

Abstract
Nuclear Dbf2p-related (NDR) kinases and associated proteins are recognized as a conserved network that regulates eukaryotic cell polarity. NDR kinases require association with MOB adaptor proteins and phosphorylation of two conserved residues in the activation segment and hydrophobic motif for activity and function. We demonstrate that the Neurospora crassa NDR kinase COT1 forms inactive dimers via a conserved N-terminal extension, which is also required for the interaction of the kinase with MOB2 to generate heterocomplexes with basal activity. Basal kinase activity also requires autophosphorylation of the COT1-MOB2 complex in the activation segment, while hydrophobic motif phosphorylation of COT1 by the germinal center kinase POD6 fully activates COT1 through induction of a conformational change. Hydrophobic motif phosphorylation is also required for plasma membrane association of the COT1-MOB2 complex. MOB2 further restricts the membrane-associated kinase complex to the hyphal apex to promote polar cell growth. These data support an integrated mechanism of NDR kinase regulation in vivo, in which kinase activation and cellular localization of COT1 are coordinated by dual phosphorylation and interaction with MOB2.
INTRODUCTION
Nuclear Dbf2p-related (NDR) kinases represent a subfamily of AGC (protein kinase A [PKA], PKG, and PKC-like) kinases. These kinases share structural similarities and require (auto)phosphorylation of a conserved Ser/Thr residue within the activation segment of the kinase domain for their activity. Phosphorylation of the activation segment leads to conformational changes that are required for basal enzymatic activity (31, 42, 45). Activation of AGC kinases also requires a second phosphorylation event or the permanent presence of an acidic residue in a hydrophobic motif that is located 45 to 60 residues C-terminal of the catalytic kinase domain in addition to phosphorylation of the activation loop (7, 33, 34). Germinal center (GC) kinases, a subfamily of Ste20-type kinases (10, 47), are involved in phosphorylation of this hydrophobic motif (9, 12, 26, 55, 57, 64). The phosphorylated motif interacts with a hydrophobic pocket in the N-terminal lobe of the kinase domain and induces/stabilizes the reconfiguration of the bilobal kinase structure (6, 13, 16, 60). Thus, both the phosphorylated activation segment and the phosphorylated hydrophobic motif are required for achieving the active conformation of the kinase (31, 42, 45, 64). Nevertheless, the chronology of these two phosphorylation events depends on the specific kinase, and either phosphorylation of the activation loop or hydrophobic motif phosphorylation can occur as the first step of kinase activation (4, 18, 54, 56).
Most AGC kinases contain functional domains other than the kinase core that are involved in regulating kinase activity and in vivo function. Important for NDR kinases are MOB coactivator proteins, which bind to the conserved N terminus of these kinases and are involved in their initial activation, presumably by promoting autophosphorylation of their activation segment (23, 27, 36, 37, 59). However, the detailed function of MOB in the initial activation step and the hierarchy of the MOB interaction with and autophosphorylation of the NDR kinase have not been clearly defined. Furthermore, phosphorylation of MOB1 by the upstream-acting GC kinase is important for promoting the MOB1-NDR interaction and kinase activation (26, 49, 59), yet this has not been shown for the interaction of an NDR kinase with MOB2-type adaptors, and the general nature of this activation mechanism is currently unclear. An additional complication arises from the finding that phosphorylation of the activation loop is critical for kinase activity but is only partially required for in vivo function, while phosphorylation of the C-terminal hydrophobic motif is essential in vivo but is not required for basal kinase activity (28, 30, 38, 64). Thus, it has not been resolved how these critical phospho residues coordinate kinase activity with MOB2 interaction and how these events are coordinated in vivo. Intriguingly, it has been shown that targeting of the kinase complex to the membrane by addition of a palmitoylation or myristoylation motif to either the NDR kinase or the MOB coadaptor increases NDR activity and phosphorylation status (23, 27, 38), but the mechanism of membrane targeting of NDR kinases and its relation with kinase activation is not understood.
The NDR kinase COT1 of the ascomycete Neurospora crassa is the founding member of this kinase family and is regulated by the GC kinase POD6 and two interacting MOB2-type proteins (37, 39, 52, 53, 62). COT1, POD6, and MOB2A/MOB2B are required for polar cell elongation but not for the establishment of polarity per se. Strains with mutations in these central COT1 complex components are viable and display hyperbranched cell growth, indicating that the COT1 pathway is essential for hyphal tip extension and is required to restrict excessive branch initiation in subapical regions of the cell. A similar branching and growth termination phenotype has been observed in neuronal cells of NDR kinase mutants (11, 12, 17, 19, 63), suggesting an evolutionarily conserved function of NDR kinases in the formation of branched cellular structures. Ser417 and Thr589 in the activation loop and hydrophobic motif of COT1, respectively, are two key regulatory phosphorylation sites that regulate polar growth and hyphal branch initiation by altering cell wall integrity and actin organization (38, 64). The interaction of COT1 with the two MOB2 proteins and activation of kinase activity require a conserved region directly preceding the kinase domain of COT1, which is sufficient for the formation of COT1-MOB2 heterodimers but is also necessary for kinase homodimerization (37). An additional, glutamine-rich N-terminal extension that is poorly conserved, but present in most fungal NDR kinases, is required for further stabilization of both types of interactions and for stimulating COT1 activity (37). COT1 lacking this region is degraded in a mob-2 background, suggesting that monomeric COT1 is unstable. Here, we dissect the mechanisms of COT1 autophosphorylation and hydrophobic motif phosphorylation and provide a multistep model for coordinating activation of COT1 with targeting of the kinase to the apex of the highly polar hyphal tip. Our results also help to explain the available data on the regulation of NDR kinases in other organisms.
MATERIALS AND METHODS
Strains, growth conditions, and microscopy.
Strains used in this study are listed in Table 1 (see also reference 40). General genetic procedures and media used in the handling of N. crassa are available through the Fungal Genetic Stock Center (FGSC; www.fgsc.net). Growth rates of fungal strains were determined by measuring radii of colonies on agar plates, starting with a well-established colony to exclude the lag phase of germination and the initial slow-growth phase of a developing colony.
Table 1.
Neurospora crassa strains used in this study
| Abbreviated genotype | Genotype | Source or reference |
|---|---|---|
| Wild-type mating-type A | 74-OR23-1A | FGSC 987 |
| Wild-type mating-type a | ORS-SL6a | FGSC 4200 |
| cot-1(ts) | cot-1(H351R) | FGSC 4066 |
| pod-6(ts) | pod-6(I310K) | Seiler et al. (53) |
| HA-pod-6 | Ppod-6-HA-pod-6; his-3 | This study |
| Δpod-6 | pod-6Δ::natR | Seiler et al. (53) |
| myc-cot-1 | Pcot-1-myc-cot-1 | Maerz et al. (37) |
| myc-cot-1(S417A) | Pcot-1-myc-cot-1(S417A) | Ziv et al. (64) |
| myc-cot-1(S417E) | Pcot-1-myc-cot-1(S4127E) | Ziv et al. (64) |
| myc-cot-1(T589A) | Pcot-1-myc-cot-1(T589A) | Ziv et al. (64) |
| myc-cot-1(T589E) | Pcot-1-myc-cot-1(T589E) | Ziv et al. (64) |
| myc-cot-1(S417A,T589E) | Pcot-1-myc-cot-1(S417A,T589E) | O. Yarden, Hebrew University of Jerusalem, Israel |
| myc-cot-1(D337A) | Pcot-1-myc-cot-1(D337A) | Maerz et al. (37) |
| myc-cot-1(D337A) + his-3 | Pcot-1-myc-cot-1(D337A) + his-3 | myc-cot-1(D337A) + FGSC 6103 |
| Δpod-6; myc-cot-1(S417A) | Pcot-1-myc-cot-1(S417A); pod-6Δ::hph | This study |
| Δpod-6; myc-cot-1(S417E) | Pcot-1-myc-cot-1(S417E); pod-6Δ::hph | This study |
| Δpod-6; myc-cot-1(T589A) | Pcot-1-myc-cot-1(T589A); pod-6Δ::hph | This study |
| Δpod-6; myc-cot-1(T589E) | Pcot-1-myc-cot-1(T589E); pod-6Δ::hph | This study |
| his-3 | his-3 | FGSC 6103 |
| pm-HA-mob-2a | Pccg-1-pm-HA-mob-2a::his-3 | This study |
| pm-HA-mob-2a; Δpod-6 | Pccg-1-pm-HA-mob-2a::his-3; pod-6Δ::hph | This study |
| pm-myc-cot-1 | Pccg-1-pm-myc-cot-1::his-3 | This study |
| pm-myc-cot-1(T589A) | Pccg-1-pm-myc-cot-1(T589A)::his-3 | This study |
| pm-myc-cot-1(T589E) | Pccg-1-pm-myc-cot-1(T589E)::his-3 | This study |
| Δpod-6; pm-myc-cot-1 | Pccg-1-pm-myc-cot-1::his-3; pod-6Δ | This study |
| Δpod-6; pm-myc-cot-1(T589A) | Pccg-1-pm-myc-cot-1(T589A)::his-3; pod-6Δ::hph | This study |
| Δpod-6; pm-myc-cot-1(T589E) | Pccg-1-pm-myc-cot-1(T589E)::his-3; pod-6Δ::hph | This study |
| Δmob-2a; Δmob-2b | mob-2aΔ::hph; mob-2bΔ::hph | Maerz et al. (37) |
| Δmob-2a; Δmob-2b; myc-cot-1 | Pcot1-myc-cot-1; mob-2aΔ::hph; mob-2bΔ::hph | Maerz et al. (37) |
| Δmob-2a; Δmob-2b; myc-cot-1(T589A) | Pcot-1-myc-cot-1(T589A); mob-2aΔ::hph; mob-2bΔ::hph | This study |
| Δmob-2a; Δmob-2b; myc-cot-1(T589E) | Pcot-1-myc-cot-1(T589E); mob-2aΔ::hph; mob-2bΔ::hph | This study |
| Δmob-2a; Δmob-2b; myc-cot-1(S417A) | Pcot-1-myc-cot-1(S417A); mob-2aΔ::hph; mob-2bΔ::hph | This study |
| Δmob-2a; Δmob-2b; myc-cot-1(S417E) | Pcot-1-myc-cot-1(S417E); mob-2aΔ::hph; mob-2bΔ::hph | This study |
| Δmob-2a; Δmob-2b; pm-myc-cot-1(T589E) | Pccg-1-pm-myc-cot-1(T589E); mob-2aΔ::hph; mob-2bΔ::hph | This study |
| Pcot-1-cot-1-GFP | Pcot-1-cot-1-gfp; his-3− | This study |
| Pccg-1-cot-1-GFP | Pccg-1-cot-1-GFP::his-3; cot-1Δ::hph | This study |
| Pcot-1-myc-cot-1; Pccg-1-cot-1-GFP | Pcot-1-myc-cot-1; Pccg-1-cot-1-GFP::his-3 | This study |
| cot-1(T589A)-GFP | Pccg-1-cot-1(T589A)-GFP::his-3 | This study |
| cot-1(T589E)-GFP | Pccg-1-cot-1(T589E)-GFP::his-3 | This study |
| cot-1(S417E)-GFP | Pccg-1-cot-1(S417A)-GFP::his-3 | This study |
| cot-1(S417A)-GFP | Pccg-1-cot-1(S417E)-GFP::his-3 | This study |
| mob-2a-GFP | Pccg-1-mob-2a-GFP::his-3 mob-2aΔ::hph | This study |
| pm-myc-cot-1-GFP | Pccg-1-pm-myc-cot-1-GFP::his-3 | This study |
| yfpc-bni-1; cot-1-yfpn | Pccg-1-yfpc-bni-1::his-3; Pgpd-1-myc-cot-1-yfpn::his-3 | This study |
| yfpc-cot-1; cot-1ΔN-yfpn | Pccg-1-yfpc-myc-cot-1::his-3; Pgpd-1-myc-cot-1ΔN-yfpn::his-3 | This study |
| myc-cot-1; cot-1-tev-GFP | Pcot-1-myc-cot-1; Pccg-1-cot-1-tev-GFP::his-3 | This study |
| myc-cot-1;cot-1-tev-GFP; HA-mob-2a | Pcot-1-myc-cot-1; Pccg-1-cot-1-tev-GFP::his-3; Pccg-1-HA-mob-2a::hph | This study |
| cot-1-tev-GFP | Pccg1-cot-1-tev-GFP::his-3 | This study |
| cot-1-tev-GFP; HA-mob-2a | Pccg-1-cot-1-tev-GFP::his-3; Pccg-1-HA-mob-2a::hph | This study |
| myc-cot-1; HA-mob-2a | Pcot-1-myc-cot-1; Pccg-1-HA-mob-2a::his-3 | Maerz et al. (37) |
| cot-1-GFP; HA-mob-2a | Pcot-1-cot-1-gfp; Pccg-1-HA-mob-2a::his-3 | This study |
| cot-1(ts); pm-cot-1(D337A)-GFP | cot-1(H351R); Pccg-1-pm-cot-1(D337A)-GFP::his-3 | This study |
| pod-6(ts); pm-cot-1(D337A)-GFP | pod-6(I310K); Pccg-1-pm-cot-1(D337A)-GFP::his-3 | This study |
| cot-1(ts); cot1(D337A)-GFP | cot-1(H351R); Pccg-1-cot-1(D337A)-GFP::his-3 | This study |
| pod-6(ts); cot1(D337A)-GFP | pod-6(I310K); Pccg-1-cot-1(D337A)-GFP::his-3 | This study |
| myc-cot-1; cot1(D337A)-GFP | Pcot-1-myc-cot-1; Pccg-1-cot-1(D337A)-GFP::his-3 | This study |
| myc-cot-1(T589E); cot-1(D337A)-GFP | Pcot-1-myc-cot-1(T589E); Pccg-1-cot-1(D337A)-GFP::his-3 | This study |
Low-magnification documentation of fungal hyphae or colonies was performed with an SZX16 stereomicroscope equipped with a Colorview III camera and CellD imaging software (Olympus). Images were further processed using Photoshop CS2 (Adobe). Fluorescence microscopy was performed as described previously (1, 32). Strains were grown on Vogel's minimal medium plates for 1 to 2 days. The plasma membrane and the Spitzenkörper structure were stained with FM4-64 (1 μg/ml). An inverted Axio Observer Z1 microscope (Zeiss) equipped with a QuantEM 512SC camera (Photometrics) and Slidebook 5.0 software (Intelligent Imaging Innovations) were used for image acquisition.
Plasmid construction and fungal expression of tagged proteins.
To generate the hemagglutinin (HA)-pod-6 fusion construct, the cosmid X1F7 (52) was used to amplify the 5′ region of pod-6 with the primers 5′-TTA CAT ATG ACT CTT ACA AGA CCA CGA GTT CC-3′ and 5′-ACC GGC GTA GTC GGG CAC GTC GTA GGG GTA GGA AGC GTA ATC AGG ACA TCG TAA GGG TAC GCC ATG TTT GCT GAC TGC CCC CGC-3′, which contain a partial sequence of the future 3× HA tag. The pod-6 gene was amplified using the primers 5′-ACC GGC GTA GTC GGG CAC GTC GTA GGG GTA GGA AGC GTA ATC AGG ACA TCG TAA GGG TAC GCC ATG TTT GCT GAC TGC CCC CGC-3′ (encompassing the second part of the HA tag) and 5′-TAT CAT ATG CTA CCT CCC TCA GAC ACT CGT G-3′. The amplified 5′ and pod-6 regions were fused via PCR by their partial overlapping HA tag sequence, generating the HA-pod-6 replacement cassette with NdeI restrictions sites at each end. The HA-pod-6 replacement cassette was excised with NdeI and transformed into a pod-6(ts); Δmus52::bar; his-3 strain. Transformants were screened for growth at 37°C and expression of HA-tagged POD6. Positive clones were backcrossed to obtain the tagged pod-6 allele in a his-3 background lacking the Δmus52::bar marker. Proper integration of the HA-pod-6 cassette at the endogenous locus was verified by Southern blot analysis.
The consensus motif for N-terminal palmitoylation (pm; M-G-C-X-X-S-A/S/T), which is present in the N. crassa Gα-subunit GNA1 and the mammalian Lck tyrosine kinase and targets the respective proteins to the fungal/animal plasma membrane, was fused to either MOB2A or COT1. HA-mob-2a was amplified from pHAN1-mob2a (37) with the primers 5′-TTG TCT AGA ATG GGC TGC GTC TGC AGC TCT AAC TAC CCA TAC GAT GTT CCA GAT TAC-3′ and 5′-GGA ATT CCT AGC TCG AGG GTG GGC C-3′, thereby introducing the palmitoylation motif and restrictions sites for XbaI and EcoRI. The resulting PCR product was cloned into pHAN1 via XbaI and EcoRI to generate pHA-pm-HA-mob-2a. To introduce the palmitoylation motif into different myc-COT1 variants, the following primers were used to amplify the gene from genomic DNA of the respective myc-cot-1 strains: 5′-TTG TCT AGA ATG GGC TGC GTC TGC AGC TCT AAC GAC AAC ACC AAC CGC CCC C-3′ and 5′-TTG GGG CCC TTA TCG GAA GTT GTT GTC G-3′. The PCR fragments were cloned into pHAN1 via the restriction sites XbaI and ApaI, thereby eliminating the HA tag present in the original plasmid. All constructs were sequenced and targeted for ectopic expression at the his-3 locus of FGSC 6103.
In order to generate C-terminal COT1-green fluorescent protein (GFP) fusion constructs, the different variants of this gene were amplified from the respective genomic DNA by using either 5′-TTG TCT AGA ATG GGC TGC GTC TGC AGC TCT AAC GAC AAC ACC AAC CGC CCC C-3′ or 5′-TCT AGA ATG GAC AAC ACC AAC CGC-3′ and 5′-GGA TCC CTC GGA AGT TGT TGT CG-3′ and into pMF272 (15) via the introduced restriction sites XbaI and BamHI and targeted for ectopic expression at the his-3 locus of FGSC 6103. To introduce a tobacco etch virus (TEV) protease cleavage site, the GFP tag was amplified from pMF272 using the following primers: 5′-ATC CCC GGG GAA AAC CTA TAC TTT CAG GGC ATC GTG AGC AAG GGC GAG G-3′ and 5′-TCG AAT TCT TAC TTG TAC AGC-3′. The obtained PCR fragment was cloned into the pMF272-COT1 fusion plasmid via the introduced SmaI and EcoRI sites, thereby replacing the original GFP tag with a TEV protease-cleavable GFP tag, and transformed into either a his-3 or a myc-cot-1; his-3 strain.
The dual expression vector for bimolecular fluorescence complementation (BiFC) localization studies was generated by amplifying the Aspergillus nidulans Pgpd promoter from pEHN1-nat (kindly provided by S. Pöggeler, Göttingen, Germany) using the primers 5′-AGC GGC CGC GAG CTC TGT ACA GTG ACC G-3′ and 5′-CTT CTG CTT GTC CAT GGT GAT GTC TGC TCA AG-3′, thereby introducing a NotI restriction site to the 5′ end of the PCR fragment. In a second step, the portion of the yfp gene encoding the C-terminal part of the protein (subsequently called yfpc) was amplified from pYFPC (3) by using 5′-GAC ATC ACC ATG GACAAG CAG AAG GGC ATC-3′ and 5′-AGC GGC CGC GAG AAT TCA GGC ATT TAA ATG CAG ATC TCT TGT ACA GCT CGT CCA TGC-3′ as primers to generate a yfpc PCR fragment that contained BglII, EcoRI, and NotI sites at its 3′ end. The sequence of the 5′ end of yfpc and the 3′ end of the Pgpd fragment were complementary, thereby enabling both fragments to be fused via fusion PCR. This fragment was digested with NotI and cloned into the NotI restriction site of pYFPN (3) to generate pBiFC for expression of N-terminally YFPC-tagged and C-terminally YFPN-tagged fusion proteins under the control of PgpdA and Pccg-1 promoters, respectively, and targeted to the his-3 locus. In order to generate the yfpn and/or yfpc cot-1 fusion construct, myc-cot-1 was amplified from genomic DNA using the following primer pairs: 5′-ACT AGT ATG GAC AAC ACC AAC C-3′ and 5′-tta att aaT CGG AAG TTG TTG TCG-3′ for generation of yfpc-cot-1 and 5′-ATT TAA ATC CAT GGA CAA CAC CAA C-3′ or 5′-ATT TAA ATC CAT GAA CTA CCA G-3′ and 5′-ATT TAA ATT TTA TCG GAA GTT GTT GTC-3′ for the cot-1-yfpn fusion construct. The alternative start primer, 5′-ATT TAA ATC CAT GAA CTA CCA G-3′ was used for amplification to generate cot-1ΔN-yfpn, an N-terminal truncated yfpn fusion construct lacking amino acids 1 to 212. The obtained PCR fragments were digested with SpeI and PacI or SwaI, respectively, and cloned into pBiFC, thereby generating plasmids for expression of either yfpc-cot-1, cot-1-yfpn, cot-1ΔN-yfpn, or combinations of them. In order to obtain a pBiFC construct for expression of cot-1-yfpn plus yfpc-bni-1, the formin gene was excised from pMF272_bni-1_sgfp (32) via SpeI and PacI and cloned into the respective sites of pBiFC containing cot-1-yfpn. The sequenced constructs were targeted for ectopic expression at the his-3 locus of FGSC 6103.
The HA-mob-2a expression cassette containing the ccg-1 promoter and the HA-tagged open reading frame was excised from pHAN1-mob2a (37) by using NotI and PstI for digestion and cloned into pCSN44, which contains a hygromycin resistance cassette. The obtained fragment was blunted using T4 DNA polymerase and cloned into the SmaI site of pCSN44. MOB2A-GFP fusion constructs were generated by amplifying the mob-2a gene from genomic DNA using the primers 5′-TCT AGA ATG GAT CCC AAT AAT GG-3′ and 5′-CCC GGG GCT CGA GGG TG-3′ and cloned into pMF272 via the XbaI and SmaI restriction sites, thereby generating pMF272-MOB2A.
A kinase-dead variant of COT1(D337A) was generated by site-directed mutagenesis of the cot-1 wild-type allele residing in a subcloning vector by using the primer pair 5′-GTT GTG CAT GCA GAG cTA TTA AGC CAG ACA AC-3′ and 5′-GTT GTC TGG CTT AAT AgC TCT GCA TGC ACA AC-3′. Integration of the point mutation was verified by sequencing. The mutated cot-1 variant and the target vectors pMF272-COT1 and pMF272-pmCOT1 were digested with MscI, and the obtained mutated cot-1 fragment was introduced into the final vectors, thereby generating pMF272-COT1(D337A) and pMF-pmCOT1(D337A). The sequenced constructs were targeted for ectopic expression at the his-3 locus of each respective strain.
Bacterial expression of MOB2A.
In order to generate a myelin basic protein (MBP)-tagged fusion construct of MOB2A for expression in Escherichia coli, cDNA of MOB2A was amplified with the primers 5′-A TGC GAA TTC ATG GAT CCC AAT AAT GGT TCG ATC-3′ and 5′-ATC GGC GGC CGC CTA GCT CGA GGG TGG GCC G-3′. The obtained PCR fragment was cloned into pMalc2x (32, 58) via the PCR-introduced EcoRI and NotI sites.
Rosetta2(DE3) cells were grown at 20°C in LB+ medium (1% NaCl, 0.8% yeast extract, 1.8% tryptone, and 0.04% glucose) to an optical density of 0.4, and protein expression was induced for 3 h with 0.1 M isopropyl-β-d-thiogalactopyranoside. Cell extracts were generated by digestion with lysozyme (1 mg/ml) for 1 h at 4°C in lysing buffer (50 mM Tris [pH 7.5], 125 mM NaCl, 5 mM MgCl2, 0.02% NP-40, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM benzamidine). MBP-MOB2A was purified using amylose resin (New England BioLabs, United Kingdom), washed twice with lysing buffer, eluted with 20 mM maltose, and stored at −20°C in 50% glycerol. Fifteen micrograms of the MBP-fusion protein was used for the POD6 kinase assay.
Kinase assays.
Purification of myc-tagged COT1 and peptide-based in vitro activity determinations were performed as described previously (37), using the peptide KKRNRRLSVA as an artificial substrate. To determine phosphorylation of the activation segment (RSRRLMAYSTVGTPDYI) and the hydrophobic motif (EESPELSLPFIGYTFKRFDNNFR), the respective peptides were used at a final concentration of 2 mM. The influence of the modified hydrophobic motif-based peptides on COT1 activity was analyzed by adding 1 mM HM-A (EESPELSLPFIGYAFKRFDNNFR) or HM-D (EESPELSLPFIGYDFKRFDNNFR) to the respective reaction mixture. An adjustment to a final concentration of 2% dimethyl sulfoxide (DMSO) was necessary to prevent precipitation of HM-A. Phosphorylated and altered amino acids are underlined. In order to examine the stimulation of COT1 kinase activity of POD6, POD6 was purified as described for the POD6 kinase activity assay below. Precipitated COT1 and POD6 were mixed in a ratio of 1:3 and washed twice with COT1 kinase buffer (20 mM Tris [pH 7.5], 10 mM MgCl2, 1 mM DTT, 1 mM benzamidine, 2 mM Na3VO4, 5 mM NaF). This mixture was preincubated at 37°C for 30 min in buffer containing 0.5 mM ATP and washed once with kinase buffer. The reaction was continued as described previously (37).
For purification of the COT1-COT1 dimer, COT1-TEV-GFP was precipitated for 2 h at 4°C on a rotation device with 5 μl/ml GFP trap (Chromotek, Germany) from cell extract obtained from frozen mycelia of the respective strains and resuspended in immunoprecipitation (IP) buffer (50 mM Tris [pH 7.4], 100 mM KCl, 10 mM MgCl2, 0.15% NP-40, 20 mM β-glycerophosphate, 2 mM Na3VO4, 5 mM NaF, 0,5 mM PMSF, 1 mM benzamidine, 2 mM EGTA, 1 mM DTT, 1 μg/ml pepstatin A, 5 μg/ml aprotinin). The GFP-trapped protein was washed twice with TEV buffer containing 50 mM Tris (pH 7.4), 100 mM KCl, 2 mM EGTA, 1 mM DTT. COT1-TEV-GFP was released from the GFP trap by digestion with TEV protease (4 U/μl) at 4°C for 6 h under constant rotation The supernatant containing released COT1-GFP was adjusted to 1 ml with IP buffer followed by myc-IP and a subsequent COT1 kinase activity assay as described previously (64), using the peptide KKRNRRLSVA as an artificial substrate.
For purification of HA-POD6, mycelial samples were frozen in liquid nitrogen, pulverized, and resuspended in POD6 IP buffer (40 mM morpholinepropanesulfonic acid [MOPS; pH 7.5], 100 mM KCl, 10 mM MgCl2, 0.15% NP-40, 20 mM β-glycerophosphate, 2 mM Na3VO4, 5 mM NaF, 0.5 mM PEFAbloc, 1 mM benzamidine, 2 mM EGTA, 1 mM DTT, 1 μg/ml pepstatin A, 5 μg/ml aprotinin, 10 μg/ml leupeptin). The samples were homogenized and centrifuged at 4,500 × g for 30 min, and the supernatant was subjected to a second centrifugation step for 60 min at 17,000 × g. The generated supernatant was incubated for 1 h at 4°C on a rotation device with 1 μl anti-HA-antibody (Sigma-Aldrich, Germany) per 1 ml cell extract. The antigen-antibody complexes were recovered using protein A-Sepharose (Amersham, United Kingdom) and washed once with POD6-IP buffer, twice with POD6-IP buffer containing 250 mM NaCl, and twice with POD6 kinase reaction buffer (15 mM MOPS [pH 7.5], 10 mM MgCl2, 1 mM DTT, 1 mM benzamidine, 2 mM Na3VO4, 5 mM NaF). To determine COT1 phosphorylation by POD6, immunoprecipitates of both kinases were mixed at a ratio of 1:3. The kinase reaction was started by resuspending the beads in 50 μl POD6 kinase buffer containing 0.5 mM ATP and 2 μCi [32P]ATP and incubated for 30 min at 37°C. Samples were centrifuged for 1 min at 12,000 × g, and the supernatant was removed. The remaining Sepharose beads were boiled in Laemmli buffer and used to determine 32P incorporation into COT1 and POD6 and equal protein levels of both proteins by SDS-PAGE/autoradiography and Western blot analysis, respectively. To examine hydrophobic motif phosphorylation of COT1 by POD6, immunoprecipitated HA-POD6 was resuspended in 30 μl POD6 kinase buffer containing 0.5 mM ATP, 1 μCi [32P]ATP, and either 1 mM HM or HM-D as substrate peptides. The kinase reaction was performed at 37°C for 30 min. The supernatant was spotted onto P81 filter discs (Whatman, United Kingdom), washed five times with 1% phosphoric acid and once with acetone, and dried. 32P incorporation into the peptides was determined by liquid scintillation counting. The remaining Sepharose beads were boiled in Laemmli buffer and used to determine equal protein levels via SDS-PAGE and Western blot experiments.
RESULTS
COT1-COT1 dimers represent the inactive pool of the NDR kinase.
COT1 and other fungal NDR kinases are able to dimerize in yeast two-hybrid assays (28, 37, 41), yet the presence and the functional significance of these kinase dimers in vivo have not been addressed. When we precipitated COT1 from a strain that coexpressed myc-COT1 from the modified endogenous locus under the control of its endogenous promoter and ectopically expressed COT1-GFP under the control of the ccg-1 promoter, we detected coprecipitation of both COT1 variants, indicating that COT1 can interact with itself in vivo (Fig. 1A). This self-association of COT1 raised the question whether COT1 dimerization is a mechanism to regulate kinase activity. To test this, we purified kinase-dead COT1(D337A)-GFP from a strain that coexpressed myc-COT1 or hyperactive myc-COT1(T589E) and examined COT1 dimer formation between inactive COT1(D337A)-GFP and wild-type COT1 and between COT1(D337A)-GFP and hyperactive myc-COT1 (Fig. 1B). While we detected a robust interaction between myc-COT1 and COT1(D337A)-GFP, the association of myc-COT1(T589E) with COT1(D337A)-GFP was significantly reduced, suggesting that the self-association of COT1 occurs between inactive kinase molecules. In a complementary experiment, we used the Pcot-1-myc-cot-1; Pccg-1-cot-1-GFP strain in which COT1-GFP can be overexpressed by growth in medium containing a poor carbon source, while expression of myc-COT1 remained constant. Kinase activity of precipitated myc-COT1 grown in 3% glycerol was reduced to 51% ± 5% (n = 3), compared to the control when myc-COT1 was precipitated from the strain grown in 2% saccharose (Fig. 1C). We concluded that the increased level of total cellular COT1 resulted in reduced myc-COT1 activity, by shifting the equilibrium toward inactive COT1-COT1 complexes. To confirm that the kinase dimer is inactive, we generated a strain that coexpressed myc-COT1 and COT1-TEV-GFP. Myc-COT1/COT1-GFP dimers were purified in a tandem purification experiment by performing anti-GFP precipitation followed by release of bound COT1 via the TEV protease cleavage site and a subsequent anti-myc precipitation step. In vitro kinase assays revealed that the COT1-COT1 precipitate was inactive, while individually precipitated COT1 displayed robust activity, irrespective of the tag used (Fig. 1D). COT1 and MOB2 are tightly associated proteins (37), and a coexpressed HA-tagged mob-2a construct coprecipitated with myc-COT1 and with COT1-GFP in the individual immunoprecipitation experiments. However after the tandem purification, no HA-MOB2A could be copurified, indicating the formation of mutually exclusive inactive COT1-COT1 homodimers or active COT1-MOB2 heterodimers, a hypothesis also implied by the fact that the COT1-COT1 and COT1-MOB2 interactions both require the N-terminal 212 amino acids of COT1.
Fig 1.

COT1 forms inactive dimers. (A) Anti-GFP immunoprecipitation of COT1 from strains coexpressing myc-cot-1 and GFP-cot-1 or the respective controls indicates the presence of COT1-COT1 dimers in vivo. (B) Myc-COT1 but not hyperactive myc-COT1(T589E) can be copurified by immunoprecipitation of kinase-dead COT1(D337A)-GFP from strains coexpressing myc-cot-1 or myc-cot-1(T589E) and cot-1(D337A)-GFP. (C) Endogenously myc-tagged COT1 was precipitated from strains coexpressing COT1-GFP under the control of the glucose-repressed ccg-1 promoter and subjected to COT1 three kinase assays. Myc-COT1 was purified from mycelia, which were grown either in liquid Vogel's minimal medium (containing 2% glucose) or in Vogel's minimal medium containing 3% glycerol as a carbon source. ccg-1-driven expression is induced by growth on poor carbon sources. Precipitated myc-COT1 (upper panel) and the expression levels of myc-COT1 (middle panel) and COT1-GFP (lower panel) in the presence of either 2% saccharose or 3% glycerol are shown. (D) COT1-COT1 complexes lack detectable in vitro kinase activity. COT1 was immunoprecipitated from the indicated strains by using either anti-GFP-trap or anti-myc antibodies, and kinase activities were determined. For tandem purification of COT1-COT1 complexes (gray bars), COT1-TEV-GFP was precipitated first and released by TEV protease cleavage, followed by anti-myc immunoprecipitation and three subsequent kinase assays using the artificial peptide KKRNRRLSVA as the substrate. An exemplary set of Western blot assays for the individual and tandem immunoprecipitation results is shown below the graph. MOB2A can be coprecipitated after the individual immunoprecipitation but not after tandem purifications. Note that the GFP fusion protein cannot be detected after TEV protease cleavage, because the cleavage site is located N-terminal of GFP. (E) Bimolecular fluorescence complementation experiments indicated that the COT1-COT1 dimer localizes as a bright dot in the distal region of the Spitzenkörper, which was costained by the lipid dye FM4-64. Control strains expressing the two YFP fragments, in which only one was fused with COT1, displayed only a weak cytosolic background signal. Additional control strains, in which COT1 was either coexpressed with the formin BNI1, which also localizes to the Spitzenkörper (32), or with an N-terminal-truncated version of COT1 (COT1ΔN; amino acids 1 to 212) that lacks the COT1 dimerization domain, did not show any specific localization.
In a final set of experiments, we determined the localization of the kinase homodimer in vivo by bimolecular fluorescence complementation microscopy (Fig. 1E). In these assays, N- and C-terminal fragments of YFP were fused with COT1 and the in vivo interaction of two kinase molecules monitored by complementation of the YFP fluorescence signal of the hybrid fluorophore. Coexpression of COT1-YFPN plus YFPC and YFPC-COT1 plus YFPN did not result in detectable YFP signals. In contrast, COT1-COT1 assemblies localized as an apical dot that was closely associated, but did not fully colocalize, with the distal region of a vesicle accumulation required for polar tip growth, called the Spitzenkörper (20, 51). To ensure that this dimerization was not the result of the high local accumulation of “sticky” YFP fragments in the Spitzenkörper region, we used BiFC constructs containing COT1 and the formin BNI1, which also localizes in this part of the hyphal tip (32). When we coexpressed COT1-YFPN plus YFPC-BNI1, no apical signal was detected. As a final control, we expressed a COT1ΔN-YFPN plus YFPC-COT1 pair, in which the N-terminal amino acids 1 to 212, which are required for the COT1-COT1 interaction in yeasttwo-hybrid assays (37), were deleted in one cot-1 construct. No apical signal of the COT1-COT1 dimer was detected in strains coexpressing these constructs. Based on these assays, we concluded that COT1 forms inactive homodimers that localize as a bright, circular dot in the hyphal apex and that partially overlap with the distal region of the Spitzenkörper.
Ser417 in the activation segment is the only autophosphorylation site of COT1.
When immunoprecipitated myc-COT1 was subjected to in vitro autophosphorylation reactions, we detected 32P incorporation in both COT1 isoforms (Fig. 2A). The substitution of an aspartic acid that is essential for catalytic activity of NDR kinases (22, 30) abolished phosphate incorporation in precipitated myc-COT1(D337A), indicating that this was indeed the result of COT1 autophosphorylation and not phosphorylation of COT1 by a copurifying kinase. Next, we used peptides covering the activation segment and hydrophobic motif of COT1 as in vitro substrates and detected phosphate incorporation in COT1(409–425), but not COT1(576–589) (Fig. 2B). Thus, COT1 autophosphorylation occurred within the activation segment and not the hydrophobic motif. To differentiate between Ser417 and the neighboring Thr418 as sites of autophosphorylation, we used strains that carried substitutions of Ser417 with alanine or glutamate of the endogenous cot-1 gene (64). As a control, we used strains mutated at Thr589 of the hydrophobic motif. In vitro autophosphorylation experiments with precipitants of these COT1 variants identified Ser417 as the only autophosphorylation site of COT1 (Fig. 2C). We also detected autophosphorylation of COT1 in vivo by probing the precipitated kinase with a phospho-Ser417-specific antibody (Fig. 2C). We concluded that Ser417 is the primary site of COT1 autophosphorylation and that phosphorylation of the activation segment is independent of Thr589 modification within the hydrophobic motif.
Fig 2.
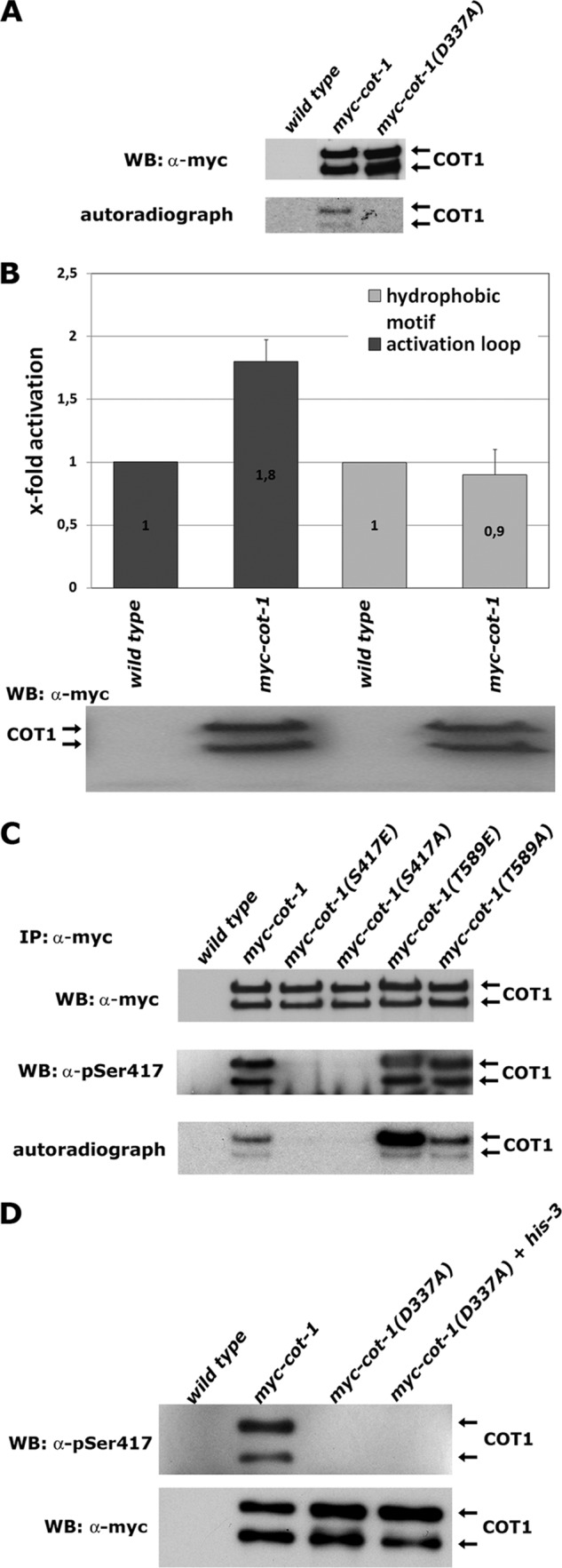
Ser417 is the only site of COT1 autophosphorylation. (A) Immunoprecipitated myc-COT1 variants from the indicated strains were probed with anti-myc antibody (upper panel) to determine equal amounts of precipitated kinase and subjected to a 32P in vitro autophosphorylation reaction (lower panel). WB, Western blotting. (B) In vitro kinase assays (performed in triplicate) with peptides covering the sequence of the activation segment (RSRRLMAYSTVGTPDYI) and hydrophobic motif (EESPELSLPFIGYTFKRFDNNFR) of COT1 as artificial substrates. Equal amounts of precipitated myc-COT1 used in the kinase reactions are shown below the graph. Phosphorylated amino acids are underlined. (C) Immunoprecipitated myc-COT1 variants from strains with the indicated genotypes were probed with anti-myc antibody (upper panel) or P-Ser417-specific antibody (middle panel) and subjected to an in vitro autophosphorylation reaction (lower panel). (D) Kinase-dead myc-COT1(D337A) was precipitated from a forced heterokaryon harboring an untagged copy of cot-1 and probed for Ser417 autophosphorylation in cis or in trans of the COT1 dimer with a phospho-Ser417-specific antibody (upper panel) or with an anti-myc antibody as a loading control (lower panel). The lack of myc-COT1(D337) phosphorylation purified from the heterokaryon indicates autophosphorylation of COT1 in cis.
Activation segment exchange is a common mechanism for autophosphorylation of kinase dimers in trans (43, 46), and we explored the possibility of autophosphorylation occurring in cis or trans for a COT1-COT1 dimer by generating forced heterokaryons in a strain expressing kinase-dead myc-COT1(D337A) with a his-3− strain harboring an untagged wild-type copy of cot-1 (Fig. 2D). No Ser417 phosphorylation was detected in COT1 precipitated from the myc-cot-1(D337A) strain or from the myc-cot-1(D337A) plus his-3− heterokaryotic strain, further indicating that the kinase dimer is inactive and strongly suggesting that Ser417 autophosphorylation in vivo is performed in cis.
COT1 is phosphorylated by the GC kinase POD6 at Thr589 within the hydrophobic motif.
Ectopic expression of myc-COT1(T589E) partially suppressed the growth defect of a conditional N. crassa pod-6(ts) strain and the corresponding Δkic1 kinase mutant in budding yeast (44, 64). In order to obtain quantitative data on the modified endogenous cot-1 locus in a pod-6 deletion background, we crossed the myc-cot-1 phosphosite alleles into a Δpod-6 strain. The tip extension rate of the Δpod-6; myc-cot-1(T589E) strain was 18% and 36% of that of the myc-cot-1 and myc-cot-1(T589E) strains, respectively (n ≥ 5), while the Δpod-6; myc-cot-1(T589A), Δpod-6; myc-cot-1(S417E), and Δpod-6; myc-cot-1(S417A) strains displayed phenotypic characteristics identical to those of the Δpod-6 (Fig. 3A) strain. Thus, acidic substitution of the hydrophobic motif phosphorylation site of COT1 only partially rescued the absence of the upstream kinase POD6.
Fig 3.

COT1 is phosphorylated by the GC kinase POD6 in the hydrophobic motif. (A) Suppression analysis of Δpod-6 in strains expressing the indicated phospho variants of COT1. Note the partial suppression of Δpod-6 by myc-cot-1(T589E). (B) myc-COT1 purified from either the myc-cot-1 or the myc-cot-1(T589A) strain was preincubated with independently HA-precipitated HA-POD6 or mock precipitate from the wild type prior to a COT1 activity assay (five replicates) using the peptide KKRNRRLSVA as the substrate. COT1, but not COT1(T589A), activity was stimulated by POD6. Equal precipitation of myc-COT1 and HA-POD6 used in these assays was confirmed by Western blot (WB) analysis. (C) Direct phosphorylation of COT1 by POD6. Anti-HA precipitates from the HA-pod-6 or a mock control were mixed with precipitated myc-COT1(S417A), myc-COT1(S417E), or control precipitate. Autophosphorylation of HA-POD6 and POD6-dependent phosphorylation of myc-COT1 are indicated. The results shown in the middle and lower panels confirm that equal amounts of precipitated kinases COT1 and POD6 were used for these assays. (D) POD6 phosphorylates COT1 at Thr589 in the hydrophobic motif. Purified HA-POD6 and a control precipitate from the wild type were used for peptide-based in vitro kinase assays. Two peptides covering the hydrophobic motif (amino acids 576 to 589) of COT1 that carried either the wild-type sequence of COT1 or a Thr589-to-aspartate substitution (termed HM and HM-D, respectively) were used as artificial POD6 substrates (experiment performed in triplicate). DMSO was used as the solvent control, and the result obtained with this control was set as 1. An immunoblot of precipitated POD6 probed with anti-HA antibody showing equal amounts of purified kinase is presented below the graph.
To test if POD6 directly phosphorylates COT1, we generated a strain in which we tagged with HA pod-6 at its endogenous locus under the control of its endogenous promoter. This strain displayed wild-type growth characteristics, confirming the full functionality of the modified kinase. Phosphate incorporation in precipitated HA-POD6 in vitro indicated that we were able to obtain active GC kinase that did undergo autophosphorylation, as described for other GC kinases (29, 48, 50). If POD6 functions upstream of COT1, we reasoned that preincubation of precipitated myc-COT1 with precipitated HA-POD6 should stimulate the in vitro activity of COT1. Consistent with this hypothesis, we observed 3.6-fold activation (±1.3-fold; n = 5) of myc-COT1 after addition of POD6 (Fig. 3B). In support of the genetic data, the activity of myc-COT1(T589A) was not stimulated by preincubation with POD6, strongly suggesting that POD6 phosphorylated Thr589 of COT1. Direct phosphorylation of COT1 by POD6 was determined by using the precipitated COT1 variants myc-COT1(S417A) and myc-COT1(S417E), which are incapable of undergoing autophosphorylation (Fig. 2C) as direct POD6 substrate (Fig. 3C). Both precipitated COT1 variants were phosphorylated by HA-POD6 but not by a mock precipitate from the wild type. The equal level of phosphate incorporation in the two-activation loop-modified COT1 variants led us to conclude that COT1 autophosphorylation is not a prerequisite for phosphorylation by POD6. To prove phosphorylation of Thr589 by POD6, we generated two peptides covering the region from 576 to 589 of COT1 that carried either the wild-type sequence of COT1 or a Thr589 to aspartate substitution (termed HM and HM-D, respectively). POD6 specifically phosphorylated HM, but not HM-D, in in vitro kinase assays (Fig. 3D), confirming that Thr589 in the hydrophobic motif of COT1 is targeted by POD6.
The interaction of COT1 with MOB2 is not regulated by POD6.
Phosphorylation of MOB1-type adaptors through upstream GC kinases regulates the affinity of the NDR kinase for its coactivators (26, 49, 59). Nevertheless, we did not observe phosphorylation of COT1-associated MOB2A and MOB2B by POD6 in our POD6 kinase assays (Fig. 3C). Thus, we tested if the interaction of myc-COT1 with the two MOB2 proteins was altered in a Δpod-6 background, but we detected wild-type levels of myc-COT1-associated MOB2A and MOB2B (Fig. 4A). To directly test for phosphorylation of MOB2 by POD6, we bacterially expressed and purified MBP-MOB2A and performed kinase assays with HA-POD6 precipitated from N. crassa. We observed strong autophosphorylation of POD6 but not phosphorylation of MOB2A, indicating that the MOB2A is not targeted by POD6 in vitro (Fig. 4B). In addition to these negative phosphorylation assays, we did not detect any physical interaction between POD6 and MOB2A or MOB2B in a yeast two-hybrid assay (Fig. 4C). Moreover, sequence analysis of fungal and animal MOB1- and MOB2-type proteins revealed that Thr12 and Thr74, the phosphomodified sites of mammalian MOB1 (2, 26, 49), are not conserved in fungal MOB2 proteins. Thr35 is absent in filamentous ascomycetes, while a homologous residue is present in MOB2 of the unicellular yeasts. In summary, these experiments strongly suggest that the interaction between COT1 and its two MOB2 adaptors is not regulated by the upstream kinase POD6.
Fig 4.
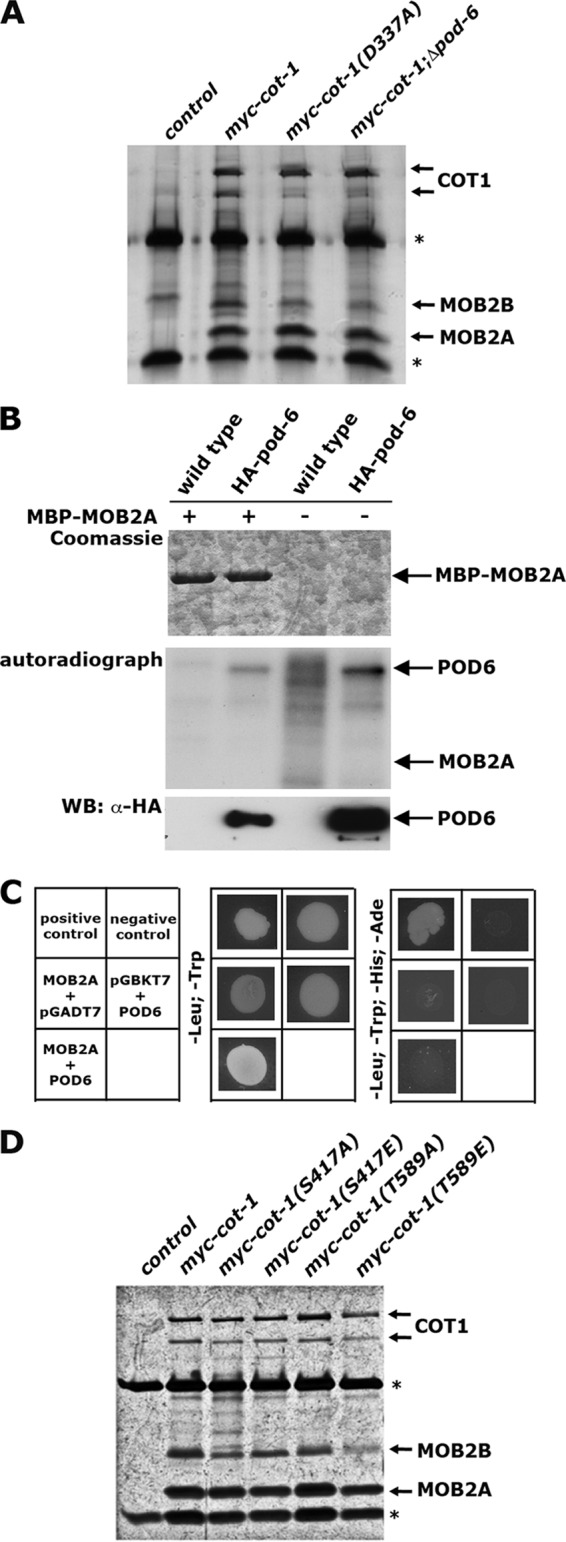
The interaction of COT1 with MOB2 is not regulated by POD6. (A) myc-COT1-associated MOB2 proteins were coprecipitated from the indicated strains by using anti-myc antibodies. The precipitants were separated by SDS-PAGE and silver stained. *, heavy and light chains of the antibody. Note that the interaction of MOB2A and MOB2B with COT1 does not depend on the kinase activity of COT1 and the presence of POD6. (B) Bacterially expressed MOB2A was purified and tested for phosphorylation by POD6 precipitated from N. crassa. No POD6-dependent phosphate incorporation in MOB2A was detected in the autoradiograph. (C) A potential physical interaction of MOB2A with POD6 was addressed in yeast two-hybrid assays. Note that these plasmids were previously used to determine interactions between COT1, POD6, and MOB2A (37). (D) myc-COT1-associated MOB2 proteins were coprecipitated from the indicated strains by using anti-myc antibodies. The precipitants were separated by SDS-PAGE and silver stained. *, heavy and light chains of the antibody. The interaction of MOB2A and MOB2B with COT1 does not depend on the phosphorylation status of COT1.
We also tested if the association of COT1 with MOB2A and MOB2B is regulated by the phospho status of COT1 or by its activity. However, we detected no significant differences in association of MOB2A and MOB2B with the Ser417- or Thr589-modified myc-COT1 phospho variants. Moreover, myc-COT1 precipitated from the kinase-dead myc-cot-1(D337A) strain that lacked 32P and P-Ser417 antibody-based autophosphorylation coprecipitated wild-type levels of MOB2A and MOB2B (Fig. 4D), indicating that COT1-MOB2 heterodimerization does not require COT1 activity and is independent of a specific phosphorylation state of COT1.
In vitro kinase activity and in vivo functionality of COT1 do not correlate.
In order to determine how these biochemical characteristics of COT1 translate into in vivo functionality, we compared in vitro kinase activities of the modified COT1 variants with the fungal growth rate of the mutant strains as a quantitative measure for in vivo functionality of the COT1 pathway (Table 2). Three distinct groups of COT1 variants with no, basal, or high activities were detected. Myc-COT1(T589E) displayed maximal kinase activity (designated as 100% activity), while myc-COT1 precipitated from the myc-cot-1(D337A), myc-cot-1; Δmob-2a; Δmob-2b, and myc-cot-1(S417A) strains displayed activities that were not significantly above background. myc-COT1 variants purified from the myc-cot-1(S417E), myc-cot-1, myc-cot-1; Δpod-6, and myc-cot-1(T589A) strains generated intermediate activities of 1 to 10% of that of the myc-cot-1(T589E) strain. We concluded that Ser417 phosphorylation in the activation segment of COT1 controls the transition between the inactive kinase and a kinase with basal activity, while phosphorylation of Thr589 leads to a fully active kinase.
Table 2.
In vitro kinase activities, autophosphorylation patterns, and relative growth rates of phosphosite-modified COT1 variants
| N. crassa abbreviated genotype | Abbreviated genotype Mean (±SD) COT1 activitya (n ≥ 3) | Ser417 autophosphorylation of COT1 |
COT1-MOB2 interaction | % relative growth rate (±SD) at 37°C (n ≥ 5) | |
|---|---|---|---|---|---|
| In vitro | In vivo | ||||
| myc-cot-1(T589E) | 100 | + | + | + | 65 ± 2 |
| myc-cot-1 (S417A,T589E) | 5–50b | − | − | + | 1 ± 1 |
| myc-cot-1(T589A) | 10 ± 4 | + | + | + | 1 ± 1 |
| myc-cot-1 | 5 ± 2 | + | + | + | 100 |
| Δpod-6; myc-cot-1 | 4 ± 1 | + | + | + | 0c |
| myc-cot-1(S417E) | 1 ± 0.5 | − | − | + | 76 ± 2 |
| myc-cot-1(S417A) | 0.04 ± 0.5 | − | − | + | 19 ± 2 |
| myc-cot-1; Δmob-2a; Δmob-2b | 0.04 ± 0.5 | − | +d | − | 0c |
| myc-cot-1; cot-1-tev-gfp | 0e | −e | −e | −e | 100 |
| myc-cot-1(D337A) | 0 | − | − | + | 0c |
Peptide-based kinase activities were determined using myc-COT1 as the reference and then recalculated relative to the myc-COT1(T589E) strain.
This COT1 variant produced highly variable kinase activities in several independent experiments, ranging from 5 to 50% of that of the myc-COT1(T589E) strain.
The indicated strains displayed phenotypes identical to those of the Δcot-1 and Δpod-6 strains. The growth rate of both deletion strains was ∼1 mm/week and is reported as 0.
Only the large isoform of COT1 was expressed and phosphorylated in a mob-2 double deletion background.
Characteristic of the COT1-COT1 dimer.
As predicted for an inactive kinase, the growth characteristics of myc-cot-1(D337A) and myc-cot-1; Δmob-2a; Δmob-2b strains mimicked those of the Δcot-1 strain, and these strains had phenotypic characteristics indistinguishable from those of the Δcot-1 strain (Table 2). However, the poor kinase activity measured for myc-COT1(S417A) [1% of myc-COT1 and 0.04% of myc-COT1(T589E)] translated into reasonable growth of the myc-cot-1(S417A) strain (19% of the growth rate of the myc-cot-1 strain). In contrast, the myc-cot-1(T589A) strain displayed an in vitro kinase activity not significantly different from that of the myc-cot-1 strain, but it displayed only 1% of the growth rate of the myc-cot-1 strain. Thus, very low levels of active COT1 are sufficient for growth if Thr589 can be phosphorylated. Moreover, COT1 purified from a Δpod-6 background was only 20% less active than the myc-COT1 control, indicating that only a minor fraction of cellular COT1 is active, similar to what has been determined for the budding yeast homolog Cbk1p (8).
Consistent with the hypothesis that activation of the kinase by Thr589 phosphorylation occurs after Ser417 autophosphorylation, we observed autophosphorylation of COT1 precipitated from myc-cot-1(T589A) and Δpod-6; myc-cot-1 strains (Table 2), indicating that binding of the MOB2A/B coactivators and Ser417 phosphorylation are independent events and that minimal in vitro COT1 activity is sufficient for efficient autophosphorylation in vivo. Thus, we tested if Thr589 phosphorylation was able to overcome the lack of phosphorylation of Ser417 by generating a strain expressing double-mutated myc-COT1(S417A,T589E). This myc-COT1 variant had pronounced activity (Table 2), indicating that Thr589 phosphorylation allows shifting of inactive COT1(S471A) into its active conformation. However, this COT1 variant produced highly variable kinase activities in several independent experiments, ranging from 5 to 50% of that of myc-COT1(T589E), suggesting that the double substitution resulted in a highly instable protein, a potential reason for its poor functionality in vivo, and the mutant displayed only 1% of the growth rate of the myc-cot-1 strain.
Maximal catalytic activity of COT1 requires a conformational change induced by hydrophobic motif phosphorylation.
The sequence analysis of COT1 and selected AGC kinases revealed the conservation of all residues involved in the interaction of the phosphorylated hydrophobic motif with the hydrophobic pocket in COT1 (Fig. 5A). Thus, we tested if a conformational change induced by hydrophobic motif phosphorylation is involved in activating COT1 by using hydrophobic motif peptides, in which we introduced Thr589 substitutions with aspartate or alanine to mimic the phosphorylated or nonphosphorylated hydrophobic motif (termed HM-D and HM-A, respectively). We observed 2-fold-increased myc-COT1 activity in the presence of HM-D, while the addition of HM-A to the kinase assay mixtures reduced myc-COT1 activity by 20% (Fig. 5B). Furthermore, myc-COT1(T589E) was not stimulated by the addition of HM-D, but the addition of HM-A reduced its activity to 80%. The opposite pattern was observed in kinase assays with myc-COT1(T589A). Addition of HM-D increased kinase activity 2.5-fold, while the addition of HM-A did not alter its activity. We concluded that the HM-D peptide induces the active state of COT1 in trans, while addition of HM-A shifts the equilibrium toward the conformation exhibiting basal kinase activity. In line with these biochemical data, we also observed that the myc-cot-1(T589A) strain but none of the other phospho site mutants, displayed temperature-sensitive growth defects (Fig. 5C), which may indicate an instable conformational state when Thr589 cannot be phosphorylated.
Fig 5.

A conformational change induced through hydrophobic motif phosphorylation is required for activation of COT1. (A) Alignment of COT1 and selected AGC kinases, emphasizing residues that are important for the interaction between the hydrophobic pocket within the small lobe of the kinase domain and the C-terminal phosphorylated hydrophobic motif (based on data from references 16 and 61). Green, residues that bind the first two Phe/Tyr of the hydrophobic motif; blue, residues that bind the phosphate of the hydrophobic motif; red, residues that bind the last Phe/Tyr of the hydrophobic motif; black, ion pair. (B) In vitro kinase activities of myc-COT1 variants precipitated from the indicated strains, determined using the peptide KKRNRRLSVA as artificial substrate. The activity of each COT1 variant was set to 100%, and the impacts of adding HM-A or HM-D peptides were determined. Results of a typical experiment are shown. To compare and quantify multiple assays, the relative activities of the indicated myc-COT1 variant from at least 3 experiments with the standard deviations relative to myc-COT1 were calculated. Due to the low kinase activity of COT1(S417A), which was barely above background (only 3% of the activity of myc-COT1), and the resulting high variability of the assay results, activity changes after incubation with the HM-A and HM-D peptides were not quantified for this variant. (C) Relative growth rates of the indicated strains at 25°C and 37°C. Note the temperature sensitivity of the myc-cot-1(T589A) strain.
Phosphorylation of Thr589 and interaction with MOB2 proteins are essential for distinct aspects of the polar localization of COT1.
Artificial membrane targeting of animal NDR kinases or of MOB1-type adaptors increases NDR activity (23, 27), but the mechanism of kinase transfer to the membrane is still unresolved. We determined if POD6 and/or the two MOB2 proteins were necessary for localizing COT1. Ectopically expressed COT1-GFP complemented the Δcot-1 deletion and labeled an apical dot that partly colocalized with the Spitzenkörper (Fig. 6A), similar to what we observed for the inactive COT1-COT1 dimer (Fig. 1E). However, COT1-GFP also localized as an apex-associated crescent in growing hyphal tips, which were not stained by the BiFC constructs, suggesting that the apical dot represents part of the inactive pool of the kinase, while the apex-associated crescent constitutes the active from of COT1. In line with this hypothesis, functional MOB2A-GFP localized in a pattern indistinguishable from that of COT1 and labeled both the Spitzenkörper-associated dot and the apical crescent (Fig. 6A). COT1(T589A)-GFP only poorly complemented Δcot-1 and resulted in a strain with a morphology and growth rate comparable to that of the myc-cot-1(T589A) strain. When we ectopically expressed COT1(T589A)-GFP in a wild-type background, we did observe a Spitzenkörper-associated dot, but no localization as an apex-associated crescent, indicating that hydrophobic motif phosphorylation is required for localization of active COT1 at the hyphal tip (Fig. 6A). In contrast, COT1(T589E)-GFP showed a localization pattern indistinguishable from that of wild-type COT1. We found the the localizations of Ser417-modified COT1-GFP constructs interesting. While COT1(S417E)-GFP displayed a near-wild-type localization, COT1(S417A)-GFP localized in a broader apical zone and a crescent that was not as confined.
Fig 6.

Membrane targeting of COT1 requires POD6-mediated phosphorylation of COT1 Thr589. (A) Localization of the indicated COT1-GFP variants, of MOB2A-GFP, and of the COT1-COT1 dimer, based on BiFC assays. The apical membrane-associated crescent observed for most constructs was abolished in strains expressing COT1(T589A)-GFP and the COT1-COT1 dimer. (B) The addition of a palmitoylation/myristoylation (pm) motif to the N terminus of COT1 allowed targeting of COT1 to the plasma membrane. (C) Temperature-sensitive mutants of cot-1(ts) and pod-6(ts) or strains expressing membrane-targeted, kinase-dead pm-COT1(D337A)-GFP were grown at room temperature and shifted to 37°C for 10 h. pm-COT(D337)-GFP did not complement the two kinase mutants. (D) Membrane targeting of the indicated myc-COT1 variants complemented the Δpod-6 strain (determined in triplicate experiments). (E) Membrane targeting of the indicated myc-COT1 strains rescued the hyperbranching defect of the Δpod-6 strain (n ≥ 120 measurements of branch intervals per strain).
In order to analyze how COT1 targets the hyphal tip, we engineered artificially membrane-targeted COT1 variants by attaching the consensus motif for N-terminal palmitoylation/myristoylation (pm; M-G-C-X-X-S-A/S/T) to the N terminus of COT1, analogous to animal MOB1 and NDR1 (23, 27). Ectopically expressed pm-COT1-GFP not only formed an apical cap at the hyphal tip but also associated with the plasma membrane along the whole hypha (Fig. 6B), confirming the suitability of the tag. In addition, abundant cytosolic spots were detected that may constitute COT1 associated with vesicular transport intermediates. As a control, we also tested if a kinase-dead version of COT1 regained functionality when targeted to the plasma membrane. However, the pm-cot-1(D337A) strain did not complement conditional cot-1(ts) and pod-6(ts) mutants (Fig. 6C), indicating that kinase activity is essential for COT1 function. In contrast, pm-myc-COT1(T589E) fully suppressed growth and branching defects of the Δpod-6 strain (Fig. 6D and E), indicating that Thr589 phosphorylation of COT1 by POD6 is a prerequisite for membrane association of COT1. Strikingly, pm-myc-COT1 and pm-myc-COT1(T589A) also fully suppressed the Δpod-6 defects. Thus, artificial targeting of the kinase to the plasma membrane in cells lacking the endogenous targeting signal (i.e., phosphorylation of Thr589 by POD6) is more important for the in vivo functionality of the NDR kinase than its Thr589 phosphorylation-dependent maximal kinase activity. As a control, we determined the kinase activities of COT1 precipitated from all strains and detected a general reduction of the pm-myc-COT1 variants of 50 to 65% relative to the nontargeted controls (Table 3). Thus, altered kinase activity cannot account for the suppressive effect of the palmitoyl-myristoyl-tagged COT1 versions in the Δpod-6 strain.
Table 3.
Comparison of in vivo growth rates and corresponding in vitro COT1 activities of selected N. crassa strains
| Abbreviated genotype | In vivo growth rate (cm/day; n ≥ 4) | % kinase activity in vitro (n ≥ 4) |
|---|---|---|
| myc-cot-1 | 4.0 ± 0.1 | 100 |
| Δpod-6; myc-cot-1 | 0 | 80 ± 23 |
| pm-myc-cot-1 | 3.9 ± 0.1 | 53 ± 8 |
| Δpod-6; pm-myc-cot-1 | 3.9 ± 0.1 | 41 ± 13 |
| myc-cot-1(T589A) | 0.4 ± 0.1 | 221 ± 158 |
| Δpod-6; myc-cot-1(T589A) | 0 | Not determined |
| pm-myc-cot-1(T589A) | 3.8 ± 0.2 | 94 ± 29 |
| Δpod-6; pm-myc-cot-1(T589A) | 3.9 ± 0.1 | 108 ± 23 |
| myc-cot-1(T589E) | 1.9 ± 0.1 | 2,320 ± 1,040 |
| Δpod-6; myc-cot-1(T589E) | 0.7 ± 0.2 | 2,650 ± 1,420 |
| pm-myc-cot-1(T589E) | 3.7 ± 0.2 | 1,550 ± 550 |
| Δpod-6; pm-myc-cot-1(T589E) | 3.9 ± 0.1 | 1,480 ± 340 |
We also observed full suppression of the Δpod-6 strain by targeting MOB2A (and thus, presumably, the COT1-MOB2 complex) to the membrane by using a pm-HA-mob-2a construct in a Δpod-6 background (Fig. 7A). To determine if the MOB2 adaptors also perform an active function in localizing COT1 in addition to their stimulatory role in the initial activation of the kinase, we crossed the phosphosite-mutated myc-cot-1 alleles in a Δmob-2a; Δmob-2b background. myc-COT1(T589E) purified from a Δmob-2a; Δmob-2b background displayed a kinase activity of 16% ± 3% of myc-COT1 from the wild type (Fig. 7B), and thus in principle enough to support good growth [e.g., compared with myc-cot-1(S417A)] (Table 2). Consistent with the increased COT1 activity of myc-COT1(T589E) from the Δmob-2a; Δmob-2b strain compared to myc-COT1 from this strain, we observed increased growth of the cot-1(T589E); Δmob-2a; Δmob-2b strain, but not of any of the remaining phosphosite-mutated strains in the Δmob-2a; Δmob-2b background, which displayed defects indistinguishable from those of the Δmob-2a; Δmob-2b strain (Fig. 7C and D). However, growth was not restricted to the hyphal apex but occurred over the whole cell surface, resulting in increased cell size and hyphal diameter and apolar morphology, indicating delocalized activity of myc-COT1(T589E). We also tested if palmitoylation of myc-COT1(T589E) further compensated the Δmob-2a; Δmob-2b defects by generating a pm-myc-cot-1(T589E); Δmob-2a; Δmob-2b strain. However, we did not observe increased growth of the Δmob-2a; Δmob-2b; pm-myc-cot-1(T589E) strain in comparison to the Δmob-2a; Δmob-2b; myc-cot-1(T589E) strain (Fig. 7C). These data suggest that the MOB2 adaptor and COT1 Thr589 phosphorylation are essential for distinct aspects of localizing COT1 and support an active function of MOB2 in restricting membrane-associated Thr589-phosphorylated COT1 to the site of active growth at the hyphal apex.
Fig 7.

MOB2 is required for polar localization of COT1. (A) Membrane targeting of HA-MOB2A by addition of a palmitoylation/myristoylation (pm) motif to the N terminus of MOB2A rescues the growth defect of the Δpod-6 strain (triplicate experiments). (B) myc-COT1 was precipitated from strains with the indicated genotypes and subjected to triplicate in vitro COT1 kinase assays. Note the increased activity of myc-COT1(T589E) compared to the myc-COT1 control. Equal amounts of precipitated myc-COT1 for the kinase reactions were confirmed by an anti-myc Western blot assay. Note the absence of the smaller COT1 isoform in strains lacking both mob-2 genes. (C) Expression of the indicated cot-1 phosphomutants in a Δmob-2a; Δmob-2b background; the increased activity of myc-COT1(T589E) in the Δmob-2a; Δmob-2b; myc-cot-1(T589E) strain allowed enhanced, but apolar, growth of the strain. Artificial targeting of myc-COT1(T589) to the membrane in a Δmob-2a; Δmob-2b; pm-myc-cot-1(T589E) strain did not further increase the functionality of the NDR kinase. (D) The hyphal diameter was quantified in strains with the indicated genotypes (≥15 measured hyphal diameters per strain).
DISCUSSION
A critical advantage of fungal models is their genetic tractability, which allows quantitative analysis of in vitro and in vivo characteristics of proteins under endogenous expression conditions. In contrast, most data obtained in animals are derived from transient-overexpression experiments. In this study, we provide a detailed analysis of the NDR kinase COT1 and its regulation through the upstream GC kinase POD6 and two MOB2 coactivator proteins in the filament-forming ascomycete N. crassa.
COT1 and other NDR kinases can dimerize in yeast two-hybrid assays (28, 37). One of the major findings of this work is that these COT1 dimers represent the inactive pool of the NDR kinase in vitro and in vivo. We showed that the association of COT1 with MOB2 is abolished in the COT1-COT1 dimer and that the formation of a MOB2-COT1 heterocomplex is essential for kinase activity. Although we cannot rule out the existence of COT1 oligomers in addition to kinase homodimers, the mutually exclusive detection of COT1 homodimers versus COT1-MOB2 heterocomplexes in our coimmunoprecipitation experiments, the BiFC results, and the fact that the same N-terminal region of COT1 is required for both types of interactions in yeast two-hybrid assays make the formation of higher-order COT1 assemblies unlikely.
COT1 autophosphorylation occurs primarily at Ser417 in the activation segment. We believe that this is one of the initial steps of COT1 activation, because we detected Ser417 phosphorylation in all strain backgrounds except the COT1 homodimer and kinase-dead COT1(D337A). The signal that regulates the transition from the inactive COT1 dimer to the partially active COT1-MOB2 heterodimer is currently unknown. The interaction between MOB2A/B and COT1 is intact in kinase-dead myc-COT1(D337A), and COT1 purified from the Δmob-2a; Δmob-2b double deletion strain was Ser417 phosphorylated in vivo. Thus, MOB2 binding and COT1 autophosphorylation are independent events. However, we speculate that autophosphorylation may be the signal for dissociation of the inactive COT1-COT1 complex, resulting in the subsequent binding of MOB2A and MOB2B. This hypothesis is consistent with results obtained with animal NDR2, showing that the replacement of Tyr32 with alanine retains intact basal kinase activity, although the interaction with hMOB1 proteins is abolished (5). Moreover, we found that COT1 is autophosphorylated in the Δmob-2a; Δmob-2b strain in vivo, despite (almost) absent in vitro kinase activity in this strain's background. Our data also indicate that autophosphorylation is tightly regulated, because the in vivo phospho-Ser417 status is similar in all tested kinase variants despite distinct in vitro autophosphorylation rates and peptide-based activities. Similarly, we observed equal labeling of both COT1 isoforms in vivo by the anti-phospho-Ser417 antibody. This contrasts with the in vitro 32P incorporation that occurred primarily in the large isoform and suggested additional levels of regulation, e.g., by phosphatases, occurring in vivo.
The interaction of MOB2 with COT1 is not regulated by the upstream kinase POD6. This is different from the regulation of NDR kinases in animal cells, in which the phosphorylation of MOB1-type proteins by the mammalian GC kinases MST1/2 or Drosophila melanogaster Hpo has been shown to control the association of MOB1 with its NDR kinase (26, 49, 59). A potential reason for this differential regulation of the fungal and animal NDR kinases may be the increased complexity of NDR kinase cassettes in higher eukaryotes. The fruit fly expresses at least three different MOB1/2-like genes and two NDR kinases, while mammals have more than five MOB1/2-like proteins and four NDR kinases (14, 21, 35). Moreover, individual components are exchangeable between different animal NDR kinase pathways, while two clearly separated NDR kinase modules exist in fungi (25, 38). For example, the two NDR kinase modules present in Drosophila are distinguishable only by their NDR kinase and the respective scaffolding protein, but each NDR kinase is activated by the same upstream kinase and can interact with the same set of MOB proteins (12, 21, 24). Consequently, regulation by the upstream GC kinase may have evolved as an exclusive mode for the regulation of the MOB1-NDR kinase interaction in higher eukaryotes.
POD6 phosphorylates COT1 at Thr589, an event that increases its in vitro kinase activity more than 20-fold. This activation is presumably driven by a conformational change induced by the interaction of the phosphorylated hydrophobic motif with a hydrophobic pocket in the N-terminal lobe, as described for other AGC kinases (16, 61). Although HA-POD6 did not show any preference for either myc-COT1(S417A) or myc-COT1(S417E) in our in vitro assays, Thr589 phosphorylation did overcome the poor kinase activity of Ser417 with alanine-modified COT1 in the myc-COT1(S417A,T589E) double mutant. Moreover, COT1 autophosphorylation occurred in the Thr589-modified kinase, supporting the view that phosphorylation of the activation loop is independent of the Thr589 status. Thus, hydrophobic motif phosphorylation likely occurs after autophosphorylation.
COT1 as well as MOB2A accumulated at the hyphal tip, forming a membrane-associated apical crescent, and partially colocalized with the Spitzenkörper as a dot-like structure. The reason for the bright dot-like accumulation and the significance of the partial colocalization with the Spitzenkörper is unclear, but it may reflect a cytosolic storage pool of inactive COT1-COT1 complexes and/or COT1 primed for activation (e.g., MOB2 binding and autophosphorylation). The fact that COT1 homodimers visualized in the BiFC experiments did not form an apical crescent but only the Spitzenkörper-associated dot strongly suggests that the tip-associated crescent likely represents the active pool of COT1, while the subapical dot-like accumulation may comprise the inactive pool of the kinase. It is striking that this apical crescent is shifted to a more diffuse accumulation within the apical region in the cot-1(S417A)-GFP strain, suggesting that apex association is possible but less efficient when COT1 cannot autophosphorylate.
Thr589 phosphorylation of COT1 controls not only its in vitro activity but also the membrane association of the kinase-MOB2 complex. The coupling of activation and localization by a single phosphorylation event seems the primary reason for the poor functionality of COT1(T589A) in vivo, because the myc-cot-1(T589A) strain is sufficiently active in vitro to allow sustained growth [e.g., compare the myc-cot-1(T589A) strain with the myc-cot-1 strain] but is a poorly growing strain. However, artificial targeting of pm-myc-COT1(T589E) to the plasma membrane is not sufficient for polar growth in a Δmob-2ai; Δmob-2b background, indicating an active function of the coactivator proteins in the localization of the NDR kinase. We speculate that hydrophobic motif phosphorylation functions as a general membrane-targeting signal and that MOB2 is required for restricting the active, membrane-associated kinase complex to the hyphal tip, which is required for polar growth.
Based on the data presented for COT1 and available knowledge on other NDR kinases, we propose the following model for COT1 activation and function (Fig. 8). Inactive COT1 is self-assembling. We propose that the transition between the inactive kinase homodimer and the COT1-MOB2 heterodimer is the first step in the activation of the kinase. This transition is regulated via competing interaction sites in the N terminus of COT1 (37) and is not dependent on phosphorylation of MOB2 by the GC kinase POD6. COT1-MOB2 heterodimerization and Ser417 autophosphorylation result in basal activity of the kinase-MOB2 complex. The subsequent phosphorylation of Thr589 through POD6 leads to a conformational change and full activation of the NDR kinase. It may also be a prerequisite for the efficient interaction with and phosphorylation of endogenous COT1 substrates. Most importantly, Thr589 phosphorylation is essential for targeting the active kinase complex to the plasma membrane. The interaction of MOB2 with COT1 may then restrict the membrane-associated kinase complex to the hyphal tip to drive polarized growth. This mechanistic coupling of kinase activation with polar localization of COT1 via hydrophobic motif phosphorylation and interaction with MOB2 explains the observed discrepancy of in vitro kinase activity and the in vivo defect of the phospho site NDR kinase variants in ours and other systems (22, 28, 30, 64) and provides an elegant example for the multilayered coordination of NDR kinase activation with cellular localization.
Fig 8.
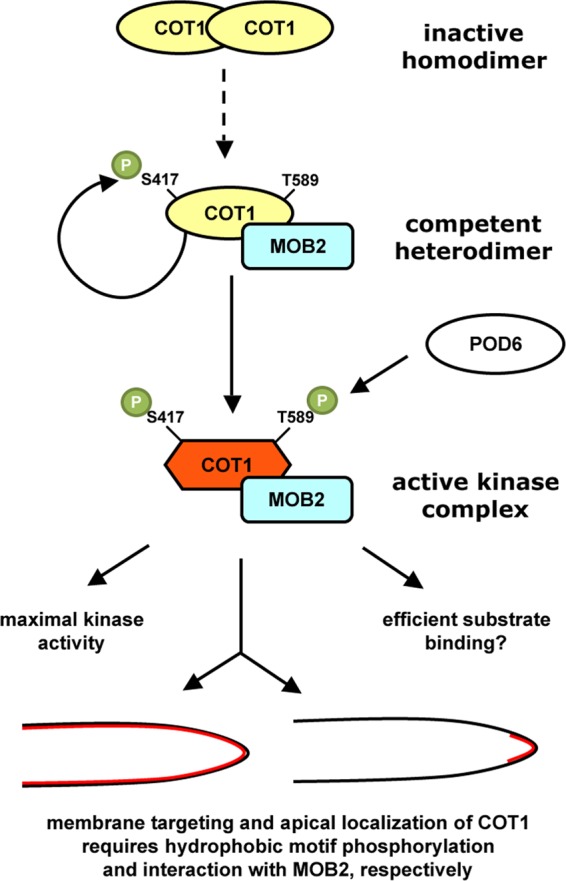
Model for activation and cortical localization of COT1. COT1 forms homodimers via a conserved N-terminal extension, which is also required for the interaction of the kinase with MOB2. These mutually exclusive states allow the formation of distinct homo- and heterocomplexes and may regulate the transition between the inactive COT homodimers and the COT1-MOB2 heterodimer. The kinase complex is fully activated upon phosphorylation of Thr589 within the hydrophobic motif by the germinal center kinase POD6, which induces a conformational change of the NDR kinase. This conformational change might alter substrate specificity and efficient substrate binding. Thr589 phosphorylation also functions as a membrane-targeting signal of COT1. While the phosphorylation of COT1's hydrophobic motif is critical for general membrane association, the MOB2 adaptor protein is required for restricting the active, membrane-associated kinase complex to the site of polar cell growth at the hyphal apex.
ACKNOWLEDGMENTS
This research project was financially supported through DFG research grants SE1054/3-2 and SE1054/4-1 and GIF grant 1059-14.3/2008.
We thank E. L. Weiss, Northwestern University, Illinois, for the kind gift of the phospho-Ser417-COT1-specific anti-phospho-Ser570-Cbk1p antibody and O. Yarden, Hebrew University of Jerusalem, Israel, for providing the myc-cot-1(S417A,T589E) strain. We acknowledge the FGSC and the Neurospora program project for strains and continuous support.
Footnotes
Published ahead of print 26 March 2012
REFERENCES
- 1. Araujo-Palomares CL, Richthammer C, Seiler S, Castro-Longoria E. 2011. Functional characterization and cellular dynamics of the CDC-42-RAC-CDC-24 module in Neurospora crassa. PLoS One 6:e27148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bao Y, et al. 2009. Roles of mammalian sterile 20-like kinase 2-dependent phosphorylations of Mps one binder 1B in the activation of nuclear Dbf2-related kinases. Genes Cells 14:1369–1381 [DOI] [PubMed] [Google Scholar]
- 3. Bardiya N, et al. 2008. Characterization of interactions between and among components of the meiotic silencing by unpaired DNA machinery in Neurospora crassa using bimolecular fluorescence complementation. Genetics 178:593–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behn-Krappa A, Newton AC. 1999. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr. Biol. 9:728–737 [DOI] [PubMed] [Google Scholar]
- 5. Bichsel SJ, Tamaskovic R, Stegert MR, Hemmings BA. 2004. Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J. Biol. Chem. 279:35228–35235 [DOI] [PubMed] [Google Scholar]
- 6. Biondi RM, et al. 2000. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 19:979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biondi RM, Nebreda AR. 2003. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 372:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brace J, Hsu J, Weiss EL. 2011. Mitotic exit control of the Saccharomyces cerevisiae Ndr/LATS kinase Cbk1 regulates daughter cell separation after cytokinesis. Mol. Cell. Biol. 31:721–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan EH, et al. 2005. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24:2076–2086 [DOI] [PubMed] [Google Scholar]
- 10. Dan I, Watanabe NM, Kusumi A. 2001. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11:220–230 [DOI] [PubMed] [Google Scholar]
- 11. Emoto K, et al. 2004. Control of dendritic branching and tiling by the Tricornered-kinase/Furry signaling pathway in Drosophila sensory neurons. Cell 119:245–256 [DOI] [PubMed] [Google Scholar]
- 12. Emoto K, Parrish JZ, Jan LY, Jan YN. 2006. The tumour suppressor Hippo acts with the NDR kinases in dendritic tiling and maintenance. Nature 443:210–213 [DOI] [PubMed] [Google Scholar]
- 13. Engel M, et al. 2006. Allosteric activation of the protein kinase PDK1 with low molecular weight compounds. EMBO J. 25:5469–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang X, Adler PN. 2010. Regulation of cell shape, wing hair initiation and the actin cytoskeleton by Trc/Fry and Wts/Mats complexes. Dev. Biol. 341:360–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freitag M, Hickey PC, Raju NB, Selker EU, Read ND. 2004. GFP as a tool to analyze the organization, dynamics and function of nuclei and microtubules in Neurospora crassa. Fungal Genet. Biol. 41:897–910 [DOI] [PubMed] [Google Scholar]
- 16. Frodin M, et al. 2002. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 21:5396–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gallegos ME, Bargmann CI. 2004. Mechanosensory neurite termination and tiling depend on SAX-2 and the SAX-1 kinase. Neuron 44:239–249 [DOI] [PubMed] [Google Scholar]
- 18. Gao T, Toker A, Newton AC. 2001. The carboxyl terminus of protein kinase C provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J. Biol. Chem. 276:19588–19596 [DOI] [PubMed] [Google Scholar]
- 19. Geng W, He B, Wang M, Adler PN. 2000. The tricornered gene, which is required for the integrity of epidermal cell extensions, encodes the Drosophila nuclear DBF2-related kinase. Genetics 156:1817–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harris SD, et al. 2005. Polarisome meets Spitzenkorper: microscopy, genetics, and genomics converge. Eukaryot. Cell 4:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He Y, et al. 2005. Drosophila Mob family proteins interact with the related tricornered (Trc) and warts (Wts) kinases. Mol. Biol. Cell 16:4139–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He Y, Fang X, Emoto K, Jan YN, Adler PN. 2005. The tricornered Ser/Thr protein kinase is regulated by phosphorylation and interacts with furry during Drosophila wing hair development. Mol. Biol. Cell 16:689–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hergovich A, Bichsel SJ, Hemmings BA. 2005. Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol. Cell. Biol. 25:8259–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hergovich A, Hemmings BA. 2009. Mammalian NDR/LATS protein kinases in hippo tumor suppressor signaling. Biofactors 35:338–345 [DOI] [PubMed] [Google Scholar]
- 25. Hergovich A, Stegert MR, Schmitz D, Hemmings BA. 2006. NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell. Biol. 7:253–264 [DOI] [PubMed] [Google Scholar]
- 26. Hirabayashi S, et al. 2008. Threonine 74 of MOB1 is a putative key phosphorylation site by MST2 to form the scaffold to activate nuclear Dbf2-related kinase 1. Oncogene 27:4281–4292 [DOI] [PubMed] [Google Scholar]
- 27. Ho LL, Wei X, Shimizu T, Lai ZC. 2010. Mob as tumor suppressor is activated at the cell membrane to control tissue growth and organ size in Drosophila. Dev. Biol. 337:274–283 [DOI] [PubMed] [Google Scholar]
- 28. Hou MC, Guertin DA, McCollum D. 2004. Initiation of cytokinesis is controlled through multiple modes of regulation of the Sid2p-Mob1p kinase complex. Mol. Cell. Biol. 24:3262–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang TY, Markley NA, Young D. 2003. Nak1, an essential germinal center (GC) kinase regulates cell morphology and growth in Schizosaccharomyces pombe. J. Biol. Chem. 278:991–997 [DOI] [PubMed] [Google Scholar]
- 30. Jansen JM, Barry MF, Yoo CK, Weiss EL. 2006. Phosphoregulation of Cbk1 is critical for RAM network control of transcription and morphogenesis. J. Cell Biol. 175:755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson LN, Noble ME, Owen DJ. 1996. Active and inactive protein kinases: structural basis for regulation. Cell 85:149–158 [DOI] [PubMed] [Google Scholar]
- 32. Justa-Schuch D, Heilig Y, Richthammer C, Seiler S. 2010. Septum formation is regulated by the RHO4-specific exchange factors BUD3 and RGF3 and by the landmark protein BUD4 in Neurospora crassa. Mol. Microbiol. 76:220–235 [DOI] [PubMed] [Google Scholar]
- 33. Keranen LM, Dutil EM, Newton AC. 1995. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 5:1394–1403 [DOI] [PubMed] [Google Scholar]
- 34. Keshwani MM, Harris TK. 2008. Kinetic mechanism of fully activated S6K1 protein kinase. J. Biol. Chem. 283:11972–11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kohler RS, Schmitz D, Cornils H, Hemmings BA, Hergovich A. 2010. Differential NDR/LATS interactions with the human MOB family reveal a negative role for human MOB2 in the regulation of human NDR kinases. Mol. Cell. Biol. 30:4507–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lai ZC, et al. 2005. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120:675–685 [DOI] [PubMed] [Google Scholar]
- 37. Maerz S, et al. 2009. Two NDR kinase-MOB complexes function as distinct modules during septum formation and tip extension in Neurospora crassa. Mol. Microbiol. 74:707–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maerz S, Seiler S. 2010. Tales of RAM and MOR: NDR kinase signaling in fungal morphogenesis. Curr. Opin. Microbiol. 13:663–671 [DOI] [PubMed] [Google Scholar]
- 39. Maerz S, et al. 2008. The nuclear Dbf2-related kinase COT1 and the mitogen-activated protein kinases MAK1 and MAK2 genetically interact to regulate filamentous growth, hyphal fusion and sexual development in Neurospora crassa. Genetics 179:1313–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCluskey K. 2003. The Fungal Genetics Stock Center: from molds to molecules. Adv. Appl. Microbiol. 52:245–262 [DOI] [PubMed] [Google Scholar]
- 41. Nelson B, et al. 2003. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell 14:3782–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nolen B, Taylor S, Ghosh G. 2004. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15:661–675 [DOI] [PubMed] [Google Scholar]
- 43. Oliver AW, Knapp S, Pearl LH. 2007. Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem. Sci. 32:351–356 [DOI] [PubMed] [Google Scholar]
- 44. Panozzo C, Bourens M, Nowacka A, Herbert CJ. 2010. Mutations in the C-terminus of the conserved NDR kinase, Cbk1p of Saccharomyces cerevisiae, make the protein independent of upstream activators. Mol. Genet. Genomics 283:111–122 [DOI] [PubMed] [Google Scholar]
- 45. Pearce LR, Komander D, Alessi DR. 2010. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11:9–22 [DOI] [PubMed] [Google Scholar]
- 46. Pike AC, et al. 2008. Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 27:704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pombo CM, et al. 2007. The GCK II and III subfamilies of the STE20 group kinases. Front. Biosci. 12:850–859 [DOI] [PubMed] [Google Scholar]
- 48. Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. 2004. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 381:453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Praskova M, Xia F, Avruch J. 2008. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18:311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Record CJ, et al. 2010. Structural comparison of human mammalian ste20-like kinases. PLoS One 5:e11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riquelme M, et al. 2011. Architecture and development of the Neurospora crassa hypha: a model cell for polarized growth. Fungal Biol. 115:446–474 [DOI] [PubMed] [Google Scholar]
- 52. Seiler S, Plamann M. 2003. The genetic basis of cellular morphogenesis in the filamentous fungus Neurospora crassa. Mol. Biol. Cell 14:4352–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Seiler S, Vogt N, Ziv C, Gorovits R, Yarden O. 2006. The STE20/germinal center kinase POD6 interacts with the NDR kinase COT1 and is involved in polar tip extension in Neurospora crassa. Mol. Biol. Cell 17:4080–4092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shah OJ, Hunter T. 2004. Critical role of T-loop and H-motif phosphorylation in the regulation of S6 kinase 1 by the tuberous sclerosis complex. J. Biol. Chem. 279:20816–20823 [DOI] [PubMed] [Google Scholar]
- 55. Stegert MR, Hergovich A, Tamaskovic R, Bichsel SJ, Hemmings BA. 2005. Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol. Cell. Biol. 25:11019–11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toker A, Newton AC. 2000. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J. Biol. Chem. 275:8271–8274 [DOI] [PubMed] [Google Scholar]
- 57. Vichalkovski A, et al. 2008. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr. Biol. 18:1889–1895 [DOI] [PubMed] [Google Scholar]
- 58. Vogt N, Seiler S. 2008. The RHO1-specific GTPase-activating protein LRG1 regulates polar tip growth in parallel to Ndr kinase signaling in Neurospora. Mol. Biol. Cell 19:4554–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wei X, Shimizu T, Lai ZC. 2007. Mob as tumor suppressor is activated by Hippo kinase for growth inhibition in Drosophila. EMBO J. 26:1772–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang J, et al. 2002. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9:940–944 [DOI] [PubMed] [Google Scholar]
- 61. Yang J, et al. 2002. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell 9:1227–1240 [DOI] [PubMed] [Google Scholar]
- 62. Yarden O, Plamann M, Ebbole DJ, Yanofsky C. 1992. cot-1, a gene required for hyphal elongation in Neurospora crassa, encodes a protein kinase. EMBO J. 11:2159–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zallen JA, Peckol EL, Tobin DM, Bargmann CI. 2000. Neuronal cell shape and neurite initiation are regulated by the Ndr kinase SAX-1, a member of the Orb6/COT-1/warts serine/threonine kinase family. Mol. Biol. Cell 11:3177–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ziv C, et al. 2009. Cell elongation and branching are regulated by differential phosphorylation states of the nuclear Dbf2-related kinase COT1 in Neurospora crassa. Mol. Microbiol. 74:974–989 [DOI] [PubMed] [Google Scholar]