

Abstract
Observational studies have been largely consistent in showing an inverse association between vitamin D and an individual’s risk of developing colorectal cancer. Vitamin D protection is further supported by a range of preclinical colon cancer models, including carcinogen, genetic and dietary models. A large number of mechanistic studies in both humans and rodents point to vitamin D preventing cancer by regulating cell proliferation. Counterbalancing this mostly positive data are the results of human intervention studies in which supplemental vitamin D was found to be ineffective for reducing colon cancer risk. One explanation for these discrepancies is the timing of vitamin D intervention. It is possible that colon lesions may progress to a stage where they become unresponsive to vitamin D. Such a somatic loss in vitamin D responsiveness bears the hallmarks of an epigenetic change. Here, we review data supporting the chemopreventive effectiveness of vitamin D and discuss how gene silencing and other molecular changes somatically acquired during colon cancer development may limit the protection that may otherwise be afforded by vitamin D via dietary intervention. Finally, we discuss how understanding the mechanisms by which vitamin D protection is lost might be used to devise strategies to enhance its chemopreventive actions.
The promise of colon cancer prevention by vitamin D
Colon cancer is the third most common type of cancer in the USA and accounts yearly for ∼11% of all cancer deaths (Center for Disease Control and American Cancer Society) (1,2). Thus, identifying strategies that reduce its incidence is critically important. Although early detection and polyp removal through screening colonoscopy has offered significant benefit (3), particularly in the distal colon, the fact remains that colon cancer continues to take a serious toll on the USA population. Identifying dietary agents and supplements that may reduce the risk of colon cancer development could offer a powerful accompaniment to screening colonoscopy. For example, high-risk individuals presenting colon lesions could be encouraged to utilize chemopreventive agents to reduce the risk of ‘interval’ cancers that develop in between examinations. Ideally, personalized chemopreventive approaches could be devised based on molecular deficiencies identified within early lesions. Finally, broad-acting chemopreventive agents could provide a level of protection for those who are unlikely to undergo screening colonoscopies.
Conclusively demonstrating that an agent has cancer-preventing activity is a difficult task. Non-steroidal anti-inflammatory agents, particularly aspirin, are probably the most well-established chemopreventive agents (4–6), but the effectiveness of many other compounds is still contested. Similarly, vitamin D, through its active metabolite 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], has shown chemopreventive activity in several clinical trials, other studies have found protection to be minimal or absent. Initial positive results came from geographical correlation studies, which showed an inverse relationship between sunlight exposure and the incidence and death rates for colorectal cancer (7). Subsequent observational studies correlated higher dietary or plasma vitamin D levels with a reduced risk of colon cancer. In an American Cancer Society cohort study, data from more than 120 000 men and women detected protection in men with the highest vitamin D intake relative to those with the lowest (8), although no effect was observed in women in this study. A smaller study from several VA centers found that vitamin D intake reduced the risk of developing a high-grade adenoma or cancer (9). A relationship between plasma vitamin D and colon cancer incidence has also been reported. A National Institutes of Health study of over 16 000 participants showed that individuals with higher vitamin D blood levels had a significantly lower risk of death related to colorectal cancer (10). Meta-analysis of published epidemiological data support this contention, with either trends toward protection or statistically significant protection observed (11). Although some studies have not detected protection by vitamin D (12,13), taken together there is sufficient positive data to consider vitamin D as a likely chemopreventive agent.
Preclinical and short-term interventions lend support
Vitamin D was first tested in carcinogen-induced rodent colon cancer models over 20 years ago. In the MNU, MNNG and DMH rat models, significant vitamin D protection has been reported (14–19). In some instances, a more pronounced protective effect was obtained using protocols that included a strong tumor-promoting agent. For example, Pence and Buddingh (20) observed protection by vitamin D in DMH-treated rats but only when colon tumors were promoted by a high fat diet containing 20% corn oil. Kawaura et al. (16,21) also reported protection in the MNU tumor model when the tumor-promoting agent lithocholic acid was included in the diet. These (and other) data support a role for vitamin D in suppressing colon tumor promotion rather than affecting earlier initiating events. A particularly interesting observation made in the DMH rodent model was reported by Lamprecht et al. (18). They found a significant reduction in vitamin D receptor (VDR) activity within the colonic mucosa 10 weeks after DMH treatment, suggesting that the ability of vitamin D to elicit protection to the colonic mucosa might become diminished under some circumstances. It should be noted, however, that some of the preclinical studies employed a synthetic form of vitamin D, 1α-hydroxyvitamin D3 [1(OH)D3; alfacalcidol] (14,15,17). Although 1(OH)D3 is efficiently converted into the active 1,25(OH)2D3 (22), it was not the form used in human intervention trials, which raises issues about the translational potential of some of the preclinical animal work. Nevertheless, 1(OH)D3 has been shown to function in a similar manner to vitamin D in the animal cancer models, supporting a common mechanism of action (14–19).
In addition to the carcinogen models described above, vitamin D protection has also been observed in a diet-induced model of colon cancer. Sporadic colon tumors can be induced by maintaining mice on a Western-style diet that is high in fat and low in vitamin D, calcium and folate (23–25). Interestingly, tumor development in this dietary model can be suppressed by the reintroduction of vitamin D and calcium (23). This effect appears to be related in part to the ability of vitamin D to suppress the hyperproliferation of colonic epithelial cells induced by the Western diet (26). Finally, the actions of vitamin D in the ApcMin/+ model have been tested. Interestingly, tumor frequency in ApcMin/+ mice is not affected by vitamin D status, but tumor burden is decreased by almost 50% (27,28). These data again underscore the ability of vitamin D to prevent tumor promotion or progression rather than act at the tumor initiation stage.
An important mechanistic conclusion drawn from the animal studies is that vitamin D probably functions in part by influencing cell turnover within the colonic mucosa. Interestingly, such effects have also been observed in humans. Several reports document changes in human colonic tissue following a 6 month intervention with vitamin D and calcium. In one report, vitamin D supplementation resulted in an altered expression pattern of the p21/WAF1 cell cycle inhibitor within the colonic crypt. Vitamin D exposure caused p21 expression to extend deeper into the crypt toward the proliferative zone (29). In this same study, vitamin D supplementation was found to reduce the number of hTert positive cells appearing in the upper region of the crypts (29). Since hTert is normally restricted to the proliferative compartment of the crypt (30), this finding suggests that vitamin D may help to ‘normalize’ deviations in colonic crypt organization that may occur during tumorigenesis. Vitamin D supplementation also favorably affected the expression of the proapoptotic protein Bax, increasing its expression within the upper regions of the crypt (31). Although the precise role of these protein expression changes in reducing colon cancer risk is not fully established, they are consistent with vitamin D protection occurring prior to the formation of a histological lesion.
Limitations of vitamin D protection—a potential loss of tissue responsiveness
Although many studies have implicated vitamin D in the prevention of colon cancer, there are notable instances in which vitamin D has failed to provide protection. Moreover, some of these failures occurred within the context of intervention trials. One such study was the Women’s Health Initiative polyp prevention trial, a randomized multicenter trial designed to determine the effects of fiber, fruits and vegetables and fat on adenoma recurrence. The dietary information obtained during this trial allowed researchers to assess the impact of dietary vitamin D on the risk of adenoma recurrence. Total vitamin D intake was not found to significantly reduce the risk of recurrent adenomas (32). Another notable failure was obtained in a large placebo-controlled trial in which a group of postmenopausal women received a daily vitamin D and calcium supplement for 7 years (33). The incidence of invasive colorectal cancer did not differ significantly between the supplementation group and the placebo group.
The lack of protection observed in the polyp prevention and placebo-controlled trials has raised a valid concern regarding vitamin D chemoprevention. However, a number of issues with respect to patient compliance and the levels of vitamin D supplementation in this trial have been raised. For example, many of the participants were already taking non-study calcium supplements (∼70%) and a multivitamin with vitamin D (∼30%) at the beginning of the trial. By the end of the study, the percentage of participants taking the assigned level of supplement was <60%, indicating a low compliance rate. Moreover, the daily vitamin D dose of 400 IU employed is below present recommendation levels of 600–800 IU per day (34). Another caveat is that these studies were probably biased toward detecting changes at later stages of cancer development. In the polyp prevention trial, a high-risk pool of patients was reassessed at 1 and 4 years for the appearance of new polyps (32). Likewise, the vitamin D intervention in the placebo-controlled trial took place over a 7 year time frame (33). These are relatively short time periods considering that the transformation of normal tissue to a polyp and then to a malignant cancer most probably requires several decades or longer. In comparison, agents that inhibit later stages of cancer development can show protection in these short-term intervention experiments. For example, non-steroidal anti-inflammatory agents can suppress later stages of colon cancer development in part because they inhibit Cox-2 overexpressed in adenomas and cancers (35–37). In contrast, vitamin D may function at earlier promotional stages of colon cancer development, making protection difficult to detect in an intervention study. It is possible that many of the participants in the intervention trials harbored colon lesions that had lost their ability to respond to vitamin D, and there is evidence in the literature for a number of specific mechanisms by which vitamin D protection can be lost. For example, expression of the key vitamin D target, VDR, can be silenced or its actions limited by increased expression of transcriptional repressors. Also, the presence of the active vitamin D metabolite, 1,25(OH)2D3, can be limited by the overexpression of catabolic enzymes. Here, we discuss the evidence for these potential cancer-promoting tissue alterations. Understanding the common mechanisms by which vitamin D sensitivity is lost is an important step toward developing approaches to enhance its chemopreventive actions.
Impact of chromatin structure on VDR activity
Much of the present data points to the high affinity VDR as being a critical mediator of vitamin D protection. Zheng et al. tested this directly and found that ApcMin/+ mice on a VDR null background developed larger tumors than wild-type controls. VDR is a member of the steroid receptor superfamily that forms a dimer with an RXR receptor and binds to the vitamin D response element (VDRE) on target genes (Figure 1). VDR has several conserved domains that serve to translate 1,25(OH)2D3 binding to gene activation. These domains include a ligand-binding domain, a DNA-binding domain and a transcriptional activation domain. Ligand binding causes a conformational change in VDR that increases: (i) its association with RXR, which in turn activates its DNA-binding activity (38,39); (ii) its interaction with the basal transcription apparatus (40–42) and (iii) its interaction with a number of coactivators, at the expense of corepressors. Although VDR may have a role in the cytoplasm and plasma membrane (43–46), most of the VDR enters the nucleus after ligand binding where it can exert profound physiological changes by activating target genes. There appears to be two general classes of VDR-binding sites in the genome: high affinity sites that are constitutively bound and lower affinity sites that are bound only in the presence of vitamin D (47). A notable feature of the vitamin D response is that the VDR gene itself is regulated by VDREs, which results in a strong positive feedback loop following vitamin D stimulation (48).
Fig. 1.
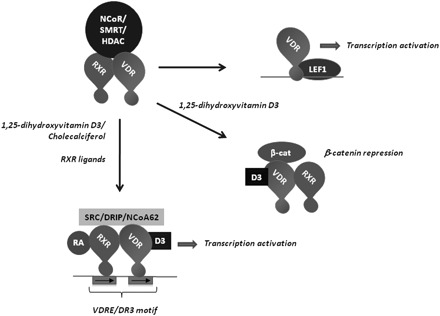
Summary of VDR complexes. Unliganded VDR can associate with RXR and the transcriptional repressors nuclear receptor corepressor, silencing mediator for retinoid or thyroid-hormone receptors and HDACs. VDR or RXR ligand binding increases VDR-RXR dimer stability and causes a replacement of transcriptional repressors for coactivators. Other VDR complexes formed include a 1,25(OH)2D3-dependent interaction with β-catenin that inhibits β-catenin activity and a 1,25(OH)2D3-independent binding to Lef1 that can activate Lef1 promoters.
In the absence of ligand, VDR interacts with the nuclear receptor corepressor and silencing mediator for retinoid or thyroid-hormone receptors corepressors, which in turn bind histone deacetylases (HDACs) that deacetylate nucleosomes, restrict chromatin accessibility and prevent gene activation (49–51). Transcriptional corepressors and HDACs can become overexpressed in cancer cells, which in turn can promote their association with VDR. A number of reports have found that the vitamin D responsiveness of prostate cancer cells is suppressed by the overexpression of corepressors and that this responsiveness can be restored either by corepressor knockdown or HDAC inhibition (52–55). Likewise, VDR may stably associate with corepressors and HDACs that are overexpressed in colon cancer cells to preclude some of the growth-regulatory effects of vitamin D (56–60). As shown in Figure 2, a VDRE-regulated luciferase reporter transfected into HT-29 cells (that do express the VDR) is not responsive to vitamin D stimulation unless an HDAC inhibitor is present. In these experiments, the HDAC inhibitor increases promoter activity and renders the promoter responsive to 1,25(OH)2D3 stimulation. These findings are consistent with histone acetylation and chromatin structure playing an important role in regulating the vitamin D sensitivity of colon cancer cells. A number of reports have shown that histone acetylation levels are generally lower in colon cancers, suggesting a less-permissive chromatin environment that would restrict gene activation by vitamin D (and other signals) (61,62).
Fig. 2.
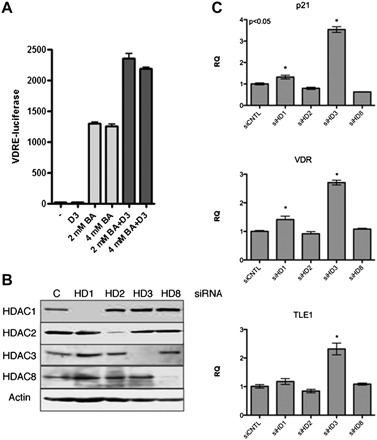
Histone deacetylases participate in VDR regulation. (A) Regulation of a VDRE-regulated luciferase gene by 1,25(OH)2D3 (D3) and the HDAC inhibitor butyrate (BA). HT29 cells were transfected with a VDRE-regulated luciferase reporter and a constitutive cytomegalovirus-regulated lacZ control plasmid. Cells were then treated overnight with the indicated concentrations of butyrate in the presence or absence of 25 nM 1,25(OH)2D3. Data show luciferase expression normalized to β-galactosidase activity. Analysis of variance indicates significant differences in expression between the control and the butyrate-treated groups and the butyrate-treated and D3 plus butyrate-treated groups. (B) Knockdown of HDAC expression in HCT116 cells following transfection with siRNA. HCT116 cells were transfected with Smart-pool siRNA (Dharmacon) with HDAC expression levels determined by immunoblotting 48 h following transfection. (C) Gene expression changes in HCT116 cells following HDAC knockdown. RNA was isolated 48 h after siRNA transfection and quantified for VDR, TLE1 and p21 expression. HDAC3 knockdown provided the highest level of induction for these three genes.
1,25(OH)2D3 binding to the VDR triggers a conformational change in the receptor’s AF-2 domain, which results in the loss of corepressor binding and the association of coactivators (63,64). Coactivator complexes that associate with liganded VDR include the DRIP complex (vitamin D receptor interacting protein complex) and NCoA62 (65–68). However, the coactivators that function at the earliest stage of gene activation by VDR are the steroid receptor coactivators (SRCs), SRC1, 2 and 3 (69). The SRC coactivators possess an intrinsic histone acetyltransferase activity and recruit other histone acetyltransferase proteins to target promoters as well (i.e. P300/CBP-associated factor and p300) (70–73). The SRC–histone acetyltransferase activity is required for steroid hormone receptors to acetylate nucleosomes at target genes to facilitate gene activation (73). The essential role of histone acetylation for gene activation by VDR is supported by the finding that activation of VDRE-regulated promoters can be suppressed if HDACs are targeted to these promoters through adjacent promoter elements (74). This latter finding further illustrates the antagonism between VDR and transcriptional-repressing HDACs in cancer cells.
In addition to being a transcriptional activator, VDR can also repress transcription in a ligand-dependent manner. Gene repression by VDR-RXR frequently entails its interaction with other promoter-bound transcription factors, preventing them from recruiting coactivators. The vitamin D-induced repression of the CYP27B1 transcription is a good example of ligand-induced gene repression by vitamin D (75). The CYP27B1 promoter includes a negative VDRE, in addition to a number of VDREs (76,77). The basic helix-loop-helix protein VDR interacting repressor binds to the negative VDRE and, on its own, stimulates CYP27B1 expression through recruitment of the p300 coactivator. However, in the presence of 1,25(OH)2D3, VDR-RXR interacts with VDR interacting repressor and prevents it from recruiting p300, while additionally enhancing its association with corepressors. In addition, VDR can repress transcription through a similar mechanism by interfering with the transcriptional activator proteins CREB and SP1 (78,79). With regard to colon cancer, the transcription factor that may be of most interest for the repressive activity of VDR is β-catenin. The VDR repression of β-catenin is discussed in more detail below.
VDR expression changes during colon carcinogenesis
Numerous reports have investigated VDR expression at different stages of colon cancer development. These reports have come to a general consensus that VDR expression is frequently increased at early stages before being lost in more advanced lesions. The loss of receptor expression is potentially linked to cellular dedifferentiation. Initial studies of colon cancer cell lines showed that well-differentiated cell lines tend to maintain higher levels of VDR expression relative to poorly differentiated lines with a greater metastatic potential (80). Studies of patient-derived colorectal carcinoma tissue extracts initially generated conflicting results (80–84). However, later studies employing histological approaches have generally shown VDR expression to be relatively low in normal epithelial tissue, increased in low-grade adenocarcinomas and then lost in metastatic cancers (85,86). Increased VDR expression may in fact occur very early in colon cancer development, as increased expression has been reported in preneoplastic aberrant crypt foci (86).
VDR repression in colon cancers—Snail, Slug and the epithelial–mesenchymal transition
The dramatic drop in VDR expression in advanced colon cancers is not the result of a genetic mutation or deletion but instead appears to involve the aberrant mobilization of a potent developmental transcriptional repression system. Specifically, the Snail transcription factor appears to play an important role in VDR repression, at least in a subset of colon cancers (87–89). Snail is a C2H2 zinc finger transcription factor that promotes mesoderm formation by blocking the expression of many non-mesoderm genes (90–92). This activity of Snail is critical for promoting mesoderm migration at gastrulation and its aberrant expression at later stages of colon cancer development similarly promotes an epithelial–mesenchymal transition and the acquisition of an invasive phenotype (Figure 3) (93,94). Evidence has been obtained that Snail, and the related protein Slug (Snail2), repress VDR transcription directly by binding regulatory elements in the VDR promoter (87–89).
Fig. 3.
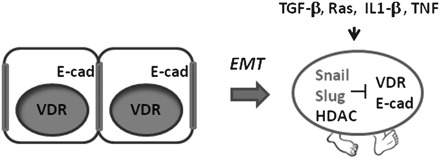
Colon cancer cell changes following an epithelial–mesenchymal transition. Normal colonocytes express the VDR and form tight junctions through E-cadherin (E-cad). Activation of Snail and Slug suppresses expression of VDR and E-cadherin and generates a transformed cell with increased motility. A number of changes in cancer cells or cancer tissue may facilitate the epithelial–mesenchymal transition (EMT), including cytokines or specific oncogene mutations (as shown).
Using colon cancer cell lines, Snail expression has been shown to confer a poorly differentiated phenotype with low VDR and E-cadherin expression (Figure 3) (89). Aberrant Snail and Slug expression in colon cancer may therefore represent an important potential mechanism for circumventing the chemopreventive actions of vitamin D. As predicted from the in vitro work, Snail and Slug expression is frequently increased in human colon cancers (roughly 50%) with an inverse relationship to VDR expression (89,95). Although the expression of these transcriptional repressors is associated with advanced lesions that have undergone an epithelial–mesenchymal transition, recent work has also suggested that adjacent normal mucosa may also express Snail and Slug (96). This latter finding raises the intriguing possibility that advanced lesions may arise from a field of cells in which transcriptional repression of VDR and other epithelial genes are being actively repressed.
Snail and Slug function by binding ‘E-box’ sequences on target genes and repress transcription through the recruitment of corepressors, including Sin3a, HDACs and the Polycomb group complex 2 (PRC2) (97–99). HDAC inhibitors and specific HDAC RNAi knockdown have been reported to increase VDR expression in some colon cancer cell lines, indicating that the HDAC containing complexes can be deployed for VDR repression (55,62). HDACs may be targeted to the VDR promoter by Snail and Slug or other promoter-associated factors. Figure 2 compares different HDAC siRNAs for their ability to stimulate VDR expression in the HCT116 colon cancer cell line. HDAC3 knockdown is found to be particularly effective at stimulating VDR expression in this cell line. In addition to HDAC-based repression mechanisms, VDR repression might also be achieved through recruitment of the Polycomb repressor complex PRC2 to the VDR promoter by Snail and Slug—PRC2 represses transcription through histone H3 methylation at lysine 27, a less reversible histone modification than acetylation (99,100). Finally, a CpG island on the VDR promoter has been reported to be hypermethylated in some breast cancer cells, opening up yet another potential means of repression (101). Understanding common mechanisms of VDR repression in colon cancers could provide important clues for enhancing the cancer preventive actions of vitamin D.
Although most of the work on the effects of Snail and Slug on colon cancer development has focused on sporadic lesions, a connection between colonic inflammation and VDR repression has recently been uncovered and suggests a possible mechanistic link between long standing ulcerative colitis (UC) and increased colorectal cancer risk. In a retrospective study of UC patients, VDR expression was found to be decreased in inflamed colonic mucosa (102). Additionally, long-term UC patients (>10 years), who were at elevated risk of developing colorectal cancer, showed significantly lower VDR expression than short-term UC patients (102). The mechanism of VDR downregulation in inflamed colonic mucosa is not known. However, inflammatory mediators such as tumor necrosis factor and transforming growth factor-β have been reported to increase Snail and Slug expression in cancers, raising the possibility that cytokine signaling may influence VDR expression in the colon (103–106) (shown schematically in Figure 3). The reduction in VDR expression directly impacts the intensity of intestinal inflammation, as indicated by studies with VDR knockout mice (107,108). A mechanism for the interplay between vitamin D, inflammatory signaling and colon cancer progression has also recently been proposed in which macrophage-generated cytokines such as interleukin-1β stimulate Wnt signaling in adjacent epithelial cells (109). Vitamin D appears to disrupt this cancer-promoting pathway in a VDR-dependent manner. The loss of VDR expression or the circumvention of this VDR-dependent blockade is a potential mechanism by which vitamin D protection might be compromised.
microRNA and potential VDR silencing
Although chromatin-based VDR repression occurs frequently in colon cancers, they certainly are not the only means of repression. VDR repression in colon cancer may also be achieved through changes in microRNA expression (110,111). A functional recognition element for miR-125b has been found in the 3′-untranslated region of human VDR messenger RNA (111). Expression of this microRNA is elevated in metastatic colon cancer and potentially contributes to the resistance of these cells to growth suppression by vitamin D (112). Knowing the frequency, timing and nature of the various VDR repression mechanisms is critical for developing approaches to optimize the effectiveness of vitamin D in colon cancer prevention.
Antagonism between VDR and Wnt signaling
A number of reports have indicated that VDR is a direct inhibitor of the Wnt signaling pathway, thus placing VDR in a critically important growth-regulatory pathway in the colon. Early work by a number of groups noted an antagonistic relationship between many nuclear receptors and the canonical Wnt/β-catenin pathway (113–115). In these studies, it was found that some nuclear receptor ligands, including vitamin D, could suppress activity of the Wnt pathway. Subsequent work showed that VDR repression of Wnt/β-catenin signaling depends on the vitamin D-dependent interaction between the VDR AF-2 transcriptional activation domain and β-catenin (116–118) (Figure 1). The stoichiometric interaction between VDR and β-catenin suggests that the capacity of VDR complex to control β-catenin activity may be limited. In normal colonic mucosa with physiological levels of β-catenin, enough VDR may be available to restrain β-catenin activity. However, under circumstances of excessive Wnt signaling, such as would occur following APC loss, β-catenin levels could overwhelm the regulatory capacity of VDR. Thus, even when VDR expression is maintained, its ability to control the growth of early neoplasms might be compromised within the context of APC loss of heterozygosity, which can occur at a relatively early stage of colon cancer development (119).
Although VDR can suppress β-catenin activity through direct binding, ‘unliganded’ VDR may in some cellular contexts actually enhance Wnt signaling though its ability to bind the Lef1 transcription factor (Figure 1). VDR binding does not bind Lef1 through its AF-2 domain but instead relies upon a region within its DNA-binding domain (Figure 1) (120). The Wnt-enhancing activity of VDR has been noted for its role in maintaining the stem cell population of the hair follicle (121). In the normal gut mucosa, β-catenin associates with Tcf4 on Wnt-regulated gene promoters, whereas other Wnt-responsive tissues employ Lef1 (122). However, colon cancers can sometimes express Lef1 in place of Tcf4 (123,124). In this case, VDR may be converted from a Wnt-signaling inhibitor to an activator, which may make vitamin D supplementation counterproductive.
RXR—an important companion
VDR must form a dimer with the RXR nuclear receptor to acquire VDRE-specific DNA-binding activity. RXR is itself a nuclear receptor and is not a silent participant in the regulation of VDRE promoters and enhancers. There have been numerous reports that RXR ligands (e.g. 9-cis-retinoic acid) can accentuate gene activation by vitamin D and may even be able to achieve some of the growth-regulatory effects of vitamin D on its own (81,125). In this regard, cell growth regulation and cancer suppression by vitamin D may be significantly influenced by dietary vitamin A. The intimate relationship between the VDR and RXR also brings up the potential importance of RXR expression in responding to vitamin D. The level of RXR expression can be modulated and is potentially subjected to epigenetic silencing. For instance, RXRA is silenced by methylation in colon tumors formed in the mouse ApcMin/+/AOM combination model (126). Although RXRs are not frequently silenced in human colon cancers, altered expression or activity of RXR could certainly impact the vitamin D responsiveness. In support of this possibility, recent results indicate that allelic variation in RXRA affects the risk of metachronous colorectal lesions (127). It has been proposed that nutrients interact as biological action packages to achieve cancer prevention and it is likely that vitamin D and vitamin A are components in one such package (128).
Other means of abrogating vitamin D protection—Cyp24A1 expression
The level of 1,25(OH)2D3 in colon tissue depends upon the activity of enzymes that catalyze its production and degradation. Circulating 25-hydroxyvitamin D3 is converted into active 1,25(OH)2D3 within the colonic mucosa by 1α-hydroxyvitamin D3 hydroxylase (Cyp27B1). 1,25(OH)2D3 signaling is then terminated by the Cyp24A1 catalyzed hydroxylation of 1,25(OH)2D3 to calcitroic acid. The expression of Cyp27B1 remains relatively constant or is modestly increased in colonic lesions (129–132) [although alterations in cellular localization have been noted (86,133)]. On the other hand, Cyp24A1 expression is increased in colon cancers, thus limiting the duration of the vitamin D signal. Initial in vitro studies reported that Cyp24A1 is highly expressed in colon cancer cell lines (131). Tissue analysis likewise revealed that the majority of adenocarcinomas express high levels of this enzyme, relative to normal tissue and precancerous lesions (130,134). A significant correlation between Cyp24A1 expression and the proliferative marker Ki-67 has also been reported (130), suggesting that vitamin D degradation may contribute to elevated cell proliferation in colon cancers. Given the potential importance of Cyp24A1 in the interference with cellular growth control by vitamin D, efforts are underway to identify and develop Cyp24A1 inhibitors (135–137). A number of interesting compounds have been identified and are presently being assessed for in vivo activity (138). These inhibitors may effectively complement vitamin D supplementation for colon cancer prevention.
Concluding remarks
Epidemiological and preclinical data over the past several decades have generated enthusiasm for vitamin D as a colon cancer preventive agent. Although intervention studies have fallen short of achieving the degree of protection anticipated from observational and preclinical studies, accumulating evidence supports the likelihood that colonic lesions may activate a number of mechanisms to evade growth regulation by vitamin D. As discussed in this review, a number of potential mechanisms have been implicated in the suppression of vitamin D signaling during colon tumor formation. These include repression of VDR expression through the activation of the Snail/Slug transcriptional repressor system as well as increased turnover of vitamin D via activation of the Cyp24A1 degradation pathway. It remains to be determined which mechanisms may be most important for limiting vitamin D protection in the colon. Clearly, by developing a better understanding of an individual’s total cancer risk profile, it may be possible to optimize cancer prevention strategies utilizing vitamin D intervention.
Funding
This work was supported in part by a grant from the National Institutes of Health R21CA125592 to C.G.; and a Diet and Health Initiative (DHI) grant from the University of Connecticut to C.G. and D.W.R.
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- SRC
steroid receptor coactivator
- VDR
vitamin D receptor
- VDRE
vitamin D response element
- UC
ulcerative colitis
References
- 1.Richardson,L.C. et al. (2011) Vital signs: colorectal cancer screening, incidence, and mortality—United States, 2002–2010. MMWR Morb. Mortal. Wkly Rep. 2011;60:884–889. [PubMed] [Google Scholar]
- 2.Edwards BK, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson RS, et al. Colorectal cancer screening. Curr. Oncol. Rep. 2009;11:482–489. doi: 10.1007/s11912-009-0065-8. [DOI] [PubMed] [Google Scholar]
- 4.Cuzick J, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 5.Chan AT, et al. Aspirin dose and duration of use and risk of colorectal cancer in men. Gastroenterology. 2008;134:21–28. doi: 10.1053/j.gastro.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawk ET, et al. Colorectal cancer prevention. J. Clin. Oncol. 2005;23:378–391. doi: 10.1200/JCO.2005.08.097. [DOI] [PubMed] [Google Scholar]
- 7.Garland CF, et al. Do sunlight and vitamin D reduce the likelihood of colon cancer? Int. J. Epidemiol. 1980;9:227–231. doi: 10.1093/ije/9.3.227. [DOI] [PubMed] [Google Scholar]
- 8.McCullough ML, et al. Calcium, vitamin D, dairy products, and risk of colorectal cancer in the Cancer Prevention Study II Nutrition Cohort (United States) Cancer Causes Control. 2003;14:1–12. doi: 10.1023/a:1022591007673. [DOI] [PubMed] [Google Scholar]
- 9.Lieberman DA, et al. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290:2959–2967. doi: 10.1001/jama.290.22.2959. [DOI] [PubMed] [Google Scholar]
- 10.Freedman DM, et al. Prospective study of serum vitamin D and cancer mortality in the United States. J. Natl Cancer Inst. 2007;99:1594–1602. doi: 10.1093/jnci/djm204. [DOI] [PubMed] [Google Scholar]
- 11.Huncharek M, et al. Colorectal cancer risk and dietary intake of calcium, vitamin D, and dairy products: a meta-analysis of 26,335 cases from 60 observational studies. Nutr. Cancer. 2009;61:47–69. doi: 10.1080/01635580802395733. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, et al. Intakes of calcium and vitamin D and risk of colorectal cancer in women. Am. J. Epidemiol. 2005;161:755–764. doi: 10.1093/aje/kwi101. [DOI] [PubMed] [Google Scholar]
- 13.Terry P, et al. Dietary calcium and vitamin D intake and risk of colorectal cancer: a prospective cohort study in women. Nutr. Cancer. 2002;43:39–46. doi: 10.1207/S15327914NC431_4. [DOI] [PubMed] [Google Scholar]
- 14.Kawaura A, et al. 1 Alpha-hydroxyvitamin D3 suppresses colonic tumorigenesis induced by repetitive intrarectal injection of N-methyl-N-nitrosourea in rats. Cancer Lett. 1990;55:149–152. doi: 10.1016/0304-3835(90)90025-s. [DOI] [PubMed] [Google Scholar]
- 15.Oda M, et al. Effects of 1 alpha-hydroxyvitamin D3 on N-methyl-N-nitrosourea-induced colonic tumorigenesis, and on fecal bile acid profiles with respect to soluble and precipitated phases in rats. Tokushima J. Exp. Med. 1990;37:75–81. [PubMed] [Google Scholar]
- 16.Kawaura A, et al. Supplemental administration of 1 alpha-hydroxyvitamin D3 inhibits promotion by intrarectal instillation of lithocholic acid in N-methyl-N-nitrosourea-induced colonic tumorigenesis in rats. Carcinogenesis. 1989;10:647–649. doi: 10.1093/carcin/10.4.647. [DOI] [PubMed] [Google Scholar]
- 17.Kawaura A, et al. Inhibitory effect of 1alpha-hydroxyvitamin D3 on N-methyl-N'-nitro-N-nitrosoguanidine-induced gastrointestinal carcinogenesis in Wistar rats. Cancer Lett. 1998;122:227–230. doi: 10.1016/s0304-3835(97)00397-2. [DOI] [PubMed] [Google Scholar]
- 18.Belleli A, et al. A protective role of 1,25-dihydroxyvitamin D3 in chemically induced rat colon carcinogenesis. Carcinogenesis. 1992;13:2293–2298. doi: 10.1093/carcin/13.12.2293. [DOI] [PubMed] [Google Scholar]
- 19.Sitrin MD, et al. Dietary calcium and vitamin D modulate 1,2-dimethylhydrazine-induced colonic carcinogenesis in the rat. Cancer Res. 1991;51:5608–5613. [PubMed] [Google Scholar]
- 20.Pence BC, et al. Inhibition of dietary fat-promoted colon carcinogenesis in rats by supplemental calcium or vitamin D3. Carcinogenesis. 1988;9:187–190. doi: 10.1093/carcin/9.1.187. [DOI] [PubMed] [Google Scholar]
- 21.Kawaura A, et al. Suppression of colonic carcinogenesis by vitamin D in rats. J. Nutr. Sci. Vitaminol. (Tokyo) 1992:331–332. doi: 10.3177/jnsv.38.special_331. [DOI] [PubMed] [Google Scholar]
- 22.Seino Y, et al. Circulating 1 alpha,25-dihydroxyvitamin D levels after a single dose of 1 alpha,25-dihydroxyvitamin D3 or 1 alpha-hydroxyvitamin D3 in normal men. Bone Miner. 1987;2:479–485. [PubMed] [Google Scholar]
- 23.Newmark HL, et al. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30:88–92. doi: 10.1093/carcin/bgn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang K, et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008;68:7803–7810. doi: 10.1158/0008-5472.CAN-08-1209. [DOI] [PubMed] [Google Scholar]
- 25.Newmark HL, et al. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis. 2001;22:1871–1875. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 26.Yang K, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J. Nutr. 2008;138:1658–1663. doi: 10.1093/jn/138.9.1658. [DOI] [PubMed] [Google Scholar]
- 27.Huerta S, et al. Intestinal polyp formation in the Apcmin mouse: effects of levels of dietary calcium and altered vitamin D homeostasis. Dig. Dis. Sci. 2003;48:870–876. doi: 10.1023/a:1023083025595. [DOI] [PubMed] [Google Scholar]
- 28.Huerta S, et al. 1alpha,25-(OH)(2)-D(3) and its synthetic analogue decrease tumor load in the Apc(min) mouse. Cancer Res. 2002;62:741–746. [PubMed] [Google Scholar]
- 29.Fedirko V, et al. Effects of vitamin D and calcium on proliferation and differentiation in normal colon mucosa: a randomized clinical trial. Cancer Epidemiol. Biomarkers Prev. 2009;18:2933–2941. doi: 10.1158/1055-9965.EPI-09-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fajkus J, et al. Changes in telomerase activity, expression and splicing in response to differentiation of normal and carcinoma colon cells. Anticancer Res. 2003;23:1605–1612. [PubMed] [Google Scholar]
- 31.Fedirko V, et al. Effects of vitamin D and calcium supplementation on markers of apoptosis in normal colon mucosa: a randomized, double-blind, placebo-controlled clinical trial. Cancer Prev. Res. (Phila) 2009;2:213–223. doi: 10.1158/1940-6207.CAPR-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartman TJ, et al. The association of calcium and vitamin D with risk of colorectal adenomas. J. Nutr. 2005;135:252–259. doi: 10.1093/jn/135.2.252. [DOI] [PubMed] [Google Scholar]
- 33.Wactawski-Wende J, et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N. Engl. J. Med. 2006;354:684–696. doi: 10.1056/NEJMoa055222. [DOI] [PubMed] [Google Scholar]
- 34.Michels KB. The women’s health initiative—curse or blessing? Int. J. Epidemiol. 2006;35:814–816. doi: 10.1093/ije/dyl133. [DOI] [PubMed] [Google Scholar]
- 35.Sinicrope FA. Targeting cyclooxygenase-2 for prevention and therapy of colorectal cancer. Mol. Carcinog. 2006;45:447–454. doi: 10.1002/mc.20232. [DOI] [PubMed] [Google Scholar]
- 36.Bertagnolli MM. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol. 2007;8:439–443. doi: 10.1016/S1470-2045(07)70139-0. [DOI] [PubMed] [Google Scholar]
- 37.Bertagnolli MM, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 38.Sone T, et al. Vitamin D receptor interaction with specific DNA. Association as a 1,25-dihydroxyvitamin D3-modulated heterodimer. J. Biol. Chem. 1991;266:23296–23305. [PubMed] [Google Scholar]
- 39.Kimmel-Jehan C, et al. Salt concentration determines 1,25-dihydroxyvitamin D3 dependency of vitamin D receptor-retinoid X receptor—vitamin D-responsive element complex formation. Arch. Biochem. Biophys. 1997;341:75–80. doi: 10.1006/abbi.1997.9952. [DOI] [PubMed] [Google Scholar]
- 40.Ing NH, et al. Members of the steroid hormone receptor superfamily interact with TFIIB (S300-II) J. Biol. Chem. 1992;267:17617–17623. [PubMed] [Google Scholar]
- 41.MacDonald PN, et al. The vitamin D receptor interacts with general transcription factor IIB. J. Biol. Chem. 1995;270:4748–4752. doi: 10.1074/jbc.270.9.4748. [DOI] [PubMed] [Google Scholar]
- 42.Blanco JC, et al. Transcription factor TFIIB and the vitamin D receptor cooperatively activate ligand-dependent transcription. Proc. Natl Acad. Sci. USA. 1995;92:1535–1539. doi: 10.1073/pnas.92.5.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizwicki MT, et al. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc. Natl Acad. Sci. USA. 2004;101:12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou LX, et al. 1 alpha,25(OH)2-vitamin D3 analog structure-function assessment of intestinal nuclear receptor occupancy with induction of calbindin-D28K. Endocrinology. 1995;136:1145–1152. doi: 10.1210/endo.136.3.7867568. [DOI] [PubMed] [Google Scholar]
- 45.Nemere I, et al. Identification of a specific binding protein for 1 alpha,25-dihydroxyvitamin D3 in basal-lateral membranes of chick intestinal epithelium and relationship to transcaltachia. J. Biol. Chem. 1994;269:23750–23756. [PubMed] [Google Scholar]
- 46.Barsony J, et al. Rapid accumulation of cyclic GMP near activated vitamin D receptors. Proc. Natl Acad. Sci. USA. 1991;88:1436–1440. doi: 10.1073/pnas.88.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramagopalan SV, et al. (2010) A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 20:1352–1360. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer MB, et al. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J. Steroid Biochem. Mol. Biol. 2010;121:136–141. doi: 10.1016/j.jsbmb.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tagami T, et al. The interaction of the vitamin D receptor with nuclear receptor corepressors and coactivators. Biochem. Biophys. Res. Commun. 1998;253:358–363. doi: 10.1006/bbrc.1998.9799. [DOI] [PubMed] [Google Scholar]
- 50.Leong GM, et al. Ski-interacting protein, a bifunctional nuclear receptor coregulator that interacts with N-CoR/SMRT and p300. Biochem. Biophys. Res. Commun. 2004;315:1070–1076. doi: 10.1016/j.bbrc.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Jimenez-Lara AM, et al. The vitamin D receptor binds in a transcriptionally inactive form and without a defined polarity on a retinoic acid response element. FASEB J. 1999;13:1073–1081. doi: 10.1096/fasebj.13.9.1073. [DOI] [PubMed] [Google Scholar]
- 52.Ting HJ, et al. Increased expression of corepressors in aggressive androgen-independent prostate cancer cells results in loss of 1alpha, 25-dihydroxyvitamin D3 responsiveness. Mol. Cancer Res. 2007;5:967–980. doi: 10.1158/1541-7786.MCR-06-0318. [DOI] [PubMed] [Google Scholar]
- 53.Ting HJ, et al. Androgen-receptor coregulators mediate the suppressive effect of androgen signals on vitamin D receptor activity. Endocrine. 2005;26:1–9. doi: 10.1385/ENDO:26:1:001. [DOI] [PubMed] [Google Scholar]
- 54.Abedin SA, et al. Elevated NCOR1 disrupts a network of dietary-sensing nuclear receptors in bladder cancer cells. Carcinogenesis. 2009;30:449–456. doi: 10.1093/carcin/bgp005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abedin SA, et al. Epigenetic corruption of VDR signalling in malignancy. Anticancer Res. 2006;26:2557–2566. [PubMed] [Google Scholar]
- 56.Weichert W, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008;14:1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 57.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 58.Spurling CC, et al. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 59.Nakagawa M, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 60.Wilson AJ, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 61.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 62.Godman CA, et al. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol. Ther. 2008;7:1570–1580. doi: 10.4161/cbt.7.10.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heery DM, et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 64.Masuyama H, et al. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol. Endocrinol. 1997;11:1507–1517. doi: 10.1210/mend.11.10.9990. [DOI] [PubMed] [Google Scholar]
- 65.Rachez C, et al. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature. 1999;398:824–828. doi: 10.1038/19783. [DOI] [PubMed] [Google Scholar]
- 66.Rachez C, et al. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 1998;12:1787–1800. doi: 10.1101/gad.12.12.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacDonald PN, et al. Emerging insights into the coactivator role of NCoA62/SKIP in Vitamin D-mediated transcription. J. Steroid Biochem. Mol. Biol. 2004;89–90:179–186. doi: 10.1016/j.jsbmb.2004.03.097. [DOI] [PubMed] [Google Scholar]
- 68.Zhang C, et al. Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J. Biol. Chem. 2003;278:35325–35336. doi: 10.1074/jbc.M305191200. [DOI] [PubMed] [Google Scholar]
- 69.McKenna NJ, et al. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 70.Lanz RB, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 71.Liu Z, et al. Sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc. Natl Acad. Sci. USA. 2001;98:12426–12431. doi: 10.1073/pnas.231474798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Utley RT, et al. Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- 73.Spencer TE, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 74.Jenster G, et al. Steroid receptor induction of gene transcription: a two-step model. Proc. Natl Acad. Sci. USA. 1997;94:7879–7884. doi: 10.1073/pnas.94.15.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colston KW, et al. Feedback regulation of vitamin D metabolism by 1,25-dihydroxycholecalciferol. Biochem. J. 1977;164:83–89. doi: 10.1042/bj1640083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turunen MM, et al. Selective use of multiple vitamin D response elements underlies the 1 alpha,25-dihydroxyvitamin D3-mediated negative regulation of the human CYP27B1 gene. Nucleic Acids Res. 2007;35:2734–2747. doi: 10.1093/nar/gkm179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murayama A, et al. Transrepression by a liganded nuclear receptor via a bHLH activator through co-regulator switching. EMBO J. 2004;23:1598–1608. doi: 10.1038/sj.emboj.7600157. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78.Yuan W, et al. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007;282:29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 79.Huang YC, et al. 1,25-dihydroxyvitamin D3 transcriptionally represses p45Skp2 expression via the Sp1 sites in human prostate cancer cells. J. Cell. Physiol. 2006;209:363–369. doi: 10.1002/jcp.20741. [DOI] [PubMed] [Google Scholar]
- 80.Evans SR, et al. Vitamin D receptor expression as a predictive marker of biological behavior in human colorectal cancer. Clin. Cancer Res. 1998;4:1591–1595. [PubMed] [Google Scholar]
- 81.Kane KF, et al. Antiproliferative responses to two human colon cancer cell lines to vitamin D3 are differently modified by 9-cis-retinoic acid. Cancer Res. 1996;56:623–632. [PubMed] [Google Scholar]
- 82.Kane KF, et al. 1,25-Dihydroxyvitamin D3 and retinoid X receptor expression in human colorectal neoplasms. Gut. 1995;36:255–258. doi: 10.1136/gut.36.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cross HS, et al. Vitamin D receptor and cytokeratin expression may be progression indicators in human colon cancer. Anticancer Res. 1996;16:2333–2337. [PubMed] [Google Scholar]
- 84.Vandewalle B, et al. 1,25-dihydroxyvitamin D3 receptors in normal and malignant human colorectal tissues. Cancer Lett. 1994;86:67–73. doi: 10.1016/0304-3835(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 85.Sheinin Y, et al. In situ mRNA hybridization analysis and immunolocalization of the vitamin D receptor in normal and carcinomatous human colonic mucosa: relation to epidermal growth factor receptor expression. Virchows Arch. 2000;437:501–507. doi: 10.1007/s004280000275. [DOI] [PubMed] [Google Scholar]
- 86.Matusiak D, et al. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1{alpha}-hydroxylase in normal and malignant human colon. Cancer Epidemiol. Biomarkers Prev. 2005;14:2370–2376. doi: 10.1158/1055-9965.EPI-05-0257. [DOI] [PubMed] [Google Scholar]
- 87.Larriba MJ, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30:1459–1468. doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 88.Larriba MJ, et al. SNAIL vs vitamin D receptor expression in colon cancer: therapeutics implications. Br. J. Cancer. 2005;92:985–989. doi: 10.1038/sj.bjc.6602484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palmer HG, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat. Med. 2004;10:917–919. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]
- 90.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 91.Batlle E, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 92.Cano A, et al. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 93.Carver EA, et al. The mouse snail gene encodes a key regulator of the epithelial–mesenchymal transition. Mol. Cell. Biol. 2001;21:8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yokoyama K, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:65–71. doi: 10.1016/s1368-8375(00)00059-2. [DOI] [PubMed] [Google Scholar]
- 95.Pena C, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum. Mol. Genet. 2005;14:3361–3370. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- 96.Pena C, et al. SNAI1 expression in colon cancer related with CDH1 and VDR downregulation in normal adjacent tissue. Oncogene. 2009;28:4375–4385. doi: 10.1038/onc.2009.285. [DOI] [PubMed] [Google Scholar]
- 97.Hajra KM, et al. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613–1618. [PubMed] [Google Scholar]
- 98.Peinado H, et al. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004;24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Herranz N, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell. Biol. 2008;28:4772–4781. doi: 10.1128/MCB.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hong S, et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl Acad. Sci. USA. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marik R, et al. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol. Ther. 2010;10:44–53. doi: 10.4161/cbt.10.1.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wada K, et al. Vitamin D receptor expression is associated with colon cancer in ulcerative colitis. Oncol. Rep. 2009;22:1021–1025. doi: 10.3892/or_00000530. [DOI] [PubMed] [Google Scholar]
- 103.Dong R, et al. Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial–mesenchymal transition of MCF-7 cells. Braz. J. Med. Biol. Res. 2007;40:1071–1078. doi: 10.1590/s0100-879x2007000800007. [DOI] [PubMed] [Google Scholar]
- 104.Zhou C, et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial–mesenchymal transition phenotype. Breast Cancer Res. 2008;10:R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zavadil J, et al. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 106.Peinado H, et al. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 107.Froicu M, et al. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol. 2007;8:5. doi: 10.1186/1471-2172-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Froicu M, et al. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006;117:310–318. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaler P, et al. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009;28:3892–3902. doi: 10.1038/onc.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Essa S, et al. VDR microRNA expression and epigenetic silencing of vitamin D signaling in melanoma cells. J. Steroid Biochem. Mol. Biol. 2010;121:110–113. doi: 10.1016/j.jsbmb.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 111.Mohri T, et al. MicroRNA regulates human vitamin D receptor. Int. J. Cancer. 2009;125:1328–1333. doi: 10.1002/ijc.24459. [DOI] [PubMed] [Google Scholar]
- 112.Baffa R, et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009;219:214–221. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 113.Mulholland DJ, et al. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2005;26:898–915. doi: 10.1210/er.2003-0034. [DOI] [PubMed] [Google Scholar]
- 114.Palmer HG, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pendas-Franco N, et al. Vitamin D and Wnt/beta-catenin pathway in colon cancer: role and regulation of DICKKOPF genes. Anticancer Res. 2008;28:2613–2623. [PubMed] [Google Scholar]
- 116.Shah S, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol. Cell. 2006;21:799–809. doi: 10.1016/j.molcel.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 117.Beildeck ME, et al. Control of TCF-4 expression by VDR and vitamin D in the mouse mammary gland and colorectal cancer cell lines. PLoS One. 2009;4:e7872.. doi: 10.1371/journal.pone.0007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zheng W, et al. Inactivation of the vitamin D receptor in APC(min/+) mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int. J. Cancer. 2012;130:10–19. doi: 10.1002/ijc.25992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Powell SM, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 120.Luderer HF, et al. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. J. Biol. Chem. 2011;286:18444–18451. doi: 10.1074/jbc.M110.188219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cianferotti L, et al. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc. Natl Acad. Sci. USA. 2007;104:9428–9433. doi: 10.1073/pnas.0702884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.de Lau W, et al. LEF1 turns over a new leaf. Nat. Genet. 2001;28:3–4. doi: 10.1038/ng0501-3. [DOI] [PubMed] [Google Scholar]
- 123.Atcha FA, et al. A new beta-catenin-dependent activation domain in T cell factor. J. Biol. Chem. 2003;278:16169–16175. doi: 10.1074/jbc.M213218200. [DOI] [PubMed] [Google Scholar]
- 124.Hovanes K, et al. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 2001;28:53–57. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- 125.Sanchez-Martinez R, et al. The retinoid X receptor ligand restores defective signalling by the vitamin D receptor. EMBO Rep. 2006;7:1030–1034. doi: 10.1038/sj.embor.7400776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Volate SR, et al. Epigenetic modulation of the retinoid X receptor alpha by green tea in the azoxymethane-Apc Min/+ mouse model of intestinal cancer. Mol. Carcinog. 2009;48:920–933. doi: 10.1002/mc.20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Egan JB, et al. Genetic polymorphisms in vitamin D receptor VDR/RXRA influence the likelihood of colon adenoma recurrence. Cancer Res. 2010;70:1496–1504. doi: 10.1158/0008-5472.CAN-09-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meyskens FL, Jr, et al. Diet and cancer: the disconnect between epidemiology and randomized clinical trials. Cancer Epidemiol. Biomarkers Prev. 2005;14:1366–1369. doi: 10.1158/1055-9965.EPI-04-0666. [DOI] [PubMed] [Google Scholar]
- 129.Cross HS, et al. Regulation of the colonic vitamin D system for prevention of tumor progression: an update. Future Oncol. 2009;5:493–507. doi: 10.2217/fon.09.22. [DOI] [PubMed] [Google Scholar]
- 130.Horvath HC, et al. The candidate oncogene CYP24A1: a potential biomarker for colorectal tumorigenesis. J. Histochem. Cytochem. 2010;58:277–285. doi: 10.1369/jhc.2009.954339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bareis P, et al. 25-hydroxy-vitamin d metabolism in human colon cancer cells during tumor progression. Biochem. Biophys. Res. Commun. 2001;285:1012–1017. doi: 10.1006/bbrc.2001.5289. [DOI] [PubMed] [Google Scholar]
- 132.Cross HS, et al. 25-Hydroxyvitamin D(3)-1alpha-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids. 2001;66:287–292. doi: 10.1016/s0039-128x(00)00153-7. [DOI] [PubMed] [Google Scholar]
- 133.Matusiak D, et al. CYP27A1 and CYP24 expression as a function of malignant transformation in the colon. J. Histochem. Cytochem. 2007;55:1257–1264. doi: 10.1369/jhc.7A7286.2007. [DOI] [PubMed] [Google Scholar]
- 134.Anderson MG, et al. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother. Pharmacol. 2006;57:234–240. doi: 10.1007/s00280-005-0059-7. [DOI] [PubMed] [Google Scholar]
- 135.Barycki R, et al. Removal of the 20-methyl group from 2-methylene-19-nor-(20S)-1alpha,25-dihydroxyvitamin D(3) (2MD) selectively eliminates bone calcium mobilization activity. Bioorg. Med. Chem. 2009;17:7658–7669. doi: 10.1016/j.bmc.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schuster I, et al. Selective inhibitors of vitamin D metabolism—new concepts and perspectives. Anticancer Res. 2006;26:2653–2668. [PubMed] [Google Scholar]
- 137.Schuster I, et al. Inhibitors of vitamin D hydroxylases: structure-activity relationships. J. Cell Biochem. 2003;88:372–380. doi: 10.1002/jcb.10365. [DOI] [PubMed] [Google Scholar]
- 138.Muindi JR, et al. CYP24A1 inhibition enhances the antitumor activity of calcitriol. Endocrinology. 2010;151:4301–4312. doi: 10.1210/en.2009-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]