7, 25.3, 24.3. MS (ESI) calculated for C24H29NO7, [M+H]+, m/z 444.20, found for [M+Na]+, m/z 465.97.
Methyl 2-(4-(2,5-dimethoxyphenyl)butanoyl)-5,8-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (40)
1H NMR (CDCl3, 400 MHz): δ = 6.70-6.65 (m, 2 H, Ar), 6.63-6.55 (m, 3 H, Ar), 5.63-5.60 & 4.72 (dd & d, 1J = 2.3 Hz, 2J = 6.8 Hz & 1J = 4.9 Hz, 1 H, CH), 5.02-4.97 & 4.56-4.53 (dd, 1J = 18.7 Hz, 1J = 181.9 Hz, 1 H, CH), 4.44-4.09 (dd, 1J = 17.0 Hz, 2J = 117.8 Hz, 1 H, CH), 3.71 (s, 3 H, CH3), 3.70 (s, 3 H, CH3), 3.69 (s, 3 H, CH3), 3.67 (s, 3 H, CH3), 3.55 (s, 3 H, CH3), 3.40-3.37 (dd, 1J = 2.2 Hz, 2J = 17.1 Hz, 1 H, CH), 2.75-2.70 (dd, 1J = 6.8 Hz, 2J = 17.0 Hz,1 H, CH), 2.66-2.53 (m, 2 H, CH2), 2.51-2.31 (m, 2 H, CH2), 1.97-1.89 (m, 2 H, CH2). 13C NMR (CDCl3, 100 MHz): δ = 173.4, 173.0, 171.6, 171.0, 153.5, 151.9 (×2), 150.6, 150.1, 149.6, 131.5, 131.2, 122.5, 122.4, 121.8, 121.1, 116.4, 116.3, 111.5, 111.3, 111.2, 108.2, 107.8, 107.6, 107.4, 55.8, 55.7, 55.4, 53.9, 52.6, 52.3, 49.6, 40.9, 38.8, 33.3, 33.0, 29.9, 29.8, 25.3, 25.1, 24.9, 24.3. MS (ESI) calculated for C25H31NO7, [M+H]+, m/z 458.22, found for [M+Na]+, m/z 480.40.
Methyl 2-(3-(2,5-dimethoxyphenyl)propanoyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (41)
1H NMR (CDCl3, 400 MHz): δ = 6.72-6.69 (m, 2 H, Ar), 6.67-6.63 (m, 1 H, Ar), 6.54 (d, 1J = 9.4 Hz, 1 H, Ar), 6.45 (s, 1 H, Ar), 5.52-5.50 & 4.87-4.85 (dd & dd, 1J = 3.0 Hz, 2J = 6.1 Hz & 1J = 2.4 Hz, 2J = 5.8 Hz, 1 H, CH), 4.88-4.54 (dd, 1J = 17.0 Hz, 2J = 120.7 Hz, 1 H, CH), 4.53-4.30 (dd, 1J = 17.2 Hz, 2J = 74.0 Hz, 1 H, CH), 3.78-3.67 (m, 12 H, 4 × CH3), 3.55 (d, 1J = 8.5 Hz, 3 H, CH3), 3.78-3.09 (m, 1 H, CH), 2.99-2.84 (m, 3 H, CH, CH2), 2.70-2.67 (m, 2 H, CH2).13C NMR (CDCl3,100 MHz): δ = 172.8, 172.7, 171.6, 171.2, 153.6 (×2), 151.8, 151.6, 148.3, 148.1 (×2), 147.8, 130.7, 130.3, 124.5, 124.1, 123.7, 122.9, 116.5 (×2), 111.7, 111.5, 111.3 (×2), 111.2, 111.1, 111.0, 109.4, 108.9, 65.8, 55.9 (×2), 55.8, 55.7, 54.9, 52.6, 52.3, 51.0, 46.9, 45.1, 43.1, 34.2, 30.3, 29.4, 27.1, 26.7, 26.5. MS (ESI) calculated for C24H29NO7, [M+H]+, m/z 444.20, found for [M+Na]+, m/z 466.04.
Methyl 2-(4-(2,5-dimethoxyphenyl)butanoyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (42)
1H NMR (CDCl3, 400 MHz): δ = 6.71-6.62 (m, 3 H, Ar), 6.55 (d, 1J = 6.16 Hz, 1 H, Ar), 6.46 (s, 1 H, Ar), 5.52-5.50 & 4.69-4.67 (dd & dd, 1J = 2.9 Hz, 2J =5.9 Hz & 1J = 2.4 Hz, 2J = 5.6 Hz, 1 H, CH), 4.86-4.82 & 4.48-4.48 (dd, 1J = 17.0 Hz, 2J = 136.0 Hz, 1 H, CH), 4.48-4.25 (dd, 1J = 16.0 Hz, 2J = 75.8 Hz, 1 H, CH), 3.78 (s, 3 H, CH3), 3.77 (s, 3 H, CH3), 3.69 (s, 3 H, CH3), 3.68 (s, 3 H, CH3), 3.54 (d, 1J = 3.2 Hz, 3 H, CH3), 3.17-3.09 (m, 1 H, CH), 3.04-2.93 (m, 1 H, CH), 2.64-2.58 (m, 2 H, CH2), 2.44-2.40 (m, 2 H, CH2), 1.97-1.89 (m, 2 H, CH2). 13C NMR (CDCl3, 100 MHz): δ =173.1, 173.0, 171.6, 171.1, 153.5, 151.9, 151.8, 148.3, 148.1, 147.8, 131.3, 131.2, 124.5, 124.1, 123.6, 122.9, 116.5, 116.4, 111.5, 111.3 (×2), 111.2 (×2), 111.0, 109.4, 109.0, 56.6, 56.0, 55.9 (×2), 55.7, 54.8, 52.6, 52.3, 50.9, 45.2, 43.1, 33.1, 33.0, 31.6, 31.3, 30.9, 30.3, 29.8 (×2), 28.9, 25.0, 24.8. MS (ESI) calculated for C25H31NO7, [M+H]+, m/z 458.22, found for [M+Na]+, m/z 479.96.
2-(2-(2,5-Dihydroxyphenyl)acetyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (37)
To a stirred solution of THIQ3CA ester 35 (0.45 gm, 1.0 mmol) in anhydrous CH2Cl2 (5 mL), BBr3 (1 M solution in CH2Cl2, 0.4 gm, 9 mmol) was added dropwise at −78 °C under nitrogen atmosphere over 10 min. After 2 h, the reaction mixture was brought to RT and allowed to equilibrate overnight. A mixture of methanol/water (1:1 v/v, 2 mL) was then added slowly at 0 °C and vigorously stirred for 10 min to quench the reaction. The reaction mixture was then concentrated and partitioned between EtOAc (25 mL) and saturated solution of NH4Cl (15 mL), and aqueous layer further washed with EtOAc (2 × 20 mL). The combined organic layer was concentrated in vacuo and the crude was purified by 0% to 25% (CH3OH:CH2Cl2) as mobile phase gradient in flash chromatography to yield 37 (75 %) as yellow solid. 1H NMR (acetone-d6, 400 MHz): δ = 6.65-6.58 (dd, 1J = 2.9 Hz, 2J = 25.1 Hz, 1 H, Ar), 6.57-6.55 (dd, 1J = 3.2 Hz, 2J = 7.3 Hz, 1 H, Ar), 6.53 (d, 1J = 14.7 Hz, 2 H, Ar), 6.47-6.44 (dd, 1J = 2.9 Hz, 1J = 8.6 Hz, 1 H, Ar), 5.20-5.18 & 5.15-5.13 (dd & dd, 1J = 3.6 Hz, 2J = 5.9 Hz & 1J = 2.6 Hz, 2J = 5.7 Hz, 1 H, CH), 4.79-3.43 (dd & dd, 2 H, CH2, and d & d & d, 2 H, CH2), 3.07-3.02 & 3.01-2.96 (dd & dd, 1J = 2.5 Hz, 2J = 15.5 Hz & 1J = 3.6 Hz, 2J = 15.6 Hz, 1H, CH), 2.88-2.84 (m, 1 H, CH). 13C NMR (acetone-d6, 100 MHz): δ = 173.3, 172.2, 151.3, 149.9, 149.5, 145.0, 144.9, 124.7, 124.6, 124.5, 124.0, 123.3, 118.5, 118.0, 117.9, 117.8, 117.7, 115.7, 115.6, 115.5, 115.2, 113.8, 113.7, 56.2, 52.6, 46.5, 43.9, 36.5, 36.1, 31.9, 30.9. MS (ESI) calculated for C18H17NO7, [M+H]+, m/z 360.11, found for [M+Na]+, m/z 382.24.
Methyl 6,7-dimethoxy-2-(4-methoxybenzyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (43)
To a stirred solution of methyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate 27 (0.1 gm, 0.4 mmol) in CH3CN (2 mL), diisopropylethylamine (DIPEA) (0.15 gm, 1.2 mmol) was slowly added. After 15 min, 4-methoxybenzyl chloride (0.078 gm, 0.5 mmol) was dissolved in CH3CN (1 mL) and was then added slowly to the reaction mixture. The reaction mixture was heated up in the microwave (CEM Discover) for 20 min at 150 °C. The reaction mixture was cooled to RT, diluted with EtOAc (10 mL) and organic layer worked up as usual to give a crude product, which was purified using 30 % (EtOAc: hexanes) in flash chromatography to yield 43 (yellow oil, 92%). 1H NMR (CDCl3, 400 MHz): δ = 7.29 (d, 1J = 8.6 Hz, 2 H, Ar), 6.87 (d, 1J = 8.6 Hz, 2 H, Ar), 6.58 (s, 1 H, Ar), 6.47 (s, 1H, Ar), 3.95 (d, 1J = 15.2 Hz, 1H, CH), 3.85 (d, 1J = 2.2 Hz, 2 H, CH2), 3.84 (s, 3 H, CH3), 3.80 (s, 3 H, CH3), 3.79 (s, 3 H, CH3), 3.77-3.75 (dd, 1J = 4.1 Hz, 2J = 6.0 Hz, 1 H, CH), 3.72 (d, 1J = 15.8 Hz, 1 H, CH), 3.68 (s, 3 H, CH3), 3.14-3.09 (dd, 1J = 6.0 Hz, 2J = 16.1 Hz, 1 H, CH), 3.05-3.00 (dd, 1J = 3.8 Hz, 2J = 16.0 Hz, 1 H, CH). 13C NMR (CDCl3, 100 MHz): δ = 173.2, 158.9, 147.5, 130.6, 130.1, 128.6, 126.1, 124.0, 113.8, 111.3, 109.3, 58.9, 58.4, 55.9, 55.8, 55.3, 51.4, 50.6, 30.9. MS (ESI) calculated for C21H25NO5, [M+H]+, m/z 372.18, found for [M+H]+, m/z 372.32.
General Procedure for Xylene-Based Dimerization of 6,7-Dimethoxy THIQ3CA Ethyl Ester
Ethyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate 28 (0.75 gm, 2.8 mmol) was dissolved in DMF:CH3CN (1:1 v/v, 8 mL) and stirred with Cs2CO3 (2.2 gm, 11.2 mmol). After 15 min, p- or m- α,α′-dibromo xylene (0.37 gm, 1.4 mmol) in CH3CN (2 mL) was slowly added to the reaction mixture and then refluxed for 12 h. The reaction mixture was cooled, partitioned between EtOAc (25 mL) and water (25 mL), and the organic layer processed in a routine manner to prepare a crude product, which was purified using 30% (EtOAc: hexanes) in flash chromatography to give the desired xylene-based dimer (44 or 45) as yellow oil in 62–66% yield.
Diethyl 2,2′-(1,3-phenylene bis(methylene)) bis(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate) (44)
1H NMR (CDCl3, 400 MHz): δ = 7.41 (s, 1 H, Ar), 7.30 (s, 3 H, Ar), 6.58 (s, 2 H, Ar), 6.46 (s, 2 H, Ar), 4.19-4.10 (m, 4 H, 2 × CH2), 33.99-3.93 (m, 6 H, 2 × CH, 2 × CH2), 3.84 (s, 6 H, 2 × CH3), 3.79 (s, 6 H, 2 × CH3), 3.76-3.72 (m, 4 H, 2 × CH2), 3.14-3.09 (dd, 1J = 6.0 Hz, 2J = 16.1 Hz, 2 H, 2 × CH), 3.05-3.00 (dd, 1J = 4.0 Hz, 2J = 16.0 Hz, 2 H, 2 × CH), 1.24 (t, 1J= 7.1 Hz, 6 H, 2 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 172.7, 147.5, 138.9, 129.4, 128.3, 127.8, 126.1, 124.1, 111.3, 109.3, 60.3, 59.2, 59.0, 55.9, 55.8, 50.7, 31.1, 14.4. MS (ESI) calculated for C36H44N2O8, [M+H]+, m/z 633.32, found for [M+H]+, m/z 633.16.
Diethyl 2,2′-(1,4-phenylene bis(methylene)) bis(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (45)
1H NMR (CDCl3, 400 MHz): δ = 7.28 (s, 4 H, Ar), 6.52 (s, 2 H, Ar), 6.40 (s, 2 H, Ar), 4.12-4.05 (m, 4 H, 2 × CH2), 3.85-3.93 (m, 6 H, 2 × CH, 2 × CH2), 3.77 (s, 6 H, 2 × CH3), 3.73 (s, 6 H, 2 × CH3), 3.60-3.70 (m, 4 H, 2 × CH2), 3.07-3.03 (dd, 1J = 3.5 Hz, 2J = 14.9 Hz, 2 H, 2 × CH), 3.00-2.95 (dd, 1J = 4.1 Hz, 2J = 16.0 Hz, 2 H, 2 × CH), 1.18 (t, 1J = 7.1 Hz, 6 H, 2 × CH3). 13C NMR (CDCl3, 100 MHz): δ = 172.7, 147.5, 138.8, 129.0, 126.0, 124.1, 111.3, 109.2, 60.4, 59.2, 58.7, 55.9, 55.8, 50.7, 30.9, 14.4. MS (ESI) calculated for C36H44N2O8, [M+H]+, m/z 633.32, found for [M+H]+, m/z 633.23.
Acknowledgments
This work was supported by grants HL090586, HL099420, and HL107152 from the National Institutes of Health. We thank Dr. Neil Scarsdale of Virginia Commonwealth University for help with NMR experiments.
Abbreviations
- AT
antithrombin
- fXa
factor Xa
- FID
free induction decay
- THIQ
1,2,3,4-tetrahydroisoquinoline
- THIQ3CA
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ueno H, Yokota K, Hoshi J, Yasue K, Hayashi M, Hase Y, Uchida I, Aisaka K, Katoh S, Cho H. J Med Chem. 2005;48:3586–3604. doi: 10.1021/jm058160e. [DOI] [PubMed] [Google Scholar]
- 2.Ueno H, Yokota K, Hoshi J, Yasue K, Hayashi M, Uchida I, Aisaka K, Hase Y, Katoh S, Cho H. Bioorg Med Chem Lett. 2005;15:185–189. doi: 10.1016/j.bmcl.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 3.Watson NS, Adams C, Belton D, Brown D, Burns-Kurtis CL, Chaudry L, Chan C, Convery MA, Davies DE, Exall AM, Harling JD, Irvine S, Irving WR, Kleanthous S, McLay IM, Pateman AJ, Patikis AN, Roethke TJ, Senger S, Stelman GJ, Toomey JR, West RI, Whittaker C, Zhou P, Young RJ. Bioorg Med Chem Lett. 2011;21:1588–1592. doi: 10.1016/j.bmcl.2011.01.129. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Feng J, Liu C, Zhang L, Jiao J, Fang H, Su L, Zhang X, Zhang J, Li M, Wang B, Xu W. Bioorg Med Chem. 2010;18:1761–1772. doi: 10.1016/j.bmc.2010.01.060. [DOI] [PubMed] [Google Scholar]
- 5.Cheng S, Zhang X, Wang W, Zhao M, Zheng M, Chang HW, Wu J, Peng S. Eur J Med Chem. 2009;44:4904–4919. doi: 10.1016/j.ejmech.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Azukizawa S, Kasai M, Takahashi K, Miike T, Kunishiro K, Kanda M, Mukai C, Shirahase H. Chem Pharm Bull. 2008;56:335–345. doi: 10.1248/cpb.56.335. [DOI] [PubMed] [Google Scholar]
- 7.Cai TB, Zou Z, Thomas JB, Brieaddy L, Navarro HA, Carroll FI. J Med Chem. 2008;51:1849–1860. doi: 10.1021/jm701344b. [DOI] [PubMed] [Google Scholar]
- 8.Desai UR. Med Res Rev. 2004;24:151–181. doi: 10.1002/med.10058. [DOI] [PubMed] [Google Scholar]
- 9.Raghuraman A, Liang A, Krishnasamy C, Lauck T, Gunnarsson GT, Desai UR. Eur J Med Chem. 2009;44:2626–2631. doi: 10.1016/j.ejmech.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Horani RA, Liang A, Desai UR. J Med Chem. 2011;54:6125–6138. doi: 10.1021/jm2008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox ED, Cook JM. Chem Rev. 1995;95:1797–1842. [Google Scholar]
- 12.Whaley WM, Govindachari TR. In: Organic Reactions. Adams R, editor. Vol. 6. Wiley; New York: 1951. pp. 74–150. [Google Scholar]
- 13.Liu JM, Young JJ, Li YJ, Sha CK. J Org Chem. 1986;51:1120–1123. [Google Scholar]
- 14.Kotha S, Sreenivasachary N. Bioorg Med Chem Lett. 2000;10:1413–1415. doi: 10.1016/s0960-894x(00)00259-6. [DOI] [PubMed] [Google Scholar]
- 15.Kotha S, Sreenivasachary N. Chem Commun. 2000:503–504. [Google Scholar]
- 16.Toda J, Matsumoto S, Saitoh T, Sano T. Chem Pharm Bull. 2000;48:91–98. doi: 10.1248/cpb.48.91. [DOI] [PubMed] [Google Scholar]
- 17.Hirsenkorn R. Tetrahedron Lett. 1991;32:1775–1778. [Google Scholar]
- 18.Kommidi H, Balasubramaniam S, Aidhen IS. Tetrahedron. 2010;66:3723–3729. [Google Scholar]
- 19.Kihara M, Kashimoto M, Kobayashi Y. Tetrahedron. 1992;48:67–78. [Google Scholar]
- 20.Gremmen C, Wanner MJ, Koomen GJ. Tetrahedron Lett. 2001;42:8885–8888. [Google Scholar]
- 21.Connors RV, Zhang AJ, Shuttleworth SJ. Tetrahedron Lett. 2002;43:6661–6663. [Google Scholar]
- 22.Miles WH, Heinsohn SK, Brennan MK, Swarr DT, Eidam PM, Gelato KA. Synthesis. 2002:1541–1545. [Google Scholar]
- 23.Nielsen TE, Diness F, Meldal M. Curr Opin Drug Discov Devel. 2003;6:801–814. [PubMed] [Google Scholar]
- 24.Pal B, Jaisankar P, Giri VS. Synth Commun. 2003;33:2339–2348. [Google Scholar]
- 25.Nielsen TE, Meldal M. J Org Chem. 2004;69:3765–3773. doi: 10.1021/jo049918p. [DOI] [PubMed] [Google Scholar]
- 26.Taylor MS, Jacobsen EN. J Am Chem Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]
- 27.Maryanoff BE, Zhang HC, Cohen JH, Turchi IJ, Maryanoff CA. Chem Rev. 2004;104:1431–1628. doi: 10.1021/cr0306182. [DOI] [PubMed] [Google Scholar]
- 28.Lesma G, Danieli B, Lodroni F, Passarella D, Sacchetti A, Silvani A. Comb Chem & High Thr Scr. 2006;9:691–701. doi: 10.2174/138620706778700134. [DOI] [PubMed] [Google Scholar]
- 29.Meutermans WDF, Alewood PF. Tetrahedron Lett. 1995;36:7709–7712. [Google Scholar]
- 30.Nicoletti M, O’Hagan D, Slawin AMZ. J Chem Soc Perkin Trans. 2002;1:116–121. [Google Scholar]
- 31.Judeh ZMA, Ching CB, Bu J, McCluskey A. Tetrahedron Lett. 2002;43:5089–5091. [Google Scholar]
- 32.Schollkopf U, Hinrchs R, Lonsky R. Angew Chem, Int Engl Ed. 1987;26:143–145. [Google Scholar]
- 33.Seebach D, Dziadulewicz E, Behrendt L, Cantoreggi S, Fitzi R. Liebigs Ann der Chem. 1989;12:1215–1232. [Google Scholar]
- 34.Kammermeier T, Lerch U, Sommer C. Synthesis. 1992:1157–1160. [Google Scholar]
- 35.Wang C, Moseberg HI. Tetrahedron Lett. 1995;36:3623–3626. [Google Scholar]
- 36.Mash EA, Williams LJ, Pfeiffer SS. Tetrahedron Lett. 1997;38:6977–6988. [Google Scholar]
- 37.Chinchilla R, Galindo N, Najera C. Synthesis. 1999:704–717. [Google Scholar]
- 38.Fukuyama T, Linton SD, Tun MM. Tetrahedron Lett. 1990;31:5989–5992. [Google Scholar]
- 39.Kazmierski WM, Urbanczyk-Lipkowska Z, Hruby VJ. J Org Chem. 1994;59:1789–1795. [Google Scholar]
- 40.Al-Horani RA, Desai UR. Tetrahedron. 2010;66:2907–2918. doi: 10.1016/j.tet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RB, Rai DK. J Phys Chem. 1965;69:3461–3462. [Google Scholar]
- 42.Knor S, Laufer B, Kessler H. J Org Chem. 2006;71:5625–5630. doi: 10.1021/jo060704c. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt U, Lieberknecht A, Wild J. Synthesis. 1984:53–60. [Google Scholar]
- 44.Enomoto T, Yasui Y, Takemoto Y. J Org Chem. 2010;75:4876–4879. doi: 10.1021/jo100788j. [DOI] [PubMed] [Google Scholar]
- 45.Un-natural α-amino acids including 4–methoxy, 2,3,4–trimethoxy, and 3,4,5–trimethoxy– phenylalanine methyl esters were in fact synthesized and fully characterized by 1H and 13C NMR. Their syntheses were achieved in quantitative yields by application of similar conditions of Horner–Wadsworth–Emmons reaction using glycine donor 3, followed by the catalytic hydrogenation reaction.
- 46.Knowles WS. Angew Chem, Int Engl Ed. 2002;41:1998–2007. [Google Scholar]
- 47.Wang W, Li H. Tetrahedron Lett. 2004;45:8479–8481. [Google Scholar]
- 48.Bringmann G, Ochse M, Michel M. Tetrahedron. 2000;56:581–585. [Google Scholar]
- 49.Kitamura M, Tsukamoto M, Takaya H, Noyori R. Bull Chem Soc Jpn. 1996;69:1695–1700. [Google Scholar]
- 50.Fraenkel G, Cava MP, Dalton DR. J Am Chem Soc. 1967;89:329–332. [Google Scholar]
- 51.de Koning CB, van Otterlo WAL, Michael JP. Tetrahedron. 2003;59:8337–8345. [Google Scholar]
- 52.Wouters J, Elassad K, Norberg B, Graulich A, Liegeois JF. Eur J Med Chem. 2010;45:3240–3244. doi: 10.1016/j.ejmech.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 53.Bishop GJ, Price BJ, Sutherland IO. J Chem Soc, Chem Commun. 1967:672–674. [Google Scholar]
- 54.Shanan-Atidi H, Bar-Eli KH. J Phys Chem. 1970;74:961–963. [Google Scholar]
- 55.Kost D, Carlson EH, Raban M. J Chem Soc, Chem Commun. 1971:656–657. [Google Scholar]
- 56.3G0 = −2.3RTlogKe, in which 3G0 is the standard energy difference; R is the gas constant; T is temperature in Kelvin and Ke is the equilibrium constant which was found to be 0.33 based on the NMR study.
- 57.Clayden J, Moran WJ, Edwards PJ, LaPlante SR. Angew Chem Int Ed. 2009;48:6398– 6401. doi: 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]