

Abstract
Ligand enrichment among top-ranking hits is a key metric of molecular docking. To avoid bias, decoys should resemble ligands physically, so that enrichment is not simply a separation of gross features, yet be chemically distinct from them, so that they are unlikely to be binders. We have assembled a directory of useful decoys (DUD), with 2950 ligands for 40 different targets. Every ligand has 36 decoy molecules that are physically similar but topologically distinct, leading to a database of 98,266 compounds. For most targets, enrichment was at least half a log better with uncorrected databases such as the MDDR than with DUD, evidence of bias in the former. These calculations also allowed forty-by-forty cross docking, where the enrichments of each ligand set could be compared for all 40 targets, enabling a specificity metric for the docking screens. DUD is freely available online as a benchmarking set for docking at http://blaster.docking.org/dud/.
Keywords: virtual screening, molecular docking, docking decoy, automation, enrichment, binding pose
Introduction
Although molecular docking screens of chemical databases are widely used for ligand discovery,1–7 the method retains important weaknesses.8–13 A testament to these is the criterion by which docking screens are evaluated: the enrichment of annotated ligands from among a large database of presumed non-binding, “decoy” molecules. In these retrospective calculations, the enrichment factor is the concentration of the annotated ligands among the top-scoring docking hits compared to their concentration throughout the entire database. Other possible metrics, such as the magnitude of the docking energies, or even monotonic rank order among the ligands, are only used occasionally and in restricted sets; in the general case they remain unreliable because of the many approximations used in docking. Thus the success of a docking screen is evaluated by its capacity to enrich the small number of known active compounds in the top ranks of a screen from among a much greater number of decoy molecules in the database.14–23
The relationship of the decoy molecules to the ligands is critical in assessing enrichment factors in docking screens. Docking scoring functions can depend on molecular size.24–26 For instance, Verdonk and colleagues27 have observed that if there are significant differences in size distribution between ligands and decoys, docking enrichments can appear to be artificially good, and the same is undoubtedly true for other physical features. The database decoys should thus resemble the physical properties of the annotated ligands well enough so that enrichment is not simply a separation of trivial physical features. The decoys nevertheless should be chemically distinct from the ligands so that they are likely to be, in fact, non-binders.
Investigators have assembled sets of ligands and presumed decoys for numerous targets and used them to evaluate docking performance based on enrichment. Rognan and colleagues made an important contribution towards this end with the introduction of a set of 990 randomly chosen molecules combined with 10 thymidine kinase (TK) and 10 estrogen receptor (ER) antagonists, which were subsequently used in several studies.15,19,20,23,28–30 More recently, Jain and colleagues introduced a set of 1,000 random drug-like compounds to complement the Rognan set, and combined those with 252 ligands from 27 protein targets to evaluate docking enrichment factors.23 Several other groups, including ourselves, have used the 100,000 molecule MDL Drug Data Report database (MDDR, Elsevier MDL, San Leandro CA) as a source of both ligands and decoys.31–34 Each of these approaches has drawbacks. None of these sets of molecules has been adjusted so that the physical properties of the ligands are matched by those of the decoys. Indeed, in both the Rognan set and its derivatives the annotated ligands and the presumed decoys differ greatly in their physical properties, making enrichment factors calculated with these sets open to bias (see Results). Whereas there is less room for bias in the MDDR database, and statistical variance is less likely here than in the smaller decoy sets, differences between ligand and decoy sets lead to significant enrichment-factor bias (see Results). The MDDR has the further disadvantage of being a non-public access database, which was an advantage of the Rognan and Jain sets.
We were interested in developing large benchmarking sets to evaluate docking screening calculations. We wanted these sets to cover a large number of proteins so as to offer a reliable view of how docking might perform on typical and interesting targets. We also wanted these sets to be publicly available, so that docking programs could be compared broadly in “apples to apples” comparisons. We wanted the database to be large enough to have decoys and ligands physically matched so as to be as free from physical and statistical features as possible. We began with 2,950 ligands for 40 different target proteins taken from the literature; each set of ligands for each protein had tens to hundreds of molecules in it. For each ligand in each set, 36 molecules were chosen from the “drug-like” subset of the ZINC database of commercially available compounds.35 Each of these 36 resembled the particular ligand in physical properties, such as molecular weight, cLogP, and number of hydrogen bonding groups, but differed from the ligand topologically. This resulted in a database of 95,316 decoy molecules whose physical properties closely matched those of the 2,950 ligands that they were chosen to counterpoint. This “directory of useful decoys” (DUD) was docked against the 40 protein targets, using an automated docking engine that required little or no user guidance.
Here we report on the enrichments resulting from this large, bias-corrected database and compare these to those from both small, uncorrected decoy sets15,23 and to the large MDDR database. Our results suggest that DUD provides a more stringent test with which to evaluate virtual screening performance. Even using the MDDR as decoys has enrichment factors half a log better than those using DUD with exactly the same docking procedure, speaking to enrichment factor biases even in large, uncorrected databases. These calculations also allowed for a forty-by-forty cross docking, where the enrichments of each ligand set were compared for all forty targets using the same background decoys. These cross-docking results suggest a specificity metric to evaluate docking screens. The usefulness of these targets, ligands and decoys as community benchmarking sets for docking will be considered here, as will the prospects for the automated docking pipeline that was deployed in these studies. The benchmarking sets, including protein structures, docking energy grids and input files, and the full DUD database are available at blaster.docking.org/dud/.
Method
Protein Target Selection and Ligand Collection
Forty protein targets were selected based on the availability of annotated ligands, crystal structures and, often, previous docking studies. We used the structure used in previous docking studies if one was available, and otherwise we used the best complex available as judged by resolution and the absence of errors. All of these proteins have ligand-bound x-ray crystal structures available in the Protein Data Bank (PDB)36, with the exception of the PDGFrb and VEGFr2 kinases. We organized these targets into six classes: nuclear hormone receptors, kinases, serine proteases, metallo-enzymes, folate enzymes, and other enzymes (Table 1). The number of ligands varies from 12 (~ 0.01% of database) to 416 (~ 0.4% of database). A total of 2,950 ligands were included overall.
Table 1.
Enrichments of the annotated ligands using the decoys in DUD for forty targets by docking. Six representative targets (in bold) are discussed in more details in the text.
| Protein category |
Protein | PDB code |
Resol- ution (Å) |
Num. of ligands |
Num. of decoys |
EFmax | EF1 | EF20 |
|---|---|---|---|---|---|---|---|---|
| Nuclear hormone receptors |
AR | 1xq2 | 1.9 | 74a,b | 2,630 | 60.2 | 33.5 | 3.8 |
| ERagonist | 1l2i | 1.9 | 67a–c | 2,361 | 29.6 | 19.2 | 4.5 | |
| ERantagonist | 3ert | 1.9 | 39a–d | 1,399 | 101.6 | 12.7 | 1.3 | |
| GR | 1m2z | 2.5 | 78a | 2,804 | 31.7 | 8.9 | 1.4 | |
| MR | 2aa2 | 1.9 | 15a | 535 | 330.0 | 46.2 | 3.7 | |
| PPARg | 1fm9 | 2.1 | 81a | 2,910 | 1.0 | 0.0 | 0.0 | |
| PR | 1sr7 | 1.9 | 27a | 967 | 2.9 | 0.0 | 2.0 | |
| RXRa | 1mvc | 1.9 | 20a | 708 | 148.5 | 24.8 | 2.2 | |
| Kinases | CDK2 | 1ckp | 2.1 | 50e,f | 1,780 | 19.8 | 13.9 | 1.4 |
| EGFr | 1m17 | 2.6 | 416g | 14,914 | 3.8 | 2.1 | 2.4 | |
| FGFr1 | 1agw | 2.4 | 118g | 4,216 | 1.0 | 0.0 | 0.2 | |
| HSP90 | 1uy6 | 1.9 | 24h | 861 | 10.8 | 8.6 | 2.0 | |
| P38 MAP | 1kv2 | 2.8 | 234g | 8,399 | 4.1 | 2.1 | 2.4 | |
| PDGFrb | Model | n/a | 157g | 5,625 | 1.2 | 0.0 | 0.6 | |
| SRC | 2src | 1.5 | 162g | 5,801 | 3.1 | 1.2 | 1.5 | |
| TK | 1kim | 2.1 | 22a,d,i | 7,85 | 63.0 | 54.0 | 5.0 | |
| VEGFr2 | 1vr2 | 2.4 | 74j | 2,647 | 2.2 | 1.3 | 1.4 | |
| Serine Proteases |
Fxa | 1f0r | 2.7 | 142e,f,k | 5,102 | 34.9 | 14.6 | 3.8 |
| Thrombin | 1ba8 | 1.8 | 65e,l,m | 2,294 | 18.3 | 13.7 | 2.9 | |
| Trypsin | 1bju | 1.8 | 43e,l | 1,545 | 22.5 | 22.5 | 2.6 | |
| Metallo- enzymes |
ACE | 1o86 | 2.0 | 49a,m | 1,728 | 141.4 | 40.4 | 3.7 |
| ADA | 1ndw | 2.0 | 23a,e | 822 | 21.5 | 12.9 | 2.4 | |
| COMT | 1h1d | 2.0 | 12a | 430 | 11.8 | 0.0 | 3.3 | |
| PDE5 | 1xp0 | 1.8 | 51f | 1,810 | 29.1 | 11.8 | 2.3 | |
| Folate enzymes |
DHFR | 3dfr | 1.7 | 201m | 7,150 | 28.7 | 21.7 | 3.5 |
| GART | 1c2t | 2.1 | 21n | 753 | 70.7 | 42.4 | 3.3 | |
| Other enzymes |
AChE | 1eve | 2.5 | 105a,e,m | 3,732 | 3.1 | 1.9 | 2.0 |
| ALR2 | 1ah3 | 2.3 | 26o | 920 | 76.2 | 38.1 | 2.3 | |
| AmpC | 1xgj | 2.0 | 21p | 734 | 23.6 | 17.1 | 4.7 | |
| COX-1 | 1p4g | 2.1 | 25i | 850 | 9.9 | 4.0 | 1.6 | |
| COX-2 | 1cx2 | 3.0 | 349c,f,m | 12,491 | 29.1 | 20.1 | 3.3 | |
| GPB | 1a8i | 1.8 | 52e,m | 1,851 | 28.6 | 22.8 | 4.1 | |
| HIVPR | 1hpx | 2.0 | 53a,e | 1,888 | 9.3 | 3.7 | 2.2 | |
| HIVRT | 1rt1 | 2.6 | 40q | 1,439 | 49.5 | 5.0 | 3.0 | |
| HMGR | 1hw8 | 2.1 | 35a,i | 1,242 | 198.0 | 33.9 | 2.1 | |
| InhA | 1p44 | 2.7 | 85r | 3,043 | 1.0 | 0.0 | 0.3 | |
| NA | 1a4g | 2.2 | 49c,e,i | 1,745 | 60.6 | 20.2 | 3.3 | |
| PARP | 1efy | 2.2 | 33s | 1,178 | 6.3 | 6.0 | 3.6 | |
| PNP | 1b8o | 1.5 | 25e,t | 884 | 158.4 | 31.7 | 4.4 | |
| SAHH | 1a7a | 2.8 | 33i | 1,159 | 120.0 | 78.0 | 5.0 | |
Annotated ligands were collected from:
KiBank67,
NCTR data set68,
Stahl data set16,
from Ref.15,
PDBbind database69,
Jorissen/Gilson data set70,
Kinase inhibitor data set32,
PubChem (http://pubchem.ncbi.nlm.nih.gov),
Jacobsson test set38,
Bohm Serine Protease inhibitor data set76,
Sutherland QSAR test set77,
Ref.78,
Ref.79,
Ref.82,
contributed by Dr. Xin He (UCSF, personal communication),
Ref.83 and
Ref.84.
Abbreviations: AR, androgen receptor; ER, estrogen receptor; GR, glucocorticoid receptor; MR, mineralocorticoid receptor; PPARg, peroxisome proliferator activated receptor gamma; PR, progesterone receptor; RXRa, retinoic X receptor alpha; CDK2, cyclin-dependent kinase 2; EGFr, epidermal growth factor receptor; FGFr1, fibroblast growth factor receptor kinase; HSP90, human heat shock protein 90; P38 MAP, P38 mitogen activated protein; PDGFrb, the platelet derived growth factor receptor kinase; SRC, tyrosine kinase SRC; TK, thymidine kinase; VEGFr2, vascular endothelial growth factor receptor; FXa, factor Xa; ACE, angiotensin-converting enzyme; ADA, Adenosine deaminase; COMT, catechol O-methyltransferase; PDE5, Phosphodiesterase 5; DHFR, dihydrofolate reductase; GART, glycinamide ribonucleotide transformylase; AChE, acetylcholinesterase; ALR2, aldose reductase; AmpC, AmpC beta-lactamase; COX-1, cyclooxygenase-1; COX-2, cyclooxygenase-2; GPB, glycogen phosphorylase beta; HIVPR, HIV protease; HIVRT, HIV reverse transcriptase; HMGR, hydroxymethylglutaryl-CoA reductase; InhA, enoyl ACP reductase; NA, Neuraminidase; PARP, poly(ADP-ribose) polymerase; PNP, purine nucleoside phosphorylase; SAHH, S-adenosyl-homocysteine hydrolase.
Most of these targets have been studied previously by experimental methods and computational approaches. Among them, estrogen receptor (ER)15,20,23,28,29,37–39 and thymidine kinase (TK)15,19,20,23,28,30,40 have been extensively used to benchmark and evaluate different docking methods and scoring functions. Enrichment studies were also published on several of the other systems, including CDK220,41, P38 MAP kinase18,20,32,39, thrombin20,31,39,41, factor Xa38,42, HIV protease18,20,40, DHFR31,40, neuraminidase39, aldose reductase31,43, HIV-RT20,30, AChE31,38, and COX-220,39,44. Where possible, we chose to use the crystal structure
DUD Generation
DUD was created as follows (Figure 1). The 2,950 annotated ligands were seeded among 3.5 million Lipinski-compliant molecules from the ZINC database of commercially available compounds (version 6, December 2005)35. Chiral annotated ligands were prepared in the correct stereochemical form if known. Feature key fingerprints were calculated using the default type 2 substructure keys of CACTVS45 and the fingerprint-based similarity analysis was performed with the program SUBSET46. Substructure keys are bit strings where 1 represents the presence of a particular functional group. Compounds with Tanimoto coefficient (Tc) less than 0.9 to any annotated ligand were selected, excluding chirality duplicates (we note that a Tc less than 0.9 for CACTVS type 2 fingerprints roughly corresponds to a Tc less than about 0.7 for the widely-used Daylight fingerprints, see Results). This reduced the ZINC compounds to 1.5 million molecules topologically dissimilar to the ligands. The program QikProp (Schrodinger, L.L.C., New York) was used to calculate 32 physical properties of all the annotated ligands and selected ZINC compounds from the previous step, and QikSim (Schrodinger, L.L.C., New York NY) was applied to prioritize ZINC compounds possessing similar properties to any of the ligands. A weight of 4 was used to emphasize the drug-like descriptors (molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds and LogP), and a weight of 1 was used for the number of important functional groups (amine, amide, amidine and carboxylic acid), and the rest of the descriptors were ignored (weight 0) during the similarity analysis procedure. Thirty-six decoy compounds were selected for each ligand, leading to a total of 95,316 decoys that were physically similar but topologically dissimilar to the 2,950 annotated ligands. The total number of decoys is less than 36 times the number of annotated ligands because some ligands had the same decoys.
Figure 1.

The schematic description of the procedure to generate DUD. Molecular weight (MW), number of hydrogen bond acceptors (HBacc), number of hydrogen bond donors (HBdon), number of rotatable bonds (RB). * We note that a Tc less than 0.9 for CACTVS type 2 fingerprints roughly corresponds to a Tc less than about 0.7 for the Daylight fingerprints.
Dockable Database Preparation
Molecules were prepared for docking using the latest version of the ZINC protocol35. Briefly, molecules were converted from 2D SDF to isomeric SMILES using OEChem (OpenEye Scientific Software, Santa Fe NM). An initial 3D structure was generated with Corina (Molecular Networks GmbH, Germany). A protonated form of each molecule at pH 7.0 was calculated with LigPrep (Schrodinger, L.L.C., New York NY) with additional protonated and tautomeric forms calculated in the range of pH 5.75 to 8.25 using modified versions of LigPrep’s parameter files. For each protonated form, we again used Corina to obtain a 3D model and then used AMSOL to calculate partial atomic charges and atomic desolvation energies.47 We used Omega (OpenEye Scientific Software, Santa Fe NM) to enumerate accessible conformations; ring conformations calculated by Corina were preserved. AMSOL and Omega results were combined into a single “flexibase” format file using our program Mol2db.33 All new parameter files used in this process (rules.txt, tautomer_list, ionizer.ini, and torlib.txt) are available in the Supplementary Material.
Overall Virtual Screening Strategy
To undertake docking screens against 40 targets, it was important to automate our procedures as much as possible (Supplementary Material, Figure S1). Most of the labor-intensive steps formerly performed manually have been automated, including most of the binding site preparation, sphere or “hot spot” generation, scoring grid calculation, docking calculation and data analysis. For simplicity, our automated procedure removes all water molecules, including structural waters, by default. We describe docking results achieved with or without expert intervention as “semi-automated” or “automated”, respectively. For both semi-automated and automated procedures, the only input requirements were a protein structure file and a specification of the protein-binding site. Expert intervention during protein structure preparation or binding site identification significantly improved the docking results for 13 out of the 40 targets (see Results). The fully automated procedure was used for all 40 targets, the semi-automated procedure being attempted only when docking enrichment from the fully automated procedure was poor.
Manual Preparation
For some targets, protein structure preparation involves steps that are challenging to automate, such as differentiating cofactors from ligands, parameterizing those cofactors, perceiving structural water molecules, identifying and parameterizing metal ions involved in ligand binding, correctly assigning the protonation state on binding site residues (e.g. histidines and cysteines), and selecting among disordered residues.
Parameterizing the cofactor is challenging to automate. Cofactors were present for the following targets: TK, SO4; DHFR, NADPH; GART, β-GAR; ALR2 and InhA, NADP+; GPB, PLP; PNP, PO4; and SAHH, NAD+. In these cases, we treated the cofactors as part of the target, manually preparing their parameters for the van der Waals (vdW) and electrostatic energy calculations. Once a parameter file has been prepared for a cofactor, the scripts can recognize it based on its PDB residue name, and becomes part of the automated procedure in future runs.
For control calculations, the crystallographic ligand was also prepared manually for docking using SYBYL48. In the case of PDGFrb kinase where no crystal structure was available and a modeled structure was used49,50, the cognate ligand was obtained from the x-ray crystal structure of c-Kit kinase, the homology modeling template. Similarly, only an uncomplexed apo structure was available for VEGFr1 kinase, so its native ligand was obtained by superimposing FGFr1 kinase, a homolog with high sequence and structural identity.
In target systems with large ligands spanning more than one pocket, it is helpful to specify that part of the ligand most intimately involved in binding. Such a fragment is presented to the automated scripts as an individual file that can be recognized as the reference state for generating the docking spheres or “hot spots”. Other special measures include manually redistributing the partial atomic charges of polar atoms in critical binding site residues to increase polarity and thus favoring polar ligands, as described previously.5,34,51
Automated Steps
The automated docking pipeline begins with the receptor structure file and its co-crystallized ligand, or a manually-curated specification of the binding site. All tasks including sphere generation, scoring grid and docking calculations, and analysis of enrichment, are driven automatically (Figure S1). The scheduling system Condor (U. Wisconsin, Madison WI) was used manage jobs on our Linux cluster.
Binding site residues are identified as those being within 12 Å of any heavy atom of the crystallographic ligand or the residues used to define the site, using the program FILT (from the DOCK3.5 distribution). The solvent-accessible molecular surface52 of the protein binding site is then calculated with the program DMS53 using a probe radius of 1.4 Å. Receptor-derived spheres are calculated using the program SPHGEN (part of the UCSF DOCK suite),54 while the ligand-derived spheres are simply generated from the positions of the heavy atoms of the crystallographic ligand, if available. If the molecular fragment file is present, the ligand-derived spheres are created from the molecular fragment instead of using the entire ligand structure. The matching spheres, required for orientation of the ligand in the binding site, are obtained by augmenting the ligand-derived spheres with receptor-derived spheres. Spheres furthest away from ligand-derived spheres, furthest from the centroid of the remaining spheres, too close to receptor atoms or too close to each other are removed iteratively until the total number of spheres is 35 or less. Spheres are labeled for chemical matching based on the hydrogen bonding properties and charged states of nearby receptor atoms.55
The scoring grids are also prepared automatically. The grid box dimensions are initially set so that the edges extend 15 Å beyond the matching spheres. The box dimensions are refined to maximize the coverage of the protein without exceeding two million grid points at a resolution of three points per Ångstrom. Polar hydrogens are added to the protein using SYBYL48. Four scoring grids are generated: an excluded volume grid using DISTMAP56, a united atom AMBER-based van der Waals potential grid using CHEMGRID56, an electrostatic potential grid using DelPhi57 and a solvent occlusion map using the program SOLVMAP (B Shoichet, unpublished results). The Delphi grid potential is calculated using a dielectric of 2 with the internal low dielectric volume determined by the protein atoms augmented by dummy atoms occupying the binding pocket, and an external dielectric of 78 for the external solvent environment. When structural waters, cofactors or metal ions are present, they are treated as part of the protein.
Docking was performed with DOCK 3.5.54, a flexible-ligand method that uses a force-field-based scoring function composed of van der Waals and electrostatic interaction energies corrected for ligand desolvation.33,47,56 The sampling of ligand orientations in DOCK3.5.54 can be varied according to several user-defined parameters, which we set to the same values for all 40 systems, as follows. The bin size for both receptor and ligand were set to 0.4 Å and the overlap bin size was set to 0.3 Å. A distance tolerance (dislim) of 1.5 Å was applied for matching the ligand onto the matching spheres, and ligand orientations were rejected if the color of a ligand-receptor pair did not match. For each ligand orientation, the conformational ensemble is filtered for steric complementarity using DISTMAP with the polar and non-polar close contact limits of 2.3 Å and 2.6 Å respectively. Ligand conformations are scored based on the total docking energy (Etot = Eele + Evdw − ΔGlig-solv), which is the sum of electrostatic (Eele) and van der Waals (Evdw) interaction energies, corrected by the partial ligand desolvation energy (ΔGlig-solv).47 Final energies are computed after 25 steps of rigid-body minimization. A single docking pose with the best total energy score is saved for each docked molecule. For ligands with multiple protonation states and tautomeric forms, only the best scoring representation is retained.
Results
A directory of useful decoys (DUD)
We have created DUD as a research tool to benchmark structure-based virtual screening. DUD contains 2950 annotated ligands for 40 diverse targets, plus 36 decoy molecules for each annotated ligand, each decoy having similar physical properties but dissimilar chemical structures to its active (Table 1). Topological dissimilarities were originally calculated using CACTVS fingerprints, but it is convenient to compare decoys and ligands using Daylight fingerprints, which are more widely used. Of the 95,316 DUD decoys, and based on standard Daylight fingerprints58, only 90 compounds are above a Tc of 0.85 to any annotated ligand, only 400 compounds are above a Tc of 0.8 and only 1,300 above a Tc of 0.7, indicating that DUD decoys are topologically dissimilar to the annotated ligands and are thus likely to be true negatives, although of course we cannot be completely sure that this is the case.59
Histograms of five physical properties (molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds and LogP) were calculated for the DUD ligands, DUD decoys, MDDR database compounds, Jain’s decoys23 and Rognan’s decoys15 (Figure 2). For each of the 40 targets, the DUD decoys are designed to match the physical properties of the specific ligands for that target. Strictly speaking, there is no reason why the amalgamation of the 40 decoy sets (the DUD decoys) should provide good decoys for each of the 40 ligand sets (the DUD actives), but in fact they typically do (see Discussion). The properties of the DUD decoys are comparable to the actives from which they were generated: they span the same ranges and have maxima at about the same place. Conversely, the uncorrected databases can differ substantially from the physical properties of the DUD ligands (figure 2). The largest differences are observed for the 1000 decoys introduced by Rognan, which differ substantially in all physical properties from the DUD ligands. The 98,000 MDDR database also differed significantly from the DUD ligands, being typically larger in every physical property (e.g., 9% larger, on average, in molecular weight and having 15% more hydrogen-bond acceptors) than the DUD ligands. The 1000 decoy set introduced by Jain was the second best-matched to the DUD ligands, after only the DUD decoys themselves, typically differing only in being slightly smaller or lower in physical properties (figure 2). Of course, there is no reason why these other decoy sets should match the physical properties of the DUD ligands, since they were not selected to match these ligands. Nor is there any reason to require them to match, unless one believes the DUD ligands to be especially representative of good physical properties, which we do not contend. What the differing physical properties among the decoy sets allows us to probe is how matched and unmatched ligand-decoy sets affect enrichment calculations in docking.
Figure 2.

The physical property distributions of the ligands and different sets of decoys. The brown line represents the annotated ligands (2,950 compounds); the blue line represents the DUD decoys (95,316 compounds); the green line represents the properties of the MDDR database (98,000 compounds); the orange line represents the Jain’s decoys (randomly selected 1,000 ZINC drug-like compounds) and the cyan line represents the Rognan’s decoys (randomly selected 990 ACD compounds).
Overall Enrichments
Virtual screening is benchmarked using two criteria: enrichment of annotated ligands among top scoring docked molecules from a database of decoys, and by the geometric fidelity of the docked poses compared to those of the experimental structures. The docking enrichment factor (EF) reflects the ability of the docking calculations to find true positives throughout the background database compared to random selection. This enrichment factor is calculated as EFsubset = {Ligandsselected/Nsubset} / {Ligandstotal/Ntotal}.47 For instance, for a given protein with 100 annotated ligands (Ligandstotal) in a database of 98,000 compounds (Ntotal), only one of the known ligands (Ligandsselected) would be expected to be found in any chosen subset of 980 molecules (Nsubset) by random selection, which corresponds to an enrichment factor of 1. The key results of docking to 40 targets are summarized in Figure 3 and Table 1. Figure 3 shows the overall profile of percentage of ligands found (y-axis) plotted as a function of the percentage of the ranked docked database (x-axis in logarithmic scale) for each system. Here, we present two different enrichments using two different background databases, namely, the “entire database” contains the 2,950 DUD ligands and the 95,316 DUD decoys (blue line), while the “own decoys” only includes the native ligands and their corresponding decoys (red line). The percentage of true ligands found by docking at any given percentage of the docking ranked database should always be greater compared to being chosen by random selection (gray line). The higher the percentage of known ligands found at a given percentage of the ranked database, the better the enrichment performance of the virtual screening. In general, the docking enrichments are poorer against the “own decoys” than against the entire database; for some targets, the enrichment difference between the entire database and the “own decoys” is dramatic (e.g. TK, PNP and SAHH).
Figure 3.



Docking enrichment plots for forty protein targets using DUD. The docking ranked database (x-axis) is plotted against the percentage of known ligands found by calculations (y-axis) at any given percentage of ranked database. Targets are listed in same order as in Table 1, and six representative systems are highlighted in light yellow (see text). The gray line represents the results expected from selecting ligands randomly; the blue line is docking enrichment against the entire DUD database (98,266 compounds), and the red line is the docking enrichment against the “own decoy” subset for any target. “Automated” represents the results achieved from the fully automated procedure; “semi-automated” represents the enrichments obtained with some expert intervention.
The enrichment results for entire-database docking may be summarized using three enrichment indicators: EFmax (maximum enrichment factor), EF1 (enrichment factor at 1% of the ranked database), and EF20 (enrichment factor at 20% of the database) (Table 1). EFmax and EF1 present the early enrichment while EF20 presents the late stage database screening. Significant enrichment is obtained for most targets, with an average EFmax of 49.1, where 20 systems have EFmax greater than or very close to 30, 10 systems have an EFmax less than 5, and only 4 out of these 10 systems fail to enrich their native ligands above random. Superior enrichments are observed in 7 diverse targets with EFmax greater than 100. On average, 17.3% and 52.9% of the known ligands can be found in the top 1% and 20% of the docking ranked database, respectively, corresponding to enrichment factors of 17.3 and 2.6. It is also notable that six out of ten targets with poor enrichment are kinases. We now take up in more detail the dependence on decoys, docking specificity via cross docking, and consider six representative targets.
1. Enrichments against DUD compared to uncorrected databases
To test the influence of decoys on docking performance, we docked exactly the same ligand sets against 12 different targets, varying only the background decoy database. We compared the 98,000 compounds of the MDDR, the 98,266 compounds of DUD, and the 1000 compound sets introduced by Rognan15 and Jain23. For comparing the performance of the large DUD and MDDR databases to the smaller Rognan and Jain sets, we used receiver operator characteristic (ROC) curves to avoid biases introduced in enrichment plots when the ratio of actives to decoys grows large.60 ROC curves plot sensitivity (Se) and specificity (Sp), where Sesubset = {Ligandsselected/Ligandstotal} and Spsubset = {(Decoystotal − Decoysselected)/Decoystotal}. We plotted the ROC curves as (1 – Sp) (i.e., % selected decoys) versus Se (i.e., % selected ligands) (Figure 4). Like an enrichment plot, the further away the ROC curve is above the diagonal, the better the docking enrichment. Docking enrichments typically followed the following trend: the Rognan decoys led to the best enrichments, followed closely by the MDDR decoys, then the Jain decoys, with the worst enrichments against DUD. Here, better enrichment means only less competitive decoys. Thus, targets that had poor or no enrichment using DUD had very respectable enrichments against the other decoy sets, using exactly the same ligands and docking protocols. To investigate whether a smaller decoy database itself introduces artificial enrichment and thus unfairly biases the smaller decoy sets, ROC curves were also generated using a randomly selected 1,000 compounds from DUD and compared with ROC curves using the entire DUD. No difference was observed (not shown), suggesting that there is little size dependent behavior using the entire DUD versus a random portion of DUD. The more competitive behavior of the DUD decoys presumably reflects their closer physical similarity to the ligands docked; indeed, the monotonic order of enrichments follows the level of dissimilarity of the decoys to the ligands, with the Rognan decoys being the most dissimilar and the Jain and DUD decoys being the most similar and correspondingly leading to the worst (i.e. most competitive) enrichments.
Figure 4.

ROC curves for twelve targets using four different background databases, DUD (blue), MDDR (green), Jain’s decoys (orange) and Rognan’s decoys (cyan). The gray line represents the results expected from random selection of ligands. The ROC curves were plotted as the Se (% selected actives) versus (1 – Sp) (% selected decoys). The same annotated ligands were used for the different background databases. Targets are listed in the same order as in Table 1.
2. Docking Specificity via Cross-Docking Simulations
We docked DUD against all 40 targets and compared the enrichment of each ligand set against each target (Table 2). We used two enrichment indicators, ETmax and ET20, to define the enrichment performance for each matrix unit, from very good (ETmax ≥ 30 and ET20 ≥ 3), good (30 > ETmax ≥ 20 and 3 > ET20 ≥ 2.5), medium (20 > ETmax ≥ 10 and 2.5 > ET20 ≥ 2) and poor (ETmax < 10 and ET20 < 2). An exception (e.g. ERantagonist) is made when one of the two enrichment indicators is well above its defined cutoff while the other is marginally below its cutoff. In these cases an averaged enrichment performance classification is assigned. Several features of the cross docking table are noteworthy. First, it is a sparse matrix, mostly white, showing that most annotated ligand sets are not highly enriched against most targets. Second, many of the diagonal elements are black or red, indicating very good or good enrichment of the target’s own ligands. Third, many of the off diagonals make sense. For example, serine protease ligands (thrombin, trypsin and factor Xa) are enriched against other serine protease targets, nuclear hormone receptor ligands are enriched against most nuclear hormone receptors, such as androgen receptor (AR), mineralocorticoid receptor (MR) and estrogen receptor (ER), and nucleoside analogs are enriched against most of the nucleoside recognizing enzymes such as thymidine kinase (TK), purine nucleoside phosphorylase (PNP) and S-adenosyl-homocysteine hydrolase (SAHH). Fourth, the kinases generally show poor enrichments, not just of their own ligands, but of all ligand lists. More generally, we note that when the cognate ligands are not enriched against a target, other ligand lists are also not enriched, giving rise to blank rows. Conversely, when its own ligands are well enriched against a target, other ligand lists are often also enriched.
Table 2.
The matrix of cross-enrichments. The color-coded table unit presents poor (white), medium (green), good (red) and very good enrichment (black). Dark boxes are drawn around related targets (Nuclear hormone receptors, kinases, serine proteases, metallo-enzymes, folate enzymes, and other enzymes).
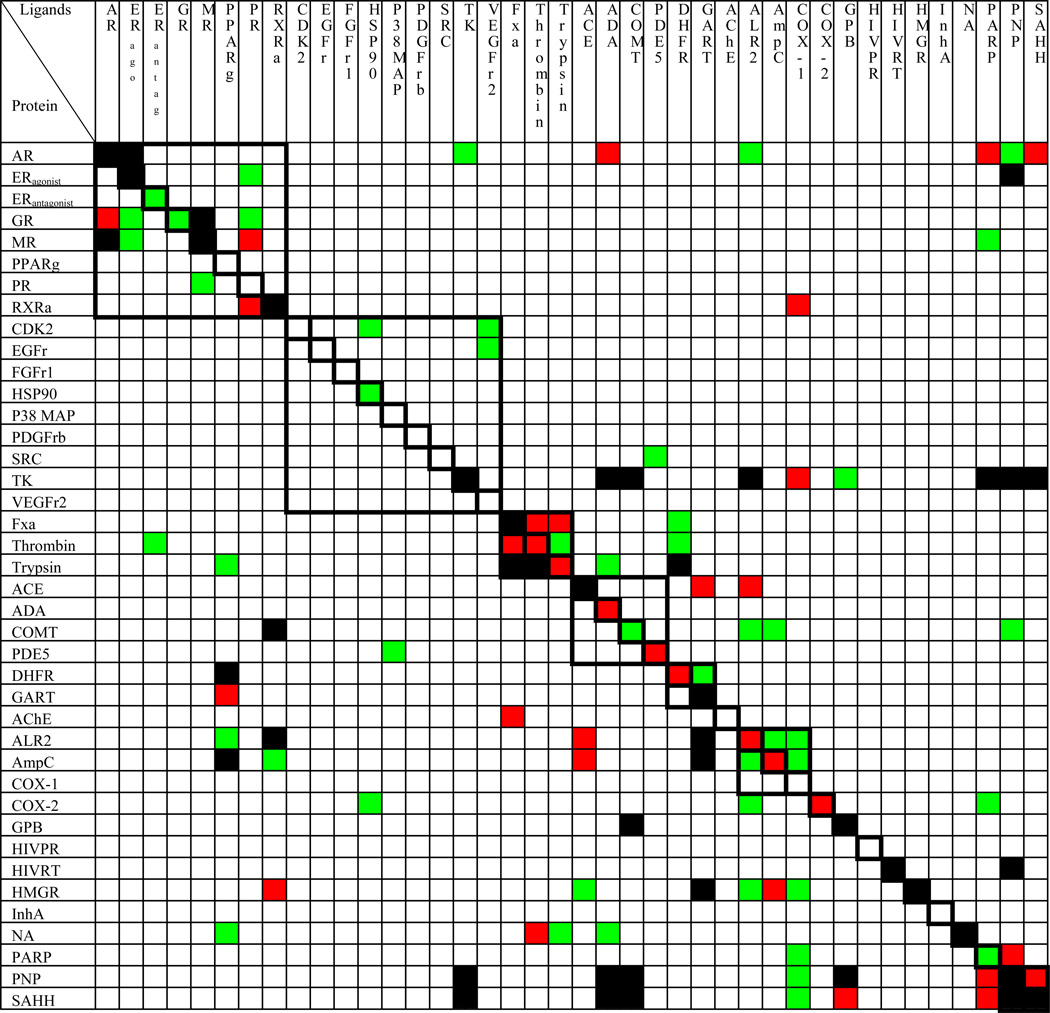 |
Very good (ETmax ≥ 30 and ET20 ≥ 3), good (30 > ETmax ≥ 20 and 3 > ET20 ≥ 2.5), medium (20 > ETmax ≥ 10 and 2.5 > ET20 ≥ 2) and poor (ETmax < 10 and ET20 < 2). The only exception is when one of the two enrichment indicators is well above the defined cutoff while the other is marginally below the defined cutoff, and then an averaged enrichment performance is assigned to compensate it.
3. Automated vs semi-automated docking
The large number of docking targets motivated us to develop a fully automated docking engine. We were able to automate most of the steps formerly performed manually, resulting in satisfactory enrichments for 24 of 40 targets. For the remaining 16 targets, we resorted to a semi-automated procedure involving expert intervention in the preparation of the receptor binding site. Thirteen of the sixteen targets with poor enrichment using the automated protocol were improved with expert intervention (Supplementary Material, Figure S4). This intervention was often trivial – we did not try very hard to maximize the docking performance, and suspect that further intervention could improve the results even more, but that was not the goal of this study.
4. Detailed Results for Six Representative Systems
We examined several representative targets in greater detail (highlighted in Table 1 and Figure 3). ER and TK were chosen based on their strong ligand enrichment as well as a substantial number of published docking studies.15,19,20,23,28–30,37–40 P38 MAP kinase was chosen to represent poorly performing protein kinases. ADA was chosen to represent targets that failed with the fully automated docking engine, but were rescued by the semi-automated procedure. ALR2 was chosen to represent targets with intermediate enrichment. InhA was chosen to represent what we consider a failure of our docking method. For these six representative systems, the docking accuracy is presented by both the enrichment performance and the ability to reproduce the binding geometry observed in the crystallographic complex structure. In assessing the docked binding geometries, we only consider the geometry of the crystallographic ligand produced as part of the overall DUD database screen without optimization; the ligand shown has been prepared as every other DUD molecule, starting from the SMILES string representation, to avoid bias. Depending on the size of the binding pocket and our sampling criteria, docking DUD takes several hours to several days per target on a single 2.8 GHz CPU (Table 3).
Table 3.
Docking statistics on six representative targets.
| Receptor | Unique molecules scored a |
Total molecules scored b |
Orientations sampled per molecule |
Conformations sampled per molecule |
Total configurations scored b |
Total time (h) c |
|---|---|---|---|---|---|---|
| ER | 97,427 | 416,990 | 1,895 | 6,543 | 2.69 × 1010 | 54.4 |
| P38 MAP | 93,887 | 294,917 | 592 | 7,875 | 8.97 × 109 | 20.1 |
| TK | 37,240 | 180,451 | 3,437 | 4,302 | 2.67 × 109 | 21.9 |
| ADE | 85,053 | 297,400 | 14,632 | 5,308 | 2.19 × 1010 | 65.5 |
| ALR2 | 98,724 | 430,313 | 4,272 | 10,109 | 1.44 × 1011 | 296.4 |
| InhA | 97,668 | 429,579 | 2,325 | 6,809 | 5.87 × 1010 | 123.5 |
. Only orientations and configurations passing the steric filter were scored.
. Some molecules were represented in the database in multiple rigid fragment, protonation and tautomeric forms.
. Scaled to reflect time on a 2800 MHz Pentium IV.
4A. Estrogen Receptor (ERantagonist)
ER is considered to be an easy docking target because of its deeply buried hydrophobic binding site and high affinity ligands.15 Consistent with this view, automated docking achieves a significant early enrichment with an EFmax of 101.6 in the top 0.1% of the ranked database. This corresponds to finding 4 ER antagonists among the top scoring 98 compounds from a screen of 98,000 compounds. From automated docking, the docked pose of the crystallographic ligand, included and prepared as part of DUD, correctly reproduces the crystallographic binding pose of 4-hydroxy-tamoxifen (Figure 5A).
Figure 5.

Binding pose predictions for docked ligands (green) superposed on crystallographic structures (colored by atom type) for six representative targets. Key hydrogen bonds are shown by yellow lines and the protein molecular surface65 is colored by atom type. Images generated with Chimera.66
As was observed for almost all targets, docking enrichments were strongly influenced by the choice of background database. For estrogen receptor, the three other decoys sets – the 990 Rognan decoys, the 1,000 Jain decoys and the 98,000 MDDR decoys all led to much better enrichments than did DUD, with the early enrichment being particularly striking (Figure 4). Since the docking parameters and the ligands were exactly the same in each calculation, the better enrichments compared to the DUD results can only mean that the other databases present easier decoys. This view is substantiated by comparing the physical properties of the ER antagonists to the decoys in each background database. In DUD, these properties are closely matched, by design. In the other decoys sets, there are substantial differences. For example, the molecular weight varies from 380 to 460 for 90% of ER antagonists. Sixty percent of DUD compounds are within this range whereas only 40%, 42% and 44% of compounds in Rognan’s set, MDDR, and Jain’s set satisfy this criterion, respectively. In addition, the ER binding site requires specific hydrogen bonding interactions whereas a large portion of Rognan’s decoys lack hydrogen bonding functionalities, further increasing the likelihood of them being easier decoys than either the MDDR or Jain’s decoys.
4B. Thymidine Kinase (TK)
TK is considered to be a difficult target for docking because of receptor flexibility, a highly exposed binding pocket, the importance of water-bridged interactions, and the low affinity of most ligands.15 Despite these drawbacks, a high overall enrichment was achieved for this target (Figure 3). From automated docking, the docked pose corresponds closely to the crystallographic structure (Figure 5B). We note that TK is among the most promiscuous targets, in that it also highly enriches non-native ligands (Table 2), where those non-ligands are either nucleoside analogs (PNP and SAHH ligands) or highly polar or charged small compounds (ADA, COMT, ALR2, and PARP ligands).
Of the 12 targets where we compared decoy sets, TK was the only one where DUD led to better enrichments than the other sets. The TK inhibitors are much smaller and more hydrophilic than most other ligand sets, and correspondingly smaller than much of DUD. The molecular weight of 70% of the TK inhibitors is between 210 and 290, but only a small portion of the background database compounds fell within this range, 5%, 9%, 10% and 19% of DUD, Jain’s set, MDDR and Rognan’s decoys, respectively. The idiosyncratic nature of the TK ligands is brought home by comparing the very good enrichment against all of DUD compared with the relatively miserable enrichment compared to the “own decoys” of the TK ligands (Figure 3, highlighted). This is one of the rare cases where the overall DUD decoys had very different physical properties from the “own decoys” of a particular target.
4C. P38 Mitogen Activated Protein Kinase (P38 MAP)
P38 MAP kinase is a challenging target for docking due to its structural flexibility. For many ligands, a new allosteric site is induced upon binding.61 Docking is also complicated by the high degree of solvent exposure and a relatively shallow, hydrophobic cleft; taken together, these features result in poor enrichment and docking geometries that miss critical interactions. Many highly ranked decoys explore irrelevant binding regions, and manual intervention had little effect. From the automated docking, the docked ligand reproduces the correct binding geometry of urea and naphthyl groups, but not the morpholino substituent on the naphthyl ring which are thought to be crucial for potency61 (Figure 5C). Similar defects were observed with other protein kinases (PDGFrb, VEGFr2, EGFr, SRC and FRFr1).
Like many other targets, the enrichment performance in decreasing order is Rognan’s set, MDDR, Jain’s set and DUD. As for TK, a molecular size dependence biases the docking enrichments. The molecular weight ranges from 320 to 410 for 75% of the P38 kinase inhibitors, whereas for DUD, Jain’s set and MDDR, 67%, 54% and 37% of molecules fall within this range, respectively, directly corresponding to their enrichment performance. Although sixty percent of Rognan’s set falls within this weight range, a lack of suitable hydrogen bonding functional groups in the set ensures the best enrichment performance.
4D. Adenosine Deaminase (ADA)
ADA is one of four metalloenzymes we targeted. Its binding pocket is large and contains a zinc ion coordinated by three histidines. No enrichment is achieved for this target via fully automated docking. After manually redistributing the partial atomic charges of the N-epsilon atoms in the ligating histidine residues to the Zn ion, which we have previously found to be important for docking to metalloenzymes34, and included one structural water in the active site, the enrichment improved significantly (Supplementary Material, Figure S4). From automated docking, the docked ligand is approximately matched with the crystallographic ligand. Although some key groups and interactions have shifted, the ligand still occupies the same space as the crystallographic ligand, inhibiting approach to the catalytic zinc (Figure 5D). Electrostatics plays an important role in these metalloenzymes. All ADA inhibitors contain hydrogen bond donors ranging from 1 to 5, whereas 95%, 75%, 69% and 45% for DUD, Jain’s set, MDDR and Rognan’s decoys do, respectively. Correspondingly, the enrichments with the MDDR and Rognan decoys are better than those with DUD and Jain’s decoys.
4E. Aldose reductase (ALR2)
ALR2 presents a solvent exposed binding surface that requires both good polar and hydrophobic complementarity between the enzyme and inhibitors. Intermediate enrichment performance is observed in ALR2 with a good early enrichment (EF1 = 38) but a rather weak enrichment at a later stage of database screening (EF20 = 2.3), which might be attributed to the conformational changes induced by the binding of different sizes of ligands. From automated docking, the docking pose reproduces the critical polar interactions within the protein binding site and overlaps with the crystallographic binding pose except for the flipping of the aromatic ring (Figure 5E). Unsurprisingly, both the molecular size and hydrogen bonding capacity of the decoys influence docking enrichment. The molecular weight ranges from 260 to 400 for 85% of the ALR2 inhibitors, whereas 72%, 68%, 66% and 45% of the DUD decoys, Jain’s set, Rognan’s decoys and the MDDR compounds do, respectively, which explains the better enrichment using MDDR. Although, molecular size alone is not adequate to distinguish Rognan’s set from the others, the absence of suitable hydrogen bonding functional groups ensures its best enrichment performance.
4F. InhA
The worst docking scenario occurs with InhA (Figure 3) where docking gives no enrichment despite reasonable docking geometries of the crystallographic ligand and related analogs (Figure 5F), a reminder that reproducing the crystallographic binding pose is a necessary but not sufficient criterion for evaluating virtual screening. The docking enrichment decreases in the order: Rognan’s set, MDDR, Jain’s set, the DUD decoys. It seems that the requirements of the presence of hydrogen bond acceptors and the steric fitness to the InhA binding site are important for ligand binding, which makes the DUD decoys the most competitive.
Considering all 40 targets, docking enrichment varied from excellent to poor, returning what we consider to be satisfactory-but-not-stellar enrichments. Typically, poor enrichments could be attributed to sampling of ligand conformations and, in the cases of kinases, undersampling those of the receptor. There were other targets for which the differentiation between ligands and strong decoys was uninspiring, reflecting failures of our physics-based scoring function. These are all important weaknesses in our docking program, and are not uncommon in the field. As important as they are, they are not the subject of this paper, which is focused on database bias and its role in evaluating docking enrichment factors.
Discussion
Three key results emerge from this study. Perhaps the one that will have the greatest pragmatic impact is the creation of the DUD database itself. DUD is composed 2950 annotated actives together with compounds having dissimilar topology but similar physical properties to the active ligands. Because of these properties, it provides a challenging, but relatively unbiased metric for evaluating docking performance. As DUD is composed entirely of compounds in the public domain, it may be used without restriction; it may be downloaded from http://blaster.docking.org/dud/. Second, by docking all ligand sets against all 40 targets, we unintentionally undertook a very large “cross docking” experiment. The specificity matrix that results from these cross-docking results suggests interesting patterns relating to docking promiscuity and level of difficulty for docking targets. Finally, it is somewhat surprising that a fully automated docking pipeline yielded reasonably good results for 24 of 40 cases, and that minor expert intervention improved docking enrichments for 13 of the remaining 16 targets.
DUD is by far the largest and most comprehensive public data set for benchmarking virtual screening programs of which we are aware. The forty targets used to create DUD offer a diverse range of binding site types: some have deeply buried hydrophobic pockets, such as estrogen receptor and COX-2; some have more open binding sites displaying both polar and apolar binding regions, such as DHFR and P38 MAP kinase; and some have highly solvent-exposed polar sites, such as thymidine kinase and neuraminidase. The receptor diversity is reflected in the ligands they recognize: some are mostly hydrophobic (e.g. the ER ligands have LogP values in the range of 3 to 8), some are highly polar (e.g. the TK ligands have LogP values between −3 to 2), some are mostly cationic (e.g. the thrombin ligands have one or two positively charged groups) and some are mostly anionic (e.g. the GART ligands typically have two negatively charged groups). This binding site diversity allows us to evaluate the robustness and generality of our docking methods with some confidence that the range of targets is representative.
Our results are consistent with the observation by Verdonk27 that enrichment depends on the background database used, and suggest that misleadingly good enrichment may result if the physical properties of the decoys are easily distinguished from those of the actives (Figure 4). It is for this reason that the uncorrected databases, using exactly the same basis ligands, consistently lead to better enrichments than docking with DUD. For this purpose, good enrichments indicate nothing other than relatively poor decoys. Even so large and diverse a database as the MDDR led to artificial improvements in enrichment factors, typically by half a log over DUD. This is not to say that DUD itself is ideal. An indication of this is the greater stringency often provided by the “own decoys”, i.e. those decoys matched only to the annotated ligands for a particular target, compared to DUD overall (compare red and blue curves in Figure 3). Indeed, it could be argued that it is the “own decoys” that should always be used, and not the amalgamated DUD. Our own view is that both “own decoy” and amalgamated DUD docking should be performed, because they present distinct challenges to the docking program. We are aware, also, that the five physical properties we used to match ligands with decoys were not comprehensive, and it is likely that other properties could usefully be included. DUD can be seen as a procedure for building a decoy database as much as an instance of one. Thus, there is probably no such thing as a perfect single decoy set for testing docking algorithms against all targets, but there certainly are better and worse ones, and the former offer better protection against artifactual performance to the unwary docker.
In docking each of 40 DUD ligand and decoy sets against all 40 targets, we unintentionally undertook a very large cross-docking experiment, and so arrived at a measure of library-scale specificity. “Cross-docking” typically investigates the specificity of a particular ligand for a particular protein conformation26,62,63 or, more rarely, the specificity of a particular ligand for a few possible targets64. It is a more subtle gauge of docking success than simply the distance to a crystallographic orientation. Correspondingly, the specificity of ligands for their cognate receptor versus the other 39 targets more stringently measures docking success than enrichment against a single target alone. Most of the diagonal elements in the cross docking matrix indicate decent to very good enrichment (Table 2), and the matrix overall is sparse, with little enrichment against off-diagonal targets. Specificity was rarely perfect, however. Often, off-diagonal promiscuity reflected similarities among the targets (squared regions along the diagonal in Table 2). Thus the nuclear hormone analogs have moderate to very good enrichments against several non-cognate nuclear hormone receptors and the serine protease inhibitors often do well against not only their own targets, but also against several of the other serine proteases that recognize similar functionality. But even this is not the full story – many targets that enriched their cognate ligands also enriched ligands of unrelated targets, typically those with similar physical properties as their own ligands. Indeed, one of the most striking features of the cross-docking matrix is that targets that had very good enrichments for their cognate ligands typically also had good enrichments against a few other ligand sets, whereas targets that had poor enrichment for their own ligands typically had no enrichment against any other sets (white rows in Table 2). These latter, white-row targets are effectively difficult targets, at least for our docking program.
Somewhat to our surprise, fully automated docking performed well in 24 of 40 cases (Figure 3, Table 1). Typically in docking, one spends a great deal of time visually inspecting the receptor site, identifying binding site hot spots, adjusting the protonation states and orientation of rotatable protons of critical binding site residues, and deciding which structural waters, cofactors or metal ions should be included in the model. This artisanal treatment becomes less feasible in larger studies, such as this one, and indeed has inhibited the proteome-level efforts in docking that are common in related fields, such as comparative modeling of protein structures49. Whereas we do not anticipate the end of artisanship in docking campaigns, after all we had to return to manual intervention in 40% of the targets, the relative success of the automated pipeline suggests that efforts to expand such treatments merit more attention.
In docking screens, one cannot expect to correctly predict binding affinity or even monotonically rank order the ligands, and so the method falls back on the weak metric of ligand enrichment. Enrichment is a weak measure of docking success because it is always measured relative to the decoys in the database. The very same docking program with the same ligands can have very good or very mediocre enrichments with worse or better decoys. This is currently something we must live with as a field. What we can do is minimize the biases inherent in enrichment factors by matching the physical properties in decoys with those of the ligands. DUD attempts to do just this, and may provide a useful benchmarking set for the field. It is available to all investigators at http://blaster.docking.org/dud/.
Supplementary Material
Acknowledgements
Supported by NIH grants GM71896 (to BKS & JJI) and GM59957 (to BKS). We thank MDL Inc. for the MDDR database and ISIS software, Schrodinger Inc for LigPrep, Xemistry GmbH for CACTVS, OpenEye (Santa Fe, NM) for OEChem, Molecular Networks GmbH for Corina, Daylight for the fingerprint and smiles toolkits. We thank Austin Kirchner and David Lorber for scripts, code and assistance in the early stages of this work, and Kaushik Raha, Alan Graves and Michael Mysinger for reading this manuscript.
Abbreviations
- DUD
directory of useful decoys
- EF
enrichment factor
- MDDR
MDL Drug Data Report
- Tc
Tanimoto coefficient
- ROC
receiver operating characteristic
- ACE
angiotensin-converting enzyme
- AChE
acetylcholinesterase
- ADA
adenosine deaminase
- ALR2
aldose reductase
- AmpC
AmpC beta-lactamase
- AR
androgen receptor
- CDK2
cyclin-dependent kinase 2
- COMT
catechol O-methyltransferase
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- DHFR
dihydrofolate reductase
- EGFr
epidermal growth factor receptor
- ER
estrogen receptor
- FGFr1
fibroblast growth factor receptor kinase
- FXa
factor Xa
- GART
glycinamide ribonucleotide transformylase
- GPB
glycogen phosphorylase beta
- GR
glucocorticoid receptor
- HIVPR
HIV protease
- HIVRT
HIV reverse transcriptase
- HMGR
hydroxymethylglutaryl-CoA reductase
- HSP90
human heat shock protein 90
- InhA
enoyl ACP reductase
- MR
mineralocorticoid receptor
- NA
neuraminidase
- P38 MAP
P38 mitogen activated protein
- PARP
poly(ADP-ribose) polymerase
- PDE5
phosphodiesterase 5
- PDGFrb
platelet derived growth factor receptor kinase
- PNP
purine nucleoside phosphorylase
- PPARg
peroxisome proliferator activated receptor gamma
- PR
progesterone receptor
- RXRa
retinoic X receptor alpha
- SAHH
S-adenosyl-homocysteine hydrolase
- SRC
tyrosine kinase SRC
- TK
thymidine kinase
- VEGFr2
vascular endothelial growth factor receptor
- ATP
adenosine-5’-triphosphate
- β-GAR
β-glycinamide ribonucleotide
- NAD(P)-(H)
nicotinamide adenine dinucleotide (phosphate)-(reduced)
- PLP
pyridoxal-5’-phosphate
Footnotes
Supporting information Available. Schematic description of the automated docking pipeline; selected property distribution historgrams and 2D depictions of typical molecules for annotated ligands and their decoys; property distribution histograms of Rognan’s ligands and decoys; enrichment plots comparing the semi-automated with the fully automated docking procedure; complete listings of the modified parameter files used. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference
- 1.DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, et al. Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc Natl Acad Sci U S A. 1990;87:6644–6648. doi: 10.1073/pnas.87.17.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoichet BK, Stroud RM, Santi DV, Kuntz ID, Perry KM. Structure-based discovery of inhibitors of thymidylate synthase. Science. 1993;259:1445–1450. doi: 10.1126/science.8451640. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Gao J, Satoh T, Friedman TM, Edling AE, et al. A computer screening approach to immunoglobulin superfamily structures and interactions: discovery of small non-peptidic CD4 inhibitors as novel immunotherapeutics. Proc. Natl. Acad. Sci. U S A. 1997;94:73–78. doi: 10.1073/pnas.94.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruneberg S, Stubbs MT, Klebe G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: strategy and experimental confirmation. J Med Chem. 2002;45:3588–3602. doi: 10.1021/jm011112j. [DOI] [PubMed] [Google Scholar]
- 5.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Camb) 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 6.Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr Identification of Non-Phosphate-Containing Small Molecular Weight Inhibitors of the Tyrosine Kinase p56 Lck SH2 Domain via in Silico Screening against the pY + 3 Binding Site. J. Med. Chem. 2004;47:3502–3511. doi: 10.1021/jm030470e. [DOI] [PubMed] [Google Scholar]
- 7.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed Engl. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 9.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 10.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez JC. High-throughput docking as a source of novel drug leads. Curr. Opin. Chem. Biol. 2004;8:1–6. doi: 10.1016/j.cbpa.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 13.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11:323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 14.Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 15.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases: 1. evaluation of different docking/scoring combinations. J. Med. Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 16.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J. Med. Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 17.Wang R, Lu Y, Fang X, Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein-ligand complexes. J Chem Inf Comput Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 18.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 19.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 20.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 21.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III Assessing scoring functions for protein-ligand interactions. J. Med. Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 22.Cummings MD, DesJarlais RL, Gibbs AC, Mohan V, Jaeger EP. Comparison of automated docking programs as virtual screening tools. J Med Chem. 2005;48:962–976. doi: 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- 23.Pham TA, Jain AN. Parameter Estimation for Scoring Protein-Ligand Interactions Using Negative Training Data. J. Med. Chem. 2005 doi: 10.1021/jm050040j. [DOI] [PubMed] [Google Scholar]
- 24.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci U S A. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Y, Huang N, Cho S, MacKerell AD., Jr Consideration of Molecular Weight during Compound Selection in Virtual Target-Based Database Screening. J. Chem. Inf. Comput. Sci. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- 26.Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 27.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, et al. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 28.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 29.Yang JM, Shen TW. A pharmacophore-based evolutionary approach for screening selective estrogen receptor modulators. Proteins. 2005;59:205–220. doi: 10.1002/prot.20387. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Li C, Gui C, Luo X, Chen K, et al. GAsDock: a new approach for rapid flexible docking based on an improved multi-population genetic algorithm. Bioorg Med Chem Lett. 2004;14:4671–4676. doi: 10.1016/j.bmcl.2004.06.091. [DOI] [PubMed] [Google Scholar]
- 31.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 32.Diller DJ, Li R. Kinases, homology models, and high throughput docking. J. Med. Chem. 2003;46:4638–4647. doi: 10.1021/jm020503a. [DOI] [PubMed] [Google Scholar]
- 33.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irwin JJ, Raushel FM, Shoichet BK. Virtual Screening against Metalloenzymes for Inhibitors and Substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 35.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. The Protein Data Bank. Nucl. Acid. Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J. Med. Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 38.Jacobsson M, Liden P, Stjernschantz E, Bostrom H, Norinder U. Improving structure-based virtual screening by multivariate analysis of scoring data. J. Med. Chem. 2003;46:5781–5789. doi: 10.1021/jm030896t. [DOI] [PubMed] [Google Scholar]
- 39.Schulz-Gasch T, Stahl M. Binding site characteristics in structure-based virtual screening: evaluation of current docking tools. J Mol Model (Online) 2003;9:47–57. doi: 10.1007/s00894-002-0112-y. [DOI] [PubMed] [Google Scholar]
- 40.Kontoyianni M, Sokol GS, McClellan LM. Evaluation of library ranking efficacy in virtual screening. J Comput Chem. 2005;26:11–22. doi: 10.1002/jcc.20141. [DOI] [PubMed] [Google Scholar]
- 41.Claussen H, Gastreich M, Apelt V, Greene J, Hindle SA, et al. The FlexX Database Docking Environment - Rational Extraction of Receptor Based Pharmacophores. Current Drug Discovery Technologies. 2004;1:49–60. doi: 10.2174/1570163043484815. [DOI] [PubMed] [Google Scholar]
- 42.Xing L, Hodgkin E, Liu Q, Sedlock D. Evaluation and application of multiple scoring functions for a virtual screening experiment. J Comput Aided Mol Des. 2004;18:333–344. doi: 10.1023/b:jcam.0000047812.39758.ab. [DOI] [PubMed] [Google Scholar]
- 43.Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft docking and multiple receptor conformations in virtual screening. J Med Chem. 2004;47:5076–5084. doi: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mozziconacci JC, Arnoult E, Bernard P, Do QT, Marot C, et al. Optimization and validation of a docking-scoring protocol; application to virtual screening for COX-2 inhibitors. J Med Chem. 2005;48:1055–1068. doi: 10.1021/jm049332v. [DOI] [PubMed] [Google Scholar]
- 45.Ihlenfeldt WD, Takahashi Y, Abe S, Sasaki S. Computation and Management of Chemical Properties in CACTVS: An Extensible Networked Approach Toward Modularity and Flexibility. J. Chem. Inf. Comput. Sci. 1994;34:109–116. [Google Scholar]
- 46.Voigt JH, Bienfait B, Wang S, Nicklaus MC. Comparison of the NCI open database with seven large chemical structural databases. J Chem Inf Comput Sci. 2001;41:702–712. doi: 10.1021/ci000150t. [DOI] [PubMed] [Google Scholar]
- 47.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 48.SYBYL. 6.7 ed. St. Louis, MO: Tripos Associates; [Google Scholar]
- 49.Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 51.Brenk R, Irwin JJ, Shoichet BK. Here Be Dragons: Docking and Screening in an Uncharted Region of Chemical Space. J Biomol Screen. 2005;10:667–674. doi: 10.1177/1087057105281047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 53.Ferrin TE, Huang CC, Jarvis LE, Langridge R. The MIDAS display system. J. Mol. Graphics. 1988;6:13–27. [Google Scholar]
- 54.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 55.Shoichet BK, Kuntz ID. Matching chemistry and shape in molecular docking. Protein Eng. 1993;6:723–732. doi: 10.1093/protein/6.7.723. [DOI] [PubMed] [Google Scholar]
- 56.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992;13:505–524. [Google Scholar]
- 57.Nicholls A, Honig B. A Rapid Finite-Difference Algorithm, Utilizing Successive over-Relaxation to Solve the Poisson-Boltzmann Equation. J. Comput. Chem. 1991;12:435–445. [Google Scholar]
- 58.Weininger D. Clustering package. Irvine CA: Daylight Chemical Information Systems; [Google Scholar]
- 59.Matter H. Selecting optimally diverse compounds from structure databases: a validation study of two-dimensional and three-dimensional molecular descriptors. J Med Chem. 1997;40:1219–1229. doi: 10.1021/jm960352+. [DOI] [PubMed] [Google Scholar]
- 60.Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. Virtual screening workflow development guided by the "receiver operating characteristic" curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005;48:2534–2547. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 61.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 62.Claussen H, Buning C, Rarey M, Lengauer T. FlexE: efficient molecular docking considering protein structure variations. J Mol Biol. 2001;308:377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 63.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 64.Sotriffer CA, Dramburg I. "In situ cross-docking" to simultaneously address multiple targets. J Med Chem. 2005;48:3122–3125. doi: 10.1021/jm050075j. [DOI] [PubMed] [Google Scholar]
- 65.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 66.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 67.Zhang J, Aizawa M, Amari S, Iwasawa Y, Nakano T, et al. Development of KiBank, a database supporting structure-based drug design. Computational Biology and Chemistry. 2004;28:401–407. doi: 10.1016/j.compbiolchem.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Fang H, Tong W, Shi LM, Blair R, Perkins R, et al. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001;14:280–294. doi: 10.1021/tx000208y. [DOI] [PubMed] [Google Scholar]
- 69.Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind database: methodologies and updates. J. Med. Chem. 2005;48:4111–4119. doi: 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- 70.Jorissen RN, Gilson MK. Virtual screening of molecular databases using a support vector machine. J. Chem. Inf. Model. 2005;45:549–561. doi: 10.1021/ci049641u. [DOI] [PubMed] [Google Scholar]
- 71.Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, et al. Structure-Activity Relationships in Purine-Based Inhibitor Binding to HSP90 Isoforms. Chemistry & Biology. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 72.Dymock BW, Barril X, Brough PA, Cansfield JE, Massey A, et al. Novel, Potent Small-Molecule Inhibitors of the Molecular Chaperone Hsp90 Discovered through Structure-Based Design. J. Med. Chem. 2005;48:4212–4215. doi: 10.1021/jm050355z. [DOI] [PubMed] [Google Scholar]
- 73.Hennequin LF, Thomas AP, Johnstone C, Stokes ES, et al. Pl inverted question marke, P. A. Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J Med Chem. 1999;42:5369–5389. doi: 10.1021/jm990345w. [DOI] [PubMed] [Google Scholar]
- 74.Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Ple PA, et al. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem. 2002;45:1300–1312. doi: 10.1021/jm011022e. [DOI] [PubMed] [Google Scholar]
- 75.Sun L, Tran N, Liang C, Tang F, Rice A, et al. Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. J Med Chem. 1999;42:5120–5130. doi: 10.1021/jm9904295. [DOI] [PubMed] [Google Scholar]
- 76.Bohm M, St rzebecher J, Klebe G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem. 1999;42:458–477. doi: 10.1021/jm981062r. [DOI] [PubMed] [Google Scholar]
- 77.Sutherland JJ, O'Brien LA, Weaver DF. A comparison of methods for modeling quantitative structure-activity relationships. J. Med. Chem. 2004;47:5541–5554. doi: 10.1021/jm0497141. [DOI] [PubMed] [Google Scholar]
- 78.Varney MD, Palmer CL, Romines WH, 3rd, Boritzki T, Margosiak SA, et al. Protein structure-based design, synthesis, and biological evaluation of 5-thia-2,6-diamino-4(3H)-oxopyrimidines: potent inhibitors of glycinamide ribonucleotide transformylase with potent cell growth inhibition. J. Med. Chem. 1997;40:2502–2524. doi: 10.1021/jm9607459. [DOI] [PubMed] [Google Scholar]
- 79.Van Zandt MC, Jones ML, Gunn DE, Geraci LS, Jones JH, et al. Discovery of 3-[(4,5,7-trifluorobenzothiazol-2-yl)methyl]indole-N-acetic acid (lidorestat) and congeners as highly potent and selective inhibitors of aldose reductase for treatment of chronic diabetic complications. J. Med. Chem. 2005;48:3141–3152. doi: 10.1021/jm0492094. [DOI] [PubMed] [Google Scholar]
- 80.Graves AP, Brenk R, Shoichet BK. Decoys for docking. J. Med. Chem. 2005;48:3714–3728. doi: 10.1021/jm0491187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tondi D, Morandi F, Bonnet R, Costi MP, Shoichet BK. Structure-based optimization of a non-beta-lactam lead results in inhibitors that do not up-regulate beta-lactamase expression in cell culture. J. Am. Chem. Soc. 2005;127:4632–4639. doi: 10.1021/ja042984o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Kang X, Kuntz ID, Kollman PA. Hierarchical Database Screenings for HIV-1 Reverse Transcriptase Using a Pharmacophore Model, Rigid Docking, Solvation Docking, and MM-PB/SA. J. Med. Chem. 2005;48:2432–2444. doi: 10.1021/jm049606e. [DOI] [PubMed] [Google Scholar]
- 83.Tikhe JG, Webber SE, Hostomsky Z, Maegley KA, Ekkers A, et al. Design, synthesis, and evaluation of 3,4-dihydro-2H-[1,4]diazepino[6,7,1-hi]indol-1-ones as inhibitors of poly(ADP-ribose) polymerase. J. Med. Chem. 2004;47:5467–5481. doi: 10.1021/jm030513r. [DOI] [PubMed] [Google Scholar]
- 84.Ealick S, Babu Y, Bugg C, Erion M, Guida W, et al. Application of Crystallographic and Modeling Methods in the Design of Purine Nucleoside Phosphorylase Inhibitors. Proc. Natl. Acad. Sci. U S A. 1991;88:11540–11544. doi: 10.1073/pnas.88.24.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.