

Background: Nucleotide metabolism is a promising therapeutic target in Trypanosoma brucei.
Results: T. brucei has two thymidine kinase sequences, domain 1 and domain 2, fused into a single open reading frame.
Conclusion: Domain 1 is catalytically inactive but improves substrate binding by domain 2.
Significance: Thymidine kinases with tandem repeats exist in many parasites and may represent an adaptive trait.
Keywords: Nucleoside, Nucleoside Nucleotide Biosynthesis, Nucleoside Nucleotide Metabolism, Parasite Metabolism, Trypanosoma brucei, African Sleeping Sickness, TK1, Tandem Protein, Thymidine Kinase
Abstract
Trypanosoma brucei causes African sleeping sickness, a disease for which existing chemotherapies are limited by their toxicity or lack of efficacy. We have found that four parasites, including T. brucei, contain genes where two or four thymidine kinase (TK) sequences are fused into a single open reading frame. The T. brucei full-length enzyme as well as its two constituent parts, domain 1 and domain 2, were separately expressed and characterized. Of potential interest for nucleoside analog development, T. brucei TK was less discriminative against purines than human TK1 with the following order of catalytic efficiencies: thymidine > deoxyuridine ≫ deoxyinosine > deoxyguanosine. Proteins from the TK1 family are generally dimers or tetramers, and the quaternary structure is linked to substrate affinity. T. brucei TK was primarily monomeric but can be considered a two-domain pseudodimer. Independent kinetic analysis of the two domains showed that only domain 2 was active. It had a similar turnover number (kcat) as the full-length enzyme but could not self-dimerize efficiently and had a 5-fold reduced thymidine/deoxyuridine affinity. Domain 1, which lacks three conserved active site residues, can therefore be considered a covalently attached structural partner that enhances substrate binding to domain 2. A consequence of the non-catalytic role of domain 1 is that its active site residues are released from evolutionary pressure, which can be advantageous for developing new catalytic functions. In addition, nearly identical 89-bp sequences present in both domains suggest that the exchange of genetic material between them can further promote evolution.
Introduction
Trypanosoma brucei is a unicellular parasite causing African sleeping sickness (1), a fatal disease that is spread by tsetse flies. In the first stage of the disease, the parasites circulate in the blood and lymph and cause an undulating fever. In the second stage of the disease, the parasites enter the central nervous system, and this eventually leads to coma and death. The disease is most often diagnosed in the second stage and, at this point, there are only two drugs that are effective, melarsoprol and eflornithine. Melarsoprol is a toxic arsenic compound with severe side effects; ∼10% of the patients acquire drug-induced encephalopathy, and 50% of these cases are fatal. Eflornithine, which is generally given in combination with nifurtimox, is only effective against T. brucei gambiense, one of two subspecies causing the disease. Nucleoside analogs have been used extensively as anticancer and antiviral agents, and many of them have been shown in clinical studies to pass the blood-brain barrier via transporters used for the uptake of natural nucleosides into the brain (2). Some of these nucleoside analogs, including cordycepin and adenine arabinoside, are active against T. brucei (3, 4). It has been shown that adenine arabinoside is phosphorylated into the corresponding nucleoside triphosphate, which causes inhibition of T. brucei nucleic acid biosynthesis, reduced ATP pools (nucleoside/nucleotide phosphorylation requires ATP), and unbalanced dNTP pools (4).
The phosphorylation of nucleoside analogs is dependent on nucleoside and deoxynucleoside kinases with substrate specificities that vary from species to species (5, 6). The specific properties of nucleoside/deoxynucleoside kinases in pathogens are interesting from a drug development perspective. Acyclovir and other nucleoside analogs used against herpes simplex virus are, for example, specifically recognized by the virus's thymidine kinase (TK)3 but not by any of the host cell's kinases. Therefore, these drugs are able to selectively target virus-infected cells (6). T. brucei has two known nucleoside/deoxynucleoside kinases, adenosine kinase and TK (Fig. 1). Studies of trypanosomes grown in the presence of different deoxynucleosides have shown that deoxyadenosine and thymidine are readily phosphorylated by the parasites, and their pools of dATP and dTTP increase under these conditions (7). The recombinant T. brucei adenosine kinase phosphorylates adenosine and deoxyadenosine as well as antitrypanosomal adenosine analogs, such as cordycepin, adenine arabinoside, and fludarabine (4). The crucial role of this enzyme in nucleoside analog activation was demonstrated in adenosine kinase knockdown T. brucei cells, which had a strongly reduced sensitivity to the nucleoside analog drugs (4, 8). Much less is known about the T. brucei TK; its activity has so far only been studied in an acetone-precipitated cell extract from the T. brucei rhodesiense subspecies (9, 10). Similarly to human thymidine kinase 1 (TK1), the partially purified T. brucei rhodesiense enzyme phosphorylates thymidine and is feedback-inhibited by dTTP. T. brucei possesses de novo dNTP synthesis pathways but has limited supplies of CDP and CTP available for de novo dCTP synthesis (7, 11). This is compensated for by having a ribonucleotide reductase that strongly prefers CDP to UDP (7, 12) and by lacking dCMP deaminase, an enzyme present in most other eukaryotes that participates in a pathway that converts dCTP to dTTP. A consequence of these dCTP-conserving strategies is that de novo dTTP synthesis can only be performed via UDP reduction. The parasites are able to compensate for this problem by acquiring dTTP via TK-mediated salvage pathways.
FIGURE 1.

Biosynthesis of dNTPs in T. brucei. Reactions catalyzed by ribonucleotide reductase (RNR), adenosine kinase (AK), and TK are highlighted. Dephosphorylation reactions, as indicated by the gray arrows, can proceed either directly from dNTPs to dNMPs or via dNDPs.
Mammalian cells have a much wider repertoire of deoxynucleoside kinases than T. brucei and are able to phosphorylate all natural deoxynucleosides found in the cytosol and mitochondria (5). In the cytosol, TK1 phosphorylates thymidine and deoxyuridine, whereas deoxycytidine kinase phosphorylates deoxycytidine, deoxyadenosine, and deoxyguanosine. In the mitochondria, thymidine kinase 2 (TK2) and deoxyguanosine kinase phosphorylate pyrimidine and purine deoxynucleosides, respectively. TK2 belongs to the same family of deoxynucleoside kinases as deoxycytidine kinase, deoxyguanosine kinase, and herpes simplex TK, whereas TK1 belongs to a separate family of enzymes consisting of TKs from a wide variety of organisms.
We have found that four parasite species, T. brucei, Trypanosoma congolense, Ascaris suum, and Trichomonas vaginalis, have unusually large deoxynucleoside kinases with two or four TK1 sequences (here called domains) in tandem within the same polypeptide. The two homologous domains forming the T. brucei TK have an overall identity of 62%, and each contains an identical stretch of 33 amino acids. The conserved stretch is 99% identical at the DNA sequence level, suggesting that it is the product of genetic exchange between the two domains. The T. brucei full-length TK and each of its two domains were independently expressed and characterized. Domain 1, which lacks three conserved residues involved in substrate binding and catalysis, was catalytically inactive. Domain 2, on the other hand, was active and had a similar turnover number (kcat) as the wild-type TK. The main difference in enzyme activity between domain 2 and the full-length enzyme was a ∼5-fold reduction in affinity for its main substrates, thymidine and deoxyuridine. In contrast to all TK1 proteins studied to date (5), domain 2 did not dimerize efficiently. Domain 1 can therefore be considered a covalently attached partner that helps domain 2 to bind its substrates more efficiently. A consequence of domain 1 losing its catalytic function and becoming a supporting partner of domain 2 is that the evolutionary pressure to keep the remaining active site residues is relieved. It is therefore not surprising that it has lost more than one of the catalytic residues, whereas most other conserved residues are retained. Mutations within the catalytic active site, as well as the ability of the two domains to exchange genetic material with each other, can be evolutionarily advantageous for the development of new protein functions over time.
EXPERIMENTAL PROCEDURES
Cloning of T. brucei TK cDNA
T. brucei TC221 cells were cultivated as described (13), and their DNA was extracted (see supplemental material). A TK gene fragment was amplified by PCR from the T. brucei DNA, labeled with 32P, and subsequently used to screen a cDNA library from procyclic T. brucei 427 (TbGARP16) (14). A positive clone was selected, and its cDNA was subcloned into a pUC18 vector to make the plasmid pUC-TbTK. The inserted gene was sequenced from both directions and was found to contain two nearly identical 89-bp stretches of DNA. To confirm that the duplicated part was not a library artifact, a TK fragment containing the two regions was amplified from the original T. brucei TC221 genomic DNA and sequenced. The PCR product was confirmed to contain both 89-bp fragments. In addition, the sequencing of this fragment and the remaining part of the T. brucei TK gene revealed that there were two alleles present in TC221 differing at five positions (see “Results”). The isolation of the TK gene from the cDNA library and the characterization of the two TK alleles in T. brucei TC221 are described in detail in the supplemental material.
Construction of His6-tagged T. brucei TK Expression Vector (see Details in Supplemental Material)
The T. brucei TK open reading frame (ORF) was amplified from pUC-TbTK, digested with NdeI and BamHI, and subcloned into a pET3a vector. The resulting plasmid (pET-TbTK) was opened with NdeI, and two annealed oligonucleotides encoding an N-terminal His6 tag were inserted. The resulting vector, pET-HisTbTK, allowed bacterial expression of His6-tagged T. brucei TK.
Subcloning of Full-length T. brucei TK, Its Two Domains, and Human TK1 into Expression Vectors Containing Fusion Partners
Full-length T. brucei TK and each of its two domains were amplified and subcloned into several variants of bacterial expression vectors (pET vector series from European Molecular Biology) containing His6-tagged fusion partners in front of the cloning cassette. The best expression yields were obtained for maltose-binding protein (MBP)-tagged domain 1 (pETM-41 derivative) and thioredoxin A (TrxA)-tagged domain 2 (pETM-20 derivative). For the full-length TK, expression yield was not improved with any of the tested fusion partner constructs as compared with that of the His6-tagged TK. However, the ability to cleave off the His6-tagged fusion partners in all constructs with tobacco etch virus (TEV) protease made Z-tagged T. brucei TK (tagged with the Z domain of staphylococcal protein A) useful as a control to confirm that the His6 tag did not affect enzyme activity (data not shown). The vectors encoding MBP-tagged domain 1, TrxA-tagged domain 2, and Z-tagged T. brucei TK were called pET-MBP-D1, pET-TrxA-D2, and pET-Z-TbTK, respectively.
The human TK1 ORF was amplified from a cDNA clone (IMAGE 2905608, Source Bioscience) and subcloned into a pETZ2-1a plasmid to create the expression vector pETZ-hTK1. The sequences of all T. brucei and human TK constructs were verified from both directions.
Expression and Purification of His6-tagged T. brucei TK
The T. brucei TK-containing vector, pET-HisTbTK, was transfected into Epicuran Coli BL21-CodonPlus (DE3) cells from Stratagene. Bacteria containing pET-HisTbTK were grown at 37 °C in 2 liters of Terrific Broth containing carbenicillin (100 mg/liter) and chloramphenicol (25 mg/liter) to an A600 nm of 1.0 under vigorous shaking. At this point, the temperature was lowered to 16 °C. After a 1-h incubation, TK production was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. After ∼15 h, the cells were harvested by centrifugation, resuspended in 60 ml of buffer A (0.3 m NaCl, 50 mm Na2SO4, 6 mm β-mercaptoethanol, and 20 mm Tris-HCl, pH 7.8), split into 15-ml aliquots, flash-frozen in liquid nitrogen, and stored at −80 °C. For each purification, frozen bacteria from one of the 15-ml aliquots were lysed with an X-press and thawed. The volume was adjusted to 30 ml with buffer A, and the cell lysate was centrifuged at 45,000 rpm for 1 h (Beckman L-90 ultracentrifuge, Ti70 rotor). The supernatant (30 ml) was loaded onto a 3-ml nickel-NTA-agarose (Qiagen) column equilibrated with buffer A. After loading the protein, the column was washed in a stepwise manner with 50 ml of 10 mm imidazole and 20 ml of 50 mm imidazole (all imidazole-containing washing and elution solutions were prepared in buffer A). TK was eluted from the column with 150 mm imidazole and collected in 2-ml fractions. The fractions containing the most protein were pooled and supplemented with 4 mm MgCl2, 1 mm EDTA, and 1 mm DTT. The protein was equilibrated into buffer B (50 mm Na2SO4, 4 mm MgCl2, 1 mm EDTA, 2 mm DTT, and 50 mm Tris-HCl, pH 7.8) by Sephadex-G25 chromatography. When sulfate-free protein was desired, the protein was dissolved and equilibrated into buffer B lacking Na2SO4. Protein concentration was assessed with the Bio-Rad Protein Assay using a standard curve made from bovine serum albumin as a reference. The purified protein was flash-frozen in liquid nitrogen and stored at −80 °C. The purification yield was generally 5 mg of T. brucei TK protein from 15 ml of frozen bacterial cells. Protein solutions containing Na2SO4 were stable through numerous freeze-thaw cycles, whereas protein in the absence of sulfate was stored in small aliquots to avoid repeated freeze-thaw cycles.
Expression and purification of Z-tagged human TK1, Z-tagged T. brucei TK, MBP-tagged domain 1, and TrxA-tagged domain 2 were in most aspects similar to that of the His6-tagged T. brucei TK protein because the fusion partners of all of these constructs contained a His6 tag. These constructs, however, were expressed in Rosetta (DE3) pLysS E. coli cells (Novagen), and additional purification steps were included to remove the His6-tagged fusion partner after TEV proteolysis (for details, see supplemental material). The purified proteins were divided into small aliquots, flash-frozen in liquid nitrogen, and stored at −80 °C. They were only thawed once to avoid any loss of enzyme activity.
TK Assay
T. brucei TK or human TK1 was incubated in 50 μl of buffer containing a 3H- or 14C-labeled nucleoside, 2 mm ATP, 5 mm MgCl2, 2 mm DTT, 100 mm potassium acetate, and 50 mm Tris-HCl, pH 7.8, at 37 °C for 10–30 min, depending on the substrate. At the end of the reaction time, the samples were incubated for 2 min at 100 °C to inactivate the enzyme, and 20 μl of each reaction mixture was then spotted onto a DE81 filter (Whatman). As described previously (15), the filters were dried, washed with ammonium formate, and incubated with HCl/KCl before scintillation counting (15). We noted that the filters should be kept away from sunlight during the drying process to minimize background counts. An alternative HPLC-based procedure was used to detect the product in assays using non-radiolabeled nucleosides. After completion of the assay and inactivation of the enzyme, each sample was diluted 100 times in water, and 20 μl was loaded onto a 50 × 4.6 mm ACE 3 C18 column (Advanced Chromatography Technologies, Aberdeen, Scotland, UK) using a flow rate of 1 ml/min. The mobile phase consisted of 7% (v/v) methanol, 2 mm tetrabutylammonium bisulfate, and 84 mm KH2PO4 adjusted to pH 6 with KOH. The outlet of the HPLC column was attached to a UV detector and a flow scintillation analyzer (Radiomatic 150TR, Packard) connected in series in order to also make it usable for assays with labeled deoxynucleosides or with [γ-32P]ATP. The enzymatic reactions were linear over time (30 min) and with protein concentrations. Similar enzyme activities were obtained in all three enzyme assay variations (labeled deoxynucleoside, labeled ATP, or non-labeled substrates).
Gel Filtration Analysis
Proteins were loaded onto a Superdex-200 column (Amersham Biosciences) using a 100-μl sample loop and a mobile phase consisting of 150 mm KCl and 50 mm Tris-HCl, pH 7.6. Equilibration between different quaternary structures of the T. brucei TK was very slow but in agreement with previous experience from studies of the human enzyme (16). Reproducible results could be obtained if the sample was preincubated at its final concentration (usually 0.1 mg/ml) at 4 °C h for 1 h, flash-frozen in liquid nitrogen, and incubated at −80 °C overnight before it was thawed and analyzed. Experiments were also performed with 2 mm ATP and 5 mm MgCl2 included in the mobile phase. Prior to analysis, the protein was preincubated with Mg-ATP at 37 °C or according to the 4 °C protocol described above. No obvious effect of ATP on the T. brucei TK quaternary structure was observed regardless of preincubation protocol. The UV detector was set to 290 nm in the experiments with ATP in order to minimize background absorbance.
RESULTS
DNA and Amino Acid Sequences of T. brucei Tandem TK
The T. brucei TK gene (Fig. 2, A and B) was isolated from a T. brucei 427 TbGARP16 cDNA library using a radiolabeled fragment of the TK gene as a probe. The T. brucei TK amino acid sequence was found to be approximately double the length of most other members in the TK1 family. In Fig. 2C, its sequence is divided into N- and C-terminal parts (domains 1 and 2), which are aligned with other TK1 proteins. Domain 1 is 62 and 42% identical to domain 2 and human TK1, respectively. Strikingly, it lacks three conserved residues considered to be important for catalysis or substrate recognition based on the human TK1 crystal structure (17). Of particular importance is Glu-98 (human numbering), which is replaced by Asn in domain 1. This residue plays a central role in all known deoxynucleoside kinases by abstracting a proton from the 5′-OH group of the deoxynucleoside substrate, thus enabling its nucleophilic attack on the γ-phosphate of ATP (5, 17). The second residue is Asp-58, which is replaced by Asn in domain 1. Asp-58 participates in the binding of thymidine by forming a hydrogen bond with the 3′-OH group of the deoxynucleoside. The third residue, Arg-60 (which, based on the sequence alignment, appears to be deleted in domain 1), is suggested to stabilize the transition state in the reaction. Residues not related to the deoxynucleoside binding sites are generally conserved in domain 1, including the ATP/GTP binding motif (P-loop) in the N terminus.
FIGURE 2.

DNA and amino acid sequences of T. brucei TK. A and B, schematic figure (A) and DNA sequence (B) of the T. brucei TK ORF with its two homologous domains shown in white and blue. Nearly identical 89-bp fragments present in the two domains are highlighted in gray (the only base that differs between the two fragments is not highlighted). The paired nucleotides (shown as one on top of the other) in A and B represent differences in the sequence between the two T. brucei TC221 alleles, TKallele 1 (top bases) and TKallele 2 (bottom bases). C, amino acid sequence alignment of T. brucei TKallele 1 N- and C-terminal parts (domains 1 and 2) with human and E. coli TK1 proteins. The human numbering scale is used. Amino acids conserved in the two T. brucei TK domains are shown in light gray, and the conserved amino acids that are substituted in domain 1 are highlighted in yellow. The two dark gray residues, A and G, are replaced by T and D in TKallele 2.
Analysis of the T. brucei cDNA sequence suggested that the two domains have exchanged genetic material relatively recently because they contain nearly identical 89-bp fragments that only differ by a single base pair (Fig. 2, A and B). This region encodes the ATP/GTP binding site. Because the high level of sequence identity is on the DNA level and at all positions, not just those that affect the encoded amino acid sequence, it was concluded that the repeat units are not conserved due to evolutionary pressure but rather must have been copied from one domain to the other at a recent time in evolution. We conclude that T. brucei TK has undergone a series of gene duplication, genetic drift, and genetic exchange events based on the presence of two homologous domains, loss of important active site residues in one of them, and a stretch of nearly identical sequence present in both.
Two TK Alleles in T. brucei TC221
To ensure that the duplicated sequence in T. brucei TK was not a library artifact, we sequenced the TK gene and verified the existence of the duplication in T. brucei TC221 genomic DNA (the T. brucei genome was not sequenced at that time). Two alleles were found in the TC221 strain (Fig. 2, A and B); one of them is identical to the one found in the cDNA library (TKallele 1), whereas the other has five base substitutions (TKallele 2) and is identical to the corresponding ORF in the T. brucei gambiense DAL972 genome project (GenBankTM accession number CBH15015.1). Interestingly, each 89-bp fragment of the second allele has a T where the first allele has a C at exactly the same position in the sequence, suggesting that the two 89-bp fragments have continued to exchange genetic information after the separation of the alleles.
Distribution of Tandem TK Sequences in Parasite Genomes
When searching GenBankTM with T. brucei TK as the query, three other parasites with two or more tandem TK1 sequences within the same ORF were found (Fig. 3A). The most extreme example was the related species T. congolense, which has a variant with four TK1 sequences in one ORF (GenBankTM accession number CCC93315.1). The other two TK variants were from the unrelated parasites A. suum (ADY41981.1) and T. vaginalis (XP001321063.1). Interestingly, tandem TKs were not found in any non-parasitic species among the top 20,000 hits from GenBankTM (representing TKs from all kingdoms of life). Phylogenetic analysis of the tandem TKs showed that the two T. brucei domains were grouped together with a 96% likelihood of having a common origin according to the bootstrap value (Fig. 3B). If the N-terminal part corresponding to the 89-bp fragment was removed from the alignment, this number was still high (84%), which supports the hypothesis that the tandem TK ORF originated from the duplication of a single-domain TK. Similar conclusions were drawn for the two A. suum domains and the four T. congolense domains, whereas the two domains of T. vaginalis ended up in separate phylogenetic groups and may have originated from the fusion of two ORFs rather than a duplication event of a single-domain TK. The amino acid sequence alignment of the TK variants and domains used to construct the phylogenetic tree is shown in supplemental Fig. S1. Similarly to domain 1 in T. brucei TK, important residues surrounding the active site are also substituted in T. congolense domain 1 (D58H, human numbering), T. congolense domain 2 (D58N and E98G), and T. vaginalis domain 2 (D58A, R60D, and G176R). In addition, T. vaginalis domain 2 also lacks many other conserved residues.
FIGURE 3.

Tandem TK proteins in parasites. A, overview of tandem TK variants in different species. B, phylogenetic analysis of TK domains with other proteins from the TK1 family. In multidomain TKs, the domains are named D1, D2, etc. Bootstrap values from 1000 replicates are shown, and the bottom bar indicates the length for 0.05 substitutions/site.
Expression, Purification, and Specific Activities of Full-length T. brucei TK, Its Two Domains, and Human TK1
The expression and purification of the full-length T. brucei TK (allele 1), domain 1, domain 2, and human TK1 are described in detail under “Experimental Procedures.” The full-length T. brucei TK was expressed as a His6-tagged protein, whereas all other proteins were expressed as fusions with N-terminal His6-tagged fusion partners (MBP, TrxA, and Z protein for domain 1, domain 2, and human TK1, respectively). For the expression of domains 1 and 2, it was not obvious exactly where to place the end of the first domain and the start of the second. Consequently, the intermediate sequence VPNGAHG was used in both constructs (Fig. 2C), with an additional N-terminal methionine in the second construct to enable cloning into the NcoI site of the pETM-20 expression vector. All expressed protein constructs were purified by nickel-NTA-agarose chromatography, and the fusion partners were subsequently removed by TEV protease cleavage and loaded onto a second nickel-NTA column. The proteins of interest were collected in the flow-through from the second column, whereas His6-tagged TEV protease, fusion partners, and non-cleaved protein remained bound to the column. SDS-PAGE analysis showed that the purification was successful for most constructs with the exception of domain 1, for which it was difficult to completely separate cleaved and non-cleaved protein in the second nickel-NTA purification step (Fig. 4). The full-length TK protein, domain 2, and human TK1 appeared as double bands. The additional bands just below the full-length T. brucei TK band were concluded to be C-terminal degradation products as judged by Western blots with an α-His6 antibody (supplemental Fig. S2). The C terminus represents a non-conserved part that varies greatly in length between TK1 proteins from different species. Accordingly, there was no obvious correlation between specific activities and the intensity of additional bands in different preparations of T. brucei TK, domain 2, and human TK1.
FIGURE 4.
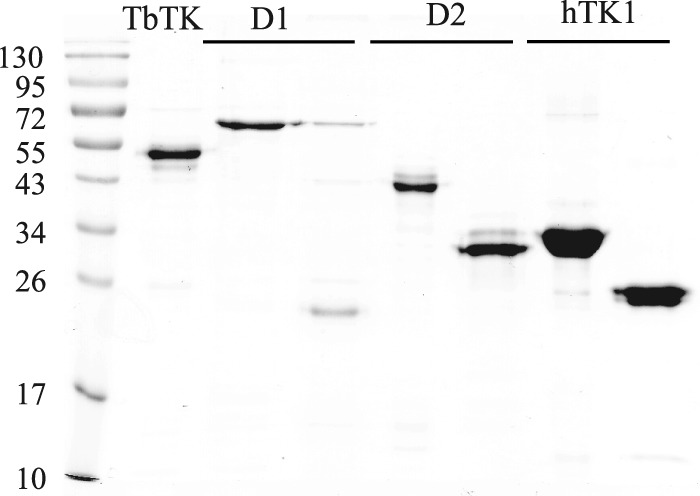
Analysis of purified TK protein constructs on a 12% SDS polyacrylamide gel. The lanes contain molecular mass marker, purified His6-tagged T. brucei TK (TbTK lane), domain 1 before and after removal of the MBP fusion partner (D1 lanes), domain 2 before and after removal of the TrxA fusion partner (D2 lanes), and human TK1 before and after removal of the Z protein fusion partner (hTK1 lanes). Theoretical molecular masses are 52 kDa (TbTK), 63 kDa (MBP-D1), 23 kDa (D1), 42 kDa (TrxA-D2), 28.5 kDa (D2), 35 kDa (Z-hTK1), and 25 kDa (hTK1). The molecular mass marker used is the PagerulerTM prestained protein ladder (catalog no. SM0671, Fermentas Life Science).
The effects of different salts, pH, and DTT on T. brucei TK activity were investigated to determine optimal storage and enzyme assay conditions. Of particular interest was sodium sulfate, which was included in all purification steps of the full-length T. brucei TK to protect the protein from precipitation. TK preparations purified in the absence or presence of sulfate had similar specific activities, but the purification yield was much higher in the latter case. Sulfate was found to inhibit T. brucei TK activity by acting as a competitive inhibitor of ATP binding (supplemental Fig. S3, A and B), but because the enzyme was generally diluted 10 times or more in a sulfate-free buffer before enzymatic analysis and only 5 μl of the resulting protein solution was used for each 50-μl assay, the effect of the storage buffer sulfate on enzyme activity was negligible. In non-standard enzyme assays (such as assays with low ATP concentrations or high protein concentrations), sulfate-free protein was used. Further optimization of enzyme activity showed that it was stimulated by DTT up to a concentration of 2 mm and weakly stimulated by salts, such as NaCl, KCl, and potassium acetate (supplemental Fig. S4). The pH optimum of the enzymatic reaction was 7.8 (supplemental Fig. S5). The standard buffer condition for enzymatic assays was consequently chosen to be 5 mm MgCl2, 2 mm DTT, 100 mm potassium acetate, and 50 mm Tris, pH 7.8. The specific activity of T. brucei TK was generally 2.8–3.6 μmol × min−1 × mg−1, using 0.5 mm thymidine as the substrate and 2 mm ATP as the phosphate donor under standard buffer conditions. Domain 1 was inactive, whereas domain 2 had a specific activity of 6–8 μmol × min−1 × mg−1. Thus, domain 2 has a similar activity (per mole of enzyme) as the full-length TK when measured at substrate concentrations high enough to saturate the enzyme. Human TK1 had a specific activity of 14 μmol × min−1 × mg−1, which is fairly similar to the previously published result of 18 μmol × min−1 × mg−1 (18).
Phosphate Donor Specificity and Feedback Inhibition of T. brucei TK Activity
Studies of ATP as the phosphate donor showed that the Km for this nucleotide was always around 0.2 mm regardless of the thymidine concentration in the assay (Table 1). This is low enough to saturate the enzyme under physiological conditions, where the ATP concentration is 1.6 mm (7). Because the thymidine concentration did not have any obvious effect on the Km of ATP, it was only used at one concentration (50 μm) in the remaining studies of phosphate donors. T. brucei TK showed a relaxed phosphate donor specificity, having nearly as high activity with CTP, UTP, and GTP as with ATP (Fig. 5A). Similar findings were previously reported for the partially purified T. brucei rhodesiense TK (9, 10). Evaluation of phosphate donors in the presence of deoxyinosine instead of thymidine as the substrate showed a relaxed phosphate donor specificity in this case as well (data not shown) but with a slightly higher Km for ATP (Table 1). For standard enzyme assay conditions, the concentration of ATP was chosen to be 2 mm, which is sufficient to have a saturating effect on enzyme activity regardless of the deoxynucleoside substrate used.
TABLE 1.
Kinetic parameters of T. brucei TK
Km and Vmax values with S.D. are calculated from fitting the Michaelis-Menten curve to a hyperbola by nonlinear regression using the GraphPad Prism software.
| Km | Vmax | kcat | kcat/Km | |
|---|---|---|---|---|
| μm | μmol × min−1 × mg−1 | s−1 | s−1 × m−1 | |
| Full-length TK | ||||
| ATP, dThd (0.5 μm)a | 180 ± 30 | 0.190 ± 0.009 | ||
| ATP, dThd (5 μm)a | 220 ± 20 | 1.9 ± 0.05 | ||
| ATP, dThd (50 μm)a | 210 ± 30 | 3.6 ± 0.10 | 3.1 | 1.5 × 104 |
| ATP, dIno (0.5 mm)a | 320 ± 60 | 0.095 ± 0.007 | ||
| dThd | 7.8 ± 0.9 | 3.6 ± 0.10 | 3.1 | 4.0 × 105 |
| 5-Fluoro-dUrd | 30 ± 4 | 4.2 ± 0.10 | 3.6 | 1.2 × 105 |
| dUrd | 80 ± 5 | 5.9 ± 0.09 | 5.1 | 6.3 × 104 |
| dCyd | 0.029b | |||
| Urd | 7800 ± 900 | 1.7 ± 0.07 | 1.5 | 190 |
| dIno | 2500 ± 300 | 0.93 ± 0.07 | 0.80 | 320 |
| dGuo | 2600 ± 200 | 0.39 ± 0.010 | 0.34 | 130 |
| dAdo | 8400 ± 700 | 0.055 ± 0.003 | 0.047 | 5.6 |
| TK Domain 2 | ||||
| ATP, dThd (50 μm)a | 98 ± 15 | 8.2 ± 0.3 | 3.9 | 4.0 × 104 |
| dThd | 46 ± 2 | 7.3 ± 0.10 | 3.5 | 7.6 × 104 |
| dUrd | 430 ± 40 | 11.2 ± 0.3 | 5.3 | 1.2 × 104 |
a Experiments to calculate kinetic constants for ATP were performed using thymidine (at a concentration of 0.5, 5 or 50 μm) or 0.5 mm deoxyinosine as a deoxynucleoside substrate. When ATP is the studied substrate, kcat and kcat/Km values are only shown in the cases where the deoxynucleoside concentration is high enough to saturate the enzyme activity.
b The value represents the enzyme activity with 10 mm substrate concentration.
FIGURE 5.

Effects of NTPs and dTTP on T. brucei TK activity. A, enzyme activities with 50 μm thymidine as substrate and different phosphate donors used at 0.2 and 2 mm concentrations. B, Lineweaver-Burk diagram of TK activity with 50 μm thymidine and varying concentration of ATP in the absence (●) or presence of 25 μm (■) or 50 μm dTTP (▴). All experiments were performed in duplicate, with the S.D. values shown by error bars.
In agreement with results from the partially purified T. brucei rhodesiense TK and other members from the TK1 family (9, 10, 19), dTTP acted as a competitive inhibitor of T. brucei TK (Ki = 4.6 ± 0.4 μm)4 with respect to ATP (Fig. 5B). The Km for ATP (0.2–0.3 mm (Table 1)) is much higher than the Ki value for dTTP. This indicates that dTTP should also be able to bind under physiological conditions, where the ATP and dTTP levels are 1.6 mm and 19 μm, respectively (7). Other dNTPs had no effect on enzyme activity, and dTTP inhibition was also observed with other deoxynucleosides as substrates (data not shown).
T. brucei TK Has Broader Substrate Specificity than Human TK1
In the presence of high substrate concentrations (10 mm), the T. brucei TK showed activity with its main substrates thymidine and deoxyuridine as well as with deoxyinosine and deoxyguanosine (Fig. 6A). The activities with the two latter substrates were ∼30 and 20%, respectively, compared with the activity with 10 mm thymidine. At the same high substrate concentrations of 10 mm, the human TK1 had ∼2-fold lower activities with the two purine substrates than the T. brucei enzyme (as a percentage relative to thymidine). However, the difference between the two enzymes was more pronounced if the deoxyinosine concentration was reduced to 0.5 mm (Fig. 6B). In fairly good agreement with the results shown in Fig. 6A, a more extensive kinetic analysis of the T. brucei TK (Table 1) yielded turnover numbers (kcat) for deoxyinosine and deoxyguanosine that were 26% and 11% of the kcat with thymidine. However, the Km values were much higher for the two purine substrates, resulting in low catalytic efficiencies (kcat/Km). Thymidine and deoxyuridine can therefore be concluded to be the main substrates of the T. brucei TK, although substrate discrimination is not as high as for human TK1. For the latter enzyme, it was not possible to reach saturation of the enzyme activity even at purine concentrations above 10 mm, and the solubility limits of the purine substrates prevented enzyme assays at substrate concentrations high enough to obtain reliable measurements of Km and kcat values. Thus, the human TK1 has a much larger difference in affinity between its best pyrimidine substrate, thymidine (Km = 0.7 μm (20)), and the best purine than the T. brucei enzyme. The broader substrate specificity of the T. brucei TK could possibly be an advantage for the development of antitrypanosomal deoxynucleoside analogs that are specifically activated by the parasite enzyme but not by its host (see “Discussion”).
FIGURE 6.

Substrate selectivity of T. brucei TK (black bars), human TK1 (white bars), and T. brucei TK domain 2 (hatched bars). Experiments were performed with 10 mm deoxynucleosides (A) or 0.5 mm deoxyinosine (B). Enzyme activities are shown as a percentage of the activity observed with 10 mm thymidine. All experiments were performed in duplicate, with the S.D. values shown by error bars.
T. brucei Domain 2 Is Enzymatically Active by Itself but Needs Domain 1 for Efficient Deoxynucleoside Substrate Binding
Kinetic analysis of domain 2 with thymidine as the substrate showed a turnover number of ∼3 s−1, which is similar to that of the full-length T. brucei TK (Table 1). Each molecule of domain 2 can therefore be concluded to be equally as active as each molecule of the full-length enzyme. Note that the similar activity of the two proteins measured in s−1 becomes a 2-fold difference when measured as μmol × min−1 × mg−1 due to their respective molecular masses of 28.5 and 52 kDa. Km value determinations revealed that domain 2 bound ATP with a 2-fold higher affinity than that of the full-length enzyme. However, deoxynucleoside substrate binding was severely compromised, with Km values for thymidine and deoxyuridine that were approximately 5 times higher than for the full-length enzyme. Domain 2 also showed low affinities for purine deoxynucleosides, which resulted in low activity at substrate concentrations of 10 mm (Fig. 6A) and the inability to measure reliable Km and kcat values within the solubility range of the substrates. It was not possible to restore deoxynucleoside affinity by adding purified domain 1 to reactions with the domain 2 protein, suggesting that the two domains need to be covalently attached for efficient substrate binding.
T. brucei Domain 2 Does Not Dimerize Efficiently
Enzymes from the TK1 family are generally dimers or tetramers (5). Gel filtration analysis of the T. brucei full-length enzyme (Fig. 7A) showed a major peak estimated to be 49 kDa, which corresponds well to a monomer (molecular mass of ∼52 kDa) and minor peaks corresponding to dimers and higher oligomers. However, because the enzyme consists of two TK sequences in tandem, it can actually be considered a pseudodimer. A corresponding analysis of domain 2 showed a major 36-kDa peak with a shoulder at ∼70 kDa, suggesting that the protein is mainly monomeric and only forms dimers to a limited extent (Fig. 7B). The theoretical molecular masses of monomers and dimers are 28.5 and 57 kDa, respectively. The small deviations from theoretical molecular masses could possibly be due to non-globular shapes. The concentration of domain 1 was too low for gel filtration analysis, but the corresponding test of the MBP-tagged domain 1 construct showed that it, similarly to domain 2, was in equilibrium between monomers and dimers but also had an additional peak of large aggregates in the void volume (data not shown). The human TK1 protein, which was used as control for the gel filtration experiments, formed a 112-kDa species (Fig. 7C), which is in agreement with previous results showing that it is mainly a tetramer (molecular mass of 100 kDa) in this protein concentration range (16). Similar results were also obtained if the proteins in Fig. 7, A and B, were analyzed in the presence of 2 mm ATP and 5 mm MgCl2 (data not shown); the full-length T. brucei TK and domain 2 remained monomeric under these conditions. The details of the Mg-ATP experiments are described under “Experimental Procedures.”
FIGURE 7.

Gel filtration analyses of full-length T. brucei TK (A), domain 2 (B), and human TK1 (C). Molecular masses shown above each peak are estimated by comparing the retention times with a standard curve (supplemental Fig. S6). Late eluting peaks (tR > 40 min) from compounds smaller than 1 kDa are not included in the chromatograms.
DISCUSSION
T. brucei infects many different mammalian hosts, and these hosts have varying nucleoside and deoxynucleoside concentrations in their bodily fluids. It is to their advantage, therefore, for the parasites to have a rapidly evolving TK sequence that allows fine tuning of the enzyme's substrate specificity over time with respect to the level of deoxynucleosides in different hosts. An advantage of having a tandem protein is that the constituent homologous parts can exchange genetic material with each other. In the T. brucei tandem TK, it is evident that a very recent transfer of an 89-bp gene fragment from one of the domains to the other has occurred. With the exception of one base pair, the DNA sequence of the fragment in each domain was identical to the other, whereas the rest of the two domains had a much lower homology (62% amino acid sequence identity).
Protozoan and metazoan endoparasites all live in a nutrient-rich environment where metabolites can be salvaged from the host, and thus they share many metabolic features. Nearly all of them lack de novo purine biosynthesis pathways, whereas pyrimidines and deoxynucleosides are generally produced through both salvage and de novo synthesis pathways (21). The finding of tandem TKs in several unrelated parasitic lineages suggests that such an arrangement may be beneficial for parasites in general. The most extreme case of tandem TKs was found in a related species, T. congolense, which had an ORF consisting of four TK sequences. Phylogenetic analysis clearly places the T. congolense and T. brucei domains into two distinct groups within the tree of African trypanosomes. It cannot be ruled out, however, that their tandem TKs have a common origin (and that the T. congolense gene has gone through a second round of duplication) because the frequency of genetic exchange will strongly affect how similar the domains within each enzyme are to each other. However, the finding of tandem TKs in the unrelated parasites T. vaginalis and A. suum strongly suggests that these repetitions have appeared several times in evolution. The two domains of the A. suum enzyme are homologous to other metazoans and therefore are not likely to have the same origin as the trypanosome TKs. In the T. vaginalis case, the two domains have very limited similarity to each other; domain 1 is homologous to animal TK1 proteins, and domain 2 is homologous to bacterial ones. It seems, therefore, that this protein has not originated from the duplication of a single TK sequence but rather from a fusion of two sequences. Regardless of mechanism, it is striking that tandem TKs have appeared at least three times in evolution and that all of them are in parasites.
T. brucei TK has a broader substrate specificity than the mammalian enzyme and is therefore of potential interest for drug discovery. Human TK1 has a more than 10,000-fold higher affinity for thymidine (Km = 0.7 μm (20)) than the purine substrates deoxyinosine and deoxyguanosine (Km > 10 mm), whereas the corresponding difference in affinity between thymidine and deoxyinosine is only ∼300-fold in the T. brucei enzyme. The lower selectivity of the T. brucei enzyme is an indication that the active site is more flexible and accommodates a wider array of substrate modifications. An ideal antitrypanosomal deoxynucleoside analog would be transported efficiently into the parasites and be a good substrate for the trypanosome TK. Potential substrate analogs can be derivatives of either deoxyinosine/deoxyguanosine or thymidine/deoxyuridine. Deoxyinosine and deoxyguanosine have an advantage over thymidine in that they can be actively transported into the parasites by way of high affinity transporters (22). They have the disadvantages, however, of having low affinities for TK and being substrates for mammalian deoxycytidine and deoxyguanosine kinases (5). Thymidine and deoxyuridine are much better substrates for TK but are believed to be mainly taken up via passive diffusion through the parasite's plasma membrane (23). The relaxed substrate specificity of the T. brucei TK is advantageous in the search for thymidine/deoxyuridine analogs with improved membrane permeability properties that are selective for the T. brucei enzyme versus human TK1.
Recombinant expression and analysis of both domains independently from each other revealed their functions. It is only domain 2 that is enzymatically active, whereas domain 1 appears to play a structural role in enhancing substrate binding affinity. In comparison with the full-length T. brucei TK, which can be regarded as a pseudodimer composed of two fused TK sequences, the recombinantly expressed domain 2 was mainly monomeric and had a 5-fold reduction in affinity for its main substrates, thymidine and deoxyuridine. The absence of domain 1 seems, therefore, to result in a loss of dimeric structure and a subsequent reduction in substrate affinity. Analyses of the human TK1 has shown that it binds thymidine much more efficiently when it exists as a tetramer rather than a dimer, with Km values of 0.7 and 15 μm, respectively (20). Our results suggest that there could be a corresponding correlation between substrate affinity and the pseudodimeric structure seen in the T. brucei case. Because domain 1 is catalytically inactive but still plays an important structural role in the holoenzyme, it is not surprising that many residues in the active site (Asp-56, Arg-60, and Glu-98) have been substituted over time, whereas conserved residues unrelated to the active site remain intact.
A consequence of having an active site with lost function in one of the domains is that this site is free to change over the course of evolution, and completely new functions may develop over time. An evolutionary advantage in the case of domain 1 is that its function as a supporting partner prevents the risk of accumulating nonsense or detrimental mutations in the non-catalytic parts of the domain. In addition, the genetic exchange mechanism means that the more rapidly evolving active site (in this case the active site in domain 1) can, in principle, help the evolution of the second active site (in domain 2) by replacing parts of its sequence to achieve new substrate specificities. This also suggests that domain 2 has the potential to help domain 1 regain activity by replacing the catalytic residues that have been lost. Continuous genetic exchange may, in this manner, result in the rapid acquisition of novel features within the tandem protein.
Supplementary Material
This work was supported by Swedish Research Council Grant 2009-5145, Swedish International Development Cooperation Agency Grant SWE-2008-069, and the Kempe Foundation.

This article contains supplemental Experimental Procedures and Figs. S1–S6.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AF395663.
The indicated value is an average of the Ki values obtained with 25 and 50 μm dTTP with the S.D. value indicated. The Ki values are calculated from the apparent Km values (Km′) obtained from non-linear regression of a Michaelis-Menten diagram by the GraphPad Prism software (using the same data as in Fig. 5B). The following formula was used for the calculations: Ki = [dTTP]/((Km′/Km) − 1).
- TK
- thymidine kinase
- TK1
- thymidine kinase 1
- TK2
- thymidine kinase 2
- MBP
- maltose-binding protein
- TrxA
- thioredoxin A
- TEV
- tobacco etch virus
- Z
- Z domain of staphylococcal protein A.
REFERENCES
- 1. Malvy D., Chappuis F. (2011) Sleeping sickness. Clin. Microbiol. Infect. 17, 986–995 [DOI] [PubMed] [Google Scholar]
- 2. Parkinson F. E., Damaraju V. L., Graham K., Yao S. Y., Baldwin S. A., Cass C. E., Young J. D. (2011) Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr. Top. Med. Chem. 11, 948–972 [DOI] [PubMed] [Google Scholar]
- 3. Rottenberg M. E., Masocha W., Ferella M., Petitto-Assis F., Goto H., Kristensson K., McCaffrey R., Wigzell H. (2005) Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. J. Infect. Dis. 192, 1658–1665 [DOI] [PubMed] [Google Scholar]
- 4. Vodnala M., Fijolek A., Rofougaran R., Mosimann M., Mäser P., Hofer A. (2008) Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei. J. Biol. Chem. 283, 5380–5388 [DOI] [PubMed] [Google Scholar]
- 5. Eriksson S., Munch-Petersen B., Johansson K., Eklund H. (2002) Structure and function of cellular deoxyribonucleoside kinases. Cell Mol. Life Sci. 59, 1327–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deville-Bonne D., El Amri C., Meyer P., Chen Y., Agrofoglio L. A., Janin J. (2010) Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation. Structural and catalytic properties. Antiviral Res. 86, 101–120 [DOI] [PubMed] [Google Scholar]
- 7. Hofer A., Ekanem J. T., Thelander L. (1998) Allosteric regulation of Trypanosoma brucei ribonucleotide reductase studied in vitro and in vivo. J. Biol. Chem. 273, 34098–34104 [DOI] [PubMed] [Google Scholar]
- 8. Lüscher A., Onal P., Schweingruber A. M., Mäser P. (2007) Adenosine kinase of Trypanosoma brucei and its role in susceptibility to adenosine antimetabolites. Antimicrob. Agents Chemother. 51, 3895–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chello P. L., Jaffe J. J. (1972) Isolation, partial purification, and properties of thymidine kinase from Trypanosoma (trypanozoon) brucei rhodesiense. J. Parasitol. 58, 298–305 [PubMed] [Google Scholar]
- 10. Chello P. L., Jaffe J. J. (1972) Comparative properties of trypanosomal and mammalian thymidine kinases. Comp. Biochem. Physiol. B 43, 543–562 [DOI] [PubMed] [Google Scholar]
- 11. Hofer A., Steverding D., Chabes A., Brun R., Thelander L. (2001) Trypanosoma brucei CTP synthetase. A target for the treatment of African sleeping sickness. Proc. Natl. Acad. Sci. U.S.A. 98, 6412–6416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hofer A., Schmidt P. P., Gräslund A., Thelander L. (1997) Cloning and characterization of the R1 and R2 subunits of ribonucleotide reductase from Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 94, 6959–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirumi H., Hirumi K. (1989) Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75, 985–989 [PubMed] [Google Scholar]
- 14. Hehl A., Pearson T. W., Barry J. D., Braun R., Roditi I. (1995) Expression of GARP, a major surface glycoprotein of Trypanosoma congolense, on the surface of Trypanosoma brucei. Characterization and use as a selectable marker. Mol. Biochem. Parasitol. 70, 45–58 [DOI] [PubMed] [Google Scholar]
- 15. Ives D. H., Durham J. P., Tucker V. S. (1969) Rapid determination of nucleoside kinase and nucleotidase activities with tritium-labeled substrates. Anal. Biochem. 28, 192–205 [DOI] [PubMed] [Google Scholar]
- 16. Munch-Petersen B. (2009) Reversible tetramerization of human TK1 to the high catalytic efficient form is induced by pyrophosphate, in addition to tripolyphosphates, or high enzyme concentration. FEBS J. 276, 571–580 [DOI] [PubMed] [Google Scholar]
- 17. Welin M., Kosinska U., Mikkelsen N. E., Carnrot C., Zhu C., Wang L., Eriksson S., Munch-Petersen B., Eklund H. (2004) Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc. Natl. Acad. Sci. U.S.A. 101, 17970–17975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berenstein D., Christensen J. F., Kristensen T., Hofbauer R., Munch-Petersen B. (2000) Valine, not methionine, is amino acid 106 in human cytosolic thymidine kinase (TK1). Impact on oligomerization, stability, and kinetic properties. J. Biol. Chem. 275, 32187–32192 [DOI] [PubMed] [Google Scholar]
- 19. Arnér E. S., Eriksson S. (1995) Mammalian deoxyribonucleoside kinases. Pharmacol. Ther. 67, 155–186 [DOI] [PubMed] [Google Scholar]
- 20. Munch-Petersen B., Tyrsted G., Cloos L. (1993) Reversible ATP-dependent transition between two forms of human cytosolic thymidine kinase with different enzymatic properties. J. Biol. Chem. 268, 15621–15625 [PubMed] [Google Scholar]
- 21. el Kouni M. H. (2003) Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol. Ther. 99, 283–309 [DOI] [PubMed] [Google Scholar]
- 22. Landfear S. M., Ullman B., Carter N. S., Sanchez M. A. (2004) Nucleoside and nucleobase transporters in parasitic protozoa. Eukaryot. Cell 3, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gudin S., Quashie N. B., Candlish D., Al-Salabi M. I., Jarvis S. M., Ranford-Cartwright L. C., de Koning H. P. (2006) Trypanosoma brucei. A survey of pyrimidine transport activities. Exp. Parasitol. 114, 118–125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.