

In Saccharomyces cerevisiae, the primary septum (PS), a functional equivalent of animal ECM, is synthesized during cytokinesis by the chitin synthase Chs2. The mitotic exit kinase Dbf2 regulates PS formation by direct phosphorylation of Chs2, which triggers its dissociation from the actomyosin ring during the late stage of cytokinesis.
Abstract
How cell cycle machinery regulates extracellular matrix (ECM) remodeling during cytokinesis remains poorly understood. In the budding yeast Saccharomyces cerevisiae, the primary septum (PS), a functional equivalent of animal ECM, is synthesized during cytokinesis by the chitin synthase Chs2. Here, we report that Dbf2, a conserved mitotic exit kinase, localizes to the division site after Chs2 and directly phosphorylates Chs2 on several residues, including Ser-217. Both phosphodeficient (chs2‑S217A) and phosphomimic (chs2‑S217D) mutations cause defects in cytokinesis, suggesting that dynamic phosphorylation–dephosphorylation of Ser-217 is critical for Chs2 function. It is striking that Chs2‑S217A constricts asymmetrically with the actomyosin ring (AMR), whereas Chs2-S217D displays little or no constriction and remains highly mobile at the division site. These data suggest that Chs2 phosphorylation by Dbf2 triggers its dissociation from the AMR during the late stage of cytokinesis. Of interest, both chs2‑S217A and chs2‑S217D mutants are robustly suppressed by increased dosage of Cyk3, a cytokinesis protein that displays Dbf2‑dependent localization and also stimulates Chs2‑mediated chitin synthesis. Thus Dbf2 regulates PS formation through at least two independent pathways: direct phosphorylation and Cyk3‑mediated activation of Chs2. Our study establishes a mechanism for direct cell cycle control of ECM remodeling during cytokinesis.
INTRODUCTION
Cytokinesis in animal and fungal cells requires spatiotemporally coordinated functions of a contractile actomyosin ring (AMR) and targeted membrane deposition. The AMR is believed to power the ingression of the plasma membrane (PM) at the division site and guide membrane deposition (Balasubramanian et al., 2004; Fang et al., 2010; Pollard, 2010), whereas membrane deposition is believed to increase surface area, as well as deliver enzymatic cargoes for localized extracellular matrix (ECM) remodeling during cytokinesis (Strickland and Burgess, 2004; Barr and Gruneberg, 2007; Fang et al., 2010). The core components of the AMR and the membrane‑trafficking machine are conserved from yeast to human (Balasubramanian et al., 2004; Pollard, 2010). Thus the budding yeast Saccharomyces cerevisiae has served as an effective model for elucidating the general mechanisms of cytokinesis.
ECM remodeling appears to play a critical role in animal cytokinesis, as evidenced by the cytokinesis failure caused by a defect in chondroitin synthesis in Caenorhabditis elegans and mice (Mizuguchi et al., 2003; Izumikawa et al., 2010). However, it remains unknown how ECM is remodeled specifically at the division site and how this process is regulated by the cell cycle machinery. In budding yeast, the primary septum (PS), a specialized cell wall structure that is synthesized by the chitin synthase Chs2 during cytokinesis (Sburlati and Cabib, 1986), is conceptually similar to animal ECM. Deletion of CHS2 causes severe defects in cytokinesis, including rapid and asymmetric AMR constriction, which underscores the importance of ECM remodeling during yeast cytokinesis (Bi, 2001; Schmidt et al., 2002; VerPlank and Li, 2005). Deletion of MYO1, which encodes the sole myosin‑II in budding yeast, also causes defects in cytokinesis and cell separation, including misoriented PS formation (Fang et al., 2010). Thus PS formation and AMR constriction are mutually dependent (Bi, 2001; Schmidt et al., 2002; VerPlank and Li, 2005), suggesting that these processes are normally coordinated to ensure efficient cytokinesis.
How PS formation is regulated by cell cycle signals remains poorly understood. During mitosis, Chs2 is synthesized and held at the endoplasmic reticulum (ER) through phosphorylation by CDK1 (Chuang and Schekman, 1996; Zhang et al., 2006; Teh et al., 2009). At the onset of cytokinesis, Chs2 is triggered to exit the ER by the mitotic exit network (MEN; Zhang et al., 2006; Teh et al., 2009; Chin et al., 2011) and is then delivered to the division site (or the bud neck) via the secretory pathway (Chuang and Schekman, 1996; VerPlank and Li, 2005). MEN consists of a Ras‑related small GTPase (Tem1), protein kinases (Cdc15, Dbf2, and its homologue Dbf20), a protein phosphatase (Cdc14), and a few regulatory proteins (Balasubramanian et al., 2004; Stegmeier and Amon, 2004). GTP‑bound Tem1 binds and recruits Cdc15 to the spindle pole bodies (SPBs; equivalent of centrosomes in animal cells), which, in turn, recruits and activates Dbf2, resulting in the release of Cdc14 from the nucleolus to the entire cell. Cdc14 then acts on a few key substrates to down‑regulate CDK1/mitotic cyclin activity, resulting in mitotic exit. Cdc14 also directly acts on Chs2 to remove the phosphorylation by CDK1, which enables Chs2 exit from the ER for subsequent transport to the division site (Chin et al., 2011). MEN may also regulate cytokinesis directly at the division site, as several MEN components, including the Cdc15 and Dbf2 kinases, localize to the division site during cytokinesis (Frenz et al., 2000; Xu et al., 2000). However, the cytokinesis targets of these kinases are largely unknown. In this study, we demonstrate that Dbf2 regulates PS formation directly by phosphorylating Chs2 at a single residue, resulting in its dissociation from the AMR during late stage of cytokinesis, and that timed phosphorylation of this residue is important for PS formation during cytokinesis.
Several proteins, including Iqg1 (the sole IQGAP in budding yeast), Inn1, Hof1 (a F‑BAR protein), and Cyk3 have been implicated in PS formation (Korinek et al., 2000; Vallen et al., 2000; Nishihama et al., 2009; Meitinger et al., 2010), but the underlying mechanism remains unknown. Iqg1 is also required for actin ring assembly (Epp and Chant, 1997; Lippincott and Li, 1998). Of interest, overexpression of Cyk3 suppresses the lethality and cytokinesis defects of iqg1∆ cells without restoring actin ring assembly, suggesting that Cyk3 functions in an AMR‑independent process—most likely septum formation—to promote cytokinesis (Korinek et al., 2000). Overexpression of Cyk3 also suppresses the cytokinesis and PS defects of inn1∆ cells (Nishihama et al., 2009) and causes the formation of ectopic PS‑like structures in wild‑type cells (Meitinger et al., 2010). These observations suggest that Cyk3 may be an activator of Chs2. In this study, we show that overexpression of Cyk3 robustly suppresses the growth and cytokinesis defects of the chs2 phosphomutants, further supporting the “activator” hypothesis. We also demonstrate that Cyk3 and Dbf2 phosphorylation define distinct mechanisms in regulating Chs2 function during cytokinesis.
RESULTS
Chs2 is a direct substrate of Dbf2 kinase in vitro
The following observations suggest that Chs2 may be a substrate of Dbf2 during cytokinesis. First, both Chs2 and Dbf2 localize to the division site during cytokinesis, although their precise timing of arrival has not been determined (Chuang and Schekman, 1996; Frenz et al., 2000). Second, Chs2 is essential for PS formation (Sburlati and Cabib, 1986; Shaw et al., 1991), whereas Dbf2 has also been implicated in the process (Hwa Lim et al., 2003; Meitinger et al., 2010). Finally, deletion analyses and in vitro chitin synthase assays suggest that an N‑terminal region, especially residues 197–230, of Chs2, which is located outside the catalytic domains (con1 and con2), plays an ill‑defined regulatory role in its activation (Figure 1A; Uchida et al., 1996), and this region contains two consensus sequences (RXXS) for Dbf2 (Mah et al., 2005), with serines 217 and 225 as the potential phosphorylation sites (Figure 1A). To determine whether Dbf2 can phosphorylate Chs2 directly, we fused the N-terminal region (residues 1–286) of Chs2 to glutathione S-transferase (GST), and subjected the fusion protein to in vitro kinase assay using recombinant Dbf2-Mob1 kinase (Mob1 is the regulatory subunit for Dbf2), which was preactivated by Cdc15 (Mah et al., 2001). Chs2 was robustly phosphorylated by preactivated Dbf2 but not by Cdc15 alone or its combination with an inactive Dbf2 (Figure 1B, left). Change of Ser-217 to alanine (S217A) decreased the phosphorylation to 73% of the wild‑type level (Figure 1, B and C). Additional change of Ser-225 to alanine (S217A, S225A) further reduced the phosphorylation to 43% (Figure 1, B and C). These data indicate that Chs2 is a direct substrate of Dbf2 in vitro and that both Ser-217 and Ser-225 are phosphorylated by Dbf2. Of importance, the same two sites were also found to be phosphorylated in vivo in studies aimed at global mapping of phosphoproteins in budding yeast, but the responsible kinase was not known (Chi et al., 2007; Dephoure and Gygi, 2011).
FIGURE 1:
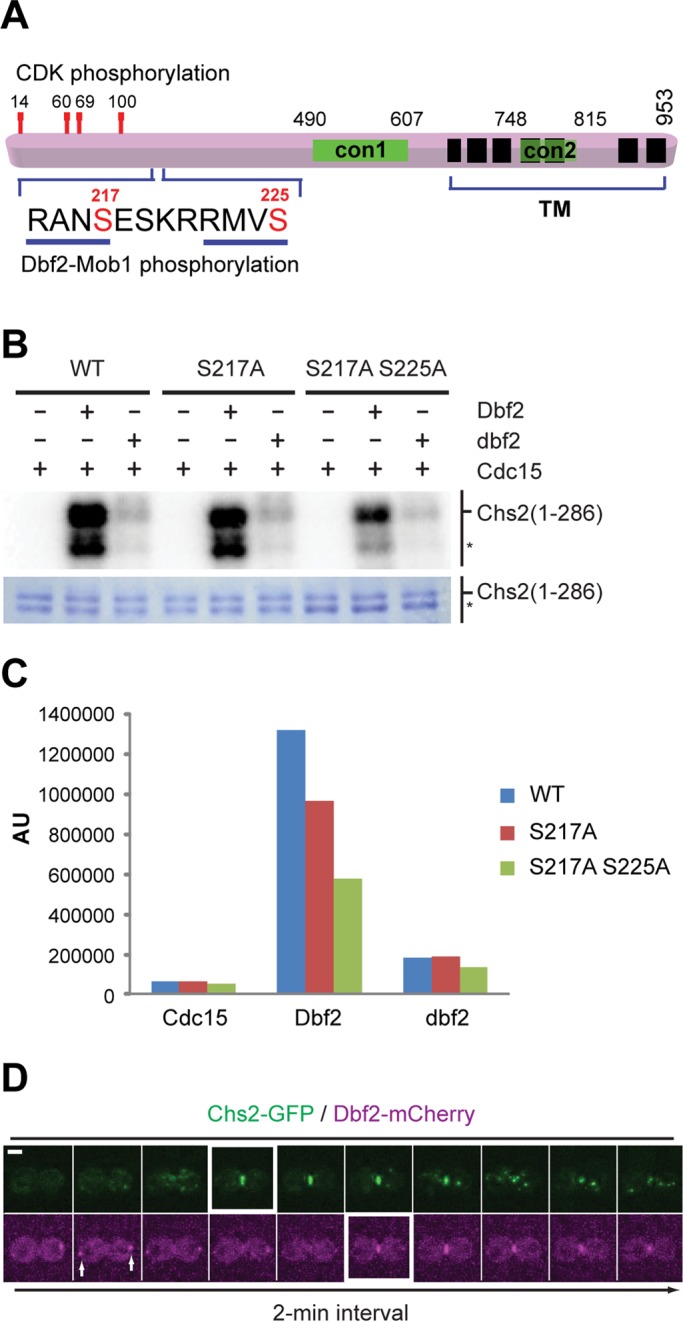
Mitotic exit kinase Dbf2 directly phosphorylates Chs2. (A) Schematic diagram of Chs2. con1 and con2, conserved regions of typical chitin synthases; TM, predicted transmembrane domains. Demonstrated and putative phosphorylation sites for CDK1 and Dbf2 kinases in the N‑terminal region of Chs2 are indicated. (B) In vitro phosphorylation of the N‑terminal region of Chs2 by Dbf2. Asterisk, degradation product of GST-Chs2 (1–286). (C) Quantification of Dbf2-mediated phosphorylation of Chs2. (D) Time-lapse analysis of a yeast cell coexpressing Chs2-GFP and Dbf2-mCherry (YO1182). Cells were grown in SC-HIS media to exponential phase at 25°C and then processed for time‑lapse microscopy. Arrows, spindle pole bodies. Scale bar, 2 μm.
Chs2 localizes to the division site before Dbf2 during cytokinesis
To determine when and where Dbf2 might phosphorylate Chs2 during cytokinesis, we performed live‑cell imaging on cells carrying both Chs2‑GFP and Dbf2‑mCherry. As expected, Chs2 was localized to the mother‑bud neck of cells at the onset of cytokinesis (Figure 1D and Supplemental Video S1; Schmidt et al., 2002; Fang et al., 2010). Consistent with a previous observation (Frenz et al., 2000), Dbf2 was first localized to the spindle pole bodies (Figure 1D, arrows) and then translocated to the bud neck during cytokinesis. Of importance, we found that the bulk of Dbf2 arrived at the neck 4 min after Chs2 (Figure 1D and Supplemental Video S1). Chs2 was generally removed from the neck region by endocytosis within 10 min after its arrival, whereas Dbf2 remained at the neck for longer time (Figure 1D). On the basis of our previous (Fang et al., 2010; Wloka et al., 2011) and present (see Figure 4D later in the paper) studies, during the first 4 min of Chs2 localization, the AMR would have constricted >50% of its initial size. Thus Dbf2 likely phosphorylates Chs2 at the neck region during late stage of cytokinesis. Of interest, the bulk accumulation and maintenance of Dbf2 at the neck appear to correlate with the onset of Chs2 endocytosis, which is marked by the appearance of Chs2–green fluorescent protein (GFP) “puncta” near the neck region.
FIGURE 4:

Chs2-S217A and Chs2-S217D display distinct dynamic behaviors during cytokinesis. (A) Localization patterns of Chs2-S217A and Chs2-S217D during the cell cycle. Cells of different strains (YO1339, YO1359, and YO1360; chs2∆ CDC3-mCherry with integrated Chs2*-GFP in at the leu2 locus) were grown in SC-LEU medium to exponential phase at 25°C and then diluted into rich media (YM-1) and grown for additional 1–2 h at 25°C before being subjected to time‑lapse analysis. (B) Three‑dimensional reconstruction of selected time points for the cells shown in A. Both the side view (top) and the en face view (bottom) of the cells are presented. The blue arrow and dashed line indicate the direction of rotation. White arrow indicates the position of Chs2*‑GFP near the end of its constriction. Scale bar, 1 μm. (C) Data acquired in A were quantified for the constriction patterns of Chs2-S217A and Chs2-S217D (WT, n = 22; S217A, n = 29; S217D, n = 41). (D) Spatial relationship between Chs2-S217A or Chs2-S217D and the actomyosin ring during cytokinesis. Cells of different strains (YO1454, YO1453, and YO1455; chs2∆ Chs2*-GFP::LEU2 with pRS314‑MYO1-C-mCherry) were cultured and imaged as in A. Arrowhead indicates the onset of AMR constriction. (E) Quantification of the durations of Chs2*-GFP, AMR contraction, and AMR disassembly during cytokinesis (WT, n = 16; S217A S225A, n = 17; S217D S225D, n = 29).
Both phosphorylation and dephosphorylation of Ser-217 are important for cytokinesis
To determine the functional consequence of Ser-217 and Ser-225 phosphorylation, C‑terminally GFP‑tagged, wild‑type, phosphodeficient (chs2-S217A, chs2‑S225A, chs2-AA [S217A S225A]) and phosphomimic (chs2-S217D, chs2‑S225D, chs2-DD [S217D S225D]) alleles of CHS2, expressed from the native CHS2 promoter, were integrated onto the chromosome of chs2Δ cells, respectively. None of the chs2 phosphomutants displayed obvious defects in cell growth or cytokinesis when cultured in minimal media (Figure 2, A, left, and B, left). However, both chs2‑S217A and chs2‑S217D mutants displayed clear defects in cell growth and cytokinesis when grown in rich media (Figure 2, A, right, and B, right). Forty-four percent of chs2‑S217A and 53% chs2‑S217D cells formed cell clusters containing three or more cell bodies, indicative of a cytokinesis defect. In comparison, none of the wild‑type cells and 66% of the chs2Δ cells formed cell clusters. Neither chs2‑S225A nor chs2‑S225D mutation caused any obvious defects in cell growth and cytokinesis even in rich media (unpublished data). In addition, the chs2‑S225 mutations only slightly enhanced the phenotypes of the chs2‑S217 mutations (Figure 2, A, right, and B, right). Together these data indicate that both phosphorylation and dephosphorylation of Ser-217 are important for cytokinesis.
FIGURE 2:
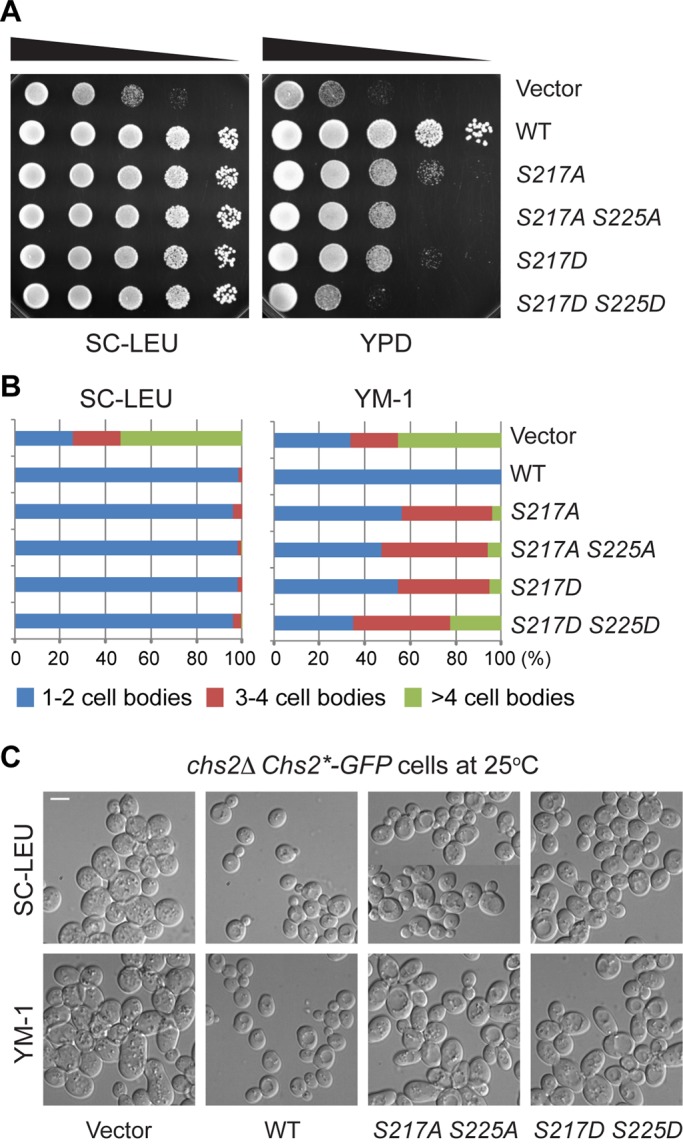
Both phosphorylation and dephosphorylation of Ser-217 are important for cell growth and division. (A) Tenfold serial dilutions of different strains (YO1338, YO1339, YO1359, YO1360, YO1450, and YO1451) containing pRS305 (Vector) or its derivatives carrying different Chs2*-GFP alleles integrated at the leu2 locus of a parent strain (chs2∆ CDC3-mCherry) were spotted onto plates containing either minimal medium (SC‑LEU) or rich medium (YPD) and grown for 2–3 d at 25°C. (B) Strains indicated in A were grown to exponential phase in liquid SC-LEU or rich (YM-1) media at 25°C. Percentages of cell clusters (counted >200 cells for each strain under each growth condition) were analyzed as indicated. (C) Representative morphologies of strains (YO1338, YO1339, YO1450, and YO1451) were visualized by DIC microscopy after grown exponentially in SC-LEU and YM-1 media at 25°C. Scale bar, 5 μm.
It is not clear why the chs2‑S217 mutants displayed media‑dependent behavior. However, media influence on the phenotypes of yeast cytokinesis mutants has been observed (Bulawa and Osmond, 1990; Nishihama et al., 2009). It is possible that the chs2‑S217 mutations have compromised the functionality of Chs2 at the division site to the extent that is still sufficient to support cytokinesis in slow‑growing minimal media but not in fast‑growing rich media. Because of the media influence, only cells grown in rich media were used for functional assays on Chs2 hereafter, unless stated otherwise.
Phosphoregulation of Chs2 is important for chitin synthesis at the division site
To explore the underlying mechanism for the cytokinesis defect caused by the chs2‑S217 mutations, we first monitored the chitin level at the division site in different strains by Calcofluor staining (Pringle, 1991). C‑terminally GFP‑tagged alleles of CHS2 (wild‑type, S217A, and S217D) under control of the native promoter and carrying a centromere‑based plasmid were introduced into a chs1Δ chs2Δ chs3Δ strain in which all chitin synthase genes in budding yeast had been deleted. The viability of the triple mutant relied solely on the introduced chs2 allele. The chitin levels at the division site of the chs2‑S217A and chs2‑S217D cells were both reduced to ∼71% of the wild‑type level (Figure 3, A and B). To ensure fair comparisons, only cells with a similar size of Chs2*‑GFP ring from different strains (Figure 3A), which is indicative of a similar cell cycle stage, were included in our quantitation. These data suggest that phosphoregulation of Chs2 at Ser-217 is important for chitin synthesis at the division site, which may account for the cytokinesis defects caused by these mutations.
FIGURE 3:
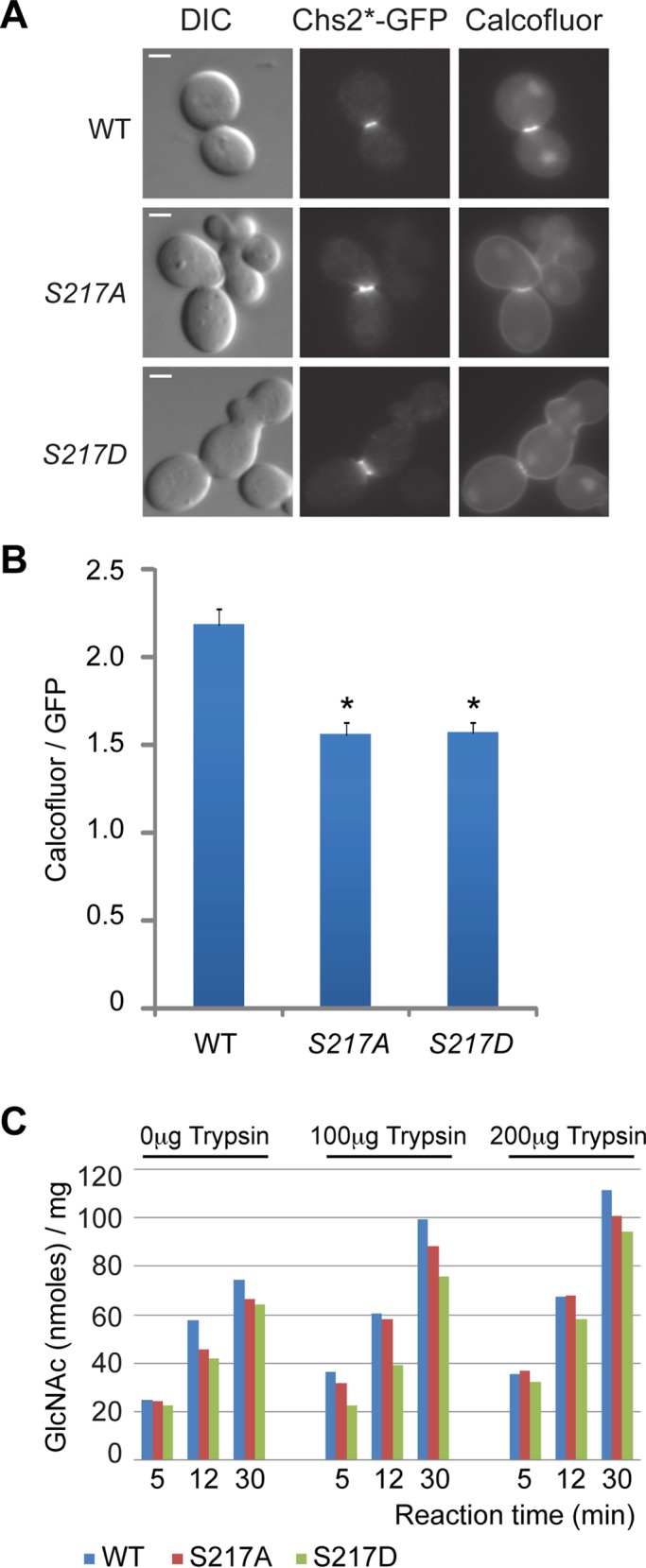
Phosphorylation and dephosphorylation of Ser-217 affect chitin synthesis at the division site. (A) Cells (YO1320, YO1324, and YO1325; chs1∆ chs2∆ chs3∆ with YCp111-Chs2*-GFP plasmids) were grown in SC-LEU medium to exponential phase at 25°C and then diluted into rich media (YM-1) and grown for additional 3 h at 25°C. Chs2*‑GFP and cellular chitin (stained with 10 mg/ml Calcofluor) were visualized by fluorescence microscopy. (B) Quantification of chitin synthesis in vivo. The ratio of Calcofluor staining intensity divided by Chs2*-GFP signal intensity was plotted for each indicated strain (WT, n = 21; S217A, n = 21; S217D, n = 26). The error bar represents SEM. *p < 0.001 in comparison to WT. (C) Cells (YO1535, YO1536, and YO1537; chs1∆ chs3∆ with YEp-pGAL1-HA3-Chs2* plasmid [2 μ, TRP1]) were grown in inducing medium (yeast nitrogen base containing 2% galactose and 1% raffinose) for 18 h at 25°C. In vitro chitin synthase II activity was measured with and without prior treatment of portions of membranes with 0, 100, and 200 μg of trypsin. Scale bar, 2 μm.
The decreased chitin synthesis at the division site could ensue as a result of decreased catalytic activity and/or altered protein expression, localization, or dynamics. To distinguish these possibilities, we first measured the chitin synthase activity of the Chs2 variants in strains lacking chitin synthase I and chitin synthase III activity. Chitin synthase II assays were done using membranes from chs1∆ chs3∆ rather than chs1∆ chs2∆ chs3∆ cells because the former are much healthier in growth and division (Shaw et al., 1991). Furthermore, the activity of chromosomally encoded Chs2 in membranes from chs1∆ chs3∆ cells grown in minimal medium is negligible (Nagahashi et al., 1995), regardless of pretreatment with trypsin, which is known to stimulate Chs2 activity in vitro (Sburlati and Cabib, 1986; Uchida et al., 1996). High‑copy plasmids expressing HA3-Chs2, HA3-Chs2-S217A, or HA3-Chs2-S217D from the inducible GAL1 promoter were introduced into chs1∆ chs3∆ cells, which were then grown in minimal medium and induced by the addition of galactose. Membranes from each strain were assayed for chitin synthase activity with or without prior treatment by trypsin and proved to have high activity (Figure 3C), whereas membranes from chs1∆ chs3∆ cells assayed in parallel incorporated background levels of radioactivity regardless of trypsin treatment (unpublished data). Trypsin treatment stimulated chitin synthase activities of all Chs2 variants to a similar extent (1.5- to 2-fold). Specific activities of membranes expressing Chs2 and Chs2-S217A were comparable, whereas the specific activity of the membranes expressing Chs2-S217D was on average ∼80% of that expressing Chs2. Similar results were obtained when cells were grown in rich medium (unpublished data). These data suggest that the defect in cytokinesis or chitin synthesis at the division site caused by the chs2‑S217 mutations may not be fully explained by the mildly decreased catalytic activity of the mutant proteins assayed at 2 mM UDP-GlcNAc.
Phosphorylation of Ser-217 regulates Chs2 dynamics at the division site
To further explore the underlying mechanism for the cytokinesis defect, we monitored the dynamics of Chs2‑S217A and Chs2‑S217D proteins at the division site. Yeast strains carrying Cdc3‑mCherry and different GFP‑tagged alleles of CHS2 (CHS2, chs2‑S217A, and chs2‑S217D as the sole source of Chs2 in the cell) were grown in rich media and imaged by three‑dimensional spin‑disk time‑lapse microscopy. Like wild-type Chs2-GFP, both Chs2-S217A-GFP and Chs2-S217D-GFP were localized to the bud neck around the onset of cytokinesis (Fang et al., 2010), which is marked by the splitting of the septin hourglass into two cortical rings (Cdc3‑mCherry; Figure 4A; Lippincott et al., 2001). The duration of Chs2-S217A-GFP and Chs2-S217D-GFP at the division is 12.8 ± 2.4 and 11.5 ± 2.0 min, respectively, slightly longer than the 10.3 ± 1.0 min for wild‑type Chs2‑GFP. It is striking that Chs2-S217A-GFP constricted asymmetrically in 79% of cells (Figure 4, B and C, and Supplemental Video S2, middle), in comparison to 100% symmetric constriction for wild‑type Chs2‑GFP (Supplemental Video S2, left); in contrast, Chs2-S217D-GFP showed little or no constriction before its disappearance from the division site in 80% of cells (Supplemental Video S2, right). As expected, the AMR (marked by the mCherry‑tagged sole myosin‑II, Myo1, in budding yeast) and Chs2‑GFP always constricted together and symmetrically in wild‑type cells (Figure 4D and Supplemental Video S3, left; Fang et al., 2010). Similarly, the AMR and Chs2‑AA‑GFP always constricted together but in an asymmetric manner (Supplemental Video S3, middle). In contrast, Chs2‑DD‑GFP failed to follow the constriction of the AMR (Supplemental Video S3, right), suggesting the uncoupling of this mutant Chs2 from the AMR. AMR constriction and disassembly appeared to be slightly longer in both Ser-217 mutants, especially in the chs2‑AA mutant, than in wild‑type cells (Figure 4E).
The uncoupling of Chs2‑S217D from the AMR is further supported by the following observations. First, despite similar profiles in protein expression (unpublished data) and the timing and duration of their localizations, Chs2‑S217D‑GFP always appeared as a broad and nonconstricting band, whereas the wild‑type Chs2‑GFP or Chs2‑S217A‑GFP appeared as a sharply focused and constricting band between the septin rings. Second, fluorescence recovery after photobleaching (FRAP) analysis indicates that wild‑type Chs2‑GFP at the division site recovers quickly after bleaching during the first ∼3 min of its localization, presumably reflecting a continuous recruitment to the division site. Chs2‑GFP is then immobilized in a myosin II–dependent manner for the rest of cytokinesis, ∼6–7 min. Using this analysis on cells with already localized Chs2 and photobleaching only a portion of the Chs2*‑GFP ring, we found that Chs2‑S217D‑GFP displayed full recovery throughout cytokinesis (Supplemental Video S4, right); in contrast, wild‑type Chs2‑GFP (Supplemental Video S4, left) and the majority of Chs2‑S217A‑GFP (Supplemental Video S4, middle) displayed no or little recovery (Figure 5, A and B). Taken together, these data suggest that phosphorylation of Chs2 by Dbf2 may trigger its dissociation from the AMR during the late stage of cytokinesis.
FIGURE 5:

Phosphomimic Chs2-S217D is highly mobile during cytokinesis. (A) FRAP analysis of Chs2‑S217A and Chs2‑S217D. Cells of different strains (YO1339, YO1359, and YO1360; chs2∆ CDC3-mCherry with integrated Chs2*-GFP at the leu2 locus) were grown in SC-LEU to exponential phase at 23°C and then diluted into rich YM-1 medium and grown for at least 3 additional hours before being subjected to FRAP analysis. (B) Data acquired in A were quantified for the recovery patterns of Chs2*-GFP (WT, n = 12; S217A, n = 28; S217D, n = 18). Arrows indicate the points of photobleaching.
Inactivation of Dbf2 causes delayed endocytic removal of Chs2 from the division site
If Chs2 were indeed a physiological substrate of Dbf2 at the division site, Chs2 would be expected to display asymmetric constriction, essentially phenocopying the chs2‑S217A mutant, when Dbf2 is inactivated. To test this hypothesis, we monitored at the nonpermissive temperature Chs2‑GFP localization in a wild‑type strain and a dbf2‑Ts (temperature‑sensitive) mutant carrying a GAL1 promoter–controlled allele of SIC1s (T5V, T33V, S76A) that encodes a stable version of the CDK1 inhibitor (Meitinger et al., 2010). Because Dbf2 inactivation simultaneously blocks both mitotic exit and cytokinesis, Sic1 overexpression was induced by adding galactose to the growth medium (see Materials and Methods for details) to force mitotic exit and trigger Chs2 translocation from the ER to the bud neck in the dbf2-mutant cells. This strategy provides the opportunity for assessing the specific effect of Dbf2 inactivation on Chs2 behavior at the division site. As expected, in wild‑type cells, Chs2‑GFP localized, constricted, and disappeared at or from the division site within 12 min (Figure 6, A and B, and Supplemental Video S5, left). In contrast, Chs2‑GFP accumulated at the division site more slowly in the dbf2-mutant than in wild‑type cells, perhaps reflecting an imperfect mitotic exit forced by Sic1 overexpression (Figure 6, A and B, and Supplemental Video S5, right). More important, Chs2‑GFP was retained at the division site of the mutant cells for >40 min with little constriction, and its signal intensity decreased gradually over time (Figure 6, A and B, and Supplemental Video S5, right). The weak signal of Chs2‑GFP at the end of our imaging, coupled with its nearly nonconstrictive behavior, prevented a clear assessment of its constriction symmetry. However, a previous EM study indicates that 81% of the dbf2 mutant cells form an asymmetric PS (Meitinger et al., 2010), suggesting that Chs2 distribution is eventually asymmetric, which lends strong support to our hypothesis.
FIGURE 7:
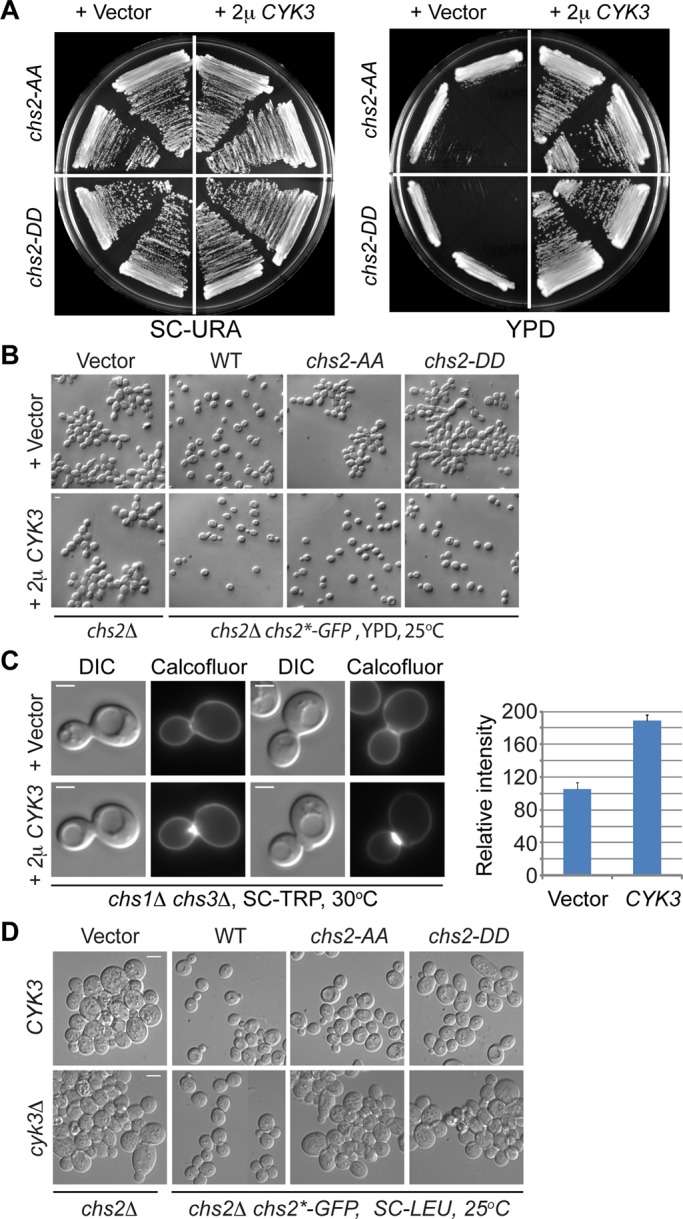
Increased dosage of Cyk3 stimulates Chs2‑dependent chitin synthesis and robustly suppresses the growth and cytokinesis defects of both chs2‑AA and chs2-DD mutants. (A) Multicopy CYK3 suppresses the growth defects of chs2‑AA and chs2‑DD. Strains (YO1543 and YO1544, top half of the plate; YO1550 and YO1551, bottom half of the plate; chs2∆ Chs2*-GFP::LEU2 CDC3-mCherry cells transformed with either an empty vector [pAG426, 2 μ, URA3] or a high‑copy CYK3 plasmid [pAG426-CYK3]) were streaked onto plates containing minimal (SC-URA) or rich (YM-1) media and grown at 25°C for 3–4 d. (B) Multicopy CYK3 suppresses the cytokinesis defects of chs2‑AA and chs2‑DD. Cells of different strains (top, YO1245, YO1246, YO1247, and YO1248; bottom, YO1213, YO1214, YO1215, and YO1216; chs2∆ leu2::LEU2 [pRS305, vector] or chs2∆ Chs2* (WT, chs2‑AA, or chs2‑DD)-GFP::LEU2 cells transformed with either an empty vector [pRS314] or a high‑copy CYK3 plasmid [pBK42]) were grown to exponential phase in rich medium (YM-1) at 25°C and then visualized by DIC microscopy. (C) Increased Cyk3 stimulates Chs2‑dependent chitin synthesis at the division site. Cells (YO1129, YO1130; chs1∆ chs3∆ cells transformed with either an empty vector [p414ADH] or a high‑copy CYK3 plasmid [pBK42]) were grown overnight at 30°C on an SC-TRP plate and then stained for cellular chitin using Calcofluor. The relative amounts of chitin in both strains (Vector, n = 14; CYK3, n = 22) were quantified and presented in the plot. The average intensity for the strain containing the “Vector” was arbitrarily set at 100. The error bar represents SEM. Scale bar, 2 μm. (D) chs2‑AA and chs2‑DD mutations display synthetically enhanced defects with cyk3Δ. Different strains (top, YO1338, YO1339, YO1450, and YO1451; chs2∆ leu2::LEU2 [pRS305, vector] and chs2∆ Chs2* (WT, chs2‑AA, or chs2‑DD)-GFP::LEU2; bottom, YO1209, YO1210, YO1211, and YO1212; chs2∆ cyk3∆ leu2::LEU2 [pRS305, vector] and chs2∆ cyk3∆ Chs2* (WT, chs2‑AA, or chs2‑DD)-GFP::LEU2) were grown to exponential phase in SC-LEU medium at 25°C and then visualized by DIC microscopy. Scale bar, 5 μm.
Similar to wild‑type Chs2, Chs2‑S217A and Chs2‑S217D accumulated at and then disappeared from the division site more slowly in the dbf2-mutant than in wild‑type cells, although Chs2‑S217D consistently seemed to disappear more quickly (Figure 6B). Together these data suggest that the endocytic removal of Chs2 is delayed in the dbf2 mutant, which is largely independent of Chs2 phosphorylation by Dbf2. This conclusion is supported by the previous observation that accumulation of actin patches at the division site, which are required for endocytosis in budding yeast, is significantly decreased in the dbf2 mutant (Corbett et al., 2006). Thus the MEN may also regulate the endocytic removal of Chs2 during cytokinesis by regulating actin‑patch polarization at the division site.
Cyk3 is a robust dosage suppressor of chs2 phosphomutants
Cyk3, a cytokinesis protein that localizes to the division site in a Dbf2‑dependent manner (Meitinger et al., 2010), has been implicated in PS formation, but the underlying mechanism remains unknown (Korinek et al., 2000; Nishihama et al., 2009; Meitinger et al., 2010). It is striking that high‑copy CYK3 robustly suppressed the growth and cytokinesis defects of both chs2-AA and chs2-DD cells (Figure 7, A and B). In contrast, high‑copy CYK3 failed to rescue chs2∆ cells (Figure 7B), suggesting that the suppression is Chs2 dependent. Of importance, high‑copy CYK3 strongly stimulated chitin synthesis at the division site of chs1Δ chs3Δ cells in which Chs2 is the sole chitin synthase (Figure 7C). Reciprocally, deletion of CYK3 exacerbated the defects of the chs2‑AA and chs2‑DD mutants even when the mutant cells were grown in minimal media (Figure 7D). Consistent with the notion of Cyk3 acting through Chs2, cyk3∆ and chs2∆ did not produce any additive or synergistic defect in growth or cytokinesis (Figure 7D). Together these data strongly suggest that Cyk3 is an activator of Chs2 in vivo and that Cyk3 and Ser-217 phosphorylation regulate Chs2 function independently during cytokinesis.
DISCUSSION
Dbf2 directly phosphorylates Chs2 to regulate its dynamics and chitin synthesis at the division site during cytokinesis
Our study demonstrated, for the first time, that Chs2 is a direct substrate of the conserved mitotic exit kinase Dbf2. Chs2 contains six putative Dbf2-consensus sites (RXXS; Ser-21, Ser-150, Ser-217, Ser-225, Ser-256, and Ser-326; Mah et al., 2005) at its N‑terminal region and two catalytically important as well as seven predicted transmembrane domains in its C‑terminal region (Figure 1A). In this study, we showed that both Ser-217 and Ser-225 are phosphorylated by Dbf2 in vitro. Of importance, the same two sites were also found to be phosphorylated in vivo in global studies of phosphoproteins in budding yeast (Chi et al., 2007; Dephoure and Gygi, 2011). Other putative Dbf2 sites and/or additional serines or threonines in the N‑terminal region of Chs2 may also be phosphorylated by Dbf2, as ∼43% of phosphorylation still remained when GST‑Chs2 (1–286, S217A S225A) was used as the substrate. However, because of the clear phenotypes displayed by the chs2‑S217 mutations, our functional analysis has thus far focused on phosphorylation at this residue. These results, together with the observations that Chs2 and Dbf2 colocalize at the division site during cytokinesis and that Dbf2 is also implicated in PS formation (Meitinger et al., 2010), make Chs2 a highly likely substrate of Dbf2 in vivo.
Our study suggests that Ser-217 phosphorylation regulates Chs2 dynamics and chitin synthesis at the division site during cytokinesis. Both the phosphodeficient and the phosphomimic mutations of Ser-217 caused little defect in the catalytic activity of Chs2 in the bulk chitin synthase assay in vitro but clearly caused a 30% reduction in chitin synthesis at the division site in vivo. Because the amount and duration of the Chs2‑S217A and Chs2‑S217D variants at the bud neck are similar to those of the wild‑type protein, these data suggest that Ser-217 is involved in fine-tuning the chitin synthase activity of Chs2. This is consistent with previous reports that the N‑terminal noncatalytic region of Chs2, including the Ser-217 residue (Figure 1A), is involved in the regulation of Chs2 activity based on in vitro chitin assays (Uchida et al., 1996; Martinez-Rucobo et al., 2009). The underlying mechanism for the involvement of Ser-217 in this regulation remains unknown. One possibility is that Ser-217 might affect the putative proteolysis of Chs2 that is believed to be required for its activation in vivo (Sburlati and Cabib, 1986; Uchida et al., 1996; Martinez-Rucobo et al., 2009). However, we note that in our experiments, the chs2-S217A and chs2-S217D mutations did not significantly affect the degree to which the in vitro Chs2 activity was stimulated by trypsin. The moderate decrease in chitin synthesis caused by both the chs2‑S217A and the chs2‑S217D mutations in vivo likely explains, at least in part, their medium‑dependent defects in cytokinesis. Presumably, cells demand more robust chitin synthesis at the division site to ensure efficient cytokinesis in the fast‑growing rich media than in the slow‑growing minimal media.
Despite their similar defects in chitin synthesis in vivo, the phosphodeficient and the phosphomimic variants of Chs2 at Ser-217 displayed strikingly different behaviors at the division site, which may also partly account for their defects in cytokinesis. Chs2‑S217A is associated with the AMR throughout cytokinesis. In contrast, Chs2‑S217D is largely uncoupled from the AMR upon its arrival at the division site. These observations suggest a simple model for the function of Ser-217 phosphorylation (Figure 8A). In wild‑type cells, “unphosphorylated” Chs2 is delivered to the division site at the onset of cytokinesis. Approximately halfway through AMR constriction, Dbf2 level reaches its peak at the bud neck, causing phosphorylation of Chs2 at Ser-217, resulting in Chs2 dissociation from the AMR. This dissociation may facilitate Chs2 inactivation and/or its endocytic removal from the division site. Persistent association of Chs2 with the AMR, as shown by Chs2‑S217A, somehow causes asymmetric distribution of the AMR components, resulting in asymmetric constriction. On the other hand, premature uncoupling of Chs2 from the AMR, as shown by Chs2‑S217D, results in broad and dynamic distribution of Chs2‑S217D between the septin rings, which presumably would lead to inefficient PS formation due to inaccurate placement of chitin. Thus timed phosphorylation of Ser-217 is important for proper PS formation and cytokinesis.
FIGURE 8:

Roles of CDK1 and MEN in the control of Chs2 localization, dynamics, and activation during cytokinesis. (A) Role of Dbf2 in regulating Chs2–AMR association during cytokinesis. (B) Multipronged regulation of Chs2 function by MEN. MEN regulates Chs2 exit from the ER by antagonizing CDK1 phosphorylation, regulates Chs2 dynamics by direct phosphorylation, and stimulates Chs2 activity through Cyk3 by a yet‑to‑be‑defined mechanism. See the text for details.
Dbf2 regulates primary septum formation through multiple pathways
Dbf2 appears to regulate PS formation at multiple levels via distinct mechanisms (Figure 8B). First, during mitosis, Chs2 is phosphorylated by CDK1 at four residues (Ser-14, Ser-60, Ser-69, and Ser-100), and this phosphorylation prevents Chs2 from prematurely exiting the ER before cytokinesis (Teh et al., 2009). During mitotic exit, Dbf2, as a part of the MEN, activates the phosphatase Cdc14, which removes the CDK1 phosphorylation from Chs2, thus promoting its exit from the ER (Chin et al., 2011). Second, we show here that Dbf2 phosphorylates Chs2 directly at the division site to regulate its chitin synthase activity and also its dissociation from the AMR. This regulatory mechanism plays an important role in cytokinesis. Third, Dbf2 regulates PS formation indirectly by controlling Cyk3 localization to the division site (Meitinger et al., 2010). We show here that increased dosage of Cyk3 stimulates Chs2‑mediated chitin synthesis and robustly suppresses chs2‑AA and chs2‑DD mutants, whereas deletion of CYK3 exacerbates the phenotypes of the chs2 mutants. These data suggest that Cyk3 is a potent activator of Chs2 in vivo and that Cyk3 and Dbf2‑mediated phosphorylation regulate Chs2 function independently during cytokinesis. Fourth, Dbf2 may regulate the endocytic removal of Chs2 from the division site by regulating actin‑patch polarization at the division site. Finally, Dbf2 is also known to phosphorylate the F‑BAR protein Hof1 (Meitinger et al., 2011), which has been implicated in PS formation and/or its coordination with the AMR during cytokinesis (Vallen et al., 2000; Meitinger et al., 2010, 2011).
It becomes increasingly clear that ECM remodeling is important for animal cytokinesis (Mizuguchi et al., 2003; Izumikawa et al., 2010). However, how the ECM is locally remodeled at the division site and how this remodeling is coupled to AMR constriction and controlled by the cell cycle machinery remain largely unknown in animal cells. In this study, we established a mechanism by which the conserved mitotic exit kinase Dbf2 directly phosphorylates Chs2 to regulate PS formation (or ECM remodeling) during cytokinesis in budding yeast. On the basis of the overall conservation in the core mechanisms of cell cycle control and cytokinesis between yeast and animal cells, we speculate that similar cell cycle regulation of ECM remodeling during cytokinesis likely exists in animal cells.
MATERIALS AND METHODS
Strains and growth media
Yeast strains used in this study are listed in Table 1. Standard culture media and genetic techniques were used (Guthrie and Fink, 1991). Escherichia coli strains DH12S (Invitrogen, Carlsbad, CA) and BL21 (DE3) (Invitrogen) were used as hosts for plasmid manipulation and recombinant protein expression, respectively. Yeast strains were grown routinely at 25°C in synthetic complete (SC) minimal medium lacking specific amino acid(s) and/or uracil or in rich medium YM-1 (Lillie and Pringle, 1980) or yeast extract/peptone/dextrose (YPD).
TABLE 1:
Yeast strains used in this study. Continued
| Strain | Genotype | Source |
|---|---|---|
| J230‑2D | a dbf2‑1 dbf20∆::TRP1 ade1 leu2 trp1 ura3 | Toyn and Johnston (1994) |
| YEF473 | a/α his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | Bi and Pringle (1996) |
| YEF473A | a his3 leu2 lys2 trp1 ura3 | Bi and Pringle (1996) |
| YEF473B | α his3 leu2 lys2 trp1 ura3 | Bi and Pringle (1996) |
| YEF2845 | As YEF473 except chs2∆::His3MX6/CHS2 | This studya |
| YEF6653 | a CHS2-GFP::His3MX6 CDC3-mCherry::URA3 | This study |
| YO790 | a chs1∆::KanMX6 chs3∆::His3MX6 | This studya |
| YO1111 | a chs1∆::NatMX6 chs3∆::His3MX6 | This study |
| YO1172 | a/α chs2∆::His3MX6/CHS2 chs3∆::KanMX6/CHS3 [pEC2, 2 μ, CHS2, URA3] | This study |
| YO1182 | a CHS2-GFP::KanMX6 DBF2-mCherry::His3MX6 | This study |
| YO1194 | α chs2∆::His3MX6 [pEC2, 2 μ, CHS2, URA3] | This study |
| YO1209 | a chs2∆::His3MX6 cyk3∆::TRP1 leu2::LEU2‑pRS305 | This study |
| YO1210 | a chs2∆::His3MX6 cyk3∆::TRP1 CHS2-GFP::KanMX6::LEU2 | This study |
| YO1211 | a chs2∆::His3MX6 cyk3∆::TRP1 chs2-S217A S225A-GFP::KanMX6::LEU2 | This study |
| YO1212 | a chs2∆::His3MX6 cyk3∆::TRP1 chs2-S217D S225D-GFP::KanMX6::LEU2 | This study |
| YO1213 | a chs2∆::His3MX6 leu2::LEU2‑pRS305 [pBK42, 2 μ, CYK3, TRP1] | This study |
| YO1214 | a chs2∆::His3MX6 CHS2-GFP::KanMX6::LEU2 [pBK42, 2 μ, CYK3, TRP1] | This study |
| YO1215 | a chs2∆::His3MX6 chs2-S217A S225A-GFP::KanMX6::LEU2 [pBK42, 2 μ, CYK3, TRP1] | This study |
| YO1216 | a chs2∆::His3MX6 chs2-S217D S225D-GFP::KanMX6::LEU2 [pBK42, 2 μ, CYK3, TRP1] | This study |
| YO1129 | a chs1∆::KanMX6 chs3∆::His3MX6 [p414ADH, CEN, TRP1] | This study |
| YO1130 | a chs1∆::KanMX6 chs3∆::His3MX6 [pBK42, 2 μ, CYK3, TRP1] | This study |
| YO1244 | a chs1∆::TRP1 chs2∆::His3MX6 chs3∆::KanMX6 [pEC2, 2 μ, CHS2, URA3] | This study |
| YO1245 | a chs2∆::His3MX6 leu2::LEU2‑pRS305 [pRS314, CEN, TRP1] | This study |
| YO1246 | a chs2∆::His3MX6 CHS2-GFP::KanMX6::LEU2 [pRS314, CEN, TRP1] | This study |
| YO1247 | a chs2∆::His3MX6 chs2-S217A S225A-GFP::KanMX6::LEU2 [pRS314, CEN, TRP1] | This study |
| YO1248 | a chs2∆::His3MX6 chs2-S217D S225D-GFP::KanMX6::LEU2 [pRS314, CEN, TRP1] | This study |
| YO1320 | a chs1∆::TRP1 chs2∆::His3MX6 chs3∆::KanMX6 [YCp111-CHS2-GFP::KanMX6, CEN, LEU2] | This study |
| YO1324 | a chs1∆::TRP1 chs2∆::His3MX6 chs3∆::KanMX6 [YCp111-CHS2-S217A‑GFP::KanMX6, CEN, LEU2] | This study |
| YO1325 | a chs1∆::TRP1 chs2∆::His3MX6 chs3∆::KanMX6 [YCp111-CHS2-S217D-GFP::KanMX6, CEN, LEU2] | This study |
| YO1338 | α chs2∆::His3MX6 CDC3-mCherry::TRP1 leu2::LEU2‑pRS305 | This study |
| YO1339 | α chs2∆::His3MX6 CDC3-mCherry::TRP1 CHS2-GFP::KanMX6::LEU2 | This study |
| YO1359 | α chs2∆::His3MX6 CDC3-mCherry::TRP1 chs2‑S217A-GFP::KanMX6::LEU2 | This study |
| YO1360 | α chs2∆::His3MX6 CDC3-mCherry::TRP1 chs2‑S217D-GFP::KanMX6::LEU2 | This study |
| YO1398 | a dbf2‑1 dbf20∆::TRP1 CDC3‑mCherry::LEU2 | This studyb |
| YO1399 | a dbf2‑1 dbf20∆::TRP1 CDC3‑mCherry::LEU2 pGAL1‑SIC1s (T5V, T33V, S76A)‑HA::URA3 [YCp111‑CHS2‑GFP::KanMX6, CEN, LEU2] | This study |
| YO1400 | a dbf2‑1 dbf20∆::TRP1 CDC3‑mCherry::LEU2 pGAL1‑SIC1s (T5V, T33V, S76A)‑HA::URA3 | This study |
| YO1410 | a dbf2‑1 dbf20∆::TRP1 CDC3‑mCherry::LEU2 pGAL1‑SIC1s (T5V, T33V, S76A)‑HA::URA3 [YCp111‑CHS2‑S217A‑GFP::KanMX6, CEN, LEU2] | This study |
| YO1411 | a dbf2‑1 dbf20∆::TRP1 CDC3‑mCherry::LEU2 pGAL1‑SIC1s (T5V, T33V, S76A)‑HA::URA3 [YCp111‑CHS2‑S217D‑GFP::KanMX6, CEN, LEU2] | This study |
| YO1450 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217A S225A-GFP::KanMX6::LEU2 | This study |
| YO1451 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217D S225D-GFP::KanMX6::LEU2 | This study |
| YO1454 | α chs2∆::His3MX6 CHS2-GFP::KanMX6::LEU2 [pRS314-MYO1-C-mCherry, CEN, TRP1] | This study |
| YO1453 | α chs2∆::His3MX6 chs2-S217A S225A-GFP::KanMX6::LEU2 [pRS314-MYO1-C-mCherry, CEN, TRP1] | This study |
| YO1455 | α chs2∆::His3MX6 chs2-S217D S225D-GFP::KanMX6::LEU2 [pRS314-MYO1-C-mCherry, CEN, TRP1] | This study |
| YO1528 | a chs1∆::NatMX6 chs3∆::His3MX6 [pRS314, CEN, TRP1] | This study |
| YO1535 | a chs1∆::NatMX6 chs3∆::His3MX6 [YEp-TRP1-pGAL1-HA3-CHS2, 2 μ, URA3] | This study |
| YO1536 | a chs1∆::NatMX6 chs3∆::His3MX6 [YEp-TRP1-pGAL1-HA3-CHS2-S217A, 2 μ, URA3] | This study |
| YO1537 | a chs1∆::NatMX6 chs3∆::His3MX6 [YEp-TRP1-pGAL1-HA3-CHS2-S217D, 2 μ, URA3] | This study |
| YO1543 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217A S225A-GFP::KanMX6::LEU2 [pAG426, 2 μ, URA3] | This study |
| YO1544 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217A S225A-GFP::KanMX6::LEU2 [pAG426-CYK3, 2 μ, URA3] | This study |
| YO1550 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217D S225D-GFP::KanMX6::LEU2 [pAG426, 2 μ, URA3] | This study |
| YO1551 | α chs2∆::HIS3 CDC3-mCherry::TRP1 chs2-S217D S225D-GFP::KanMX6::LEU2 [pAG426-CYK3, 2 μ, URA3] | This study |
aAll YEF and YO strains except YO1398‑1411 are derived from YEF473, YEF473A, or YEF473B.
bDerived from J230‑2D.
Constructions of plasmids and yeast strains
Common plasmid vectors used in this study include YCplac111 (CEN, LEU2), pRS314 (CEN, TRP1), pRS316 (CEN, URA3), pRS305 (integrative, LEU2), and YIplac204 (integrative, TRP1). Plasmids p414ADH (CEN, TRP1; Mumberg et al., 1995) and pBK42 (2μ, CYK3, TRP1; Korinek et al., 2000) were described previously. Plasmid YIplac211‑GAL1‑SIC1s (T5V, T33V, S76A)‑HA (integrative, URA3; Meitinger et al., 2010; kindly supplied by Gislene Pereira, German Cancer Research Center, Heidelberg, Germany), carrying a GAL1 promoter–controlled allele of SIC1s that encodes a stabilized protein, was cut with EcoRV and integrated at the ura3 locus of the recipient strain. Plasmids YIplac128‑CDC3‑mCherry (integrative, LEU2; Gao et al., 2007), YIplac211‑CDC3‑mCherry (integrative, URA3; Fang et al., 2010), and YIplac204-CDC3-mCherry (integrative, TRP1; Wloka et al., 2011), carrying mCherry‑tagged CDC3, were digested with BglII and integrated at the CDC3 locus of the recipient strains. Plasmid pGEX5X‑CHS2‑N was constructed by cloning a BamHI- and SalI-digested (both sites were introduced in PCR primers) CHS2 fragment encoding amino acids 1–286 into the corresponding sites of pGEX5X-1 (GE Healthcare, Pittsburgh, PA). YEp-pGAL1-HA3-CHS2 was constructed by inserting a 1.7-kb PCR fragment carrying TRP1‑pGAL1‑HA3 (Longtine et al., 1998) in front of CHS2 on plasmid pEC2 (2 μ, CHS2, URA3; Ford et al., 1996). The QuikChange Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) was used to generate phosphodeficient mutations (S217A and/or S225A) or phosphomimic mutations (S217D and/or S225D) in CHS2 using plasmid YCp111-CHS2-GFP‑KanMX6 as the template (Wloka et al., 2011). Wild‑type and the phosphomutant alleles of CHS2 were then subcloned into the integrative vector pRS305 (LEU2), as described previously (Wloka et al., 2011). The resulting plasmid pRS305‑CHS2*‑GFP was digested with AflII and integrated at the leu2 locus of the recipient strains. Plasmid pAG24‑CYK3 was constructed by transferring CYK3 from the tiling library plasmid pGP564-CYK3 (2 μ, LEU2; Open Biosystems, Lafayette, CO) onto the Gateway vector pAG24 (2 μ, URA3) using Gateway LR Clonase II (Invitrogen, Grand Island, NY).
Microscopy
A computer-controlled Eclipse 800 microscope (Nikon, Tokyo, Japan) and a high-resolution charge-coupled device camera (model C4742-95; Hamamatsu Photonics, Bridgewater, NJ) were used to visualize cell morphology, Calcofluor‑stained chitin (Pringle, 1991), and CHS2*‑GFP (wild-type [WT] or phosphomutant alleles of CHS2) by differential interference contrast (DIC) and fluorescence microscopy, respectively. The images were acquired using Image-Pro Plus version 7.0 (Media Cybernetics, Bethesda, MD).
For live‑cell imaging of yeast strains carrying a plasmid, cells were grown at 25°C in SC media lacking a specific amino acid to select for the presence of a plasmid. For imaging yeast strains carrying a chs2 phosphomutation, cells were first grown in SC‑LEU medium to keep them healthy and then washed twice with rich medium YM-1 without carbon source. Cells were then incubated further for 1–3 h at 25°C before being concentrated by centrifugation and spotted on a slab of YM-1 medium containing 2% agarose for live‑cell imaging. For live‑cell imaging at 37°C (Figure 6), the wild‑type and the dbf2‑1 dbf20∆ strains were grown in liquid YM‑1 media containing 2% raffinose at 25°C to the exponential phase and then shifted to 37°C for additional 3 h. A 2.0‑ml culture was then transferred to a 3.5‑mm dish coated with polylysine (MatTek, Ashland, MA), which was incubated in the 37°C chamber of the microscope for 20 min to let the cells settle down. A 1.5‑ml culture was then removed from the dish, which was followed by the addition of a 1.5‑ml prewarmed YM-1 medium containing 2% raffinose, 3% galactose, and 1.5% low‑melting agarose. Images were acquired at 25 or 37°C on a spinning disk confocal microscope equipped with a Yokogawa CSU 10 scan head combined with an Olympus IX 71 microscope (Olympus, Tokyo, Japan) and an Olympus 100× objective (1.4 numerical aperture, Plan S-Apo oil immersion). Acquisition and hardware were controlled by MetaMorph, version 7.7 (Molecular Devices, Downingtown, PA). A Hamamatsu ImagEM EMCCD camera (model C9100-13, Bridgewater, NJ) was used for capture. Diode lasers for excitation (488 nm for GFP and 561 nm for mCherry/RFP) were housed in a launch constructed by Spectral Applied Research (Richmond Hill, Canada). Images were taken every minute with z‑stacks ranging from 11 × 0.3 to 12 × 0.5 μm. A maximum projection was generated with MetaMorph, version 7.7, or ImageJ (1.45b; National Institutes of Health, Bethesda, MD). For quantification of relative chitin levels in different chs2 phosphomutants (Figure 3), the ratio between Calcofluor signal intensity at the bud neck was divided by Chs2-GFP signal intensity at the bud neck.
FIGURE 6:
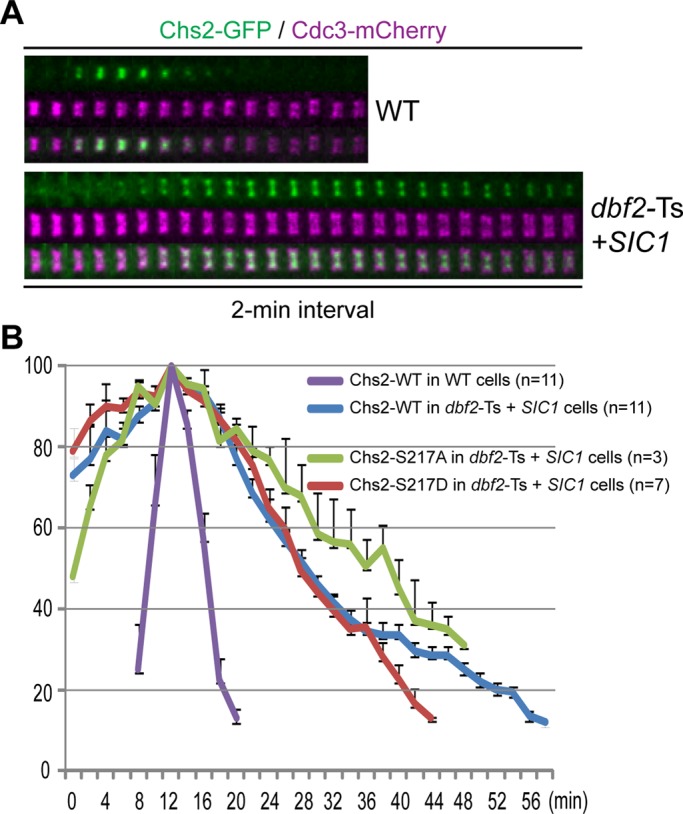
Inactivation of Dbf2 prolongs Chs2 localization at the division site. (A) Chs2 disappears from the division site slowly in a dbf2 mutant. Wild‑type (WT, CHS2‑GFP CDC3‑mCherry; YEF6653) and the dbf2‑Ts mutant (dbf2‑1 dbf20∆ CDC3‑mCherry, CHS2‑GFP, pGAL1‑SIC1s; YO1399) cells were grown under conditions to inactivate Dbf2 and also overexpress Sic1 to force mitotic exit in the mutant cells (see Materials and Methods for details) and then analyzed by time‑lapse microscopy. (B) Endocytic removal of Chs2 is delayed in the dbf2 mutant. Quantification of Chs2*‑GFP disappearance from the division site was performed on the time‑lapse data acquired in A, as well as those acquired from strains YO1410 (dbf2‑1 dbf20∆ CDC3‑mCherry, chs2‑S217A‑GFP, pGAL1‑SIC1s) and YO1411 (dbf2‑1 dbf20∆ CDC3‑mCherry, chs2‑S217D‑GFP, pGAL1‑SIC1s). The average of the peak fluorescence of Chs2*‑GFP for each strain was set at 100. The error bar represents SEM.
For the experiments involving FRAP, cells were grown to exponential phase in SC-HIS media at 23°C. Cells were then centrifuged and resuspended in 10 ml YM-1 medium and grown for at least another 3 h in a 50‑ml flask at 25°C in a water‑bath shaker. One milliliter of this culture was taken to concentrate the cells by centrifugation. Cells were then spotted on YM-1 medium containing 2% agarose for FRAP and imaging analysis. FRAP was performed using a MicroPoint computer-controlled ablation system (Photonic Instruments, St. Charles, IL) consisting of a nitrogen-pumped dye laser (wavelength, 435 nm) controlled by MetaMorph. In some cases, sequential photobleaching was applied (Figure 5A, S217D). Images were captured and processed using the same microscope, camera, and software as described earlier. Images were taken every 20 s with a z-stack consisting of 12 × 0.4 μm steps.
Kinase assay
GST-Chs2(1-286; WT, S217A, or S217D) fusion proteins were expressed in the E. coli strain BL21 (DE3) and purified as described previously (Fang et al., 2010). Dbf2-dependent phosphorylation of GST-Chs2(1-286) was performed as described previously (Mohl et al., 2009).
In vitro assay for Chs2 activity
Cells (YO1528, YO1535, YO1536, and YO1537) were grown for 24 h at 30°C in 30 ml of SC medium supplemented with tryptophan and lysine. Cells were sedimented by centrifugation and then resuspended in 250 ml 0.67% (wt/vol) yeast nitrogen base containing 2% (wt/vol) galactose, 1% (wt/vol) raffinose, tryptophan, and lysine. These cultures were incubated for 18 h at 25°C. Mixed membranes were prepared as described previously (Orlean, 1987) with several slight modifications. Induced cultures were harvested, and cells were washed once with 12 ml of cold water and once with 10 ml of cold 30 mM Tris-HCl (pH 7.5). All subsequent steps were carried out on ice. Cells were broken by vortexing with glass beads in 30 mM Tris-HCl (pH 7.5), the beads were then washed with 30 mM Tris-HCl (pH 7.5), and cell walls and unbroken cells were sedimented by low-speed centrifugation. The pellet was washed once with 30 mM Tris-HCl (pH 7.5), and the combined wall-free supernatants (10 ml) were sedimented by centrifugation at 100,000 × g for 45 min at 4°C. Mixed membranes were then homogenized in cold 30 mM Tris-HCl (pH 7.5) and then sedimented again at 100,000 × g. The washed membranes were homogenized in 0.9 ml of cold 30 mM Tris-HCl (pH 7.5) containing 33% (vol/vol) glycerol and then frozen at –80°C. Protein contents of membranes were determined using the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA). Trypsin treatment was carried out on 100-μl portions of mixed membranes by adding 3 μl of solutions containing 100 or 200 μg of trypsin (Sigma‑Aldrich, St. Louis, MO) in 30 mM Tris-HCl (pH 7.5). After incubation at 30°C for 10 min, trypsin digestion was stopped by adding 3 μl of solutions containing an amount of soybean trypsin inhibitor (Sigma‑Aldrich) double the amount of trypsin used. As no-trypsin controls, samples of membranes were incubated in parallel with buffer alone for 10 min at 30°C, after which 3 μl of a solution containing 200 μg of trypsin inhibitor was added. The treated membranes were then placed on ice and assayed forthwith. Assay mixtures contained, in a final volume of 50 μl, 2 mM UDP GlcNAc, UDP-[14C(U)] GlcNAc (specific activity, 300 mCi/mmol, 61,000 cpm per incubation [American Radiolabeled Chemicals, St. Louis, MO]), 32 mM GlcNAc, and 2.5 mM cobalt acetate. Reactions were started by the addition of 20 μl of trypsin-treated or control membranes (typically containing ∼400 μg of membrane proteins) and terminated by the addition of 1 ml of cold 10% (wt/vol) trichloroacetic acid. Insoluble radioactivity was collected by filtration on GF/C discs, and the filters were washed as described previously (Orlean, 1987).
Supplementary Material
Acknowledgments
We thank Aaron Gitler and Gislene Pereira for providing plasmids and the members of the Bi, Deshaies, and Orlean laboratories for discussions. This work was supported by National Institutes of Health Grants GM59216 and GM87365 (to E.B.), a fellowship from the Boehringer Ingelheim Fonds (to C.W.), and Taiwan Merit Scholarship TMS-094-1-A-026 (to K.J.C.). R.D. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations used:
- AMR
actomyosin ring
- ECM
extracellular matrix
- PS
primary septum
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-01-0033) on May 9, 2012.
REFERENCES
- Balasubramanian MK, Bi E, Glotzer M. Comparative analysis of cytokinesis in budding yeast, fission yeast and animal cells. Curr Biol. 2004;14:R806–R818. doi: 10.1016/j.cub.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131:847–860. doi: 10.1016/j.cell.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Bi E. Cytokinesis in budding yeast: the relationship between actomyosin ring function and septum formation. Cell Struct Funct. 2001;26:529–537. doi: 10.1247/csf.26.529. [DOI] [PubMed] [Google Scholar]
- Bi E, Pringle JR. ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa CE, Osmond BC. Chitin synthase I and chitin synthase II are not required for chitin synthesis in vivo in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1990;87:7424–7428. doi: 10.1073/pnas.87.19.7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci USA. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CF, Bennett AM, Ma WK, Hall MC, Yeong FM. Dependence of Chs2 ER export on dephosphorylation by cytoplasmic Cdc14 ensures that septum formation follows mitosis. Mol Biol Cell. 2011;23:45–58. doi: 10.1091/mbc.E11-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JS, Schekman RW. Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p. J Cell Biol. 1996;135:597–610. doi: 10.1083/jcb.135.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett M, Xiong Y, Boyne JR, Wright DJ, Munro E, Price C. IQGAP and mitotic exit network (MEN) proteins are required for cytokinesis and re-polarization of the actin cytoskeleton in the budding yeast, Saccharomyces cerevisiae. Eur J Cell Biol. 2006;85:1201–1215. doi: 10.1016/j.ejcb.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Gygi SP. A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods. 2011;54:379–386. doi: 10.1016/j.ymeth.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp JA, Chant J. An IQGAP-related protein controls actin-ring formation and cytokinesis in yeast. Curr Biol. 1997;7:921–929. doi: 10.1016/s0960-9822(06)00411-8. [DOI] [PubMed] [Google Scholar]
- Fang X, Luo J, Nishihama R, Wloka C, Dravis C, Travaglia M, Iwase M, Vallen EA, Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin-II. J Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford RA, Shaw JA, Cabib E. Yeast chitin synthases 1 and 2 consist of a non-homologous and dispensable N-terminal region and of a homologous moiety essential for function. Mol Gen Genet. 1996;252:420–428. doi: 10.1007/BF02173007. [DOI] [PubMed] [Google Scholar]
- Frenz LM, Lee SE, Fesquet D, Johnston LH. The budding yeast Dbf2 protein kinase localises to the centrosome and moves to the bud neck in late mitosis. J Cell Sci. 2000;113:3399–3408. doi: 10.1242/jcs.113.19.3399. [DOI] [PubMed] [Google Scholar]
- Gao XD, Sperber LM, Kane SA, Tong Z, Hin Yan Tong A, Boone C, Bi E. Sequential and distinct roles of the cadherin domain-containing protein Axl2p in cell polarization in yeast cell cycle. Mol Biol Cell. 2007;18:2542–2560. doi: 10.1091/mbc.E06-09-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR, editors. Guide to Yeast Genetics and Molecular Biology. New York: Academic Press; 1991. [Google Scholar]
- Hwa Lim H, Yeong FM, Surana U. Inactivation of mitotic kinase triggers translocation of MEN components to mother-daughter neck in yeast. Mol Biol Cell. 2003;14:4734–4743. doi: 10.1091/mbc.E03-04-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumikawa T, Kanagawa N, Watamoto Y, Okada M, Saeki M, Sakano M, Sugahara K, Sugihara K, Asano M, Kitagawa H. Impairment of embryonic cell division and glycosaminoglycan biosynthesis in glucuronyltransferase-I-deficient mice. J Biol Chem. 2010;285:12190–12196. doi: 10.1074/jbc.M110.100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek WS, Bi E, Epp JA, Wang L, Ho J, Chant J. Cyk3, a novel SH3-domain protein, affects cytokinesis in yeast. Curr Biol. 2000;10:947–950. doi: 10.1016/s0960-9822(00)00626-6. [DOI] [PubMed] [Google Scholar]
- Lillie SH, Pringle JR. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bacteriol. 1980;143:1384–1394. doi: 10.1128/jb.143.3.1384-1394.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott J, Li R. Sequential assembly of myosin II, an IQGAP-like protein, and filamentous actin to a ring structure involved in budding yeast cytokinesis. J Cell Biol. 1998;140:355–366. doi: 10.1083/jcb.140.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott J, Shannon KB, Shou W, Deshaies RJ, Li R. The Tem1 small GTPase controls actomyosin and septin dynamics during cytokinesis. J Cell Sci. 2001;114:1379–1386. doi: 10.1242/jcs.114.7.1379. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, DeMarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Mah AS, Elia AE, Devgan G, Ptacek J, Schutkowski M, Snyder M, Yaffe MB, Deshaies RJ. Substrate specificity analysis of protein kinase complex Dbf2-Mob1 by peptide library and proteome array screening. BMC Biochem. 2005;6:22. doi: 10.1186/1471-2091-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah AS, Jang J, Deshaies RJ. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc Natl Acad Sci USA. 2001;98:7325–7330. doi: 10.1073/pnas.141098998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Eckhardt-Strelau L, Terwisscha van Scheltinga AC. Yeast chitin synthase 2 activity is modulated by proteolysis and phosphorylation. Biochem J. 2009;417:547–554. doi: 10.1042/BJ20081475. [DOI] [PubMed] [Google Scholar]
- Meitinger F, Boehm ME, Hofmann A, Hub B, Zentgraf H, Lehmann WD, Pereira G. Phosphorylation-dependent regulation of the F-BAR protein Hof1 during cytokinesis. Genes Dev. 2011;25:875–888. doi: 10.1101/gad.622411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Petrova B, Mancini Lombardi I, Bertazzi DT, Hub B, Zentgraf H, Pereira G. Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J Cell Sci. 2010;123:1851–1861. doi: 10.1242/jcs.063891. [DOI] [PubMed] [Google Scholar]
- Mizuguchi S, Uyama T, Kitagawa H, Nomura KH, Dejima K, Gengyo-Ando K, Mitani S, Sugahara K, Nomura K. Chondroitin proteoglycans are involved in cell division of Caenorhabditis elegans. Nature. 2003;423:443–448. doi: 10.1038/nature01635. [DOI] [PubMed] [Google Scholar]
- Mohl DA, Huddleston MJ, Collingwood TS, Annan RS, Deshaies RJ. Dbf2-Mob1 drives relocalization of protein phosphatase Cdc14 to the cytoplasm during exit from mitosis. J Cell Biol. 2009;184:527–539. doi: 10.1083/jcb.200812022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Nagahashi S, Sudoh M, Ono N, Sawada R, Yamaguchi E, Uchida Y, Mio T, Takagi M, Arisawa M, Yamada-Okabe H. Characterization of chitin synthase 2 of Saccharomyces cerevisiae. Implication of two highly conserved domains as possible catalytic sites. J Biol Chem. 1995;270:13961–13967. doi: 10.1074/jbc.270.23.13961. [DOI] [PubMed] [Google Scholar]
- Nishihama R, et al. Role of Inn1 and its interactions with Hof1 and Cyk3 in promoting cleavage furrow and septum formation in S. cerevisiae. J Cell Biol. 2009;185:995–1012. doi: 10.1083/jcb.200903125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlean P. Two chitin synthases in Saccharomyces cerevisiae. J Biol Chem. 1987;262:5732–5739. [PubMed] [Google Scholar]
- Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010;22:50–56. doi: 10.1016/j.ceb.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR. Staining of bud scars and other cell wall chitin with Calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- Sburlati A, Cabib E. Chitin synthetase 2, a presumptive participant in septum formation in Saccharomyces cerevisiae. J Biol Chem. 1986;261:15147–15152. [PubMed] [Google Scholar]
- Schmidt M, Bowers B, Varma A, Roh D-H, Cabib E. In budding yeast, contraction of the actomyosin ring and formation of the primary septum at cytokinesis depend on each other. J Cell Sci. 2002;115:293–302. doi: 10.1242/jcs.115.2.293. [DOI] [PubMed] [Google Scholar]
- Shaw JA, Mol PC, Bowers B, Silverman SJ, Valdivieso MH, Duran A, Cabib E. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J Cell Biol. 1991;114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. 2004;38:203–232. doi: 10.1146/annurev.genet.38.072902.093051. [DOI] [PubMed] [Google Scholar]
- Strickland LI, Burgess DR. Pathways for membrane trafficking during cytokinesis. Trends Cell Biol. 2004;14:115–118. doi: 10.1016/j.tcb.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Teh EM, Chai CC, Yeong FM. Retention of Chs2p in the ER requires N-terminal CDK1-phosphorylation sites. Cell Cycle. 2009;8:2964–2974. [PubMed] [Google Scholar]
- Toyn JH, Johnston LH. The Dbf2 and Dbf20 protein kinases of budding yeast are activated after the metaphase to anaphase cell cycle transition. EMBO J. 1994;13:1103–1113. doi: 10.1002/j.1460-2075.1994.tb06359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Shimmi O, Sudoh M, Arisawa M, Yamada-Okabe H. Characterization of chitin synthase 2 of Saccharomyces cerevisiae. II: Both full size and processed enzymes are active for chitin synthesis. J Biochem. 1996;119:659–666. doi: 10.1093/oxfordjournals.jbchem.a021293. [DOI] [PubMed] [Google Scholar]
- Vallen EA, Caviston J, Bi E. Roles of Hof1p, Bni1p, Bnr1p, and Myo1p in cytokinesis in Saccharomyces cerevisiae. Mol Biol Cell. 2000;11:593–611. doi: 10.1091/mbc.11.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank L, Li R. Cell cycle-regulated trafficking of Chs2 controls actomyosin ring stability during cytokinesis. Mol Biol Cell. 2005;16:2529–2543. doi: 10.1091/mbc.E04-12-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C, Nishihama R, Onishi M, Oh Y, Hanna J, Pringle JR, Krauß M, Bi E. Evidence that septin diffusion barriers are dispensable for cytokinesis in budding yeast. Biol Chem. 2011;392:813–829. doi: 10.1515/BC.2011.083. [DOI] [PubMed] [Google Scholar]
- Xu S, Huang HK, Kaiser P, Latterich M, Hunter T. Phosphorylation and spindle pole body localization of the Cdc15p mitotic regulatory protein kinase in budding yeast. Curr Biol. 2000;10:329–332. doi: 10.1016/s0960-9822(00)00382-1. [DOI] [PubMed] [Google Scholar]
- Zhang G, Kashimshetty R, Ng KE, Tan HB, Yeong FM. Exit from mitosis triggers Chs2p transport from the endoplasmic reticulum to mother-daughter neck via the secretory pathway in budding yeast. J Cell Biol. 2006;174:207–220. doi: 10.1083/jcb.200604094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.