

Abstract
Nanostructured materials based on colloidal particles embedded in a polymer network are used in a variety of applications ranging from nanocomposite rubbers to organic-inorganic hybrid solar cells. Further, polymer-network-mediated colloidal interactions are highly relevant to biological studies whereby polymer hydrogels are commonly employed to probe the mechanical response of living cells, which can determine their biological function in physiological environments. The performance of nanomaterials crucially relies upon the spatial organization of the colloidal particles within the polymer network that depends, in turn, on the effective interactions between the particles in the medium. Existing models based on nonlocal equilibrium thermodynamics fail to clarify the nature of these interactions, precluding the way toward the rational design of polymer-composite materials. In this article, we present a predictive analytical theory of these interactions based on a coarse-grained model for polymer networks. We apply the theory to the case of colloids partially embedded in cross-linked polymer substrates and clarify the origin of attractive interactions recently observed experimentally. Monte Carlo simulation results that quantitatively confirm the theoretical predictions are also presented.
Keywords: polymer nanocomposites, polymer bridging, colloidal aggregation, depletion force
Incorporation of colloidal micro and nanoparticles in cross-linked polymeric materials can be used to tune their structural and electronic properties. For example, introducing inorganic nanoparticles is a well-established technique to reinforce rubbers. The mechanical properties of the material will strongly depend on the size and conformation of colloidal aggregates (1–5), and, in the last instance, on the polymer-network-mediated colloidal interactions. Semiconductor nanocolloids can be incorporated in polymer-based organic photovoltaic devices to improve efficiency (6–10), therefore, controlling the aggregation of colloids is needed to optimize the performances of materials for renewable energy production. For these reasons, there is an urgent need of tools to understand and control colloidal interactions in polymer networks, which is currently a bottleneck in the rational design of functional nanostructured materials.
Besides technological applications, polymeric cross-linked hydrogels are often adopted as model systems to study changes in motility, differentiation, and morphology of living cells in response to variations in the compliance of their environment (11–18). Recently, experimental evidence of short-range attractive interactions between silica colloids deposited on extremely soft polyacrylamide hydrogels have been reported. However, knowledge about the origin of these interactions is still missing (19).
Colloidal interactions mediated by nonadsorbing polymers in dilute solutions have been successfully described in terms of depletion by the theory of Asakura and Oosawa (20). Scientific investigation then turned to the effect induced by adsorbing polymers forming coronae around colloidal particles (21–25). The main practical motivation being the clarification of the factors leading to net repulsion between colloids in solution (steric stabilization). However, the possibility of attractive polymer-mediated interactions other than depletion was predicted under certain conditions (i.e., partially saturated colloid surface). Thereby, the idea of polymer bridging was introduced to explain such effects (24, 26, 27). In more recent years, extensive numerical studies have been devoted to clarify the origin of effective interactions between nanoparticles dispersed in concentrated polymer solutions or melts, also for the case of adsorbing polymers (28–38).
In those limits the mixture can be treated globally using the tools of equilibrium statistical mechanics, which is justified as long as the polymers are not cross-linked and their concentration is far away from the glassy regime. However, when the nanoparticles are embedded in a polymer network the system as a whole is intrinsically far from equilibrium. Under these conditions, the origin of the effective attraction between colloids remains unclear, a major obstacle to its understanding being the inapplicability of nonlocal equilibrium theories.
In the present article, we develop a local statistical mechanical approach to the problem of colloids partially or totally embedded in cross-linked polymers starting from basic laws of polymer physics. We implement a coarse-grained model and derive a fully analytical expression of intercolloidal forces assuming local thermodynamic equilibrium on length scales smaller than the network mesh-size. Our theory provides a physically transparent description of the effects without trivial fitting parameters. We test this approach on the problem of interactions between colloids partially embedded in the surface of soft cross-linked networks. The analytical results are in excellent quantitative agreement with Monte Carlo simulations and reproduce available experimental data (19). Therefore, our theory opens up the way for the rational design of nanocomposite materials with tailored colloidal interactions and microstructure. Moreover, it provides a basic and clear understanding of the interplay between entropic and energetic contributions determining these interactions, which is an important theoretical result on its own with ramifications in biophysics and surface science.
In this paper, first we introduce the coarse grained model for the colloid-polymer network system made of silica colloids and polyacrylamide (PAA), describe the analytical derivation of the force arising between two colloids partially embedded in the network, and analyze the predictions. Secondly, we present a set of Monte Carlo simulations based on the same system and compare the numerical results with the analytical theory.
Coarse-Grained Model
Polymer coils in dilute and semidilute solution can be successfully modeled as blobs repelling each other with a Gaussian potential of amplitude ≈2kBT and range comparable to the radius of gyration Rg of the coil (39–41). Similar approaches have been applied to polymer melts and blends (42, 43). In this work, an analogous coarse-graining technique is adopted to model cross-linked networks describing each chain connecting two cross-linking points as a soft blob (44). We denote with N the average number of monomers per chain and with b the monomer size. The Flory radius for a self-avoiding random walk (SARW) RF = bN3/5 (22, 45) is used to set the range of blob-blob repulsion.
Given that Rg ≈ RF/2, we approximate the Gaussian blob-blob repulsion with a square shoulder potential
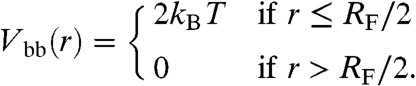 |
[1] |
The free energy of stretching the end-to-end distance of a SARW is (45, 46)
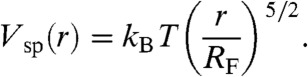 |
[2] |
In our coarse-grained picture we assume that each blob is linked to a set of nearest-neighbors by means of anharmonic springs described by Eq. 2.
Experiments show that the short-range attraction between silica colloids deposited on PAA gels is related to a colloid-gel attraction that keeps the colloids confined and partially embedded in the matrix. This effect is a consequence of attractive dispersion interactions between acrylamide monomers and silica surface. We estimate the energy of the van der Waals interaction between an acrylamide monomer and a silica colloid as -δkBT where δ ≈ 0.1. This estimate takes into account the hydrophilic repulsion between silica and acrylamide and treats the colloid surface as a flat wall (SI Appendix). We emphasize that our description is general and independent of the particular microscopic origin of the underlying polymer-colloid attraction. Hence, an appropriate value of δ can be worked out also for electrostatic, hydrophobic or other microscopic monomer-colloid attractions.
The thickness of the polymer layer adhering to a weakly adsorbing wall is ξads = bδ-3/2 (22, 45) and the free energy of adsorption per chain is ϵads = -kBTNδ5/2 (22, 45). In the colloid limit, that is for σ≫RF/2, where σ is the diameter of the colloid, ξads and ϵads can be taken as the range and the amplitude of an effective blob-colloid interaction. We use
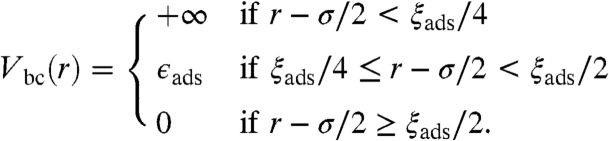 |
[3] |
The repulsive hard-core accounts for the entropic penalty of squeezing the blob on the colloid surface. In the limit of high cross-linking density (small N) or low monomer-colloid affinity (small δ) the unphysical condition of ξads > RF might occur. To avoid this inconsistency, the range of blob-colloid interaction in Eq. 3 can be redefined as min(ξads,RF). For the cases presented in this work, 0.1σ < RF/2 < 0.15σ. Because ξads ≤ RF the approximation of colloid limit is acceptable. If RF ≈ σ, Eq. 3 would need to be modified to account for the curvature of the colloid surface, for instance by means of the Derjaguin approximation (47, 48).
Different scalings, other than the SARW, could be easily implemented. For instance the ideal chain scaling (22, 45), suitable to describe a polymer network in Θ-solvent conditions, would lead to changes in the range of the blob-blob repulsion and the range and the depth of the blob-colloid interaction. It would also imply the replacement of the potential in Eq. 2 with a harmonic spring. The results of these modifications are discussed in SI Appendix.
The scaling laws we used neglect prefactors of order unity. A more accurate estimation of the blob-blob and the blob-colloid interactions can be done depending on the details of the experimental systems under consideration.
Using the scaling concepts of polymer physics we model the network-colloids system at the local level as a binary mixture of spheres interacting with square well potentials. All the relevant physical aspects are embedded in the parameters σ, b, δ, and N. The number of monomers per chain N controls the size of the blobs as well as the stiffness of the network and can be tuned experimentally by changing the concentration of cross-linkers. The range and strength of the blob-colloid interaction is regulated by δ which can be tuned by manipulating the surface chemistry of the colloids. Both δ and N influence the blob-colloid adhesion energy.
Analytical Calculation of Intercolloidal Forces
Fiocco et al. (49) have recently shown that in a binary mixture of hard spheres interacting with square well/shoulder potentials, the effective force between two particles of species 1 (big) isotropically surrounded by particles of species 2 (small) can be calculated as long as the concentration profile of the small spheres around the big spheres is known. Exploiting our blob model, we adapt this approach to calculate analytically the forces induced between two colloids by the network, specifically addressing the case of colloids partially penetrating the surface of soft cross-linked hydrogels. It is straightforward to then extend the calculation to the general case of colloids fully immersed in the matrix.
Three main issues should be considered: (i) Blobs cannot be treated as a fluid in equilibrium as they are connected in a network by anharmonic springs, thus unable to fully explore the configurational space; (ii) the blob-blob interaction is not a simple hard-sphere repulsion but the combination of the potential Vsp (Eq. 2) and the soft shoulder repulsion (Eq. 1); and (iii) in this problem, colloids are not uniformly surrounded by blobs but sedimented on the surface of the network and partially embedded in it.
Point (i) can be tackled by making a local equilibrium approximation. At length scales small enough that collisional physics dominates, this approximation enables the calculation of local physical quantities, even in systems that are macroscopically out of equilibrium. In our case, blobs can be treated as freely fluctuating, on a length scale given by the average mesh size of the gel, which is ≈RF, augmented by the range of thermal oscillation of the anharmonic springs, also ≈RF. Intercolloidal forces can be computed adapting the approach of reference (49) for surface-to-surface distances R - σ ≲ 2RF. Point (ii) can be fixed by treating the blobs as a nonideal gas with a second-order expansion in the bulk density of the network, as shown below. Point (iii) requires the introduction of the parameter l quantifying the penetration depth of the colloid within the network (Fig. 1A), which can be measured experimentally by atomic force microscopy or evaluated by computer simulations. In the limit l > σ the theory recovers the case of fully immersed particles. The dependence of l upon N can be qualitatively assessed by using theories that consider the balance between adhesion and substrate deformation à la Hertz, such as the Johnson–Kendall–Roberts theory (47). However, these theories provide quantitatively reliable estimates only in the limit of l ≪ σ, which is far from being satisfied in the present case.
Fig. 1.

Theoretical model for polymer-network-mediated colloidal interactions. (A) Schematic layout of two colloids with diameter σ (dark blue circles) at distance R = |R| partially embedded in a polymer substrate delimited by the horizontal dashed line. The dashed circle of radius |S| = σbc delimits the range of blob-colloid hard-core repulsion whereas attractive blob-colloid interaction of energy -ϵ is active in the annular region of internal radius σbc and external radius |Sλ| = λσbc. (B and C) Theoretical colloid-colloid effective potentials computed for N = 1,000, b = 0.005σ, and δ = 0.1 for different values of l using the ideal gas approximation for distribution of the blobs in Eq. 5 (B) and applying the second-order correction for blob-blob interactions in Eq. 6 (C). (D) Pair potentials computed assuming ϵ = 0. The legend applies to B, C, and D.
We define the radius of the blob-colloid hard repulsion σbc = σ/2 + ξads/4, the range of the blob-colloid attraction as λσbc = σbc + ξads/4 = σ/2 + ξads/2, and we use the shortened notation ϵads ≡ ϵ. The force between two colloids can be calculated as
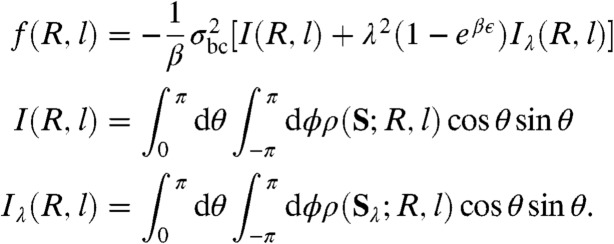 |
[4] |
In the above equation ρ(r,R,l) is the density of blobs given a center-to-center distance between the colloids R = |R| and a penetration depth l. The vectors S and Sλ are shown in Fig. 1A and β = 1/kBT. The integrations are carried out in spherical coordinates with main axis parallel to R and centered around one of the colloids. Eq. 4 is derived step-by-step in SI Appendix.
Approximating the blobs as an ideal gas, the density can be written as
| [5] |
where ρ0 is the bulk density of blobs. Vbcc(r; R,l) is the blob-colloid potential energy landscape in the presence of the two colloids. Using Eq. 5, f(R,l) can be evaluated analytically (SI Appendix). The intercolloidal polymer-mediated effective pair potentials V(R,l) can be worked out by integration and is plotted in Fig. 1B for a set of penetration depths. V(R,l) is purely attractive and always zero for R - σ > ξads. Because RF≥ξads, the local equilibrium approximation is always valid within the range of V(R,l).
Our theory allows us to clearly identify the physical effects behind V(R,l). Each colloid is surrounded by a spherical shell (corona) with high polymer density ρ′ = ρ0e-βϵ, sketched as the outer annular region in Fig. 1A. When the shells surrounding the two colloids overlap, a region of higher polymer density ρ′′ = ρ′2 = e-2βϵ appears. In this volume, a polymer blob adheres to the surface of both colloids, forming a bridge and thereby lowering the overall free energy of the system. The bigger the bridging region, the stronger the attraction. Only the portions of the bridging region that are immersed in the network contribute to the interaction, which explains the reduction in amplitude and range of V(r,l) when the penetration depth of the colloids l decreases.
However, the amplitude of the colloid-colloid attraction, computed using realistic values of N, b, and σ heavily overestimates experimental measurements reported in ref. 19. This overestimation is not surprising, because Eq. 5 neglects blob-blob interactions that effectively account for the steric repulsion between polymer coronae surrounding the colloids (24). A more reasonable result is obtained correcting locally Eq. 5 for blob-blob interactions. We treat the blobs as a gas of Brownian hard spheres with effective volume v and expand the osmotic pressure Π(ρ) to the second order in ρ. Then, we use the general relation between an external potential V(r) and Π(ρ) to extract ρ in the limit vρ0 ≪ 1. Following the derivation presented in SI Appendix we obtain
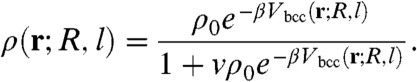 |
[6] |
The use of Eq. 6 instead of Eq. 5 also allows the analytical calculation of the force in Eq. 4.
The effective hard-sphere volume of the blobs is related to the second virial coefficient
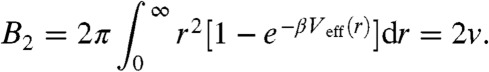 |
[7] |
Without considering anharmonic springs connecting the blobs, the effective blob-blob potential Veff(r) would reduce to Eq. 1 and Eq. 7 would give 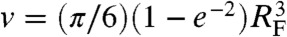 . However, anharmonic springs reduce the effective hard-sphere volume. A simple argument to account for this effect is the following: two blobs linked by the potential in Eq. 2 are confined within a distance ≈RF with an average energy ≈kBT. Thus, confinement simply reduces the amplitude of the repulsion in Eq. 1 from 2kBT to kBT. The resulting effective volume is
. However, anharmonic springs reduce the effective hard-sphere volume. A simple argument to account for this effect is the following: two blobs linked by the potential in Eq. 2 are confined within a distance ≈RF with an average energy ≈kBT. Thus, confinement simply reduces the amplitude of the repulsion in Eq. 1 from 2kBT to kBT. The resulting effective volume is
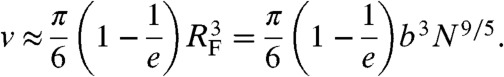 |
[8] |
The colloid-colloid pair potentials derived using Eqs. 4, 6, and 8 are shown in Fig. 1C. Using the same parameters as for curves in Fig. 1B, we find a substantial reduction in the depth of the attractive wells that now agree with experimental observations. This effect is explained with the reduction of polymer density in the bridging region by a factor (1 + vρ0e-2)-1 when Eq. 6 is used instead of Eq. 5 to account for blob-blob repulsion. Physically, correcting for blob-blob repulsion restores the disjoining osmotic pressure between polymer coronae coating the particles (24), which partially hinders the bridging.
Consistent with pioneering theoretical analysis of colloidal stability by de Gennes (22) and Pincus (24), our model accounts for the interplay between attractive energetic contributions and steric repulsion. Also, we recover the net repulsive behavior in the V(R,l) upon increasing the blob density (24). In this case the corona surrounding the colloids is saturated and no bridging can occur. The interaction is then dominated by steric repulsion between polymer coronae, which is effectively accounted via the blob-blob repulsion. Predictions by our theory in this regime are shown in SI Appendix.
In Fig. 1D we show V(R,l) computed using the same parameters as in Fig. 1C but with δ = 0. In this regime there is no polymer adsorption, and V(R,l) reduces to the typical shape of depletion interaction potentials. However, the amplitude of the attraction is experimentally negligible, indicating that entropy driven intercolloidal attraction plays a marginal role in networks of nonadsorbing polymers. The interaction is dominated by bridging or steric repulsion in networks of adsorbing polymers.
Consistent with our results, short-range attraction between colloids has been described by the polymer reference interaction site model (PRISM) integral equations theory (29, 30) and confirmed by extensive computer simulations studies (31–38) for the case of equilibrium dense polymer solutions and melts. In particular, Hooper and Schweizer (29, 30) describe four types of polymer-mediated colloidal forces, including contact depletion interaction, direct polymer bridging, and steric repulsion. Our analytical theory recovers those situations with extended applicability to the case of nonequilibrium cross-linked polymer networks with fully or partially embedded colloids.
Monte Carlo Simulations
A set of Monte Carlo simulations in the canonical ensemble based on our blob model were performed to check the predictions of our theory. We use blob-blob repulsion as described by Eq. 1. Each blob is linked to six nearest-neighbors using the potential in Eq. 2. The network is initially arranged in a simple cubic lattice of cell parameter RF spanning the simulation box in x and y direction, with periodic boundary conditions. Blobs constituting the bottom layer are confined to the plane z = 0 by a potential Vsp(z) = kBT(z/RF)5/2. Colloids interact with blobs through the potential in Eq. 3 and with other colloids via hard-sphere repulsion. Gravity of realistic modulus pushes the colloids toward the surface of the network. Full details about the simulations are provided in SI Appendix. A typical snapshot of the system is shown in Fig. 2A.
Fig. 2.

Monte Carlo simulations of colloids partially embedded in a polymer network. (A) Snapshot of the system. The two colloids are represented by big light green beads. Smaller beads represent blobs: bottom layer (red), bulk layers (light blue), and top layer of the network (green). (B) Density profile ρ(z) of the blobs in the direction z perpendicular to the surface of the network in presence of a single colloid. Here, z = 0 corresponds to the center of the colloid. Symbols indicate simulation results; solid lines are fitting curves using Eq. 9. (C) Penetration of single colloids as a function of N. (D) Color maps of the blob-density distribution around single colloids at different N. The section is taken from the vertical plane passing through the equator of the colloid. (E) Color map of the blob-density distribution around two colloids at different center-to-center distance R and N = 1,000. Sections are taken from planes Σ and μ, as schematically displayed in F. All the simulation are run using b = 0.005σ and δ = 0.1. In D and E the density is normalized to the maximum of each panel. The legend applies to both.
To calculate theoretical pair-potentials and compare them with simulated ones, we need to determine the penetration depth of the colloids l and the bulk density of the network ρ0. We run a set of simulations of single colloids at different values of N and measure the density profile ρ(z) of the polymer gel using the center of the colloid as a reference. The density ρ(z) is evaluated sampling the blobs distribution far away from the colloid to avoid perturbations following the deformation of the network. Curves of ρ(z), shown in Fig. 2B, are fitted using the Fermi function:
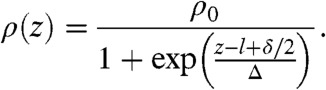 |
[9] |
The parameter Δ determines the width of the network interface that, as would happen in experimental situations, is not sharp. Fluctuations in ρ(z) are a consequence of the layered structure of the network and are more evident for small N, when the network is stiffer. The bulk density ρ0 increases with decreasing N due to the reduction of RF as well as because of the increasing stiffness of anharmonic springs which causes the gel to shrink. As shown Fig. 2C, l monotonically increases with N due to the softening of the network and the increasing of ϵ. We emphasize that the penetration is only due to colloid-blob adhesion. Gravity alone is not enough to cause any significant penetration.
In Fig. 2D we show color maps of the blobs density distribution ρ(x,z)|y=0 for different values of N, with the plane y = 0 passing through the diameter of the sphere. As expected, a corona of high monomer density surrounds the surface of the colloid embedded in the network.
In Fig. 2E we show blob-distribution maps for a two-colloids system built upon changing the colloid-colloid distance R by taking sections through the planes μ and Σ as sketched in Fig. 3F. It is clear that when the accumulation shells surrounding single colloids overlap, the region of high polymer density appears to form a bridge between the particles. Combining sections Σ and μ, we can reconstruct the shape of the bridging volume as a function of R. At short distance, it assumes a semitoroidal shape due to the exclusion of blobs from the central, narrow part of the gap between the colloids. Upon increasing R, the radius of the semitorus decreases until the gap between the colloids is wide enough to fit blobs and the bridging region becomes semiellipsoidal. At even larger distances, the accumulation shells surrounding single colloids do not overlap and the bridge does not form.
Fig. 3.

Theoretical predictions, simulations, and experimental measurements of effective pair interactions between colloids partially embedded in cross-linked polymer networks. Simulations (symbols) and theoretical calculations (solid lines) were carried out using b = 0.005σ and δ = 0.1. Here, ρ0 and l were extracted from simulations. Corrective parameter for blob-blob repulsion has been computed according to Eq. 8. Experimental data are taken from ref. 19.
We computed the pair-potential from simulations sampling the distance between two colloids and using umbrella sampling to improve efficiency (50). We compare simulated curves with theoretical predictions calculated according to Eqs. 4, 6, and 8 using ρ0 and l as extracted by simulations. The results are displayed in Fig. 3.
We see quantitative agreement between theoretical predictions and simulations concerning the depth and the range of the attractive interactions. The oscillatory behavior found in the simulations is explained as the effect of nonlocal blob-blob correlations, which are not accounted for in Eq. 6. A possible way to describe these effects consists in evaluating the blob-colloid radial distribution function numerically by means of integral equations theory and derive ρ(r; R,l) using superposition approximation (49).
Both simulations and theoretical model successfully describe the experimental pair-potentials between silica colloids deposited on PAA cross-linked gels (19).
Conclusions
In summary, we presented an analytical coarse-grained theory predicting the interactions between colloidal particles partially or fully embedded in cross-linked polymer networks, in excellent quantitative agreement with Monte Carlo simulations.
We tested the theory on the problem of interactions between silica colloids deposited on a cross-linked polymer gel for which experimental observations are available (19). We are able to explain the physical origin of these interactions, which are of considerable interest due to the widespread use of colloid-filled polymer networks in materials and energy (e.g., super-rigid rubbers, composite solar cells, etc.) and also in investigating the biomechanical response of living cells (11–18). Simulated and analytically derived pair-potentials reproduce existing experimental data (19) successfully and clarify the emergence of a net attraction between otherwise repulsive colloidal particles as a consequence of the local increase of polymer density in the gap between the two approaching colloids. The theory shows that this restructuring of the polymer is mainly driven by energetic terms related to the van der Waals attraction between monomer and colloid (which can be as weak as 0.1 kBT to produce attractions of order 2 kBT), whereas entropic effects (e.g., depletion) play a comparatively minor role.
In the limit of colloids deposited on cross-linked polymer substrates, the interaction is regulated by the penetration depth of the colloid, which ultimately depends on the cross-linking density of the network.
Our theory has no trivial fitting parameters and thus provides a quantitative tool for the synthesis of polymer-colloid nanocomposites where the colloid-colloid interactions and the colloid network structure inside the polymer network can be finely tuned and engineered by varying the cross-linkers density as the control parameter. This framework will have a deep impact on the rational design of nanostructured materials in engineering, biomedical applications, and energy-related materials.
Supplementary Material
Acknowledgments.
The authors acknowledge Daan Frenkel, Jean-Pierre Hansen, Seth Fraden, Eugene Terentjev, Carlo Pierleoni, and Jure Dobnikar for useful discussions. Simulations have been performed using the Darwin Supercomputer of the University of Cambridge High Performance Computing Service as well as CamGRID resources. This work has been supported by the Marie Curie Training Network ITN-COMPLOIDS no. 234810 and by the Oppenheimer Fund.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202171109/-/DCSupplemental.
References
- 1.Heinrich G, Klüppel M. Recent Advances in the Theory of Filler Networking in Elastomers. Vol. 160. Berlin/Heidelberg: Springer; 2002. pp. 1–44. [Google Scholar]
- 2.Heinrich G, Klüppel M, Vilgis T. Reinforcement of elastomers. Curr Opin Solid State Mater Sci. 2002;6:195–203. [Google Scholar]
- 3.Klüppel M, Schuster RH, Heinrich G. Structure and properties of reinforcing fractal filler networks in elastomers. Rubber Chem Technol. 1997;70:243–255. [Google Scholar]
- 4.Donnet J-B. Black and white fillers and tire compound. Rubber Chem Technol. 1998;71:323–341. [Google Scholar]
- 5.Kohls DJ, Beaucage G. Rational design of reinforced rubber. Curr Opin Solid State Mater Sci. 2002;6:183–194. [Google Scholar]
- 6.Coakley KM, McGehee MD. Conjugated polymer photovoltaic cells. Chem Mater. 2004;16:4533–4542. [Google Scholar]
- 7.Arango AC, Carter SA, Brock PJ. Charge transfer in photovoltaics consisting of interpenetrating networks of conjugated polymer and TiO2 nanoparticles. Appl Phys Lett. 1999;74:1698–1700. [Google Scholar]
- 8.Huynh WU, Dittmer JJ, Alivisatos AP. Hybrid nanorod-polymer solar cells. Science. 2002;295:2425–2437. doi: 10.1126/science.1069156. [DOI] [PubMed] [Google Scholar]
- 9.Beek WJE, Wienk MM, Janssen RAJ. Efficient hybrid solar cells from zinc oxide nanoparticles and conjugated polymer. Adv Mater. 2004;16:1009–1013. [Google Scholar]
- 10.Gur I, Fromer NA, Chen C-P, Kanaras AG, Alivisatos AP. Hybrid solar cells with prescribed nanoscale morphologies based on hyperbranched semiconductor nanocrystals. Nano Lett. 2007;7:409–414. doi: 10.1021/nl062660t. [DOI] [PubMed] [Google Scholar]
- 11.Discher DE, Janmey P, Wang Y-L. Tissue cells feel and respond to stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 12.Rehfeldt F, Engler AJ, Eckhardt A, Ahmed F, Discher DE. Cell responses to the mechanical microenvironment-implications for regenerative medicine and drug delivery. Adv Drug Delivery Rev. 2007;59:1329–1339. doi: 10.1016/j.addr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zajac AL, Discher DE. Cell differentiation through tissue elasticity-coupled, myosin-driven remodeling. Curr Opin Cell Biol. 2008;20:609–615. doi: 10.1016/j.ceb.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bischofs IB, Schwartz US. Cell organization in soft media due to active mechanosensing. Proc Natl Acad Sci USA. 2003;100:9274–9279. doi: 10.1073/pnas.1233544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moshayedi P, et al. Mechanosensitivity of astrocytes on optimized polyacrylamide gels analyzed by quantitative morphometry. J Phys Condens Matter. 2010;22:194114. doi: 10.1088/0953-8984/22/19/194114. [DOI] [PubMed] [Google Scholar]
- 16.Bershadsky A, Kozov M, Geiger B. Adhesion mediated mechanosensitivity: A time to experiment, and a time to theorize. Curr Opin Cell Biol. 2006;18:472–481. doi: 10.1016/j.ceb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Ulrich TA, Pardo EMD, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, mobility and proliferation of glioma cells. Cancer Res. 2009;69:4167–4173. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engler A, et al. Substrate compliance versus ligand density in cell on gel responses. Biophys J. 2004;86:617–628. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Michele L, et al. Interactions between colloids induced by a soft cross-linked polymer substrate. Phys Rev Lett. 2011;107:136101–136105. doi: 10.1103/PhysRevLett.107.136101. [DOI] [PubMed] [Google Scholar]
- 20.Asakura S, Oosawa F. On interaction between two bodies immersed in a solution of macromolecules. J Chem Phys. 1954;22:1255–1256. [Google Scholar]
- 21.de Gennes PG. Polymers at an interface. 2. Interaction between two plates carrying adsorbed polymer layers. Macromolecules. 1982;15:492–500. [Google Scholar]
- 22.de Gennes PG. Scaling Concepts in Polymer Physics. Ithaca, NY: Cornell University Press; 1979. [Google Scholar]
- 23.Pincus PA, Sandroff CJ, Witten TA. Polymer adsorption on colloidal particles. J Phys France. 1984;45:725–729. [Google Scholar]
- 24.Pincus P. Interactions between polymers and colloidal particles. In: Gonzalez AE, Varea C, editors. Lectures on Thermodynamics and Statistical Mechanics. Singapore: World Scientific; 1988. [Google Scholar]
- 25.Zaccone A, Crassous JJ, Beri B, Ballauff M. Quantifying the reversible association of thermosensitive nanoparticles. Phys Rev Lett. 107:168303. doi: 10.1103/PhysRevLett.107.168303. [DOI] [PubMed] [Google Scholar]
- 26.Wong K, Cabane B, Duplessix R. Interparticle distances in flocs. J Colloid Interface Sci. 1988;123:466–481. [Google Scholar]
- 27.Cabane B, Duplessix R. Decoration of semidilute polymer solutions with surfactant micelles. J Phys. 1987;48:651–662. [Google Scholar]
- 28.Hooper JB, Schweizer K, Desai T, Koshy R, Keblinski P. Structure, surface excess and effective interactions in polymer nanocomposites melts and concentrated solutions. J Chem Phys. 2004;121:6986–6997. doi: 10.1063/1.1790831. [DOI] [PubMed] [Google Scholar]
- 29.Hooper JB, Schweizer K. Contact aggregation, bridging, and steric stabilization in dense polymer-particle mixtures. Macromolecules. 2005;38:8858–8869. [Google Scholar]
- 30.Hooper JB, Schweizer KS. Theory of phase separation in polymer nanocomposites. Macromolecules. 2006;39:5133–5142. [Google Scholar]
- 31.Marla KT, Meredith JC. Nanoscale colloids in a freely adsorbing polymer solution: A Monte Carlo simulations study. Langmuir. 2004;20:1501–1510. doi: 10.1021/la035028d. [DOI] [PubMed] [Google Scholar]
- 32.Marla KT, Meredith JC. Simulations of interaction forces between nanoparticles in the presence of Lennard–Jones polymers: Freely adsorbing homopolymer modifiers. Langmuir. 2005;21:487–497. doi: 10.1021/la0483461. [DOI] [PubMed] [Google Scholar]
- 33.Surve M, Pryamitsyn V, Ganesan V. Nanoparticles in solution of adsorbing polymers: Pair interactions, percolations and phase behavior. Langmuir. 2006;22:969–981. doi: 10.1021/la052422y. [DOI] [PubMed] [Google Scholar]
- 34.Surve M, Pryamitsyn V, Ganesan V. Depletion and pair interactions of proteins in polymer solutions. J Chem Phys. 2005;122:154901. doi: 10.1063/1.1872772. [DOI] [PubMed] [Google Scholar]
- 35.Surve M, Pryamitsyn V, Ganesan V. Polymer bridged gels of nanoparticles in solution of adsorbing polymers. J Chem Phys. 2006;125:064903. doi: 10.1063/1.2241150. [DOI] [PubMed] [Google Scholar]
- 36.Khounlavong L, Pryamitsyn V, Ganesan V. Many-body interactions and coarse-grained simulations of structure of nanoparticle-polymer melt mixtures. J Chem Phys. 2010;133:144904. doi: 10.1063/1.3484940. [DOI] [PubMed] [Google Scholar]
- 37.Patel N, Egorov SA. Interactions between nanocolloidal particles in polymer solutions: Effect of attractive interactions. J Chem Phys. 2005;123:144916. doi: 10.1063/1.2049275. [DOI] [PubMed] [Google Scholar]
- 38.Brukhno A, Jönsson B, Akesson T, Vorontsov-Velyaminov PN. Depletion and bridging forces in polymer systems: Monte Carlo simulations at constant chemical potential. J Chem Phys. 2000;113:5493–5501. [Google Scholar]
- 39.Louis AA, Bolhuis PG, Hansen JP, Meijer EJ. Can polymer coils be modeled as “soft colloids”? Phys Rev Lett. 2000;85:2522–2525. doi: 10.1103/PhysRevLett.85.2522. [DOI] [PubMed] [Google Scholar]
- 40.Bolhuis P, Louis A, Hansen J, Meijer E. Accurate effective pair potentials for polymer solutions. J Chem Phys. 2001;114:4296–4311. [Google Scholar]
- 41.Bolhuis PG, Louis AA. How to derive and parametrize effective potentials in colloid-polymer mixtures. Macromolecules. 2002;35:1860–1869. [Google Scholar]
- 42.Murat M, Kremer K. From many monomers to many polymers: Soft ellipsoid model for polymer melts and mixtures. J Chem Phys. 1998;108:4340–4348. [Google Scholar]
- 43.Yatsenko G, Sambriski E, Guenza M. Coarse grained description of polymer blends as interacting soft colloidal particles. J Chem Phys. 2005;122:054907. doi: 10.1063/1.1835271. [DOI] [PubMed] [Google Scholar]
- 44.Craig A, Terentjev EM. Stretching of globular polymers. ii. Macroscopic cross-linked networks. J Chem Phys. 2005;122:194902. doi: 10.1063/1.1898214. [DOI] [PubMed] [Google Scholar]
- 45.Rubinstein M, Colby RH. Polymer Physics. Oxford: Oxford University Press; 2003. [Google Scholar]
- 46.Pincus P. Excluded volume effects and stretched polymer chains. Macromolecules. 1976;9:386–388. [Google Scholar]
- 47.Israelachvili JN. Intermolecular and Surface Forces: Revised. 3rd Ed. London: Academic Press; 2011. [Google Scholar]
- 48.Derjaguin B. Analysis of friction and adhesion, IV. The theory of the adhesion of small particles. Colloid Polym Sci. 1934;69:155–164. [Google Scholar]
- 49.Fiocco D, Pastore G, Foffi G. Effective forces in square well and square shoulder fluids. J Phys Chem B. 2010;114:12085–12095. doi: 10.1021/jp105145x. [DOI] [PubMed] [Google Scholar]
- 50.Frenkel D, Smit B. Understanding Molecular Simulations. San Diego: Academic Press; 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.