

Abstract
Motivation: Inferring the underlying regulatory pathways within a gene interaction network is a fundamental problem in Systems Biology to help understand the complex interactions and the regulation and flow of information within a system-of-interest. Given a weighted gene network and a gene in this network, the goal of an inference algorithm is to identify the potential regulatory pathways passing through this gene.
Results: In a departure from previous approaches that largely rely on the random walk model, we propose a novel single-source k-shortest paths based algorithm to address this inference problem. An important element of our approach is to explicitly account for and enhance the diversity of paths discovered by our algorithm. The intuition here is that diversity in paths can help enrich different functions and thereby better position one to understand the underlying system-of-interest. Results on the yeast gene network demonstrate the utility of the proposed approach over extant state-of-the-art inference algorithms. Beyond utility, our algorithm achieves a significant speedup over these baselines.
Availability: All data and codes are freely available upon request.
Contact: srini@cse.ohio-state.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
With advances in high-throughput experimental technologies, we are now witnessing a revolutionary change in our ability to measure and store various forms of interaction data (e.g. protein–protein, protein–DNA/RNA, protein–metabolites and genetic interactions) for different organisms. The central objective in Systems Biology is to fuse and analyze the diverse molecular, cellular, tissue-level and higher level data sources to deduce how sub systems and whole organisms work from these network of interactions.
A node in such a network is usually a gene or its corresponding protein, and an edge in a gene network represents a protein–protein interaction (PPI) or a transcription factor (TF)–DNA binding. A major challenge here is to identify the underlying regulatory pathways, each of which is a chain of interacting genes within a network. A regulatory pathway begins with a causal gene and ends at a target gene, and each gene in a pathway can either activate or deactivate some functions of its neighboring genes. The uncovering of potential pathways across genes helps biologists understand the cellular functions of each gene and protein. As noted earlier, a range of experimental methods such as two-hybrid analysis have been developed to uncover a large amount of interactions between genes and proteins, and gene knock-out experiments and some studies (Bader et al., 2004; Mering et al., 2002) have also computed the confidence level associated with an interaction. On the basis of discovery that genes with high centrality in a gene network usually have higher essentiality, lethality and pleiotropy (Hahn and Kern, 2005; Jeong et al., 2001), several research works (Bebek and Yang, 2007; Froehlich et al., 2007; Scott et al., 2006; Suthram et al., 2008; Tu et al., 2006; Vaske et al., 2009) have focused on inferring the regulatory pathways on a number of organisms such as fly, yeast and human.
Given a weighted directed gene network and a specific gene, we seek to develop efficient algorithms for the following sub problems: (i) UnknownCausal: if the given gene is a target gene, infer possible causal genes and their pathways; (ii) UnknownTarget: if the given gene is a causal gene, infer possible target genes and their pathways. Since, given a target gene, some biological experiments such as expression quantitative trait loci (eQTL) analysis can locate an approximate location of a causal gene and thus can provide candidate causal genes, another sub problem is (iii) CandidateCausal: given a causal gene, infer the most likely causal gene among the candidates.
Recently, approaches based on the random walk model have been suggested as a means to address this problem. A random walk typically starts from the given gene, walks through several nodes, and terminates according to some pre-defined parameters, such as walk length and edge weights. Generally, the number of visits to a node across multiple walks from a given gene gives a measure of influence or importance, which in turn provides a measure of how likely that node is involved in a pathway containing the given gene. Tu et al. (2006) first proposed this model to address this inference problem with an additional requirement, rooted in domain knowledge, that the random walk could visit any node at most once. Suthram et al. (2008) and Missiuro et al. (2009) proposed the electrical circuit model as an analogy of gene networks, in which each edge is a resistor and its conductance is proportional to the edge's weight. This circuit model can be solved according to Kirchhoff's and Ohm's laws, and the amount of current flowing through each node can be interpreted as the possibility of this node being involved in a pathway. It has been shown that this circuit model can be interpreted as the random walk model (Doyle and Snell, 1984), but it cannot directly apply on directed edges. The information flow model (Stojmirović and {Yu}, 2009) used a similar approach to topic sensitive PageRank (Haveliwala, 2003), whereas the electrical circuit model (Suthram et al., 2008) and PageRank-based method (Voevodski et al., 2009) can be seen as the special cases of the information model. As we discuss later, these approaches have some important limitations that we seek to overcome.
Specifically, in a clear departure from past work, here we discuss a novel approach to solving this inference problem by adapting the k shortest paths algorithm to directly point out potential pathways. In this article, we only discuss simple paths, in which a node is not allowed to appear more than one time. Intuitively, this agrees with the prevailing belief among domain scientists that a regulatory pathway is unlikely to repeatedly pass a node. The classic k shortest paths simple paths algorithm is due to (Yen, 1971). It executes O(n) times Dijkstra algorithm to generate candidate paths for each of the k shortest paths, so its time complexity is O(kn(m+nlogn)), where n is the number of nodes and m is the number of edges. Recently, Hershberger et al. (2007) classified the candidate paths into different classes and adopted a replacement-edge strategy for each class, and Gao et al. (2010) used the transformed graph with side cost to improve the efficiency of Yen's algorithm, but both of them have the same worst-case time complexity and space complexity as Yen's algorithm. All the algorithms discussed earlier address the single-pair problem, which is to find the k shortest paths between a pair of nodes in a weighted graph. However, in our inference problem, we need to calculate the k shortest paths from the given node to each other node to approximate the potentiality of each gene being involved in a pathway. For this reason, we introduce a single-source k shortest paths algorithm.
Our algorithm is also based on Dijkstra algorithm, moreover adopting a data structure called {pseudo tree} to reduce the space storing all single-source k shortest paths. We show that an exact algorithm to solve this problem has high complexity and therefore we propose a heuristic approximation algorithm that appears to work well in practice—the single-source k-shortest paths algorithm. We note that the k-shortest paths may often have high overlap (low diversity, since may share a large number of edges) and one of our objectives is to explicitly identify diverse shortest paths. The domain intuition here is that, diverse paths between two genes might help one uncover non-redundant transduction pathways which are of interest to domain scientists. To accommodate this additional requirement, we propose a variant of our basic approach—a single-source k diverse shortest paths algorithm.
Following best practices in silico validation procedures outlined by previous research on this problem (Beyer et al., 2006; Suthram et al., 2008; Tu et al., 2006), we demonstrate the efficacy and efficiency of the proposed approaches to inferring regulatory pathways on the Yeast gene network. Specifically, on the CandidateCausal sub-problem, our shortest paths algorithm achieves significantly higher accuracy than extant state-of-the-art approaches (Stojmirović and {Yu}, 2009; Tu et al., 2006), and on the UnknownCausal and UnknownTarget sub problems, the importance values assigned by our shortest path variant that explicitly accounts for diversity in paths, not only achieves higher enrichment but also enriches different functions. In terms of scalability, when compared with the exact single-pair k-shortest paths algorithm (Yen, 1971), as well as some recent variants (Malviya et al., 2011), on the yeast gene network, our method achieves up to two orders of magnitude speedup. We should also note that while our approach relies on a simple heuristic approximation procedure, we empirically observe no difference in quality when compared with the exact algorithm.
2 PROBLEM DEFINITION
Let G=(V, E) denote a gene network, which is a weighted directed graph, where V is the set of nodes (genes or proteins), E is the set of directed edges (interactions) and n=|V|, m=|E|. Each edge e∈E denoted by (ga, gb), ga, gb∈V, represents the direction of interaction from ga to gb. If an interaction is undirected, e.g. a PPI, the corresponding edge is bidirectional. w(e)∈[0, 1] is the weight of an edge e, and the weight represents the confidence level of the interaction. We introduce how to generate the weight in Section 5.1.
Given a weighted graph as a gene network and a gene in this network, we seek to solve the following three sub problems: (i) UnknownCausal: infer possible causal genes if the given gene is a target gene; (ii) UnknownTarget: infer possible target genes if the given gene is a causal gene; and (iii) CandidateCausal: infer the true causal gene in a given set of candidate causal genes, if the given gene is a target gene. The target gene and the causal gene are denoted by gt and gc, respectively, and the set of candidate gene is denoted by C, where gc∈C and C⊂V.
The goal for CandidateCausal is to correctly select the true causal gene from C. An algorithm for solving UnknownCausal and UnknownTarget is to give an importance value to each node except the given node, where higher importance value indicates a higher chance that a gene is involved in a pathway containing the given gene, and the goal is to assign the potential causal genes or potential target genes and the nodes in the pathways higher importance values than other genes. The importance value of a gene ga is denoted by Imp(ga).
3 REVIEW OF THE RANDOM WALK MODEL
We begin by reviewing the information flow model introduced by Stojmirović and Yu (2011), since other algorithms based on the random walk model can be thought of as specific instantiations of the information flow model. The information flow model first constructs a n×n transition matrix P by normalizing the adjacent matrix A of the graph G, where Aij=w((gi, gj)) and Pij=αAij/∑kAik.α∈(0, 1) is called ‘damping factor’. When a random walk reaches a node gi, the walk would terminate at gi with probability 1−α and go to another node gj with probability Pij in the next step. A random walk starts from a source node and once the walk reaches a sink node, the walk immediately terminates. The walk simulates the information flow in gene networks: the flow starts from the causal gene (source node), follows the pathway and finally reaches the target gene (sink node).
The random walk model has two serious problems. The first problem is that the normalization of edge weights is lossy. Figure 1(a) shows an example. Given the source node S, it is apparent that Imp(A)>Imp(C)=Imp(D) in the right graph, but in the left graph, it is not clear whether C and D is more likely to be involved in the pathway starting at the source node than A. However, both graphs generate the same transition matrix, as shown in the middle graph, so the transition matrix cannot fairly reflect the original weights. One approach to solving this problem is to use different damping factors for different nodes. If the sum of the weights of all outgoing edges for a node is higher, α should be increased to decrease the chance a walk terminates at this node, but there is no intuitive approach to tuning it. Another approach is using the electrical circuit model, but it is difficult to set the voltage to fairly reflect the information flow.
Fig. 1.
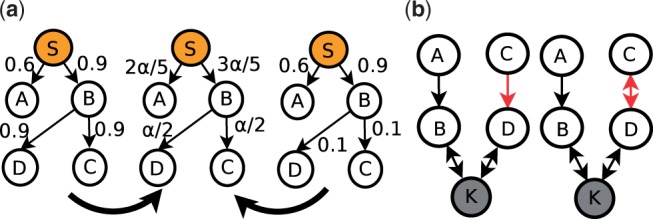
Two problems of the random walk model. (a) Normalization of edge weights is lossy. (b) The walks repeatedly go through the bidirectional edge
The second problem is that a walk can repeatedly go through the same bidirectional edge. As shown in Figure 1(b), in which every edge has the same weight, the right graph is similar to the left graph but the edge (B, D) is bidirectional. Given the sink node K, it is intuitive that Imp(A)=Imp(C) in both graphs. However, the random walk model assigns Imp(A) a higher value than Imp(C) in the right graph since walks starting from C might go back to C before termination. This problem can be solved by forcing the random walks to visit only unvisited nodes, but this in turn leads to other problems [e.g. restrictive walks and scalability (Suthram et al., 2008)]. Another possible solution for this problem is to predict the direction of each bidirectional edge before random walks start, but both directions are sometimes used by different regulatory pathways. In this work, we seek to leverage the the single-source k-shortest simple paths algorithm to address these issues.
4 SINGLE-SOURCE K-SHORTEST PATHS
4.1 Preliminaries and key intuition
The random walks simulate the information flow in gene networks. The walks visit genes connected by higher weighted edges more frequently than genes connected by lower weighted edges, and distant genes from the given gene tend to be assigned lower importance values, agreeing with domain intuition. To capture similar intuitions when using the shortest path variants, we first convert the weight of an edge into a distance as follows: d(e)=−log(w(e))+c (We add a constant c, typically set to 1, because each edge should have a minimum distance, similar to a Laplacian correction.), where d(*) denotes the distance of an edge or a path. Then, the importance value of a gene ga is defined as 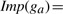 where P1,P2,…, Pk are k-shortest paths from the given gene to ga, With this transformation, it is obvious that genes having shorter distances to the given gene of interest will be assigned greater importance values. We also note that the electrical circuit model has an equivalent representation w.r.t. the k-shortest paths algorithm (it takes all paths into accounts) as pointed out by a recent study (Missiuro et al., 2009).
where P1,P2,…, Pk are k-shortest paths from the given gene to ga, With this transformation, it is obvious that genes having shorter distances to the given gene of interest will be assigned greater importance values. We also note that the electrical circuit model has an equivalent representation w.r.t. the k-shortest paths algorithm (it takes all paths into accounts) as pointed out by a recent study (Missiuro et al., 2009).
A regulatory pathway, which is from a causal gene to a target gene, might involve in multiple paths instead of a single path (Suthram et al., 2008; Tu et al., 2006), so the k-shortest paths is able to directly point out these potential regulatory pathways. A regulatory pathway might not pass a gene more than once, so we only discuss simple paths, in which a loop is not allowed. Most importantly, since the k-shortest paths algorithm does not normalize edge weights and it can be directly applied on directed edges, it does not have the problems of the random walk model.
The inference problem is now a single-source k-shortest paths problem, in which the starting node is the given causal gene when solving UnknownTarget or the given target gene when solving UnknownCausal and CandidateCausal. Since the original direction of edges regulates information from the causal gene to the target gene, the direction of all edges should be reversed first when solving UnknownCausal and CandidateCausal. To find paths from the target gene to the causal gene. Additionally, in CandidateCausal, we select the candidate causal gene with the highest importance value as our predicted causal gene. To the best of our knowledge, the state-of-the-art k-shortest paths algorithms only address the single-pair problem and their worst-case time complexity on a directed graph is O(kn(m+nlogn)) (Gao et al., 2010). Thus, it is time consuming to run a single-pair algorithm n−1 times to solve the single-source problem, and thus a single-source algorithm should be applied to this inference problem.
4.2 Single-source k-shortest paths algorithm
To store all k-shortest paths, we adopt a data structure called pseudo tree. The previous definition (Gao et al., 2010) of a pseudo tree is for single-pair problem, so it only stores k-shortest paths from the starting node to the destination node. We expand this definition to store all k-shortest paths from the starting node to each other node.
Definition 1: —
Given all top k-shortest paths from the starting node to each other node, a pseudo tree stores all paths in a tree structure. If k=1, the pseudo tree is equivalent to the shortest path tree; as k>1, all 2nd to k-th shortest paths are iteratively merged into the pseudo tree by sharing the longest common prefix path.
Definition 2: —
A tree-path is a path from the root to another node in a pseudo tree.
An example is given in Figure 2, where all top three shortest paths from S are represented by the pseudo tree. Note that because every top k-shortest path should be a simple path, no nodes repeatedly appear in a tree path. Let A⇒B denotes a path from a node A to another node B through more than one edges, and A→B denotes the path from A to B through exact one edge (A, B). P1+P2 denotes a path formed by concatenating P1–P2. Most of prefix paths of a top k-shortest paths are also top k-shortest paths; however, some prefix paths are not. For example, the path S→A→C→D in Figure 2 is a prefix path of the third shortest path from S to E, S→A→C→D→E, but it is not a top three shortest path from S to D.
Fig. 2.

A graph and its corresponding pseudo tree. k=3. The node with a dash circle is a dummy node
Definition 3: —
Path nodes and dummy nodes. A tree-path to a path node is a top-k shortest path from the root to this path node, while a tree-path to a dummy node is not.
Therefore, in the above example, we call the node D in the tree-path S→A→C→D→E a dummy node and all other nodes except S path nodes. We will show that a complete pseudo tree might contain a huge number of dummy nodes, so we develop a heuristic algorithm to construct a pseudo tree with only path nodes. First, we discuss what topological conditions can generate dummy nodes.
Theorem 1: —
If a top k-shortest path from the root S to a node A contains a dummy node B, let the sub-path from the dummy node to A be PBA, then for each top k-shortest path from S to B, denoted by PSB, either PSB contains a node in PBA besides B or there exists a top k-shortest path from S to A using PSB as a prefix path.
proof. —
Let a top k-shortest path from S to A containing a dummy node B be P and the subpath of P before and after the dummy node be P1 and P2, respectively (P=P1+P2). We prove it by showing that if there exists a top k-shortest path from S to B, denoted by P′1, where P′1 does not contain any node in P2 except B and no top k-shortest path from S to A uses P′1 as a prefix path, then such P does not exist.
Since P′1 does not contain any node in P2 except B, we can build a new path from S to A: P′1+P2; additionally, P′1 is a top k-shortest path from S to B and P1 is not, so d(P)>d(P′1+P2). Since no top k-shortest path from S to A uses P′1 as a prefix path, P′1+P2 is not a top k-shortest path from S to A. Therefore, P cannot be a top k-shortest path from S to A either, so such P does not exist. ▪
For example, for the dummy node D in S→A→C→D→E in Figure 2, two top three shortest paths from S to D, S→B→D and S→B→E→D, contain B, and the other one, S→A→D, is a prefix path of the 2nd shortest path from S to B.
Theorem 2: —
Given a graph G, the pseudo tree for the k-shortest paths contains at most O(n2k) nodes.
proof. —
According to Definitions 1 and 3, a pseudo tree should contain exactly (n−1)k path nodes in order to indicate k-shortest paths from the starting node to each other node. Note that the pseudo tree must contain the shortest path tree with the root S, and all n−1 path nodes in the shortest path tree do not require any dummy nodes. However, each of other (n−1)(k−1) path nodes might requires at most n−2 dummy nodes, so the upper bound of the number of dummy nodes is (n−1)(n−2)(k−1), and the total number of nodes is O(n2k). ▪
Figure 3 shows an example with O(n2k) dummy nodes. There are four types of nodes besides the starting node in this example, Ai, Bi, Ci and Pij, and the number of nodes of each type is q, q, k, (q−1)(k−1), respectively, q∈O(n) (recall that k is a very small number). We construct all edges in the following procedure: (i) for 2≤i≤q, Ai has one outgoing edge (Ai, Ai−1) and one incoming edge (Bq, Ai); (ii) for 1≤i≤q−1, Bi has one outgoing edge (Bi, Bi+1); (iii) every Ci has two edges (A1, Ci) and (Ci, B1); (iv) every Pij has one incoming edge (Ai, Pij) and one outgoing edge (Pij, B1); (v) The source node S has one outgoing edge to Aq. The distances of all edges can be ignored except that d(Ai, Pij)+d(Pij, B1)=10i, 2≤i≤q, 1≤j≤k−1. The (top−1) shortest path from S to Ai is S→Aq⇒Ai, and k shortest paths from S to Bi are S→Aq→Aq−1⇒A1→Cj→B1⇒Bi, 1≤j≤k. Since all k shortest paths from S to Bq contain all A nodes, we cannot form the 2nd to k-th shortest paths from S to Ai by using any of them as a prefix path. Thereby, the 2nd to k−th shortest paths from S to Ai are S→Aq⇒Ai+1→Pi+1,j→B1⇒Bq→Ai, 1≤j≤k. All B nodes in these paths are dummy nodes, so each Ai, 1≤i≤q−1, requires (k−1)q dummy nodes. The total number of dummy nodes in this pseudo tree is therefore at least q(q−1)(k−1)∈O(n2k).
Fig. 3.
An example with O(n2k) dummy nodes. (a) The graph. Each bold arrow represents k−1 paths. (b) The pseudo tree. The red box contains total (k−1)q dummy nodes in the paths from S to Aq−1
Since the number of dummy nodes in some cases is extremely large, an exact algorithm to construct the complete pseudo tree would be too time-consuming. As shown in Section 5.4, dummy nodes are actually very rare in real networks, so we adopt a heuristic algorithm that builds a pseudo tree T consisting of only path nodes. Our algorithm to build the pseudo tree is similar to the Dijkstra's algorithm: we add a node to the tree each time. Let Nx denote a node in graph G and N′x denote one of the corresponding path nodes to a node Nx∈G. Note that a pseudo tree can have at most k path nodes corresponding to a single node in the graph. Starting from the root S, we iteratively expand the pseudo tree by adding a new path node N′b and an edge (N′a, N′b) to the pseudo tree, where N′a∈T and (Na, Nb)∈E; thereby, a new tree−path S⇒N′a→N′b is formed. We require that: (i) the tree−path S⇒N′a does not contain N′b, since the new tree-path should be a simple path; (ii) T has at most k−1 N′b; and (iii) the new tree-path should have the smallest distance among all new possible paths through an expandable edge, i.e.
Thereby, the new tree-path S ⇒ N′a → N′b is a top-k shortest paths from S to Nb containing only path nodes.
The pseudo code is shown in Algorithm 1. The min priority queue pq stores entries with the format <N′x, (Nx,Ny), dis>, where N′x is a path node in T, (Nx, Ny) is an expandable edge, and dis is equal to d(S⇒N′x)+d((Nx,Ny)). pq always pops out an entry with the minimal dis among all entries. count(Nx) is the total number of nodes N′x in T plus the number of entries containing an edge to Nx in the priority queue. At the initial phase (lines 1–4), the starting node S becomes the root in T and all edges from S are inserted into pq. We then iteratively pick up an entry from pq and add the corresponding edge to T (lines 5–7). Each time we add a new edge (N′a, N′b) to T, we find all possible expandable paths S ⇒ N′b → N′c, which have less distances than the current k-th shortest path from S to Nc (line 8). We then insert these paths into pq (line 11) or update the entry (line 13) depending on whether count(Nc) is already equal to k. The whole procedure is finished when there are no entries in pq.
We can further constrain the length of a tree-path (the height of the pseudo tree) to be less than a constant h. Since a regulatory pathway usually involves in a limit number of genes, h should be set to the maximum number of genes in a regulatory pathway. Once the new path node added in line 7 is in the hth level in the pseudo tree, the following code (lines 8–14) would not be executed.

Theorem 3: —
The time complexity of Algorithm 1 is O(nklog(nk)+mk(h+k)).
Proof. —
Count(Nx) in Algorithm 1 ensures that each node is inserted into the priority queue at most k times. This guarantees that the number of nodes in the final pseudo tree plus the number of entries in the priority queue is always less than or equal to nk, and the number of times of insertion is at most nk. Therefore, the time complexity of insertion into the priority queue is O(nklog(nk)), The time complexity of an update in the priority queue is O(1) if the Fibonacci heap is adopted, and we need to update an entry at most 2mk times since each edge might cause an update if one of its incident vertices is poped out as Na in line 6. Moreover, we need O(h) time to check whether Nc is not in the tree−path S⇒N′a (line 8) and O(k) time to update the sorted array (line 14). The maximal number of times we need to execute above two operations is mk. Hence, the total time complexity of Algorithm 1 is O(nklog(nk)+mk(h+k)). ▪
Note that if we execute n−1 times Yen's algorithm, the time complexity is O(nhk(m+nlogn)), so our algorithms provides a faster solution.
4.3 Diverse k-short paths
The k-shortest paths are usually very similar to each other because replacing few edges in the shortest path can yield several short paths. Thus, if we only take into account distance, we might overemphasize on these similar paths. Studying the diverse path might identify other more interesting regulatory pathways, so we introduce a single-source diverse k-short paths algorithm.
We define the diversity of a path as the number of edges in this path but not appearing in any already found paths divided by the total number of edges in the path. Formally, if we already find κ < k diverse paths P1, P2,… Pκ, the diversity of a new candidate path Pnew is:
Note that 0≤div(P)≤1, and if there are no found paths, the diversity is always 1. A path P is considered as a diverse path if and only if div(P)≥λ, where λ is a user-specified threshold. The larger λ is, the more diverse the found k paths are, but the total distance of the found k diverse paths would be larger.
The most intuitive approach to find k diverse short paths is executing Algorithm \ref{alg:heuristicK} to find k′ > k-shortest paths from the starting node to each other node, and for each node, we try to select k diverse paths among the k′ shortest paths. However, if the diverse paths are not enough, we need to repeat the procedure until k diverse paths are found for every node. Since the time complexity of Algorithm 1 is not linear in k, and there is no intuitive way to set an appropriate increasing amount of k, this procedure is time-consuming.
Instead, we adopt a strategy that we remove some but not all edges appearing in the pseudo tree from the graph and then re-run Algorithm 1 with the new graph until all k diverse paths are found for every node. We tend to remove edges that appear in a large number of k-shortest paths to yield diverse short paths while destroying the topology of the original graph as little as possible. Moreover, the removed edges should not be in the first few levels of the pseudo tree since they are usually used as a prefix path for several short paths. For these two reasons, we simply count the number of times that an edge appears in the constructed pseudo tree and remove edges which frequently appear in the pseudo tree. Since each edge must appear at most k times in the pseudo tree, We set the probability of an edge e being removed to count(e)/k if count(e)≥k/2, where count(e) is the number of times that e appears in the pseudo tree; otherwise, the probability is 0.
The procedure of our single-source k-shortest diverse paths algorithm is described as follows: we first execute Algorithm 1, and then for each node, we examine the diversities of k shortest paths in the increasing order of their distances. If the diversity of a path exceeds the threshold λ, the path is identified as a short diverse path. If the diverse paths are not enough (<k) for some nodes, we remove edges from the graph according to the probability described earlier. Then, we repeat the procedure until k diverse paths are found for every node or less than m/n edges are removed in the last iteration. Eventually, the importance value of each node is calculated as in Section 4.2.
5 EXPERIMENTS
5.1 Dataset preprocessing
We report results on the yeast gene network. The overview of the generation procedure of our yeast gene network and gold standards is shown in Figure 4, and follows best-practice in silico validation outlined by prior research (Beyer et al., 2006; Suthram et al., 2008; Tu et al., 2006). The gold standards were extracted from the knock-out compendium elucidated by Rosetta (Hughes et al., 2000). The deleted genes in the experiments were assumed to be causal genes and the highly perturbed P-value < 10−4, genes were seen as target genes. The set of candidate causal genes for each gold standard, generated by simulating eQTL region, is from (Tu et al., 2006). Using this procedure, 1728 gold standards were eventually extracted. Each gold standard consists of a causal gene, a target gene and 10 candidate causal genes, and when solving the three sub problems, UnknownCausal, UnknownTarget and CandidateCausal, the input is the target gene, the causal gene and the target gene with 10 candidate causal genes, respectively. The causal gene is also used to evaluate the accuracy in CandidateCausal.
Fig. 4.

Overview of our inference framework
To construct a weighted gene network, the TF–DNA bindings and PPIs were first combined into an unweighted network. TF–DNA binding data is from Beyer et al. (2006), and only the bindings whose likelihood scores are higher than 6.0 were used, resulting in a total of 4842 TF--DNA bindings. PPI data is from BioGRID (Stark et al., 2006), and only physical interactions found by low-throughput experiments were used since these interactions have higher precision (Paccanaro et al., 2005). Following this process, we constructed an unweighted network with 4304 nodes and 18 413 edges. We then used the Pearson correlation coefficient of the gene expression level in Rosetta compendium to weight edges. The conditions of gene expression we selected are based on (Tu et al., 2006).
Since a gene which is likely to be involved in a regulatory pathway involving the given gene tends to have higher expression correlation with the given gene, edges (ga, gb) and (gb, ga) are both assigned to a weight
in UnknownCausal and CandidateCausal, where gt is the target gene, and in UnknownTarget, the causal gene gc replaces gt. Note that we assigned different weights to an edge for different gold standards and for different sub problems. We do not use two incident genes' expression correlation, since a ‘date hub’, which is a node with high degree but the average gene expression correlation with its neighbors is low (Han et al., 2004; Jin et al., 2007), is more likely to be involved in a pathway than other node. The minimum weight ε is applied since the expression levels of the genes in a pathway do not all necessarily correlate with the given gene. We set ε to 0.15, the maximal height of the pseudo tree h to 15 in the following experiments. We found that k>10 does not generate results with significantly different quality from k=10. Therefore, k is varied from 1 to 10 in this article to examine the trade-off between efficiency and quality of the result. The experiments were performed on a machine with Intel core i5 650 and 16GB of main memory. The information flow model (Stojmirović and Yu, 2011) was implemented in MATLAB, and Tu's algorithm (Tu et al. 2006), Yen's algorithm (source code is from http://code.google.com/p/k-shortest-paths/) (Yen, 1971), Y-STATISTICAL (Malviya et al. 2011) and our algorithm were all implemented in Java.
5.2 Candidatecausal
The goal of CandidateCausal is to correctly pick the true causal gene from the ten candidate causal genes. The accuracy is calculated by dividing the number of gold standards in which the algorithm correctly chooses the true causal gene by the total number of gold standards. Table 1 shows the prediction accuracy, time to compute, in CandidateCausal and the P-values of the paired Binomial test, which examines the significance of the difference between the prediction results of our k-shortest paths algorithm (top line) and the other algorithms we evaluate. Y-STATISTICAL is too slow to process all gold standards in feasible time, as shown in Section 5.4, so we do not report it. The computation time is the average on each gold standard. The values of the damping factor α in the information model do not affect the accuracy significantly in the suggested range (0.55–0.99): the accuracies range from 29–30%, so we just show two results with α=0.55 and α=0.85. Note that since each gold standard has 10 candidates, a purely random strategy would be expected to yield a predictive accuracy of around 10%. Both our approach and the state of-the-art baseline candidates perform much better but accuracy is limited because the gold standards extracted from the knock out compendium are noisy, and the gold standards themselves represent only the domain knowledge available to biologists currently which is known to be incomplete. In terms of results, our approach represents a significant improvement over the previous state-of-the-art as measured by the paired binomial test.
Table 1.
Accuracy and computation time of CandidateCausal
| Algorithm | Accuracy (%) | P-value | Time(s) |
|---|---|---|---|
| k=10 shortest paths (s.p.) | 34.03 | − | 0.1 |
| k=10, λ=0.5 diverse s.p. | 34.08 | 0.2265 | 1.43 |
| k=1 s.p. | 30.38 | 1.09*e-6 | 0.01 |
| IF model (α=0.55) | 30.09 | 1.52*e-4 | 38.02 |
| IF model (α=0.85) | 29.11 | 4.76*e-6 | 38.42 |
| Tu's | 22.34 | 2.46*e-23 | 0.1 |
The diverse short paths algorithm with different λ yields very close accuracy and thus only λ=0.5 is shown here.
We find that for this problem the diverse paths algorithm evaluated on different values of λ cannot significantly enhance the prediction accuracy; nevertheless, we will show that the diverse paths can improve the enrichment levels and enrich different biological functions in Section 5.5. We should reiterate that in comparison to the random-walk-based approaches, not only do we see a clear benefit in terms of accuracy (and in some cases speed) but it is also much easier to directly identify the potential regulatory pathways by the k shortest paths algorithm.
5.3 UnknownCausal and UnknownTarget
For the next set of experiments, we used SaddleSum (Stojmirović and Yu, 2010) to evaluate the enrichment levels of the GO terms (Ashburner et al., 2000) for the results of UnknownCausal and UnknownTarget of a gene set V. In the result, each gene gi∈V is assigned an importance value Imp(gi). SaddleSum is a state-of-the-art software to approximate the probability of randomly picking |Vf| genes from V whose sum of the importance values exceeds ∑gi∈Vf Imp(gi) for each function f, where Vf⊆V denotes the set of all genes annotated with the function f. For each function, a e-value is reported by SaddleSum, which is the probability after applying the Bonferroni correction to the function's P-value. If the e-value of a function f generated from SaddleSum is less, it is more likely that the pathways involving the given gene regulates the property of f. To exclude high-level GO terms, we only examine the GO terms which less than 100 genes in the yeast gene network have. The resulting set, denoted by GOall, contains 1764 GO terms. The subset of GOall in which GO terms are annotated with the given gene is denoted by GOann. Note that different gold standards have different GOann. GOann is used to verify our result; on the other hand, GOall is used to point out potential GO terms which might associate with the given gene.
The lowest e-value among GOann is used to evaluate the function enrichment for each gold standard. These e-values of top gold standards are sorted and shown in Figure 5(a) and (b) for UnknownCausal and UnknownTarget, respectively. The damping factor α in the information model is 0.99 here, because it generates the lowest e-values among all suggested values, 0.55, 0.85 and 0.99 (Stojmirović and Yu, 2011). In UnknownCausal, because the given target genes are usually annotated with less GO terms, less gold standards have been found to be enriched than in UnknownTarget. In both UnknownCausal and UnknownTarget, an obvious trend is that the lowest e-values decrease when k increases. In UnknownCausal, the e-values obtained by the shortest path algorithm (k=1) are close to the information flow model and Tu's method, but when more paths are considered (k≥3), k-shortest} paths algorithm outperforms the information model and Tu's method. Similar results are observed in UnknownTarget. When k is increased from 5 to 10, the improvement is less because the 6th–10th paths are unimportant for regulatory pathways.
Fig. 5.

Evaluation using GOann: (a/b) The sorted e-values of the top GO term. (a) UnknownCausal using different k, (b) UnknownTarget using different k. The information model uses α=0.99. (c) The sorted maximal differences of e-values in each gold standard in UnknownTarget between the shortest paths and the diverse short paths using different λ. k=5
In Figure 6(a) and (b), GOall is used instead. Again larger k can more significantly enrich GO terms. Two obvious differences are: (i) The e-values are much smaller, especially in UnknownCausal, and almost all gold standards can have an enriched GO term, because more GO terms are considered. (ii) The improvement from k=5 to k=10 is apparent, possibly because potential GO terms are considered and thus more potential paths are useful to identify these pathways.
Fig. 6.

Evaluation using GOall. (a) UnknownCausal using different k, (b) UnknownTarget using different k and (c) Maximal differences. The description is the same as Figure 5
The results illustrate that due to weight normalization and other problems stated in Section 3, the information flow model and Tu's method cannot achieve higher enrichment than our shortest paths algorithm, although both methods consider all possible paths. These results are promising in that they agree with domain insights that a causal gene may regulate the target gene across three to five paths and that there is often an in-built redundancy within such pathways.
5.4 Diversity, importance value and efficiency
In this section, we report the comparison of diversity and efficiency. Since Tu's method and information flow model are not diverse path algorithms, we compare our single-source k diverse paths algorithm with a recently proposed heuristic approach—Y-STATISTICAL (Malviya et al., 2011), which is a single-pair k diverse paths algorithm. Y-STATISTICAL generates a path in each iteration and removes an edge in the last found path with a probability parameter Pr. The higher Pr is, the more diverse found k paths are. When Pr in Y-STATISTICAL is 0, Y-STATISTICAL is the same as Yen's algorithm.
Let P1, P2,… Pk be the k paths found by our original approach and |Pi| is the number of edges in Pi. We define k-paths diversity as the ratio of distinct edges in k paths, i.e.
Figure 7(a) presents the average importance value and the average k-paths diversity of the found paths from the starting nodes GGC1 to each other node. Here, we only show the result on one starting node, because as discussed later, Y-STATISTICAL (or Yen's algorithm) is too time consuming to generate all outputs for all starting nodes, but we empirically found that the comparisons on other starting nodes we have tested are very similar. Since for some nodes, the number of found diverse paths is smaller than k, we cannot always calculate the total distance of k paths (would go to infinity), and thus the importance value is used instead. Note that a higher importance value indicates that the found paths have lower distances and hence is preferred. We varied Pr in Y-STATISTICAL and λ in our diverse paths algorithm from 0 to 1 with intervals 0.1 in Figure 7.
Fig. 7.
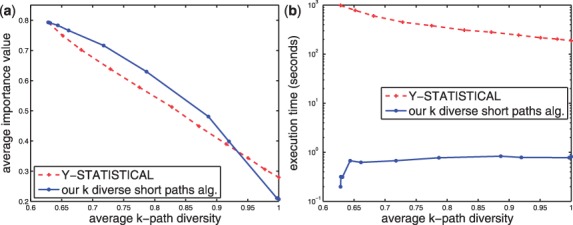
The comparison between our algorithm and Y-STATISTICAL on our yeast gene network. The starting node is GGC1. k = 5. (a) The average importance value vs. the average k-paths diversity and (b) Execution time vs. the average k-paths diversity
As mentioned earlier, when the k paths are required to be more diverse, the total distance of the k paths increases, and therefore the importance value decreases. As can be seen in the leftmost point in Figure 7(a), our heuristic k-shortest paths algorithm generates the same average importance value and the same average diversity as Yen's algorithm. We further compared the k paths found by our heuristic algorithm with the k-shortest paths found by Yen's algorithm for each node and found that all k paths are equivalent. In other words, no dummy nodes exist in the pseudo tree of the yeast gene network with the starting node GGC1, so our heuristic algorithm can generate the same paths as the exact algorithm.
If the k paths are requested to be more diverse, our diverse paths algorithm can find shorter paths than Y-STATISTICAL when the k-paths diversity is lower than 0.9. This is because Y-STATISTICAL might remove several edges in the first few levels in the pseudo tree while these edges are usually used to compose short diverse paths. Our algorithm tends to preserve these paths so it can identify shorter paths when the diversity required is relatively small. Note that extremely large diversity values will lead to less meaningful results from a utility standpoint.
Another advantage of our single-source algorithm is that it can find all k-shortest paths from the starting node to every other node, while it is very time consuming to execute n−1 times a single-pair algorithm. As shown in Figure 7(b), when k=5, the execution time (including loading the graph) of our heuristic single-source k shortest paths algorithm in yeast gene network is less than 0.3 sec, whereas Yen's algorithm costs average about 800 sec. As Y-STATISTICAL removes more edges, the execution time of Y-STATISTICAL is decreased and as λ in our k diverse paths algorithm increases, our algorithm needs more iterations to generate k paths for each node. However, our k diverse paths algorithm is still at least 100x faster than Y-STATISTICAL using any value of λ and Pr.
5.5 Diversity and enriched functions
In this section, we show that using different λ, the importance values generated by the k diverse paths algorithm can identify different potential regulatory pathways with different functions. Since more gold standards are enriched isn UnknownTarget than in UnknownCausal, we focused on UnknownTarget in this section. Figure 5(c) reveals the maximal difference of the log e-value (The e-value is set to 1 if the term is not significantly enriched (>0.01).) among all GO terms in GOann in each gold standard between the diverse k-shortest paths and the k-shortest paths algorithm (dif = log(Evalueλ=0)−log(Evalueλ>0)). Supplementary Table 1 presents the number of gold standards which have at least one significantly improved (dif≥2) GO term in GOann. Although as presented in Supplementary Figure 1, the diverse k short paths algorithm cannot decrease the lowest e-value, the diverse k short paths algorithm is able to enrich different GO terms with lower e-values. When λ=0.25, the maximal difference of e-value and the number of gold standards having an improved term are limited simply because the k diverse short paths are very similar to k shortest paths. However, if λ≥0.75, the diverse paths can enrich different functions in a large number of gold standards as the maximal difference is larger. Generally, λ=0.75 or 1.0 is able to identify a large number of different potential enriched functions and pathways, whereas λ≤0.25 produces similar paths to shortest paths and thus is relatively useless. We also show the maximal difference of the log e-value among all GO terms in GOall in Figure 6(c). The apparent difference in GOall is that the maximal difference using λ=1.0 is smaller than using λ=0.75. This is possibly because as all GO terms are considered, requiring completely diverse paths results in less interesting pathways. Therefore, λ=0.75 is a better setting to discover different functions which might annotate the given gene.
Table 2 shows some enriched GO terms whose e-values are significantly different using different λ, given starting node is RTS1. Examples with other starting nodes can be found in Supplementary Table 2. Not surprisingly, the e-values obtained using λ=0 are usually very closed to the e-values obtained using λ=0.25 since the found paths are very similar. In contrast, when λ≥0.5, different sets of enriched GO terms can be identified. For example, given the starting node RTS1, protein serine/threonine phosphatase activity is most significantly enriched using λ=0. On the other hand, using λ=0.75 can most confidently identify the pathways regulating regulation of mitotic cell cycle. As these GO terms have been found to annotate RTS1 (Chan and Amon, 2009; Wei et al., 2001), the result verifies that our algorithm is able to identify a number of regulatory pathways. Some GO terms not in GOann are also shown in Table 2 and Supplementary Table 2 to point out potential GO terms which might be associated with the given gene. For example, RTS1 might associate with spindle pole body and TOR signaling cascade according to the e-values found by different λ.
Table 2.
Case study: the e-values of some enriched GO terms found by k=5 diverse short paths algorithm with different λ, given the starting node RTS1
| GO number | Function name | λ=0 | λ=0.25 | λ=0.5 | λ=0.75 | λ=1.0 | Reference |
|---|---|---|---|---|---|---|---|
| GO:0000075a | Cell cycle checkpoint | 4.55E-6 | 6.43E-6 | 1.75E-8 | 1.11E-11 | 5.27E-6 | (Chan and Amon, 2009) |
| GO:0004721a | Phosphoprotein phosphatase activity | 1.29E-5 | 4.38E-4 | – | – | – | (Wei et al., 2001) |
| GO:0004722a | Protein serine/threonine phosphatase activity | 7.89E-7 | 1.56E-6 | 1.51E-5 | – | – | (Wei et al., 2001) |
| GO:0005816 | Spindle pole body | – | – | 1.15E-4 | 1.53E-6 | – | – |
| GO:0007062a | Sister chromatid cohesion | – | – | 9.33E-3 | – | – | (Riedel et al., 2006) |
| GO:0007346a | regulation of mitotic cell cycle | 3.89E-5 | 1.34E-5 | 1.01E-8 | 6.65E-13 | 4.44E-6 | (Chan and Amon, 2009) |
| GO:0031929 | TOR signaling cascade | 2.67E-8 | 1.44E-8 | 1.44E-6 | – | 7.71E-3 | – |
aThese GO terms are in GOann, i.e. they annotate RTS1.
E-values larger than 0.01 are denoted by ‘−’, representing insignificance.
Figure 8 visualizes the network consisting of the genes with the highest importance values using the starting node RTS1 when λ=0. The top 10 genes when λ=0 are PPH21 > RRD2 > SGO1 > TPD3 > TAP24 > PPH22 > SIT4 > RRD1 > TOR2 > CDC55. The top 10 ranked genes when λ=1.0 and their corresponding found paths are shown in Supplementary Table 3. It can be found that when λ is increased from 0 to 1.0, the order of several genes is significantly changed. For example, CDC55 becomes the 5th, and ZDS2, which is ranked 11th, becomes 7th. The change of importance values results in the different enriched GO terms as discussed earlier.
Fig. 8.
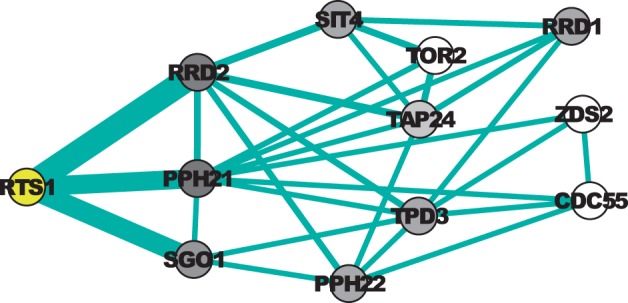
The top 10 genes when λ = 0 and their main pathways, given the starting node RTS1. The weight of each edge is reported by its thickness. Darker nodes have higher importance values when λ = 1.0. k=5
6 CONCLUSIONS
We proposed a heuristic single-source k-shortest paths algorithm and a single-source k diverse short paths algorithm to address the pathway inference problem in gene networks. We pointed out that an exact single-source k-shortest paths algorithm is practically infeasible, so a heuristic algorithm is adopted. A series of experiments was conducted to show that our algorithm can identify pathways with higher potentiality than current methods based on the random walk model, and requiring the paths to be more diverse can further uncover other potential regulated functions. Furthermore, our heuristic algorithm can achieve a huge speedup than the previous single-pair shortest paths algorithm while the found paths are equivalent in our yeast gene network.
In the future, we would like to study the structure of the pseudo tree because a number of sub paths still repeatedly appear in the tree, and thus it is possible to develop a compressed data structure.
Supplementary Material
ACKNOWLEDGEMENT
We thank Prof. Kai Tan (Iowa) and Nusrat S. Islam (Ohio St.) for initial discussions related to this work.
Funding: This work is supported by CCF-0702587, IIS-0917070 and IIS-1141828 from National Science Foundation.
REFERENCES
- Ashburner M., et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader J.S., et al. Gaining confidence in high-throughput protein interaction networks. Nat Biotechnol. 2004;22:78–85. doi: 10.1038/nbt924. [DOI] [PubMed] [Google Scholar]
- Bebek G., Yang J. Pathfinder: mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinformatics. 2007;8:335. doi: 10.1186/1471-2105-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer A., et al. Integrated Assessment and Prediction of Transcription Factor Binding. PLoS Comput Biol. 2006;2:e70. doi: 10.1371/journal.pcbi.0020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L., Amon A. The protein phosphatase 2a functions in the spindle position checkpoint by regulating the checkpoint kinase kin4. Genes Dev. 2009;23:1639–1649. doi: 10.1101/gad.1804609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P.G., Snell J.L. Random Walks and Electric Networks. Vol. 22. DC, USA: Mathematical Association of America Washington; 1984. [Google Scholar]
- Froehlich H., et al. Large scale statistical inference of signaling pathways from rnai and microarray data. BMC Bioinformatics. 2007;8:386. doi: 10.1186/1471-2105-8-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., et al. Fast top-k simple shortest paths discovery in graphs. CIKM. 2010:509–518. [Google Scholar]
- Hahn M.W., Kern A.D. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 2005;22:803. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- Han J.D.J., et al. Evidence for dynamically organized modularity in the yeast protein--protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- Haveliwala T.H. Topic-sensitive pagerank: A context-sensitive ranking algorithm for web search. IEEE Transactions on Knowledge and Data Engineering. 2003;15:784–796. [Google Scholar]
- Hershberger J., et al. Finding thekshortest simple paths: A new algorithm and its implementation. TALG. 2007;3:45. [Google Scholar]
- Hughes T.R., et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Jeong H., et al. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Jin G., et al. Hubs with network motifs organize modularity dynamically in the protein-protein interaction network of yeast. PLoS One. 2007;2:e1207. doi: 10.1371/journal.pone.0001207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malviya N., et al. A continuous query system for dynamic route planning. ICDE. 2011:792–803. [Google Scholar]
- Mering C.V., et al. Comparative assessment of large-scale data sets of protein–protein interactions. Nature. 2002;417:399–404. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- Missiuro P.V., et al. Information flow analysis of interactome networks. PLoS Comput Biol. 2009;5:e1000350. doi: 10.1371/journal.pcbi.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccanaro A., et al. Inferring protein–protein interactions using interaction network topologies. IJCNN. 2005;1:161–166. [Google Scholar]
- Riedel C., et al. Protein phosphatase 2a protects centromeric sister chromatid cohesion during meiosis i. Nature. 2006;441:53–61. doi: 10.1038/nature04664. [DOI] [PubMed] [Google Scholar]
- Scott J., et al. Efficient algorithms for detecting signaling pathways in protein interaction networks. J Comput. Biol. 2006;13:133–144. doi: 10.1089/cmb.2006.13.133. [DOI] [PubMed] [Google Scholar]
- Stark C., et al. Biogrid: a general repository for interaction datasets. Nucl. Acids Res. 2006;34((Suppl_1)):D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojmirovié A., Yu Y. ITM probe: analyzing information flow in protein networks. Bioinformatics. 2009;25:2447. doi: 10.1093/bioinformatics/btp398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojmirovié A., Yu Y. Robust and accurate data enrichment statistics via distribution function of sum of weights. Bioinformatics. 2010;26:2752. doi: 10.1093/bioinformatics/btq511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojmirovié A., Yu Y. Information flow in interaction networks II: channels, path lengths and potentials. ArXiv e-prints. 2011 doi: 10.1089/cmb.2010.0228. (0901.0287 (v3)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S., et al. eQED: an efficient method for interpreting eqtl associations using protein networks. Mol. Syst. Biol. 2008;4:162. doi: 10.1038/msb.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z., et al. An integrative approach for causal gene identification and gene regulatory pathway inference. Bioinformatics. 2006;22:489–496. doi: 10.1093/bioinformatics/btl234. [DOI] [PubMed] [Google Scholar]
- Vaske C.J., et al. A factor graph nested effects model to identify networks from genetic perturbations. PLoS Comput Biol. 2009;5:e1000274. doi: 10.1371/journal.pcbi.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voevodski K., et al. Spectral affinity in protein networks. BMC Syst. Biol. 2009;3:112. doi: 10.1186/1752-0509-3-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H., et al. Carboxymethylation of the pp2a catalytic subunit insaccharomyces cerevisiae is required for efficient interaction with the b-type subunits cdc55p and rts1p. J. Biol. Chem. 2001;276:1570–1577. doi: 10.1074/jbc.M008694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen J.Y. Finding thekshortest loopless paths in a network. Management Science. 1971;17:712–716. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.