

Abstract
A simple model is presented that describes general features of protein folding, in good agreement with experimental results and detailed all-atom simulations. Starting from microscopic physics, and with no free parameters, this model predicts that protein folding occurs remarkably quickly because native-like states are kinetic hubs. A hub-like network arises naturally out of microscopic physical concerns, specifically the kinetic longevity of native contacts during a search of globular conformations. The model predicts folding times scaling as τf ~ eξN in the number of residues, but because the model shows ξ is small, the folding times are much faster than Levinthal’s approximation. Importantly, the folding timescale is found to be small due to the topology and structure of the network. We show explicitly how our model agrees with generic experimental features of the folding process, including the scaling of τf with N, two-state thermodynamics, a sharp peak in CV, and native-state fluctuations.
Keywords: Protein Folding, Kinetic Hub, Master Equation, Markov State Model, Analytical, Folding Time
Introduction
Understanding how proteins fold remains one of the great outstanding questions of biophysics. Since Anfinsen demonstrated that proteins adopt one unique structure with overwhelming probability, physical insight into this last step of the central dogma has remained elusive.1 An adequate physical picture of protein folding must resolve Levinthal’s paradox: were folding a random search in conformation space, it would take an astronomical time for proteins to fold.2,3 We know biology must employ physics to reduce the folding time to biologically relevant timescales - the question is, how?
This intriguing question has lead to many theories attempting to explain folding.4 The “classical view” was that proteins likely follow a specific step-wise path, progressing from extended, unfolded structures to the native state, gaining native structure along the way.5,6 In the late 1980s and early 1990s, this view of folding was called into question by computational models, primarily lattice models, that demonstrated that folding could occur via many heterogeneous pathways over a single free energy barrier.5–9 This was consistent with two key experimental observations that came to light at the same time. First, many single domain proteins fold in a two-state manner, in a single cooperative step with first order kinetics.7,10 Second, high resolution experimental techniques, especially NMR-based hydrogen exchange, showed many timescales during folding, indicative of heterogeneity.5,11
These realizations lead to the “new view” of protein folding, which dictated that proteins could fold via many independent, parallel pathways that might be highly heterogeneous.5,6,12 This theory was later expanded by directly considering the free energy landscapes of proteins. Specifically it was realized that parallel paths leading to quick folding must result from a significant energy gradient biased towards a single, dominant global minimum, the native state. Further, the free energy landscape must be minimally frustrated, that is, as smooth and “funnel-like” as possible.8,9,13–16 Any bumps or dead-ends would slow folding, so this theory dictated that biology must have evolved sequences that eliminated these barriers. Amino acid sequences would be designed such that contacts in the native conformation would be energetically favorable, and non-native contacts would interact weakly, or even be unfavorable, removing kinetics traps in the landscape.9
While energy landscape theories have been successful in explaining some features of folding, they rely on assuming the structure of a landscape, rather than describing its physical origin. Here, we present a new model that connects kinetics directly to microscopic, residue-level physics, that allows us to expand the conclusions that can be reached from simple models of folding. Rather than beginning with thermodynamical considerations and attempting to derive kinetics from an energy landscape, we begin by considering kinetics. This has a number of advantages. Primarily, we need not find a reaction coordinate to describe the kinetics of our model, which not only simplifies modeling but prevents errors that might occur by projecting onto poor reaction coordinates.17,18 Furthermore, our kinetic model allows for direct comparison to experiment and simulation, allowing for theoretical verification and falsifiable predictions.
Inspired by recent results from all-atom molecular simulation, we have adopted the view that protein kinetics can be concisely represented as a set of states and the rates of exchange between those states. Known as master equation models, or more recently Markov State models (MSMs),19–21 these models consist of a state space and transition matrix characterizing the rates of exchange between each state. This is equivalent to a graphical model where nodes represent protein conformations, and edges represent rates. The representation is powerful enough to describe complex phenomena while simple enough for mathematical investigation. Further, the concept of metastable states and rates is also familiar to both physicists and chemists, and therefore provides an appealing foundation for understanding protein folding.
All-atom simulations have recently provided an empirical view of how proteins could fold. Computer technology and analysis methodology have progressed to the stage where, to date, the folding kinetics of many proteins have been described in atomic detail, with sizes up to 86 residues and folding times of ~10 milliseconds.22–29 First realized by Rao and Caflisch,30 and later expanded upon by Bowman and Pande,31 a key generality of MSMs parametrized from all-atom simulations is the hub-like nature of these models. That is, when representing protein kinetics as a graph, the native state is highly connected and central in the network, and the connectivity of states increases with native content.24,27 This means that from any non-native state, there is a direct route to the native state involving few “hops” between nodes. This suggested that any arbitrary structure might be able fold in a small, finite time, since it would be close to the native state in a kinetic sense based on the network topology alone.
After this development, Pande derived a model showing how, if non-native interactions in proteins were favorable, a hub-like structure could emerge in folding kinetics32 Further, such a hub did not result if non-native contacts were unfavorable. In that work, all non-native states were equivalent, and thus the model was limited in scope. Here, we build on that work. We adopt the view of microscopic kinetics derived previously, but expand the state space to include a diverse set of states with various degrees of native content. The resulting model mirrors all-atom MD simulation, displaying a topology with strong hub-like features around the native state.
Further, we show how a hub-like topology provides an explanation for why proteins fold quickly. The model reproduces essential experimental observations, such as the scaling of the folding rate with chain length, the correct energy-gap structure predicted by previous theory and demonstrated by experiment, and a peak in the heat capacity at the melting temperature. Our simple model is not the only attempt to explain folding rates,8,12,16,33–48 nor are we the only ones to have described protein folding as a hub-like network.30,39,49 However, our model is new in its ability to predict protein folding times without fit parameters.
We begin by outlining the theory. This first section should provide a non-mathematical introduction for a general audience, and provide a basic understanding of the model that will be useful context for the rest of the manuscript. The reader uninterested in mathematical details should be able to read this section and skip to the discussion. Following this, we begin constructing the model by recalling the results and notation of previous work. Then we define a state space describing protein conformations, and calculate some interesting thermodynamic properties of that space. Next, we derive the kinetics of proteins from the state space, with an emphasis on the folding process. Finally, we discuss the implications of our model, and draw comparisons to experiment and simulation. Some mathematical detail has been relegated to the supplemental information in the interest of conciseness and readability.
Model Summary
In what follows, we consider protein kinetics as a set of states and the rates of exchange between those states, and investigate the dynamics that arise from these considerations. From this kind of model, many interesting new insights emerge.
To make this approach tractable, we make a number of physical approximations, enumerated here.
In light of the mounting evidence from experiment,50–54 simulation (see SI, Figure S1),55,56 and theory57 that, in the absence of denaturant, unfolded proteins are globular, we consider only protein conformations that are globular. While extended conformations of proteins are certainly interesting, here we restrict ourselves to single-domain proteins in the globular phase.
Protein conformations can be accurately represented as contact maps.
The energetics of this map can be captured by only two kinds of interactions, native and non-native contacts. Native contacts are by definition more energetically favorable than non-native contacts.
Each contact map represents a metastable state - that is, each map is energetically stable enough such that transitions between individual maps will be much faster than times spent in an arbitrary contact geometry. This makes the dynamics approximately Markovian, since degrees of freedom orthogonal to the residue-residue contacts will rapidly equilibrate.
Finally, states are either “kinetically connected” and can interconvert at some rate that will be the same for all pairs of connected states, or cannot directly interconvert.
In what follows we also make a few mathematical approximations, but will be clear to note where these are introduced.
From this simplified start, we can deduce a number of interesting things. First, since all contacts can be effectively sorted into native and non-native types, fact both are attractive follows from the globular assumption. To overcome the entropy of extended conformations, polymer theory leads us to conclude that if the non-native states of proteins are in fact globular (in the absence of denaturant), non-native contacts must be favored with respect to solvent contacts. Moreover, the contact energies used here are derived through a consensus of experiment, simulation, and previous theory (see SI).
Based on these energetics, we show that the heat capacity exhibits a first-order transition between non-native and native states at one unique temperature, Tm, which is in the appropriate range of protein melting temperatures.
Next, we calculate the kinetics of the model. A natural consequence of the model is that native states have many more kinetic neighbors than non-native states, resulting naturally in a kinetic hub. Further, this hub has a remarkable structure, that of a scale-free network, where the number of connections of each state follows a power law.
The scale-free structure of the network determines certain dynamical properties of the entire system. It is possible to show that the longest timescale in the system must be fast - much faster than a random search through conformations. This folding rate scales exponentially in chain length, τf ~ eξN, but nonetheless folding occurs remarkably quickly because ξ is small. This ensures that the rate of folding is much faster than the estimate of Levinthal, and is in good agreement with experimental data.
A simple picture emerges from this model, where proteins fold by searching through globular conformations for their native state. This search is largely random, but is aided by the fact that native contacts are more favorable than non-native contacts, a fact that has been predicted to lead to fast folding.36,37 This means that native structure is statistically persistent - native contacts can break during the search, but are more likely to be retained than non-native contacts. As long as native contacts are strong enough, this alone is enough to ensure that proteins fold quickly in a cooperative manner.
Formalism
Here, we briefly recall the notation and key results of previous work,32 providing a pedagogical introduction to the formalism used. We represent a protein state as a contact map Ci j ∈ {0, 1}, a binary value for each residue pair {i j} indicating if residues i and j are in contact or not. Each contact, when formed, contributes some favorable (negative) energy. One of the key aspects of this model is that non-native contacts have a favorable energy contribution (denoted εNN), while native contacts (εN) are more favorable. This is captured by the microscopic Hamiltonian, for some arbitrary state α,
where the superscripts of Ci j represent either state α or the native state, N.
Here, we have written ε to represent an average potential of interaction. Consider the physical reaction of taking a residue from being solvent exposed (denoted R(aq)) to in contact with another residue (R2(protein)), and its associated free energy change ΔF,
where φ to captures the free energy change upon contact formation, and σ to represents the entropy cost of loop closure. To be clear, φ represents enthalpic and entropic effects intrinsic to the contact, where σ = ΔSloop/k > 0 accounts only for the entropy gained by loop formation, capturing configurational entropy of the protein and associated solvent. σ is taken to be constant for all residues, consistent with previous work,32,58 and is estimated in what follows from precise caliometric experiments (σ ≈ 4.0 ± 3.0 cal mol−1 K−1).59 This simplifies the reaction taking a non-native contact to a native contact
where the loop entropy terms cancel. These equations assume two-body interactions are dominant, i.e. the residue-pair formation is approximately independent of other residues. While this will introduce some error, since e.g. contacts amongst adjacent residues will be loosely correlated, we can justify this approximation by noting that three-body interactions will still be much less important than three body interactions.
To a first order approximation we consider the effective potential of this process to be independent of temperature. Then, for native and non-native contacts,
where ε represents a unitless interaction energy. Additionally, it will be convenient to specify the “excess” energy of a native contact, i.e. εx = εN − εNN and φx = φN − φNN.
This microscopic Hamiltonian allows us to not only define states, but rates of transition between those states. We assume that the transition from one state α to some other state β must pass through a transition state that breaks all contacts not shared by these two structures, i.e. .32 The free energy of the transition state allows one to compute the a rate for α → β using Kramer’s approximation32
where k̃ is the microscopic rate of interconversion. Now, equipped with a way to define states and derive the rates connecting them, we have a sufficient formalism to describe protein kinetics.
Let us calculate the free energies (F) of the reactant and transition states, and the associated barrier for the reaction α → β
where qα,β is the number of contacts in common between α and β, and other q variables with superscripts represent analogous values (with N always denoting the native contact map). Also note that the free energy for β would be exactly analogous to the energy for α, but does not explicitly enter the Kramer’s approximation. Further, one can see that the only place the loop entropy enters into our picture is in the transition state. Physically, this represents compact structures interconverting between each other via states with short free loops.
From this model, it is possible to intuitively see how hub-like kinetics could arise. To transition between two states, one must break all the contacts those states do not have in common. Since native contacts have a more favorable energy, breaking native contacts raises the energy of the transition state and slows the reaction, compared to breaking non-native contacts. However, if the product structure has a high number of native contacts, then there is a good chance of retaining those contacts in the transition state, lowering the barrier and speeding conversion to that native-like state. At the same time, the favorable nature of non-native contacts prevents rapid rearrangement of non-native structure. This leads to hub-like characteristics, where the interconversion between arbitrary non-native states is intrinsically slow. This process favors kinetic connections to native-like states, a feature that will be a key component of the model detailed below.
Model and Theory
Given the contact map formalism, and the corresponding method of calculating rates between states, we have all the tools needed to describe protein folding. Next, we must define a set of relevant states to consider, and then analyze the global kinetic properties resulting from the dynamics between these microstates.
Defining a State Space
To begin, we consider only states that are globular. While the issue of collapse has been a debated topic in the protein folding field, recent results from experiment,50–54 theory,57 and simulation55,56 all indicate that non-specific collapse precedes folding in the absence of any denaturant. The total number of contacts in all-atom explicit water MD simulations of protein folding show that the distribution of contacts is peaked around the native value, and that there are few conformations with few or no contacts, even in the unfolded state (see SI Figure 1).
We take the globular approximation to mean all states have the same number of total contacts and the loop entropy of each state is equivalent. This implies that all states of the model, independent of their degree of native content, consist of conformations with the same number of contacts as the native state. We denote this number of contacts by q; notice q = qα,α = qN,N for any state α. Following this notation, the free energy of a state is equal only to its internal energy, a function of the fraction of native contacts, Fα = Eα = εxqα,N + εNNq.
Now, we calculate the entropy of our state space by simply counting the number of possible contact maps, with each unique map corresponding to a unique state. We write the entropy of the system, S, for each possible number of native contacts, n, assuming that contacts can be formed between any two residues separated by at least 3 residue links. For a chain of length N residues
Where Ω(n) is the number of states with n native contacts. The first binomial bracket represents the number of ways to choose q–n non-native contacts from the contact map (omitting the q native contact positions), and the second bracket is the way to choose n native contacts from q possible native contacts. For conciseness, we define aΩ ≡ (N − 3)2/2. Therefore, for a chain of N ≈ 100 residues, aΩ is a constant of order 104.
This estimate of Ω(n) in its general form has two issues. First, it is somewhat intractable analytically, involving many factorials. Second, it certainly over counts the number of states, some of which will not be allowed due to e.g. excluded volume effects. Through a series of mathematical approximations, we derive (supplemental information)
where c is a constant of
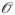 (1). Numerical results verify this approximation captures the scaling of the function, and this result is consistent with the exponential scaling estimated by Levinthal-like arguments. Further, exponential scaling is consistent with previous estimates from polymer theory and other simple models.47,60 This state space definition results in a huge number of states, the total state count is ΩT = Σn Ω(n) ~
(10200).
(1). Numerical results verify this approximation captures the scaling of the function, and this result is consistent with the exponential scaling estimated by Levinthal-like arguments. Further, exponential scaling is consistent with previous estimates from polymer theory and other simple models.47,60 This state space definition results in a huge number of states, the total state count is ΩT = Σn Ω(n) ~
(10200).
The free energy of this state space projected onto the fraction of native contacts, Q = n/q, is
Below a certain temperature Tm = −φx/kc, the native state (Q = 1) has the lowest free energy, and is therefore highly populated. Above that temperature, highly non-native states (Q = 0) will have the most population and this population will be distributed amongst the many states in this regime. It is clear that this temperature corresponds to the folding phase transition, but does not say anything about how that transition occurs.
Thermodynamics
From this state space, it is straightforward to calculate thermodynamic properties of the model. We can write the partition function in terms of the degeneracy of states with n native contacts, gn, and the energy of those states (Eα = εxqα,N + εNN q), obtaining
where the last step is evaluated as a difference of geometric sums. Note that e(c−εx)(q+1) ≫ 1, such that we can simplify the above by dropping the unity term in the numerator. Making this approximation, and recalling from statistical mechanics that
we obtain
This function has a singularity at Tm = −φx/kc, at the melting temperature (Figure 1). Previously, both theory and experiment7,61 have indicated folding is a first-order phase transition, consistent with this observation. Note that this value of Tm matches exactly the one obtained from our free energy profile in the previous section.
Figure 1.

Thermodynamic properties of the protein folding model. (Left) Free energy profiles along the order parameter Q (fraction of native contacts). There is no free energy barrier, but at low temperature folded states are populated (solid line), whereas at higher temperatures unfolded states become more populated (dashed lines). (Right) Heat capacity as a function of temperature. For a typical 100 residue protein, c ≈ 7, and Tm ≈ 370 K. There is a singularity corresponding to a highly cooperative first-order phase transition at Tm. The free energy profiles here verify this corresponds to the folding/unfolding transition.
Estimation of Tm, based on typical values of φx (see SI), indicates that it is near 370K for a typical 100-residue protein. Interestingly, CV does not depend directly on the non-native or native contact energies, but is a function only of the excess native energy εx, and scales only weakly with chain length (c ~ logN2, see supplemental information). The lack of strong scaling is consistent with experimental observations,62 which show no detectable correlation of Tm with length, stability, or other common thermodynamic or kinetic parameters. We hypothesize from this model that protein melting temperatures are likely dominated by specific structural and physical features of individual proteins, rather than chain length, thermodynamic stability, or other general features.
Master Equation Approach
Now, we consider the statistics of transitions between states. The task at hand is taking the microscopic definition of kinetics provided by the Kramer’s approximation, , and applying it to the entire state space. To do this, we employ a Markovian master equation, a technique recently used to understand molecular simulations.19–21,63 Under the master equation formalism, kinetics are defined by a rate matrix, K = {ki j}, where each ki j represents the rate of transfer from state i to state j.
The master equation is simply an extension of first-order kinetics to many states, describing the time evolution of a set of populations, Pi(t), corresponding to the probability of being in some state i at time t
This formalism is very powerful, and able to capture complex phenomena in a straightforward manner. The theory of master equations is quite advanced, and many excellent reviews are available detailing recent advances.19–21,63 Here, we restate many old results that are especially pertinent to our derivations.
In general, K represents a weighted graphical model, where nodes represent the conformations the protein could adopt (i.e. each node is a contact map), and the edges represent the rates of exchange between each node. To simplify analytical computations, here we will derive an unweighted model describing the topology of the network kinetics, and then weight the model to recover thermodynamic properties. The unweighted rate matrix, KSYM is symmetric and is the opposite of the graph Laplacian L
where D = diag(di) is the diagonal matrix of degrees (the number of connection a node has to other nodes) and Ai j ∈ {0, 1} is the adjacency matrix, a sparse symmetric square matrix with a 1 if i j share an edge, and 0 otherwise.
Our task then is to derive A. If one state converts to another at a rate faster than some critical rate, kc, then we say those states are kinetically connected, and those vertices are connected by an edge (Ai j = 1). Otherwise, set Ai j = 0. It is worth mentioning that this equal weighting of edges in KSYM is consistent with what has been observed in detailed simulation, most significantly work showing that the edge weights are robust to perturbation.64
This simplified representation will give us a symmetrical matrix we denote KSYM for emphasis. The eigenvectors of the symmetric matrix do not represent the dynamics of the actual, unsymmetric master equation based on K (see e.g.63). However, it is required by detailed balance that K has a symmetric form
where Peq = diag[Pi(t = ∞)] is a diagonal matrix of the equilibrium populations, which we have from our previous considerations of thermodynamics. Peq is also the eigenvector corresponding to the first eigenvalue of K, λ1 = 0, and represents the stationary distribution
All the dynamical processes are given by the left eigenvectors, ψn, n ≥ 2
and the timescales of these dynamics are given by the eigenvalues, λn. Specifically, there are n timescales τn/τ0 = λn, where τ0 is a constant depending on the unit of time used. Note that the eigenvalues of K and KSYM are identical, only their eigenvectors are distinct.
We proceed by evaluating the simple case of the unweighted model by statistically analyzing which states should be connected, and then set ki j = 1 for those states, and 0 otherwise, giving KSYM. We could then recover the exact kinetics by weighting this matrix by Peq. However, since in what follows we never calculate an eigenvector for the system, but only the eigenvalues, we work exclusively with KSYM.
Kinetic Topology of the State Space
Now, from the microscopic Kramer’s equation we compute which states should be kinetically close, and investigate the resulting master equation. As mentioned previously, we are considering a simplified representation of the kinetics on our state space where two states are either “kinetically close” and rapidly interconvert, or are distant and exchange slowly. The precise timescales determining these regimes, as we will soon see, are not important. If two states are kinetically close, we connect them by an edge in our graph, and increment each of their degrees.
To determine the kinetic degree of some state β, consider states all {α} with a certain number of contacts differing from β. Denote this distance Δq ≡ q − qα,β, which is the number of contacts that are broken during the transition α → β. The rate of interconversion from kαβ will be a monotonically decreasing function of Δq. This means that to differentiate between kinetically close and distant structures, we need to define some cutoff rate, kc, such that if kαβ ≥ kc the states are kinetically connected, and if kαβ < kc they are not.
Because we are considering only an unweighed model, the topology of the network is the sole determinant of the timescales involved. One concise, computationally friendly way to describe this topology is the network degree distribution - that is, the distribution of the number of connections each node has. The degree distribution alone ignores correlations amongst specific nodes, and thus captures only one aspect of the topology, but because we have no direct access to this information in our current model, we take the ansatz that degree correlations are negligible, and therefore that the degree distribution contains all relevant information about the network topology.
This degree will simply be the number of states in the model that are kinetically close. Let us compute the degree of state β, which will be representative of the degree distribution for any state with qβ,N native contacts
where
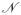 (Δq) is a function counting the number of structures within Δq broken contacts of β, and θ(·) is the Heaviside step function asserting that the states are kinetically close. This expression simply counts the number of states in our model satisfying the condition kα,β ≥ kc.
(Δq) is a function counting the number of structures within Δq broken contacts of β, and θ(·) is the Heaviside step function asserting that the states are kinetically close. This expression simply counts the number of states in our model satisfying the condition kα,β ≥ kc.
To evaluate this, recall our rate for α → β
where we have taken the linear approximation qα,β,N ≈ bqβ,N, which will be valid if native contacts are distributed in some regular way in contact space. This approximation is mathematically and physically motivated in the supplemental information, and the value for b ≈
(10−1) is derived.
The expression for kα,β shows explicitly how the rate is faster when entering a highly native state (bigger qβ,N) and slow when leaving a native-like state (bigger qα,N). Now, for each qα,N, we find some Δqmax satisfying kα,β ≥ kc for all Δq ≤ Δqmax
where in the last line, we have defined a new value, E ≡ εx/(εNN + σ ). Note that as long as εx < 0 and εNN + σ < 0, i.e. both native and non-native contacts are sufficiently favorable, E > 0. Our choice of kc should not matter to the final solution, and we will see proof of this later. Therefore, choose the simplest kc, specifically kc = k̃ such that the last term here drops out.
It follows from the way we choose Δqmax that we can re-write our degree evaluation with specific definite bounds to enforce the Heaviside function
where to lighten the notation we have written n = qα,N. Now, we must evaluate
(Δq). Physically, this is the total number of states kinetically accessible when one state breaks and re-forms Δq contacts. Strict combinatorial evaluation, while possible, will surely over-count this number without explicit consideration of, e.g. excluded volume effects and the restriction that nearby residues must form nearby contacts due to the polymer backbone.
We can estimate the number of accessible states from a simple physical argument. Let the value z represent the number of choices each residue in the loop has to form a new contact. Then, if each residue broken has approximately z choices that allow the other residues to form their contacts, we obtain
or exponential scaling in the number of possible conformations with number of broken contacts.
We can estimate z as follows. Contacts will almost certainly be broken on the surface of the protein, where loops can form. Locally, this surface will be approximately planar. Empirically the coordination number of residue-residue contacts is about 6,65,66 consistent with traditional folding lattice models.42,67–69 This means that we can approximate the surface of the protein as a lattice with coordination number 6, and envision a loop settling on this surface. Consider now one residue on the lattice surface, an anchor site that is the beginning of the loop. Now place a residue adjacent to this one on the lattice. Clearly, there are four options. Now, place another residue adjacent the residue just placed. There will only be three options for this residue, and all residues after that one will have three, two, or one placement possibilities (Figure 2). Therefore, we conclude that 1 < z < 4, but is most likely about 2, considering steric and energetic restrains. Luckily, we will see that the final folding rate scales only linearly with z, while growing exponentially with other factors.
Figure 2.

Illustration of the process used to estimate z. Black dots represent placed residues on a plane of lattice sites, red dots represent potential sites for the next residue along a chain. (1) through (4) show the sequential placement of four residues, demonstrating situations where two, three, and four sites are available for the next residue. These considerations lead to an estimate of 1 < z < 3.
With the expression for
, evaluation of dβ is straightforward. Note dβ is a function of qβ,N only. Writing δ ≡ bEqβ,N to further lighten notation, we obtain
where we have dropped constant values.
This expression can be simplified even further. Note zE(q+1) ≪ −1, so we can write
giving us directly the number of kinetic connections state β has. Notice from the evaluation of Ω(n) that the probability of picking a state with qβ,N native contacts uniformly at random is P(qβ,N) ∝ exp[c(q − qβ,N)]. The one-to-one correspondence between qβ,N and dβ, simple substitution gives
Revealing that the topology of our model exhibits a power-law degree distribution with exponent μ = c(bE logz)−1. This results in a native hub. This is a key result describing the structure of protein kinetics in a discrete configuration space. Our work up to this point has shown that this space has a hub-like structure, from here we will see if this structure has any consequences for the system kinetics.
One can see that, if E < 0, then native states with large qβ,N will have connections to many states (large Δq) and therefore have high degrees. If E > 0, (non-native contacts become energetically unfavorable) a phase transition occurs where μ becomes negative, and we get many non-native states that have high degree. The model presented here does not accurately represent this regime, since the lack of attractive non-native interactions will cause the unfolded state to consist of coil-like (as opposed to globular) conformations. We hope to examine this regime in future work, especially considering the importance of coil-like conformations of proteins under denaturing conditions.
Graphs with power-law degree distribution, commonly known as scale-free networks,70 are well studied, because they appear in many real world networks (e.g. WWW, Internet, protein interactions, etc.) and have many interesting properties. In what follows, we focus on the eigenspectrum of this structure, which reveals the dynamics involved in protein folding.
The Kinetic Spectrum
Here, we evaluate the first dynamical eigenvalue of the graph Laplacian, and use it to calculate the folding time. The Laplacian has n eigenvalues λ1 = 0 < λ2 < … < λn that describe the timescales of the dynamical processes of the system. These eigenvalues will be distributed according to an eigenspectrum specific to the graph in question. We are especially interested in λ2, which corresponds to the slowest relaxation in the kinetic system - in the case of proteins, we assume this corresponds to folding. This eigenvalue is the inverse of the folding timescale, λ2 = τ0/τf. Therefore, showing λ2 is large is equivalent to showing that proteins fold quickly.
It has been shown that λ2 depends primarily on the nodes with the smallest degrees, dmin.72 There exist proven bounds73–75 on the value of λ2 in terms of dmin for the large-graph limit, which we recall in the supplemental information and present here
where V is the number of nodes in the graph, and m is some constant. Since both these bounds are linear, they indicate that λ2 ~ dmin. Numerical simulations of uncorrelated scale-free matrices confirm linear scaling (Figure 4). Using the expression
Figure 4.

The scaling of the first dynamical eigenvalue λ2 as a function of minimum degree dmin in scale-free graphs. Presented are the results for 103 node and 104 node graphs, in blue and green, respectively. Graphs were simulated so as to have uncorrelated degrees, using the modified configuration model of Catanzaro, Boguna, and Pastor-Satorras,71 with parameter μ = 3. Graphs with μ = 2, μ = 2.5, and μ = 5 were also investigated and showed similar results. Our presented bounds, both upper and lower (m = 0.3), are plotted as dashed lines. The lower bound is shifted down by 1, for clear illustration (it is not applicable for dmin = 1, where there is no guaranteed giant component).
and recalling δ ∝ qβ,N, we can see that the minimum degree is obtained when qβ,N is minimized. Taking qβ,N = 0, we have
then, applying our scaling λ2 ~ dmin we obtain
where τf is the folding rate and we have noted that the number of contacts is expected to scale linearly with chain length, q = ρNN. Here we have denoted the contact density, simply a constant of proportionality, by ρN. Empirical data suggest ρN ≈ 0.6 (SI, Figure S2). Estimates of the energetic parameters (εN, εNN and σ) from simulation,23,24,27 theory,36,37,44 and experiment59,65,66 yield values of εN ≈ −3, εNN ≈ −2, and σ ≈ 1 (see SI). From these approximations, the exponential factor is ξ ≈
(10−1). This functional form is consistent with the derivation of Zwanzig,47 who used a different model to arrive at a similar result.
The computation of τf is the central result of our model. It shows explicitly why proteins fold quickly, and how the folding time scales as a function of chain length. The fact that scale-free networks have a spectral edge that is bounded away from λ1 = 0, the stationary distribution, means that the first dynamical process occurs very quickly, much more quickly than the naive prediction of Levinthal. Furthermore, though we have derived exponential scaling in folding time as a function of chain length, the exponential constant is very small, so the scaling is quite weak. This shows how proteins could grow to moderate chain lengths (103 residues) before reaching biologically intractable folding times.
Discussion
We have presented a model that is quite simple - it represents an attempt at a minimal description of protein kinetics. Despite this simplicity, interesting and subtle features emerge. The master equation predicts that the native state is a kinetic hub, highly connected to many diverse nodes, while non-native states interconvert slowly, similar to results from detailed all-atom simulations. This hub has a regular structure, that of a scale-free network, the topology of which dictates the timescales of dynamics in the system.
Considering the physical origins of the hub, a simple picture of folding emerges. The model depicts protein folding as a search through globular structures, converting from globule to globule through transition states with short loops. Native contacts form heterogeneously and stochastically, and once these favorable contacts are formed, they are likely to persist, since it is energetically costly to break them. We call this tendency statistical persistence. Eventually, the globule accrues more and more native content, until it is completely folded. While searching through globular states, the protein hops between compact conformations that involve only moderate rearrangements, forming temporary short loops that then collapse again (Figure 6).
Figure 6.

Exemplar pathway, taken from highly-travelled pathway in all-atom explicit water simulations of lambda repressor, and a representation of the MSM built from the simulations, illustrating the process captured by the simple model. (A) Protein folding occurs as globular structures interconvert, slowly gaining native content (orange) in a stochastic manner. (B) This process leads to a kinetic hub that dictates the timescales of the system. Represented here is the network of an MSM for this protein. Reproduced with modification from Bowman and Pande (2010),24 with permission.
One interesting point is that favorable non-native contacts play a key role in this picture. These contacts ensure that unfolded states are compact globules. Further, these non-native contacts are directly responsible for the strong hub-like features of the model and the fast folding predicted (recall without favorable non-native contacts, E > 0. Without favorable non-native contacts, the model breaks down. The properties of a model similar to this one in the unfavorable non-native regime is still an open question, and it is difficult to speculate on what such a model would look like.
Analysis of these kinetics predicts that the folding time scales weakly exponentially with chain length, τf ∝ eξN, with ξ ≈
(10−1), in strong agreement with experiment (Figure 5), though chain length is certainly not the only variable affecting folding time. Though exponential scaling might be thought as a return to Levinthal’s paradox, the model explicitly shows that the exponential constant is small. We hypothesize that exponential scaling may provide an explanation for why protein domain sizes cover less than two orders of magnitude (10–1000 residues) and why large proteins often need chaperones to fold.77
Figure 5.

Theoretical prediction of the folding rate compared with 78 experimental values, with no fit parameters. (A) The folding time scaling as a function of chain length (red solid line), for the estimated value of the exponential scaling parameter, ξ = 0.14 (z = 2, φNN = 1.0 kcal mol−1, φN = 1.5 kcal mol−1, ΔSloop = 3.5 cal mol−1 K−1). For comparison, a least-squares fit gives ξ = 0.10 (R2 = 0.53). The dashed black lines show extremal values of ξ, the estimated value increased and decreased by 50%. (B) The same data, comparing theory and experiment, but including the specific number of contacts of each protein in the native state, q, rather than the average contact density ρN. This results in a negligible difference in the model’s approximation quality. Black line is exact comparison, dashed lines indicate two orders of magnitude variation, which might be accounted for e.g. mutations and temperature variations, factors not explicitly included in the model. Data from Ref.76 The severe outlier in the bottom right of (B) is α3D (PDB: 2A3D), a de novo designed protein.
Our model shines light on a number of previous experimental and theoretical observations, which we review here. Interestingly, the thermodynamics of the model support two-state behavior, exhibiting a highly cooperative transition between native and non-native states at the melting temperature (Tm). This is surprising considering that the model also exhibits many timescales arising from heterogeneous pathways. The discrepancy may indicate why the folding process appears to be quite simple - either two or three state - when viewed with a low-resolution experimental probe, but appears complex when examined with high resolution techniques. An example is the small protein villin,78 a traditional two-state folder that exhibits complex kinetics when viewed at high resolution, either in experiment,79 or simulation.26 The apparent two-state nature of proteins is likely augmented by the fact that, while the timescales and mechanisms of folding might be heterogeneous, the thermodynamics can still dictate strong two-state behavior.80–83
We should note that currently whether or not this model predicts two-state behavior in a kinetic sense remains an open question. Experiments show that many (but not all) proteins not only exhibit two dominant thermodynamic states, but single exponential relaxation between those states. In our current formalism, this would take the form of a separation of timescales - i.e., the third eigenvalue should be much larger than the second λ3 ≫ λ2. This does not follow immediately from the topology alone, suggesting that something deeper is at play.
The hub-like nature of the model predicts folding pathways should be highly heterogenous, without any clear global transition state. While the issue of heterogeneity is not a resolved topic,22,25,29,38,84 our model is consistent with experimental observations indicative of heterogeneity, such as the fact that the vast majority of φ-values are intermediate between 0 and 1.85–88 Furthermore, heterogeneity was a key insight of energy landscape theory, which proposed that multiple pathways helped to resolve the Levinathal paradox.6,8,9 Our model not only agrees with this aspect of that theory, but explicitly shows how parallel pathways arise from microscopic considerations, and how they lower the folding time.
Moreover, our model predicts that a few native-like states will be accessible at equilibrium, and that these states will retain a high degree of native content (many native contacts), but be considerably higher in free energy than the native state (Figure 3). These states have been observed in such experiments such as equilibrium hydrogen-deuterium exchange38,89 and have been previously predicted by simple theories and simulations.12,61
Figure 3.

Spectra of the model for typical proteins. (A) The energy spectrum of the states. An exponential density leads to a large energy gap in the spectrum, previously predicted to be important for the stability of folded structures.12 Left panel is the density of states, right is a spectrum of 1000 states drawn at random from that density for illustration. (B, left panel) Density of states of a typical scale-free eigenspectrum. Plotted is the average over 580 simulations of uncorrelated scale-free networks with 104 nodes, μ = 3.0, dmin = 5. The first timescale is necessarily bounded away from long times by the spectral edge at λc (arrow), leading to fast folding. Also shown are (right panel) the longest 50 timescales (1/λ) from the first simulation, plotted on a log-scale. (C) and (D) represent the energetic and kinetic spectra of MSMs parametrized from explicit water all-atom MD simulations,23,24,27 for comparison.
The model’s key feature - the kinetic network topology - could be directly observable in experiments. We predict that, under native conditions, a single molecule experiment able to distinguish the native state and any two or more non-native states, as well as the rates of interconversion between them, should observe many more transitions from non-native states to the native state than between non-native states. At the time of publication, we are unaware of any such experiment. Observation of this kind of behavior for a large number of states on a diverse set of systems would, in our opinion, constitute good evidence of the kinetic hub theory derived here.
Conclusions
By treating kinetics directly, we derived a simple model that captures the essential features of protein folding kinetics and thermodynamics. The model exhibits qualitative fidelity with experiment and simulation, and explicitly shows how native and non-native residue contacts could lead to a kinetic hub displaying timescales appropriate for protein folding. Despite these successes, our model is quite different from how protein folding has been previously viewed. We emphasize that there is no global transition state in the model. Folding kinetics are a consequence of the scale-free properties of the hub, rather than from the crossing of one dominant free energy barrier.
Why does our model look so different from previous theory? There are two main reasons. First, we have dealt directly with kinetics, rather than energy landscapes. This allows us to retain the full dimensionality of conformation space, without having to project onto a reaction coordinate. In high-dimensional systems, these projections can lead to significant errors.17,18 Second, here we have not presumed the principle of minimal frustration. Indeed, we see that attractive non-native contacts are an important part of the model, and that without them the kinetic hub is not present. The experimentally supported prediction that water is a poor solvent for protein chains indicates that attractive non-native contacts are appropriate.50–57
While fundamentally different from previous theory, this model captures nearly all the key qualitative features of protein folding kinetics, and we hope it will provide a foundation for understanding those kinetics in future studies of protein folding.
Supplementary Material
Acknowledgments
The history and literature of protein folding is extensive. We would like to acknowledge the fact that many scholars have contributed work to the field, and while we did our best to cite previous work, we may have regrettably missed the contributions of some. We would like to thank Christian Schwantes, Gregory Bowman, Alexander Samukhin, Jesus Izaguirre and Ken Dill for insightful discussions. Thanks also to Gregory Bowman, Kyle Beauchamp, and Vincent Voelz for generously sharing data. TJL was supported by an NSF GRF. This study was made possible through funding from the NSF (MCB-0954714) and NIH (R01-GM062868).
Footnotes
This material is available free of charge via the Internet at http://pubs.acs.org/.
Supporting Information Available
Supplementary information includes (1) the detailed derivation of Ω(n), (2) the approximation of qα,β,N, (3) the derivation of the bounds on λ2, (4) a histogram showing collapse in protein folding simulations, (5) a brief discussion of the scaling of the number of residue-residue contacts with chain length, and (6) details of the estimation of contact energies and entropies.
References
- 1.Anfinsen C. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Levinthal C. J Medical Phys. 1968;65:44–45. [Google Scholar]
- 3.Levinthal C. Mossbauer Spectroscopy in Biological Systems. 1969:22–24. [Google Scholar]
- 4.Dill KA, Ozkan S, Shell M, Weikl TR. Ann Rev Biophys. 2008;9:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldwin R. J Biomol NMR. 1995;5:103–109. doi: 10.1007/BF00208801. [DOI] [PubMed] [Google Scholar]
- 6.Dill K, Chan H. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 7.Shakhnovich EI, Finkelstein AV. Biopolymers. 1989;28:1667–80. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 8.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Ann Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 9.Onuchic JN, Wolynes PG. Current opinion in structural biology. 2004;14:70–5. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Jackson SE, Fersht AR. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 11.Radford S, Dobson C. Nature. 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 12.Sali A, Shakhnovich E, Karplus M. Nature. 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 13.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Proteins. 1995;21:167–95. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 14.Bryngelson J, Wolynes PG. Biopolymers. 1990:177–188. [Google Scholar]
- 15.Bryngelson JD, Wolynes PG. J Phys Chem. 1989:6902–6915. [Google Scholar]
- 16.Bryngelson JD, Wolynes PG. Biophysics. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du R, Pande VS, Grosberg AY, Tanaka T, Shakhnovich ES. J Chem Phys. 1998;108:334. [Google Scholar]
- 18.Chandler D. Finding Transition Pathways: Throwing Ropes Over Rough Mountain Passes, in the Dark. Classical and Quantum Dynamics in Condensed Phase Simulations. 1998:51–66. [Google Scholar]
- 19.Noé F, Fischer S. Curr Op Struct Biol. 2008;18:154–62. doi: 10.1016/j.sbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Prinz JH, Keller B, Noé F. Phy Chem Chem Phys. 2011;13:16912–27. doi: 10.1039/c1cp21258c. [DOI] [PubMed] [Google Scholar]
- 21.Prinz JH, Wu H, Sarich M, Keller B, Senne M, Held M, Chodera JD, Schütte C, Noé F. The Journal of Chemical Physics. 2011;134:174105. doi: 10.1063/1.3565032. [DOI] [PubMed] [Google Scholar]
- 22.Noél’ F, Schütte C, Vanden-Eijnden E, Reich L, Weikl TR. Proceedings of the National Academy of Sciences. 2009;106:19011–19016. doi: 10.1073/pnas.0905466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voelz Va, Bowman GR, Beauchamp K, Pande VS. J Am Chem Soc. 2010;132:1526–8. doi: 10.1021/ja9090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman GR, Voelz VA, Pande VS. J Am Chem Soc. 2010:12–15. doi: 10.1021/ja9090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 26.Beauchamp K, Ensign D, Das R, Pande V. Proc Natl Acad Sci. 2011;108:12734. doi: 10.1073/pnas.1010880108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lane TJ, Bowman GR, Beauchamp K, Voelz VA, Pande VS. J Am Chem Soc. 2011;133:18413–9. doi: 10.1021/ja207470h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voelz VA, Jager M, Yao S, Chen Y, Zhu L, Waldauer SA, Bowman GR, Friedrichs M, Bakajin O, Lapidus LJ, Weiss S, Pande VS. J Am Chem Soc. 2012 doi: 10.1021/ja302528z. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 30.Rao F, Caflisch A. J Mol Biol. 2004;342:299–306. doi: 10.1016/j.jmb.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 31.Bowman G, Pande V. Proc Natl Acad Sci. 2010;107:10890. doi: 10.1073/pnas.1003962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pande VS. Phys Rev Lett. 2010;105:1–4. doi: 10.1103/PhysRevLett.105.198101. [DOI] [PubMed] [Google Scholar]
- 33.Plaxco KW, Simons KT, Baker D. J Mol Biol. 1998;277:985–94. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 34.Plaxco KW, Simons KT, Ruczinski I, Baker D. Biochemistry. 2000;39:11177–11183. doi: 10.1021/bi000200n. [DOI] [PubMed] [Google Scholar]
- 35.Muñoz V, Eaton WA. Proc Natl Acad Sci. 1999;96:11311–6. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linse S, Linse B. J Am Chem Soc. 2007;129:8481–6. doi: 10.1021/ja070386e. [DOI] [PubMed] [Google Scholar]
- 37.Zwanzig R, Szabo A, Bagchi B. Proc Natl Acad Sci. 1992;89:20. doi: 10.1073/pnas.89.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maity H, Maity M, Krishna MMG, Mayne L, Englander SW. Proc Natl Acad Sci. 2005;102:4741–6. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravasz E, Gnanakaran S, Toroczkai Z. Arxiv preprint arXiv 0705 0912. 2007:1–15. [Google Scholar]
- 40.Bruscolini P, Pelizzola A. Phys Rev Lett. 2002;88:1–4. doi: 10.1103/PhysRevLett.88.089601. [DOI] [PubMed] [Google Scholar]
- 41.Chan HS, Dill KA. Proteins. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 42.Dill KA, Bromberg S, Yue K, Fiebig KM, Yee DP, Thomas PD, Chan HS. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh K, Ozkan SB, Dill KA. J Am Chem Soc. 2007;129:11920–7. doi: 10.1021/ja066785b. [DOI] [PubMed] [Google Scholar]
- 44.Kubelka J, Henry ER, Cellmer T, Hofrichter J, Eaton WA. Proc Natl Acad Sci. 2008;105:18655–62. doi: 10.1073/pnas.0808600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Portman J, Takada S, Wolynes P. Phys Rev Lett. 1998;81:5237–5240. [Google Scholar]
- 46.Shakhnovich EI, Gutin aM. Biophys Chem. 1989;34:187–99. doi: 10.1016/0301-4622(89)80058-4. [DOI] [PubMed] [Google Scholar]
- 47.Zwanzig R. Proc Natl Acad Sci. 1995;92:9801–9804. doi: 10.1073/pnas.92.21.9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thirumalai D. J Phys (France) 1995;5:1457–1467. [Google Scholar]
- 49.Gfeller D, De Los Rios P, Caflisch A, Rao F. Proc Natl Acad Sci. 2007;104:1817–22. doi: 10.1073/pnas.0608099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoffmann A, Kane A, Nettels D, Hertzog DE, Baumgärtel P, Lengefeld J, Reichardt G, Horsley DA, Seckler R, Bakajin O, Schuler B. Proc Natl Acad Sci. 2007;104:105–10. doi: 10.1073/pnas.0604353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Best RB, Merchant KA, Gopich IV, Schuler B, Bax A, Eaton Wa. Proc Natl Acad Sci. 2007;104:18964–9. doi: 10.1073/pnas.0709567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuler B, Lipman E, Eaton W. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 53.Ziv G, Haran G. J Am Chem Soc. 2009;131:2942–7. doi: 10.1021/ja808305u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waldauer SA, Bakajin O, Lapidus LJ. Proc Natl Acad Sci. 2010;107:13713–7. doi: 10.1073/pnas.1005415107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowman GR, Pande VS. 2011 In Preparation. [Google Scholar]
- 56.Voelz VA, Singh VR, Wedemeyer WJ, Lapidus LJ, Pande VS. J Am Chem Soc. 2010;132:4702–9. doi: 10.1021/ja908369h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dill KA, Shortle D. Ann Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 58.Pande VS, Grosberg AYu, Tanaka T. Fold Des. 1997;2:109–14. doi: 10.1016/s1359-0278(97)00015-1. [DOI] [PubMed] [Google Scholar]
- 59.Makhatadze G, Privalov P. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 60.Flory P. Statistical mechanics of chain molecules. Interscience Publishers; 1969. [Google Scholar]
- 61.Shakhnovich EI. Curr Op Struct Biol. 1997;7:29–40. doi: 10.1016/s0959-440x(97)80005-x. [DOI] [PubMed] [Google Scholar]
- 62.Franzosa E, Lynagh K, Xia Y. Experimental Standard Conditions of Enzyme Characterizations. 2010:99–106. [Google Scholar]
- 63.Buchete NV, Hummer G. The J Phys Chem B. 2008;112:6057–69. doi: 10.1021/jp0761665. [DOI] [PubMed] [Google Scholar]
- 64.Weber J, Pande V. Biophys J. 2012;102:859–867. doi: 10.1016/j.bpj.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyazawa S, Jernigan RL. J Mol Biol. 1996;256:623–44. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 66.Miyazawa S, Jernigan RL. Macromolecules. 1985:534–552. [Google Scholar]
- 67.Cieplak M, Hoang T, Li M. Phys Rev Lett. 1999;83:1684–1687. [Google Scholar]
- 68.Gutin AM, Abkevich VI, Shakhnovich EI. Phys Rev Lett. 1996;77:5433–5436. doi: 10.1103/PhysRevLett.77.5433. [DOI] [PubMed] [Google Scholar]
- 69.Leopold PE, Montal M, Onuchic JN. Proc Natl Acad Sci. 1992;89:8721–5. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barabási A, Albert R. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 71.Catanzaro M, Boguñá M, Pastor-Satorras R. Phys Rev E. 2005;71:1–4. doi: 10.1103/PhysRevE.71.027103. [DOI] [PubMed] [Google Scholar]
- 72.Samukhin AN, Dorogovtsev SN, Mendes JFF. Phys Rev E. 2008;77:1–19. [Google Scholar]
- 73.Mohar B. Graph Combinator. 1991;7:53–64. [Google Scholar]
- 74.Cohen R, Havlin S. Phys Rev Lett. 2003;90:5–8. doi: 10.1103/PhysRevLett.90.058701. [DOI] [PubMed] [Google Scholar]
- 75.Bollobas B, Riordan O. Combinatorica. 2004;24:5–34. [Google Scholar]
- 76.Ouyang Z, Liang J. Prot Sci. 2008;17:1256–1263. doi: 10.1110/ps.034660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hartl FU, Hayer-Hartl M. Nat Struct Biol. 2009;16:574–81. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 78.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. J Mol Biol. 2006;359:546–53. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 79.Reiner A, Henklein P, Kiefhaber T. Proc Natl Acad Sci. 2010;107:4955–60. doi: 10.1073/pnas.0910001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pirchi M, Ziv G, Riven I, Cohen SS, Zohar N, Barak Y, Haran G. Nat Commun. 2011;2:493. doi: 10.1038/ncomms1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sridevi K, Lakshmikanth GS, Krishnamoorthy G, Udgaonkar JB. J Mol Biol. 2004;337:699–711. doi: 10.1016/j.jmb.2003.12.083. [DOI] [PubMed] [Google Scholar]
- 82.Rhoades E, Cohen M, Schuler B, Haran G. J Am Chem Soc. 2004;126:14686–7. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 83.Wright CF, Lindorff-Larsen K, Randles LG, Clarke J. Nat Struct Biol. 2003;10:658–62. doi: 10.1038/nsb947. [DOI] [PubMed] [Google Scholar]
- 84.Englander SW, Mayne L, Krishna MMG. Quart Rev Biophys. 2007;40:287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sánchez IE, Kiefhaber T. J Mol Biol. 2003;325:367–376. doi: 10.1016/s0022-2836(02)01230-5. [DOI] [PubMed] [Google Scholar]
- 86.Li L, Mirny La, Shakhnovich EI. Nat Struct Biol. 2000;7:336–42. doi: 10.1038/74111. [DOI] [PubMed] [Google Scholar]
- 87.Ozkan SB, Bahar I, Dill Ka. Nat Struct Biol. 2001;8:765–9. doi: 10.1038/nsb0901-765. [DOI] [PubMed] [Google Scholar]
- 88.Vendruscolo M, Paci E, Dobson CM, Karplus M. Nature. 2001;409:641–5. doi: 10.1038/35054591. [DOI] [PubMed] [Google Scholar]
- 89.Baldwin AJ, Kay LE. Nat Chem Biol. 2009;5:808–14. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.