

Background: In BK channels, Ca2+ and voltage sensors are allosterically connected to the pore.
Results: We optically resolved voltage sensor rearrangements, initiated by Ca2+ binding to the intracellular domains RCK1 and RCK2. Impairing the RCK2 abolished this allosteric effect.
Conclusion: The RCK2 Ca2+ sensor is required for the allosteric facilitation of voltage sensor activation.
Significance: RCK1 and RCK2 Ca2+ sensors are not functionally homologous.
Keywords: Allosteric Regulation, Cooperativity, Electrophysiology, Fluorescence, Potassium Channels, Slo1, UV Photolysis, Voltage Clamp Fluorometry
Abstract
Large conductance voltage- and Ca2+-activated K+ (BK) channels are potent regulators of cellular processes including neuronal firing, synaptic transmission, cochlear hair cell tuning, insulin release, and smooth muscle tone. Their unique activation pathway relies on structurally distinct regulatory domains including one transmembrane voltage-sensing domain (VSD) and two intracellular high affinity Ca2+-sensing sites per subunit (located in the RCK1 and RCK2 domains). Four pairs of RCK1 and RCK2 domains form a Ca2+-sensing apparatus known as the “gating ring.” The allosteric interplay between voltage- and Ca2+-sensing apparati is a fundamental mechanism of BK channel function. Using voltage-clamp fluorometry and UV photolysis of intracellular caged Ca2+, we optically resolved VSD activation prompted by Ca2+ binding to the gating ring. The sudden increase of intracellular Ca2+ concentration ([Ca2+]i) induced a hyperpolarizing shift in the voltage dependence of both channel opening and VSD activation, reported by a fluorophore labeling position 202, located in the upper side of the S4 transmembrane segment. The neutralization of the Ca2+ sensor located in the RCK2 domain abolished the effect of [Ca2+]i increase on the VSD rearrangements. On the other hand, the mutation of RCK1 residues involved in Ca2+ sensing did not prevent the effect of Ca2+ release on the VSD, revealing a functionally distinct interaction between RCK1 and RCK2 and the VSD. A statistical-mechanical model quantifies the complex thermodynamics interplay between Ca2+ association in two distinct sites, voltage sensor activation, and BK channel opening.
Introduction
Allostery is an intrinsic property of all proteins that allows distant sites to be energetically coupled, resulting in the mutual and efficient regulation of many biological processes (1, 2). Allostery can originate from binding events, covalent modifications, mutations, or changes in the environment. In ion channels, the gating mechanism(s) rely on the allosteric coupling between the pore and other modulatory domains, specialized to sense different stimuli, such as changes in membrane potential, temperature, mechanical stress, and the concentration of signaling molecules. Although the available atomic structures of ion channels or their functional domains are rapidly increasing, the mechanisms underlying allostery in these proteins are far from being fully understood. Several studies have provided experimental evidence of cooperative interactions in K+ channels (3–8). An example of a highly allosteric membrane protein is the large conductance, voltage- and Ca2+-activated (BK, Slo1) K+ channel. BK channel open probability (Po) is controlled by cooperative interactions between its four transmembrane voltage-sensing domains (VSDs),2 an intracellular multiligand-sensing domain (gating ring), and the pore (6, 9–12) (Fig. 1A). Functional BK channels are tetrameric proteins (13), each subunit composed of seven transmembrane segments (S0–S6) and a large intracellular C terminus (14). As typical of voltage-activated K+ channels (15), the transmembrane region of BKchannels includes helices S5-S6, which contribute to the central, K+-selective pore domain and a VSD including the charge-bearing segments S2-S3-S4 (16–20) (Fig. 1A). BK sensitivity to Ca2+ and other intracellular ligands (Mg2+, H+, CO, Heme, etc.) is conferred by the large intracellular C-terminal region (10, 11, 21), which encompasses two tandem regulator of conductance of K domains, RCK1 and RCK2 (22–29) (Fig. 1). RCK1 includes residues involved in both high affinity Ca2+ sensing, such as Asp-362/Asp-367 (30), Met-513 (31), and Glu-535 (32), and low affinity divalent cation sensing (Glu-374 and Glu-399) (30, 33–35). RCK2 encompasses a high affinity Ca2+-binding site (the “calcium bowl”) consisting of five consecutive Asp residues (Asp-894–Asp-898) (36–38) (Fig. 1B). Recently resolved crystal structures of this intracellular domain (27–29) provided details of its structural organization, consisting of four pairs of RCK domains that assemble into a tetrameric complex (“gating ring”) akin to those of bacterial MthK channels (23) (Fig. 1B). Our recent work has shown that the purified BK gating ring selectively responds to Ca2+ and Mg2+ under physiologically relevant conditions, undergoing metal-driven conformational rearrangements, thus fulfilling its role as a chemomechanical transducer (39). Considering the highly allosteric nature of BK channel activation, these Ca2+-induced conformational changes are expected to propagate to the transmembrane functional domains. In thermodynamic terms, the activation of Ca2+ sensors must alter the equilibrium isotherms of the pore and the VSD.
FIGURE 1.

BK channel putative structure and sensing domains. A, left panel, side view of a putative structural model of the BK channel, composed of the BK gating ring structure (Protein Data Bank code 3NAF, white) (28), associated with the transmembrane portion of the KV1.2–2.1 paddle chimera (Protein Data Bank code 2R9R) (67), and S0 ideal α helices positioned according to Liu et al. (68) as previously (51). The pore domain is shown in gray, whereas VSD helices are shown in orange, except S4 (purple). TMRM fluorophores conjugated with Cys-202 at the extracellular portions of S4 are shown in red. Right panel, top view of the gating ring structure overlaid by the “footprint” of the KV1.2–2.1 transmembrane domains. B, side and top views of the human BK channel cytosolic gating ring apparatus (Protein Data Bank code 3NAF) (28) with its constituent domains RCK1 (green) and RCK2 (light blue). Asp-362 and Asp-367, which are involved in the RCK1 Ca2+ sensitivity, are shown in red. The calcium bowl (Asp-894–Asp-898) binding site in RCK2 is shown in blue. The approximate position of the phospholipid membrane is marked by the dashed line. Note that Asp-362/Asp-367 and the calcium bowl are potentially in close proximity to transmembrane voltage-sensing and pore domains.
In the present work, we tested these premises and attempted to experimentally investigate whether the Ca2+-induced conformational changes of the gating ring at the intracellular portion of the channel can propagate to, and be resolved as, structural rearrangements of the VSD. We combined the voltage-clamp fluorometry technique (40–42) with the UV photolysis of a Ca2+ cage compound, DM-Nitrophen (43–46), to simultaneously trigger voltage- and Ca2+-dependent activations of BK channels. We found that rapid intracellular Ca2+ release produced structural rearrangements that not only increased channel Po but also perturbed the conformation of the S4 helix of the BK VSD. In addition, impairing Ca2+ sensing in RCK2 (calcium bowl neutralization), but not in RCK1, abolished the effect of the elevation of intracellular Ca2+ concentration ([Ca2+]i) on VSD activation, underlining a functional difference of the two Ca2+ sensors. These findings were included in a statistical-mechanical allosteric model of BK channel activation, based on that by Horrigan and Aldrich (6) but expanded to take into account that the two high affinity Ca2+ sensors of the human BK channel are not functionally homologous. The fitting of experimental data to this new model allowed quantification of the distinct allosteric contribution of the two high affinity Ca2+-sensing sites in the gating ring to VSD activation and pore opening, resolving the network of allosteric interactions underlying voltage- and Ca2+-dependent BK channel activation.
EXPERIMENTAL PROCEDURES
Molecular Biology
The human Slo1 clone (U11058) (47) without extracellular Cys (C14S/C141S/C277S) was used. Background mutations also included R207Q and W203V to increase Po (17, 18) and fluorescence signal (20), respectively. A single Cys was substituted in position 202 in the short extracellular S3-S4 linker (S202C) for site-directed fluorescence labeling (19, 20, 48) (Fig. 1A). Throughout the paper, this clone is referred to as pseudo-WT (pWT). The two high affinity Ca2+ sensors were separately neutralized in the D362A/D367A (RCK1) and the D894N/D895N/D896N/D897N/D898N (RCK2/calcium bowl) mutants. We also used the noninactivating Shaker K+ channel (Sh-IR, #M17211). For site-directed fluorescent labeling, we introduced a unique Cys in the S3-S4 linker (M356C) (40, 41). Single point mutations were generated with QuikChange site-directed mutagenesis kit (Stratagene, CA) and confirmed by sequencing. The cDNAs were translated to cRNAs in vitro (mMESSAGE MACHINE; Ambion, Austin, TX) and stored at −80 °C.
Oocyte Preparation
Xenopus laevis (NASCO, Modesto, CA) oocytes (stages V–VI) were prepared as previously described (49) and then injected with 50 nl of cRNA solution (0.01–0.1 μg/μl) using a Drummond nano-injector. The injected oocytes were maintained at 18 °C in an amphibian saline solution supplemented with 50 μg/ml gentamycin (Invitrogen), 200 μm DTT, and 10 μm EDTA. 3–6 days after injection, the oocytes were labeled with 10 μm membrane-impermeable, thiol-reactive fluorophore, tetramethylrhodamine-5′-maleimide (TMRM) (Molecular Probes, Eugene, OR) in a depolarizing K+ solution (120 mm K-MES, 2 mm Ca(MES)2, and 10 mm HEPES, pH 7). TMRM stock (100 mm) was dissolved in Me2SO and stored at −20 °C. The oocytes were then thoroughly rinsed in a dye-free solution and injected with 100 nl of DM-Nitrophen precomplexed with Ca2+(2.5 mm final concentration in the oocyte), prior to voltage clamp fluorometry.
Electrophysiological Techniques
Voltage Clamp Fluorometry
The cut open oocyte vaseline gap technique (50) is a low noise, fast clamp technique. Changes in fluorescence signal and ionic currents were simultaneously measured from the same area of membrane isolated by the top chamber (20, 42). The optical setup consists of a Zeiss Axioscope FS microscope with filters appropriate for TMRM (Omega Optical, Brattleboro, VT). The light source is a 100 W microscope halogen lamp. A TTL-triggered Uniblitz VS 25 shutter (Vincent Associates, Rochester, NY) is mounted on the excitation light path. The objective (Olympus LUMPlanFl, 40×, water immersion) has a numerical aperture of 0.8 and a working distance of 3.3 mm (Olympus Optical). A Dagan Photomax 200 system is used for the amplification of the photocurrent and background fluorescence subtraction. A Xenon flash lamp system JML-C2 (Rapp Opto-electronik GmbH, Hamburg, Germany) delivering high energy UV flashes of adjustable intensity was used for the photolysis of caged Ca2+. An external trigger synchronized UV flashes with voltage clamp and optical recordings. A water-immersed quartz light guide was positioned ∼0.5 mm away from the oocyte upper dome.
Voltage Clamp Fluorometry Recording Solutions
The external solution contained 60 mm Na-MES, 50 mm K-MES, 2 mm Ca(MES)2, 10 mm Na-HEPES (pH 7.0). The internal solution contained 120 mm potassium glutamate, 10 mm HEPES (pH 7.0). The solution for the intracellular pipette was 2700 mm Na-MES, 10 mm NaCl. Low access resistance to the oocyte interior was obtained by permeabilizing the oocyte with 0.1% saponin dissolved in the internal solution.
Analysis
The experimental data were analyzed with a customized program developed in our division and using fitting routines in Microsoft Excel. The data for the membrane conductance (G(V)) and the fluorescence (F(V)) curves were calculated from ionic current and fluorescence recordings, by averaging ∼200 points (sampling frequency 50 μs/point) up to 2 ms before and 20 ms after Ca2+-releasing UV flashes. The G(V) curves were calculated by dividing the current-voltage relationships (I-V curves) by the driving force (Vm − EK), where Vm is the membrane potential, and EK is the equilibrium potential for K+, estimated using the Nernst equation. F(V) and G(V) data points were fitted to one Boltzmann distribution of the following form,
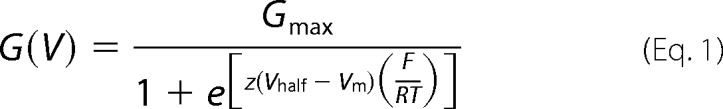 |
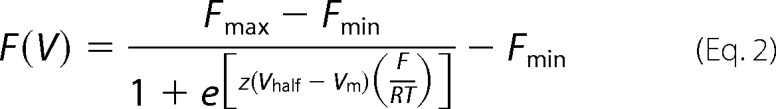 |
where Gmax and Fmax are the maximal G and F; Fmin is the minimal F; z is the effective valence of the distribution; Vhalf is the half-activating potential; Vm is the membrane potential; and F, R, and T are the usual thermodynamic values. The allosteric model is detailed in the supplemental data.
Patch Clamp
Membrane patches from Xenopus oocytes in the inside-out configuration were perfused with bath solutions containing 115 mm K-MES, 5 mm KCl, 5 mm HEDTA, 10 mm HEPES, pH 7 (supplemental Fig. S1A). [Ca2+] was varied by adding CaCl2. The free [Ca2+] was theoretically calculated with WEBMAXC v2.10 and then measured using a Ca2+ electrode (WPI, Sarasota, FL). The borosilicate glass pipettes (WPI) were filled with the bath solution at the lowest free [Ca2+]. The holding potential was 0 mV. The data were filtered to one-fifth of the sampling frequency. G(V) curves for different BK clones (supplemental Fig. S1B) at different free [Ca2+]i were constructed as described above. Plotting Vhalf against [Ca2+] produced calibration curves for the three BK clones used (supplemental Fig. S1C). To estimate the free [Ca2+]i before and after UV photolysis in the voltage clamp fluorometry experiments, we used the relation between GVhalf and free [Ca2+] in supplemental Fig. S1C.
Statistical Analysis
Scatter plots of ΔVhalf versus ΔkTln[Ca2+]i were tested for monotonicity using Spearman's rank order correlation coefficient (ρ) using the statistical software SigmaStat (Aspire Software, Ashburn, VA). Analysis using Pearson's coefficient for linear dependence yielded similarly significant measures of correlation and therefore are not included. ΔVhalf data were normalized to unitless quantities by multiplying by the fitted values for charge displacement (zL for ΔGVhalf and 4zJ for ΔFVhalf) and dividing by the change in chemical potential for calcium Δμ = ΔkTln[Ca2+]i. The collective mean of these normalized quantities, which can be interpreted as a measure of thermodynamic linkage between Ca2+- and voltage-sensing elements of the channel, was assessed for nonzero value using Student's t test. Of a total of 36 initially considered experiments, six were excluded either because: (i) Δμ did not exceed a post hoc threshold of 8 meV established to minimize error from insufficient calcium release (five experiments); or (ii) [Ca2+]i was near the positive saturated range (∼103 μm and higher), limiting the effect on ΔVhalf (1 experiment). Including these experiments introduced outliers in the normalized data points but did not affect statistical outcomes.
RESULTS
Ca2+ Binding to BK Gating Ring Induces Structural Rearrangements in VSD
The human BK channel (Slo1) VSD has been studied using voltage clamp fluorometry and shown to undergo voltage-dependent molecular rearrangements (19, 20, 48, 51). In this work, BK channels were engineered to allow for specific fluorescent labeling of the extracellular side of the S4 transmembrane segment at position 202 (this BK construct is referred to as pseudo WT or pWT throughout this paper). As we have previously shown, TMRM labeling position 202 reports conformational changes related to BK VSD activation (19, 20, 48). Also, the TMRM fluorescence reported from the adjacent position 201 shares an almost identical voltage dependence with that of gating currents (Q(V) curve) (20).
To probe for Ca2+- induced conformational changes of the VSD, Xenopus oocytes expressing pWT BK (prelabeled with TMRM) were injected with DM-Nitrophen, a caged Ca2+ compound that undergoes UV photolysis, releasing Ca2+. Thus, under voltage clamp (using the cut-open oocyte vaseline gap technique (50) modified for epifluorescence measurement (40–42)), we induced rapid increase of [Ca2+]i by delivering UV flashes focused on the upper exposed oocyte membrane. Simultaneous recordings of K+ currents and fluorescence emissions from an oocyte expressing pWT channels are shown in Fig. 2A. Ca2+ release was induced by UV flash triggered 60 ms after the onset of each depolarizing pulse. The rapid elevation of [Ca2+]i increased the BK channel Po, manifested by a downward or upward deflection of the ionic current traces (according to the K+ reversal potential, −20 mV) (Fig. 2A, black traces). Notably, UV-induced Ca2+ uncaging also induced an increase of the simultaneously recorded TMRM fluorescence emission (Fig. 2A, red traces), suggesting that the Ca2+-dependent activation of BK channels involves VSD conformational changes. The voltage dependence of the fluorescence deflections (F(V)) and macroscopic K+ conductance (G(V)) were calculated from the fluorescence and ionic current recordings, 2 ms before and 20 ms after Ca2+-releasing UV flashes (Fig. 2B). The rapid increase in [Ca2+]i produced a hyperpolarizing shift of the G(V) curve (ΔGVhalf ≈ −50 mV) associated with a smaller, but well resolved, translation of the F(V) curve in the same direction (ΔFVhalf ≈ −25 mV). Note that the Ca2+ release induced a crossover of G(V) and F(V) curves (Fig. 2B), evidence for BK channels undergoing conformational transitions while in the open state (5, 6, 16, 52).
FIGURE 2.

Intracellular Ca2+ photo release induces conformational changes of the VSD of BK channels. A, K+ current and fluorescence traces simultaneously recorded during depolarizations to the indicated potentials from an oocyte expressing pWT channels are shown superimposed. Caged Ca2+ release was triggered 60 ms after the onset of depolarization by UV light flashes delivered onto the oocyte. In this experiment [Ca2+]i increased from 10 to 84 μm. Ionic current and fluorescence signals increased following UV flashes. The photodiode amplifier was blanked for 3 ms during the UV flash to prevent overload. B, normalized fluorescence (F) and K+ conductance (G) data from the experiment in A, before and after UV flashes, were fit to a single Boltzmann distribution. Both curves were leftward-shifted at high [Ca2+]i: before UV, GVhalf = 0 mV and FVhalf = −37 mV; after UV, GVhalf = −51 mV and FVhalf = −63 mV. Note the crossing of G(V) and F(V) curves after Ca2+ release, consistent with the view that Ca2+ can open BK channels at membrane potentials where VSDs are not activated (5, 6, 16, 52).
The effects observed in BK channels were genuinely produced by Ca2+, as confirmed by the lack of detectable changes in ionic current and fluorescence recordings when Ca2+-insensitive Shaker K+ channels, fluorescently labeled to resolve their S4 movements, were similarly subjected to UV-induced Ca2+ release (supplemental Fig. S2, A and B). Moreover, ionic current and fluorescence recordings from pWT BK channels expressed in oocytes not injected with DM-Nitrophen were unaffected by UV flashes (supplemental Fig. S2, C and D). The reproducibility of Ca2+ photo release during sequential pulses is demonstrated in supplemental Fig. S3, A and B, which also shows that [Ca2+]i returned to its basal level in less than 1 s (supplemental Fig. S3C).
To estimate the local intracellular free [Ca2+]i before and after UV photolysis, we exploited the BK intrinsic Ca2+ sensing properties (i.e. the dependence of the half activation potential GVhalf on [Ca2+]i), as previously utilized in motor neurons and hair cells (53, 54). GVhalf versus free [Ca2+] calibration curves were constructed from excised patch experiments (see “Experimental Procedures” and supplemental Fig. S1). Note that in the pWT background, Ca2+ sensitivity was reduced by the well documented mutations in RCK1 and RCK2 domains, recapitulating the effect observed in WT BK channels (30, 36).
The Calcium Bowl Is Required for Ca2+-mediated Effect on VSD Movements
Two high affinity Ca2+-sensing sites are located within the BK gating ring ligand binding apparatus: one, in the RCK2 domain, coordinates Ca2+ via the five Asp residues (Asp-894–Asp-898) comprising the calcium bowl (27, 29, 36); the other, in RCK1, includes residue Asp-367, which is critical for Ca2+ sensing (30) (its Ca2+-coordinating role has been suggested (32) but not yet experimentally demonstrated (25, 27, 29)). These Ca2+-sensing sites differ in several respects, including: structure and relative positions within the gating ring and RCK domains (Fig. 1) (27–29); affinity and selectivity for divalent cations (34); apparent voltage dependence of Ca2+ binding (55); and role in epilepsy/dyskinesia-causing BK channel mutants (56). Given these established differences, we asked whether the two high affinity Ca2+ sensors also exert different contributions to the pore and VSD activation. To assess the role of these modules in the allosteric interaction between the gating ring and the VSD, the two high affinity Ca2+-sensing sites were separately neutralized by introducing mutations D362A/D367A (in RCK1) (30) and D894N/D895N/D896N/D897N/D898N (in RCK2) (36). In individual experiments measuring D362A/D367A channels, following UV flash photolysis, the rapid increase of [Ca2+]i induced a detectable shift toward more negative potentials in both the G(V) and F(V) curves. The shift was in the same direction as was observed in pWT channels, but smaller (Fig. 3, A and B), suggesting that residues Asp-362/Asp-367 are not necessary to mediate the propagation of Ca2+-induced gating ring rearrangements to the VSD.
FIGURE 3.

RCK1 and RCK2 are not functionally equivalent Ca2+ sensors. A, K+ current and fluorescence traces simultaneously recorded during depolarizations to the indicated potentials from a representative oocyte expressing BK channels with neutralized high affinity Ca2+ sensing in RCK1 (D362A/D367A) are shown superimposed. Caged Ca2+ release was triggered 35 ms after the onset of depolarization by challenging the oocyte with UV light to produce [Ca2+]i increase from 137 to 320 μm. As in the pWT channels, ionic current and fluorescence increased following UV flash. The photodiode amplifier was blanked for 3 ms during the UV flash to prevent overload. B, normalized fluorescence (F) and K+ conductance (G) data from the experiment in A, before and after UV flashes, were fit to a single Boltzmann distribution. Both curves were leftward-shifted at high [Ca2+]i: before UV, GVhalf = −21 mV and FVhalf = −85 mV; after UV, GVhalf = −35 mV and FVhalf = −101 mV. C, as in A, from an oocyte expressing BK channels with neutralized calcium bowl in RCK2 (D894N/D895N/D896N/D897N/D898N, D894–898N). Following UV flash, [Ca2+]i increased from 105 to 210 μm. Only ionic currents were affected by Ca2+ uncaging. D, as in B, for data from calcium bowl mutant in C. Before UV, GVhalf = 3 mV and FVhalf = −79 mV; after UV, GVhalf = −26 mV and FVhalf = −78 mV.
On the other hand, in D894N/D895N/D896N/D897N/D898N channels, there was no obvious effect of intracellular Ca2+ release on the F(V) curve (Fig. 3, C and D). Still, the Po of this mutant was increased by the elevation of [Ca2+]i, as demonstrated by the leftward-shifted G(V) curve following Ca2+ release (Fig. 3D). The absence of detectable Ca2+-induced VSD conformational changes excludes a direct effect of Ca2+ on the BK VSD (e.g., mediated by a charge screening effect), implying a functional, allosteric coupling between the calcium bowl and VSD.
We analyzed results from all experiments, as depicted in Fig. 4. The Ca2+-induced shift of the half-activation potential of G(V) curves (ΔGVhalf) was consistently associated with a shift of the F(V) curves (ΔFVhalf) in the same direction, but only with an intact calcium bowl (Fig. 4A). To quantify the dependence of observed shifts in G(V) and F(V) curves upon Ca2+ release in pWT and mutants channels, we performed a statistical analysis as described under “Experimental Procedures.” We found a strong monotonic correlation between Ca2+ release (ΔkTln[Ca2+]) and G(V) and F(V) voltage shifts in pWT and in the D362A/D367A mutant (Fig. 4, B and C). This was also true for the D894N/D895N/D896N/D897N/D898N mutant in the case of zL*GVhalf (p < 0.001), but not in the case of 4zJ*ΔF − Vhalf (p = 0.705). Furthermore, normalizing the voltage shifts by the change in Ca2+ chemical potential (Δμ) abolished the correlation between shifts of FVhalf and GVhalf as shown by theclustering of the data points (Fig. 4, E–G). We interpreted these data as a demonstration of the obligatory dependence of FVhalf and GVhalf on [Ca2+]. The normalized voltage shift was statistically different from zero for all channels in the case of G(V) and also for pWT and RCK1 mutant D362A/D367A in the case of F(V) (Fig. 4, E and F), but there was no significantly detectable shift in 4zJ*ΔFVhalf/Δμ in the case of RCK2 mutant D894N/D895N/D896N/D897N/D898N (Fig. 4G). Thus, with the exception of the RCK2 mutant, an increase in intracellular [Ca2+] consistently produced a leftward shift in the activation curves (ΔGVhalf) associated with a statistically significant shift of the voltage sensor activation (ΔFVhalf) in the same direction.
FIGURE 4.

Compilation of experiments measuring shifts in GVhalf and FVhalf in response to calcium release: an intact calcium bowl is necessary to mediate the allosteric interaction between the VSD and the gating ring. A, scatter plot of ΔFVhalf versus ΔGVhalf for all experiments meeting inclusion criteria described under “Experimental Procedures.” B–D, changes in activation energy produced by UV-flash triggered release (zL*ΔGVhalf and 4zJ*ΔFVhalf) plotted against ΔkTln[Ca]. A strong monotonic correlation exists between calcium release and G(V) and F(V) voltage shifts in pWT BK (zL*GVhalf: ρ = −1.0, p < 0.001; 4zJ*FVhalf: ρ = −0.915, p < 0.001) and in the D362A/D367A mutant (zL*GVhalf: ρ = −0.868, p < 0.001; 4zJ*FVhalf: ρ = −0.787, p = 0.002). This is also true for the D894N/D895N/D896N/D897N/D898N (D894–898N) mutant in the case of zL*ΔGVhalf (ρ = −0.970, p < 0.001), but not in the case of 4zJ*ΔFVhalf (ρ = 0.146, p = 0.705). E–G, normalizing the voltage shifts by dividing by the change in calcium chemical potential eliminated the correlation between ΔFVhalf and ΔGVhalf (pWT: ρ = 0.079, p = 0.811; D362A/D367A: ρ = 0.336, p = 0.296; D894N/D895N/D896N/D897N/D898N: ρ = 0.144, p = 0.705). The rectangles superimposed onto the scatter points indicate the 95% confidence interval for the mean value in both axes. The only case where the normalized voltage shift was not statistically different from zero was for D894N/D895N/D896N/D897N/D898N ΔFVhalf (p = 0.27; otherwise p < 0.001).
These results suggest that RCK1 and RCK2 employ different allosteric pathways to bring about Ca2+-mediated activation. To quantify the mechanistic interpretation of the experimental results, we fit the data with an allosteric statistical-mechanical model.
An Allosteric Model of BK Channel That Accounts for Presence of Two High Affinity Ca2+ Sensors per Subunit
The experimental data suggest that Ca2+ binding to RCK1 and RCK2 high affinity Ca2+ sensors results in different effects on BK activation. To interpret our data in the context of an allosteric scheme, we constructed a statistical-mechanical model inspired by the equilibrium Horrigan and Aldrich model of BK channel activation (6). The Horrigan and Aldrich model describes the allosteric interactions linking voltage sensing, Ca2+ binding, and pore opening in BK channels. We expanded on the Horrigan and Aldrich model to include two Ca2+-sensing domains per subunit (Fig. 5 and supplemental data), each capable of interacting with the Pore (allosteric factors C1 and C2), with the VSD (allosteric factors E1 and E2), and with each other (allosteric factor F). The normalized G(V) and F(V) data obtained at two different [Ca2+] (before and after UV photolysis) from pWT, RCK1 mutant, and RCK2 mutant channels were simultaneously fit to the model (Fig. 6 and Table 1): F(V) data were fit to the model prediction of VSD activation, whereas G(V) data were fit with Po, as previously (19). As a further constraint, we simultaneously fit averaged values of GVhalf as a function of [Ca2+] for the three BK channel clones (supplemental Fig. S1C and Fig. 6D). In fitting the mutant channels, we minimized the number of parameters needed to obtain good fits while constraining the remaining variables to their pWT equivalent. Because it appears that RCK1 retains its high affinity Ca2+ binding properties when D362/D367 are neutralized (25), Kd1 in the RCK1 mutant was constrained to be equal to the corresponding pWT value. Intersubunit interactions between BK regulatory domains were forbidden; previous work has indicated that such interactions do not play a major role (57).
FIGURE 5.

An allosteric model of BK channel activation. The schematic representation of the model includes a pore domain and three regulatory domains: the VSD and two Ca2+-sensing domains (RCK1 and RCK2) per subunit. Intrinsic gating between closed (C) and open (O) states of the pore domain is specified by the voltage-dependent equilibrium constant L; the VSD can be found either in the resting (R) or active (A) state according to equilibrium constant J; Ca2+ sensors can exist in unbound or Ca2+-bound states according to equilibrium constants K1 and K2. D, C1, C2, E1, E2, and F are allosteric factors (see supplemental data).
FIGURE 6.

The allosteric model for BK channel activation fits experimental data from three different BK channel mutants. The experimental GV and FV data for pWT (A), RCK1 mutant D362A/D367A (B), and RCK2 mutant D894N/D895N/D896N/D897N/D898N (D894–898N, C) were fitted simultaneously together with the data points describing the Ca2+ dependence of the GVhalf (D). The continuous lines are the predictions of the BK allosteric model presented in Fig. 5 (experimental data are as in Figs. 2 and 3, and supplemental Fig. S1C). The fitted parameters are listed in Table 1.
TABLE 1.
Fitting parameters for the model predictions shown in Fig. 6
For the mutants only parameters different from pWT are reported.
| Domain or Interaction | Parameters | Ca2+-sensing mutations |
||
|---|---|---|---|---|
| pWT | D362/367A | D894N/D895N/D896N/D897N/D898N | ||
| Pore | zL (e0) | 1.00 | ||
| VL (mV) | 307 | |||
| S4 | zJ (e0) | 0.60 | ||
| VJ (mV) | 20 | |||
| RCK1 | Kd1 (μm) | 10.03 | ||
| RCK2 | Kd2 (μm) | 2.98 | 4820 | |
| S4-Pore | D | 3.34 | ||
| RCK1-Pore | C1 | 2.84 | 1.00 | |
| RCK1-S4 | E1 | 10.00 | ||
| RCK2-Pore | C2 | 10.41 | ||
| RCK2-S4 | E2 | 2.28 | 0.70 | |
| RCK1-RCK2 | F | 0.02 | 0.06 | 11.60 |
The best set of fitting parameters that accounts for the experimental data is reported in Table 1. The valence of the VSD charge is zJ = 0.6 e0, similar to what has been reported for the R207Q BK channel (18). The opening of the channel (C–O transition) is also intrinsically voltage-dependent (zL = 1 e0), in general agreement with previous works (6, 18). During the simultaneous fitting, the intrinsic Ca2+ dissociation constants for RCK1 (Kd1) and RCK2 (Kd2) were constrained to within the range reported by a recent study (55), resulting in Kd1 = 10.03 μm and Kd2 = 2.98 μm for pWT channels (Fig. 6A). We also found that, in the Ca2+-bound state, RCK1 strongly stabilized VSD activation (E1 = 10) and, to a lesser extent, the conducting state (C1 = 2.84). On the other hand, RCK2 efficiently facilitated pore operation (C2 = 10.41) and exerted a positive cooperativity on VSD activation (E2 = 2.28). A negative cooperativity was exhibited between the two Ca2+-sensing domains (F = 0.02), as also suggested by a previous investigation (55).
In the RCK1 mutant channels (Fig. 6B and Table 1), D362A/D367A mutations abolished the RCK1-Pore cooperativity (C1 = 1) and slightly affected the allosteric factor F (from 0.02 to 0.06), leaving unaltered the RCK1-VSD interaction. In agreement with the consolidated knowledge that the neutralization of the calcium bowl (D894N/D895N/D896N/D897N/D898N) impairs high affinity Ca2+ sensing (36) by reducing Ca2+ binding affinity (24, 37, 38, 58), the model predicted a marked increase in Kd2 in the D894N/D895N/D896N/D897N/D898N mutant (Kd2 = 4820 μm) (Fig. 6C and Table 1). The allosteric interaction RCK2-Pore (C2) did not vary compared with the pWT channel. Instead, a negative cooperativity was found between RCK2 and VSD (E2 = 0.7). In addition, RCK1-RCK2 interaction becomes stronger (F = 11.6).
DISCUSSION
BK channels combine properties of both voltage- and ligand-gated ion channels, a feature that allows them to integrate information on membrane potential and [Ca2+]i to control key physiological processes such as the smooth muscle tone, cochlear hair cell tuning, neuronal excitability, neurotransmitter release, insulin secretion, and oxygen sensing (10, 11, 59–63). The interaction between voltage and Ca2+ sensing mechanisms was proposed for native BK channels reconstituted in lipid bilayer (52) and further investigated in cloned BK channels, demonstrating the allosteric nature of the dual activation mechanism (6, 9, 12, 55, 64). In this work, the interplay between transmembrane VSD and intracellular gating ring with the BK pore was investigated using voltage-clamp fluorometry combined with UV flash photolysis of DM-Nitrophen to elicit a fast, reversible, and reproducible increase in intracellular free [Ca2+] (Fig. 2 and supplemental Figs. S2 and S3). In the human BK channel (Slo1), we provided experimental evidence that the rapid increase of [Ca2+]i induces a conformational change that, originating in the gating ring, propagates to the VSD, facilitating its activation (Figs. 2A and 4). Indeed, the purified BK gating ring in solution can undergo Ca2+-driven structural rearrangements, resulting in an overall decrease of its hydrodynamic radius (39). Likely, both RCK1 and RCK2 contribute to the Ca2+-induced rearrangements of the gating ring, based on the sensor/transducer properties of isolated RCK1 and RCK2 homomers (24, 25).
Moreover, a physical proximity of VSD and gating ring is suggested by their mutual coordination of Mg2+ (10, 11, 35, 65). Atomic structures of the BK gating ring (27–29), have pinpointed the locations of the calcium bowl (in RCK2) and Asp-362/Asp-367 residues (in RCK1) that, in the whole channel, could face the intracellular portion of the VSD (Fig. 1).
Using a statistical-mechanical model inspired by the Horrigan and Aldrich model (6) but accounting for two distinct Ca2+ sensors (Fig. 5), we quantified the allosteric interactions between pore, VSD, and the two RCK domains. In pWT channels, the model predicted salient differences in the allosteric coupling between RCK1 and RCK2 with the VSD and the pore (Table 1), suggesting that they are functionally separable, despite their structural homology. RCK1 and RCK2 are allosterically coupled by negative cooperativity, such that the activation of one domain disfavors the activation of the other, recapitulating the findings of Sweet and Cox (55). The functional impairment of Ca2+ sensing either in RCK1 or in RCK2 highlighted their functional diversity. In D362A/D367A mutants, a rapid increase in [Ca2+]i significantly facilitated pore and VSD activation (Figs. 3A and 4), as indicated by the consistent leftward shifts of both G(V) and F(V) curves at the highest [Ca2+]i (Figs. 3B and 4). This effect was likely mediated by intact calcium bowls in RCK2 domains. The effect of the D362A/D367A is largely accounted for by the loss of cooperativity between RCK1 and the pore (allosteric factor C1) (Table 1 and Fig. 6B). It is interesting that the model can account for the effects of the D362A/D367A mutation without reducing the intrinsic Ca2+ affinity of the RCK1 domain (Kd1). This prediction would be in agreement with the lack of direct experimental evidence that Asp-362/Asp-367 residues are essential for Ca2+ coordination. However, a potential Ca2+-binding site involving Asp-367 and other residues has been proposed based on electrophysiological evidence (30, 34, 55) and inference from the gating ring crystal structure (28, 32). Interestingly, the neutralization of calcium bowl abolished the Ca2+-induced facilitation of the VSD activation; indeed, the F(V) curves were not significantly modified by the Ca2+ release (Figs. 3D and 4). To account for these experimental data, three model parameters were considerably altered compared with the corresponding pWT values (Table 1), suggesting that the neutralization of the five Asp residues not only abolished a high affinity Ca2+-binding site (calcium bowl) but also altered the energetics of gating ring operation, likely because of a structural perturbation.
Recent work has pointed out an important functional difference between RCK1 and RCK2: Ca2+ binding to the RCK1 domain (but not to RCK2) is voltage-dependent (55). To test the validity of our model prediction, we computed the steady-state occupancy of the Ca2+-bound state for RCK1 and RCK2. We found that, for a wide range of membrane potentials, the occupancy of RCK1 Ca2+-bound state was voltage-dependent, whereas RCK2 Ca2+ binding was weakly affected by membrane potential (supplemental Fig. S4). Thus, despite their structural homology, RCK1 and RCK2 domains provide different contribution to the Ca2+-dependent activation of BK channels.
The importance of achieving a deeper understanding of the BK channel biophysical properties resides in its involvement in several diseases, such as hypertension, epilepsy, schizophrenia, and diabetes (11, 62, 63, 66). In particular, elucidating the mechanisms underlying the functional coupling between VSD and the two distinct Ca2+ sensors is fundamental for the physiology and pathophysiology of excitable cells. For instance, the Slo1 epilepsy/dyskinesia-causing mutation affects Ca2+-dependent activation originating from the Ca2+-binding site in RCK1, but not in RCK2, by altering the coupling mechanism between Ca2+ binding and gate opening (56).
In summary, we have directly probed the allosteric nature of BK channel Ca2+-dependent activation by optically tracking VSD movements perturbed by Ca2+ binding to the gating ring, under voltage clamp. By investigating channels impaired in high affinity Ca2+-sensing sites, we revealed that the two BK Ca2+ sensors are not functionally equivalent: although both contribute to Ca2+-dependent channel activation, the efficiency of the allosteric interactions appears different. A functional calcium bowl (RCK2) is required to observe the propagation of Ca2+-induced “wave” of rearrangements from the cytoplasmic portion to the transmembrane VSD. The results were fit with a statistical-mechanical allosteric model to quantify the cooperative interactions among the prominent BK regulatory domains. Although this approach was successful in dissecting the allosteric contributions of high affinity Ca2+-sensing domains to BK channel activation, it did not explore the role of low affinity Ca2+-sensing sites (30, 33–35), because the experimental [Ca2+] range was limited to prevent their significant engagement. Nevertheless, it would be of great interest to further investigate their allosteric contribution to BK channel operation to better understand the complex mechanism of BK channel activation.
Acknowledgment
The Slo1 clone was a kind gift from Ligia Toro.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM082289 (to R. O.). This work was also supported by funds from the Laubisch Foundation (to R. O.) and American Heart Association Postdoctoral Fellowship (Western States Affiliate) 11POST7140046 (to A. P.).

This article contains supplemental data and Figs. S1–S4.
- VSD
- voltage-sensing domain
- pWT
- pseudo-WT
- TMRM
- tetramethylrhodamine-5′-maleimide.
REFERENCES
- 1. del Sol A., Tsai C. J., Ma B., Nussinov R. (2009) The origin of allosteric functional modulation. Multiple pre-existing pathways. Structure 17, 1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goodey N. M., Benkovic S. J. (2008) Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 4, 474–482 [DOI] [PubMed] [Google Scholar]
- 3. Piper D. R., Rupp J., Sachse F. B., Sanguinetti M. C., Tristani-Firouzi M. (2008) Cooperative interactions between R531 and acidic residues in the voltage sensing module of hERG1 channels. Cell Physiol. Biochem. 21, 37–46 [DOI] [PubMed] [Google Scholar]
- 4. Zandany N., Ovadia M., Orr I., Yifrach O. (2008) Direct analysis of cooperativity in multisubunit allosteric proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 11697–11702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magleby K. L. (2003) Gating mechanism of BK (Slo1) channels. So near, yet so far. J. Gen. Physiol. 121, 81–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horrigan F. T., Aldrich R. W. (2002) Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120, 267–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taraska J. W., Zagotta W. N. (2007) Structural dynamics in the gating ring of cyclic nucleotide-gated ion channels. Nat. Struct. Mol. Biol. 14, 854–860 [DOI] [PubMed] [Google Scholar]
- 8. Altomare C., Bucchi A., Camatini E., Baruscotti M., Viscomi C., Moroni A., DiFrancesco D. (2001) Integrated allosteric model of voltage gating of HCN channels. J. Gen. Physiol. 117, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Latorre R., Morera F. J., Zaelzer C. (2010) Allosteric interactions and the modular nature of the voltage- and Ca2+-activated (BK) channel. J. Physiol. 588, 3141–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou S., Heinemann S. H., Hoshi T. (2009) Modulation of BKCa channel gating by endogenous signaling molecules. Physiology 24, 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee U. S., Cui J. (2010) BK channel activation. Structural and functional insights. Trends Neurosci. 33, 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rothberg B. S., Magleby K. L. (2000) Voltage and Ca2+ activation of single large-conductance Ca2+-activated K+ channels described by a two-tiered allosteric gating mechanism. J. Gen. Physiol. 116, 75–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen K. Z., Lagrutta A., Davies N. W., Standen N. B., Adelman J. P., North R. A. (1994) Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes. Evidence for tetrameric channel formation. Pflugers Arch. 426, 440–445 [DOI] [PubMed] [Google Scholar]
- 14. Wallner M., Meera P., Toro L. (1996) Determinant for β-subunit regulation in high-conductance voltage-activated and Ca2+-sensitive K+ channels. An additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. U.S.A. 93, 14922–14927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Armstrong C. M. (2003) Voltage-gated K channels. Sci. STKE. 2003, re10. [DOI] [PubMed] [Google Scholar]
- 16. Stefani E., Ottolia M., Noceti F., Olcese R., Wallner M., Latorre R., Toro L. (1997) Voltage-controlled gating in a large conductance Ca2+-sensitive K+channel (hslo). Proc. Natl. Acad. Sci. U.S.A. 94, 5427–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Díaz L., Meera P., Amigo J., Stefani E., Alvarez O., Toro L., Latorre R. (1998) Role of the S4 segment in a voltage-dependent calcium-sensitive potassium (hSlo) channel. J. Biol. Chem. 273, 32430–32436 [DOI] [PubMed] [Google Scholar]
- 18. Ma Z., Lou X. J., Horrigan F. T. (2006) Role of charged residues in the S1–S4 voltage sensor of BK channels. J. Gen. Physiol. 127, 309–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pantazis A., Gudzenko V., Savalli N., Sigg D., Olcese R. (2010) Operation of the voltage sensor of a human voltage- and Ca2+-activated K+ channel. Proc. Natl. Acad. Sci. U.S.A. 107, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Savalli N., Kondratiev A., Toro L., Olcese R. (2006) Voltage-dependent conformational changes in human Ca2+- and voltage-activated K+ channel, revealed by voltage-clamp fluorometry. Proc. Natl. Acad. Sci. U.S.A. 103, 12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu R., Alioua A., Kumar Y., Eghbali M., Stefani E., Toro L. (2006) MaxiK channel partners. Physiological impact. J. Physiol. 570, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang Y., Pico A., Cadene M., Chait B. T., MacKinnon R. (2001) Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron 29, 593–601 [DOI] [PubMed] [Google Scholar]
- 23. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–522 [DOI] [PubMed] [Google Scholar]
- 24. Yusifov T., Savalli N., Gandhi C. S., Ottolia M., Olcese R. (2008) The RCK2 domain of the human BKCa channel is a calcium sensor. Proc. Natl. Acad. Sci. U.S.A. 105, 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yusifov T., Javaherian A. D., Pantazis A., Gandhi C. S., Olcese R. (2010) The RCK1 domain of the human BKCa channel transduces Ca2+ binding into structural rearrangements. J. Gen. Physiol. 136, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roosild T. P., Lê K. T., Choe S. (2004) Cytoplasmic gatekeepers of K+-channel flux. A structural perspective. Trends Biochem. Sci. 29, 39–45 [DOI] [PubMed] [Google Scholar]
- 27. Yuan P., Leonetti M. D., Pico A. R., Hsiung Y., MacKinnon R. (2010) Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science 329, 182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y., Yang Y., Ye S., Jiang Y. (2010) Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature 466, 393–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan P., Leonetti M. D., Hsiung Y., MacKinnon R. (2012) Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 481, 94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia X. M., Zeng X., Lingle C. J. (2002) Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature 418, 880–884 [DOI] [PubMed] [Google Scholar]
- 31. Bao L., Rapin A. M., Holmstrand E. C., Cox D. H. (2002) Elimination of the BK(Ca) channel's high-affinity Ca2+ sensitivity. J. Gen. Physiol. 120, 173–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang G., Huang S. Y., Yang J., Shi J., Yang X., Moller A., Zou X., Cui J. (2010) Ion sensing in the RCK1 domain of BK channels. Proc. Natl. Acad. Sci. U.S.A. 107, 18700–18705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi J., Krishnamoorthy G., Yang Y., Hu L., Chaturvedi N., Harilal D., Qin J., Cui J. (2002) Mechanism of magnesium activation of calcium-activated potassium channels. Nature 418, 876–880 [DOI] [PubMed] [Google Scholar]
- 34. Zeng X. H., Xia X. M., Lingle C. J. (2005) Divalent cation sensitivity of BK channel activation supports the existence of three distinct binding sites. J. Gen. Physiol. 125, 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H., Shi J., Zhang G., Yang J., Delaloye K., Cui J. (2008) Activation of Slo1 BK channels by Mg2+ coordinated between the voltage sensor and RCK1 domains. Nat. Struct. Mol. Biol. 15, 1152–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schreiber M., Salkoff L. (1997) A novel calcium-sensing domain in the BK channel. Biophys. J. 73, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bian S., Favre I., Moczydlowski E. (2001) Ca2+-binding activity of a COOH-terminal fragment of the Drosophila BK channel involved in Ca2+-dependent activation. Proc. Natl. Acad. Sci. U.S.A. 98, 4776–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bao L., Kaldany C., Holmstrand E. C., Cox D. H. (2004) Mapping the BKCa channel's “Ca2+ bowl.” Side-chains essential for Ca2+ sensing. J. Gen. Physiol. 123, 475–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Javaherian A. D., Yusifov T., Pantazis A., Franklin S., Gandhi C. S., Olcese R. (2011) Metal-driven operation of the human large-conductance voltage- and Ca2+-dependent potassium channel (BK) gating ring apparatus. J. Biol. Chem. 286, 20701–20709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mannuzzu L. M., Moronne M. M., Isacoff E. Y. (1996) Direct physical measure of conformational rearrangement underlying potassium channel gating. Science 271, 213–216 [DOI] [PubMed] [Google Scholar]
- 41. Cha A., Bezanilla F. (1997) Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron 19, 1127–1140 [DOI] [PubMed] [Google Scholar]
- 42. Gandhi C. S., Olcese R. (2008) The voltage-clamp fluorometry technique. In Methods in Molecular Biology, Potassium Channels (Lippiat J. D., ed) Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 43. Escobar A. L., Velez P., Kim A. M., Cifuentes F., Fill M., Vergara J. L. (1997) Kinetic properties of DM-nitrophen and calcium indicators. Rapid transient response to flash photolysis. Pflugers Arch. 434, 615–631 [DOI] [PubMed] [Google Scholar]
- 44. Ellis-Davies G. C. (2003) Development and application of caged calcium. Methods Enzymol. 360, 226–238 [DOI] [PubMed] [Google Scholar]
- 45. Faas G. C., Karacs K., Vergara J. L., Mody I. (2005) Kinetic properties of DM-nitrophen binding to calcium and magnesium. Biophys. J. 88, 4421–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ellis-Davies G. C. (2007) Caged compounds. Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 4, 619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wallner M., Meera P., Ottolia M., Kaczorowski G. J., Latorre R., Garcia M. L., Stefani E., Toro L. (1995) Characterization of and modulation by a β-subunit of a human maxi KCa channel cloned from myometrium. Receptors Channels 3, 185–199 [PubMed] [Google Scholar]
- 48. Savalli N., Kondratiev A., de Quintana S. B., Toro L., Olcese R. (2007) Modes of operation of the BKCa channel β2 subunit. J. Gen. Physiol. 130, 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haug T., Sigg D., Ciani S., Toro L., Stefani E., Olcese R. (2004) Regulation of K+ flow by a ring of negative charges in the outer pore of BKCa channels. Part I: Aspartate 292 modulates K+ conduction by external surface charge effect. J. Gen. Physiol. 124, 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stefani E., Bezanilla F. (1998) Cut-open oocyte voltage-clamp technique. Methods Enzymol. 293, 300–318 [DOI] [PubMed] [Google Scholar]
- 51. Pantazis A., Kohanteb A. P., Olcese R. (2010) Relative motion of transmembrane segments S0 and S4 during voltage sensor activation in the human BK(Ca) channel. J. Gen. Physiol. 136, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Moczydlowski E., Latorre R. (1983) Gating kinetics of Ca2+-activated K+ channels from rat muscle incorporated into planar lipid bilayers. Evidence for two voltage-dependent Ca2+ binding reactions. J. Gen. Physiol. 82, 511–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun X. P., Yazejian B., Grinnell A. D. (2004) Electrophysiological properties of BK channels in Xenopus motor nerve terminals. J. Physiol. 557, 207–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sy T., Grinnell A. D., Peskoff A., Yazejian B. (2010) Monitoring transient Ca2+ dynamics with large-conductance Ca2+-dependent K+ channels at active zones in frog saccular hair cells. Neuroscience 165, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sweet T. B., Cox D. H. (2008) Measurements of the BKCa channel's high-affinity Ca2+ binding constants. Effects of membrane voltage. J. Gen. Physiol. 132, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang J., Krishnamoorthy G., Saxena A., Zhang G., Shi J., Yang H., Delaloye K., Sept D., Cui J. (2010) An epilepsy/dyskinesia-associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron 66, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Niu X., Magleby K. L. (2002) Stepwise contribution of each subunit to the cooperative activation of BK channels by Ca2+. Proc. Natl. Acad. Sci. U.S.A. 99, 11441–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sheng J. Z., Weljie A., Sy L., Ling S., Vogel H. J., Braun A. P. (2005) Homology modeling identifies C-terminal residues that contribute to the Ca2+ sensitivity of a BKCa channel. Biophys. J. 89, 3079–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fettiplace R., Fuchs P. A. (1999) Mechanisms of hair cell tuning. Annu. Rev. Physiol. 61, 809–834 [DOI] [PubMed] [Google Scholar]
- 60. Hu H., Shao L. R., Chavoshy S., Gu N., Trieb M., Behrens R., Laake P., Pongs O., Knaus H. G., Ottersen O. P., Storm J. F. (2001) Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J. Neurosci. 21, 9585–9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Salkoff L., Butler A., Ferreira G., Santi C., Wei A. (2006) High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931 [DOI] [PubMed] [Google Scholar]
- 62. Braun M., Ramracheya R., Bengtsson M., Zhang Q., Karanauskaite J., Partridge C., Johnson P. R., Rorsman P. (2008) Voltage-gated ion channels in human pancreatic β-cells. Electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628 [DOI] [PubMed] [Google Scholar]
- 63. Wu R. S., Marx S. O. (2010) The BK potassium channel in the vascular smooth muscle and kidney. α- and β-subunits. Kidney Int. 78, 963–974 [DOI] [PubMed] [Google Scholar]
- 64. Cui J., Yang H., Lee U. S. (2009) Molecular mechanisms of BK channel activation. Cell Mol. Life Sci. 66, 852–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Horrigan F. T., Ma Z. (2008) Mg2+ enhances voltage sensor/gate coupling in BK channels. J. Gen. Physiol. 131, 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Du W., Bautista J. F., Yang H., Diez-Sampedro A., You S. A., Wang L., Kotagal P., Lüders H. O., Shi J., Cui J., Richerson G. B., Wang Q. K. (2005) Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738 [DOI] [PubMed] [Google Scholar]
- 67. Long S. B., Tao X., Campbell E. B., MacKinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382 [DOI] [PubMed] [Google Scholar]
- 68. Liu G., Niu X., Wu R. S., Chudasama N., Yao Y., Jin X., Weinberg R., Zakharov S. I., Motoike H., Marx S. O., Karlin A. (2010) Location of modulatory beta subunits in BK potassium channels. J. Gen. Physiol. 135, 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]