

Abstract
Motivation: One of the fundamental questions in genetics study is to identify functional DNA variants that are responsible to a disease or phenotype of interest. Results from large-scale genetics studies, such as genome-wide association studies (GWAS), and the availability of high-throughput sequencing technologies provide opportunities in identifying causal variants. Despite the technical advances, informatics methodologies need to be developed to prioritize thousands of variants for potential causative effects.
Results: We present regSNPs, an informatics strategy that integrates several established bioinformatics tools, for prioritizing regulatory SNPs, i.e. the SNPs in the promoter regions that potentially affect phenotype through changing transcription of downstream genes. Comparing to existing tools, regSNPs has two distinct features. It considers degenerative features of binding motifs by calculating the differences on the binding affinity caused by the candidate variants and integrates potential phenotypic effects of various transcription factors. When tested by using the disease-causing variants documented in the Human Gene Mutation Database, regSNPs showed mixed performance on various diseases. regSNPs predicted three SNPs that can potentially affect bone density in a region detected in an earlier linkage study. Potential effects of one of the variants were validated using luciferase reporter assay.
Contact: yunliu@iupui.edu
Supplementary information: Supplementary data are available at Bioinformatics online
1 INTRODUCTION
A key goal in human genetics is to identify the functional DNA variants that give rise to phenotypic differences among individuals. Recent studies of complex diseases and phenotypes have tended to focus on genome-wide association studies (GWAS) employing hundreds of thousands of single nucleotide polymorphisms (SNPs). GWAS target common DNA variants, which could either directly contribute to the clinical phenotype or provide an indirect proxy for functional variants, which are in linkage disequilibrium (LD) with the SNP being tested. Distinguishing between direct, mechanistic contributions emanating from the functional variants themselves and indirect associations resulting from LD is challenging and improved methods are needed. One feasible solution is to catalog all DNA variants in the LD region of the association, both common and rare, by utilizing next-generation sequencing (NGS) technology.
The large number of variants that will be identified generates an urgent need for bioinformatics and computational approaches capable of prioritizing the variants most likely to underlie the observed association, for further biological testing. Non-synonymous substitutions within coding regions directly affect protein structure and are likely to affect protein function; a variety of algorithms, including PolyPhen (Ramensky et al., 2002), SIFT (Ng and Henikoff, 2003), TopoSNP (Stitziel et al., 2004), PMUT (Ferrer-Costa et al., 2005), LS-SNP (Karchin et al., 2005), SNPeffect v2.0 (Reumers et al., 2006), SNPs3D (Yue et al., 2006) and PolyDoms (Jegga et al., 2007), were designed to identify functional non-synonymous substitutions. Synonymous coding variants may also exert phenotypic effect by influencing the conformation, splicing and stability of pre-mRNAs or by altering the expression level of a given protein (Capon et al., 2004; Hunt et al., 2009; Kimchi-Sarfaty et al., 2007). Coding sequences make up <2% of the human genome (Elgar and Vavouri, 2008). The regulatory component of the human genome is much less well defined but based upon conservation is 2 to 3 times larger than the coding region (Cooper et al., 2010). Regulatory variants can affect the transcription initiation rate (Mertens et al., 2009), microRNA binding and protein expression (Nicoloso et al., 2010), but are far more difficult to identify. It should therefore come as no surprise that many of the variants associated with common, complex disease by GWAS do not alter coding sequences but rather occur within the non-coding regions of genes or intergenic regions (Chen et al., 2010; Dickson et al., 2010; Glinskii et al., 2009; Hindorff et al., 2009; Johnson and O'Donnell, 2009).
Tools such as FASTSNP (Yuan et al., 2006), PupaSuite (Conde et al., 2006) and SNPlogic (Pico et al., 2009), have been designed to identify SNPs residing in known transcription factor binding sites (TFBSs), and exonic splice enhancers/silencers. Lee and Shatkay (2009) developed a scoring system for SNP prioritization that combines results from multiple independent prediction tools, using a probabilistic framework. The predictions made by these methods are not phenotype-specific, since they are based only upon our knowledge of the binding characteristics of known trans-acting regulatory factors. Nica et al. (2010) recently proposed a novel method for the prioritization of causal SNPs that employs an empirical methodology that accounts for local LD structure and integrates expression quantitative trait loci (eQTLs) and GWAS results in order to reveal the subset of association signals that are due to cis eQTLs. However, this algorithm does not consider sequence features of protein–DNA binding sites, and requires gene expression data, which is not always available for a given tissue and, more importantly, in the right biological context.
To address these limitations, we present a bioinformatics approach, regSNPs, which prioritizes transcriptional regulatory SNPs in LD regions detected by GWAS or linkage studies by integrating existing bioinformatics tools. We tested our model by assessing its ability to identify mutations logged in the Human Gene Mutation Database (HGMD) (Stenson et al., 2009), a comprehensive collection of gene mutations underlying or associated with human inherited disease. The accuracy of our approach varies among diseases, with the area under the curve (AUC) of the receiver operating characteristics (ROC) curve ranging from 53.7% in schizophrenia to 77.9% in breast cancer. We applied our method to a region on chromosome 1q that had previously been found through linkage and association analysis to harbor genetic factors that contribute to the variation of femoral neck and lumbar spine bone mineral density (BMD) in premenopausal white women (Ichikawa et al., 2008). Three SNPs were selected as likely to be functional, with an estimated false discovery rate (FDR) <25%. They were then further tested for influence on transcription in vitro. Our results suggest that the proposed approach should be well suited for prioritizing transcriptional regulatory SNPs within the chromosomal regions found to be associated with a disease or phenotype.
2 METHODS
2.1 Data source and materials
SNPs in HapMap release 27 derived from Utah residents with ancestry from Northern and Western Europe (CEU) were used. Promoter regions were defined as between upstream 3000 bp and downstream 1000 bp from transcriptional start sites of genes based on RefSeq annotation. A total of 96 069 promoter SNPs were identified; 10 000 were randomly selected as the background when generating the empirical P-values for the significance of effects on binding affinity of each transcription factor. While applying regSNPs to a BMD-related region, 51 promoter SNPs were analyzed.
The TRANSFAC 9.2 database (Wingender et al., 1996), which contains information on experimentally validated human TFBSs, was used as the source of binding information for known transcription factors. We selected 323 of the 459 human transcription factors for further study, because the rest of them do not have detailed sequences for TFBSs. These yielded 301 TFBSs. A negative control set of 10 000 promoter sequences in the human genome was randomly selected to represent putatively non-binding sequences for each TFBS.
The OMIM database (McKusick, 2007) and the HGMD 2009.1 database (Stenson et al., 2009) provide disease-related genes and mutations, respectively; both contain experimental validated data. For the purposes of analysis, we selected 13 disease states with at least two related genes in OMIM and at least nine promoter mutations documented in HGMD for model evaluation.
2.2 Evaluating the effect of a SNP on the binding affinity of a transcription factor
For a given SNP V, we estimated its effect on the binding of a particular transcription factor tf, denoted by the matching score Stf(V), by evaluating the binding affinity differences between reference and alternative alleles; the binding affinity was calculated using the position weight matrices (PWMs) documented in the TRANSFAC database using previously published methods (Wang et al., 2008).
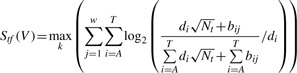 |
(1) |
where w is the width (base pair) of the binding site, k represents the index of the 2w potential binding sites that contain the candidate variants on both the positive and negative strands. Nt is the total number of experimentally validated binding sequences for each TFBS in the TRANSFAC database; bij is the number of counts of the i-th nucleotide at the j-th position in the PWM; and di represents the percentage of the i-th nucleotide (A, C, G or T) in the human genome.
For each TFBS, we calculated the distributions of matching scores for both binding (fb,tf) and non-binding (fn,tf) events, based on the matching scores with the experimentally determined binding sites (documented in the TRANSFAC database) and randomly selected genomic sequences. The potential for a specific variant to change binding affinity of a TF was estimated as:
| (2) |
where SVR and SVA denotes the matching scores [defined in Equation (1)] of the specific transcription factor (tf) binding sites on the reference and alternative alleles, respectively. Equation (2) therefore represents the logarithmic odds ratio that two alleles fell within different distributions (binding or non-binding). A positive and negative ΔSV implies that the alternative allele will result in a gain or loss of binding affinity, respectively. For each TF binding site, a P-value Ptf(V) was further calculated for each variant, by calculating the percentage of 10 000 randomly selected HapMap (Gibbs et al., 2003) promoter SNPs that have |ΔStf| scores greater than the variant |ΔSV| being evaluated.
2.3 Final ranking estimation for each variant
We combined the rankings from the previous two steps of regSNPs to derive a final score, PS(V), for each SNP; this score weights the likelihood that the observed SNP affects the binding of a transcription factor as well as the likelihood that the transcription factors are important in the corresponding phenotype.
| (3) |
where i represents all the transcription factors in the TRANSFAC database, Ptfi(V) indicates the significance of effects on binding affinity of transcription factor i, while PE(tfi) denotes the ranking scores, as the output of Endeavour, a knowledge-learning tool for gene prioritization (Aerts et al., 2006), to represent the significance of prioritized gene (transcription factor i). A lower PS score implies a stronger relationship between the candidate SNP and the disease/phenotype being studied.
2.4 ROC curve of each disease
One thousand iterations, using a different negative set of randomly sampled regulatory SNPs were generated for each of the 13 disease states (e.g. diabetes) under study. For each iteration, we first ranked all candidate variants (both experimentally validated and randomly selected) by their final PS scores [Equation (3)]. Then, we used a range of different thresholds, ranking SNPs/mutations from the lowest to highest PS scores, to select the positive mutations (scores lower than the threshold) which are recognized by regSNPs as being causally related to disease as well as negative mutations (scores higher than the threshold). In this way, one threshold can generate one pair of specificity–sensitivity values which we then used to plot the ROC curve. The AUC of the ROC is an average derived from those 1000 iterations.
2.5 FDR calculation
A P-value was calculated for each of the 51 promoter SNPs based on their priority score PS(V) [Equation (3)], using permutation analysis of 1000 iterations. In each iteration, priority scores PSr(V) of 51 randomly selected promoter SNPs were calculated. This composes a priority score distribution for non-functional SNPs. A P-value was calculated for each of the 51 SNPs by using the percentile of their priority scores in the null distribution. FDR was further calculated using Benjamini–Hochberg methods (Benjamini and Hochberg, 1995).
2.6 Luciferase reporter assay
The 2-kb region upstream of SLC39A1 exon 1 was amplified from International HapMap Project DNA samples NA07345 (AA at rs6661009) and NA12248 (CC at rs6661009) (TT and GG in the orientation of the gene), using primers tagged with restriction sites (underlined) [forward: 5′-GATC GAATTCCTTGAGCCCAAGATGTTGAGG (EcoRI) and reverse: 5′-GATCGAGCTCGAACAGCCAAACTGTCTCCG (SacI)]. The amplicons were then cloned into the EcoRI/SacI restriction sites of the pGLuc-Basic vector (New England Biolabs, Ipswich, MA, USA). The 2-kb region upstream of TPM3 exon 1 was amplified from NA12874 (GG at rs11265251), using nested PCR. First, a 2.3-kb fragment harboring the 2-kb region was amplified, using primers [forward: 5′-GATC GAATTCTCGTGATCCACCTGCCTCAG (EcoRI) and reverse: 5′-GATCGAGCTCGTGCCCACCCAGCTACTGCT (SacI)]. Then, this first reaction was used to amplify the 2-kb region, using nested primers [forward: 5′-GATC GAGCTCCAGGTGTGCACCACCACACCCG (SacI) and reverse: 5′-GATC GAGCTCGTCCCTCTGCCGCGCCCT (SacI)]. The amplicons were then cloned into the SacI restriction site of the pGLuc-Basic vector. The A allele of rs11265251 was created by site-directed mutagenesis, using the PCR overlap extension method (Higuchi et al., 1988; Ho et al., 1989). All inserts were sequenced to confirm that the nucleotides at rs6661009 and rs11265251 were the only differences introduced between the two SLC39A1 and TPM3 constructs, respectively.
Human embryonic kidney cells (HEK-293, American Type Culture Collection, Manassas, VA) were cultured in DMEM/F12 (1:1) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (Sigma-Aldrich, St Louis, MO, USA), sodium pyruvate (1 mM), l-Glutamine (2 mM), Penicillin (100 U/ml) and Streptomycin (100 μg/ml). 4 × 105cells were plated into each well of a 24-well plate and after 24 h, transfected with 800 ng pGLuc-Basic vector, SCL39A1 or TPM3 constructs. In addition, 80 ng pSV40-CLuc control plasmid (New England Biolabs) was co-transfected to allow normalization of transfection efficiency. Transfections were performed in triplicate, using Lipofectamine 2000 (Invitrogen). Media collected at 24-h post-transfection (20 μl) were assayed in triplicate for luciferase activity in Centro LB 960 Luminometer (Berthold Technologies, Oak Ridge, TN, USA), using the BioLux Cypridina Luciferase Assay Kit and the BioLux Gaussia Luciferase Assay kit (New England Biolabs). Data from three independent experiments were analyzed by paired Student's t-tests. P<0.05 were considered statistically significant.
3 RESULTS
3.1 Principles of SNP prioritization by regSNPs
We propose a novel, integrative approach that employs a combination of existing bioinformatics tools to prioritize known SNPs found in or around the promoters (defined as −3000 to +1000 bp from the transcriptional start site, see Section 2) of high priority candidate genes related to a specific phenotype or disease. Our strategy contains three major steps (Fig. 1):
Fig. 1.
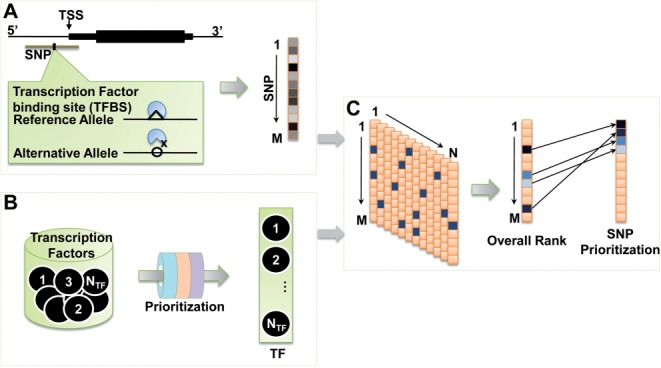
Procedures of prioritization by regSNPs. In the first step (A), the candidate SNPs are prioritized by their effects on the binding affinity of each transcription factor. Second (B), a set of training genes, collected from OMIM or Ingenuity, is used to prioritize the genome-wide transcription factors with respect to the studied phenotype/disease, using Endeavour. In the third step (C), overall rankings, characterizing the transcription regulatory roles of candidate SNPs in phenotype/disease, are calculated based on the integration of ranks of SNPs in each transcription factor (step 1) and ranks of transcription factors to phenotype/disease (Step 2). In principle, if one SNP highly alters the binding of one transcription factor which is highly related to the phenotype/disease being studied, the SNP should be prioritized as one regulatory SNP with strong confidence. TSS: transcription start site
3.1.1 Test whether the SNP is within a target sequence of a known transcription factor and prioritize its effect on binding affinity
If a SNP falls within a TFBS, we computed the binding affinities of both the reference and alternative alleles to the transcription factor, as measured by the matching scores (SVR and SVA) calculated as Equation (1) (Section 2), based on the sum of the logarithmic transformations of the frequencies of each nucleotide in a known binding site that was documented as a PWM in the TRANSFAC database (Wingender et al., 1996). This is a standard estimation of protein–DNA binding affinity. For each TFBS, we calculated the distributions of matching scores for both binding and non-binding events (Fig. 2A and Section 2). Intuitively, if one specific SNP were predicted to cause the matching score to shift from within the non-binding distribution to within the binding distribution, or vice versa, then this SNP could contribute to the gain or loss of binding affinity, respectively. Based on this principle, we calculated the effect of a SNP on the binding affinity of a transcription factor (Δ S(V) calculated as Equation (2) in Section 2), and subsequently compared the effect to 10000 randomly selected negative SNPs, which are not known to influence transcription factor binding, to calculate a P-value (see Section 2) for quantifying the likelihood that the observed SNP would change the protein-DNA binding affinity of a particular transcription factor (Fig. 2B).
Fig. 2.
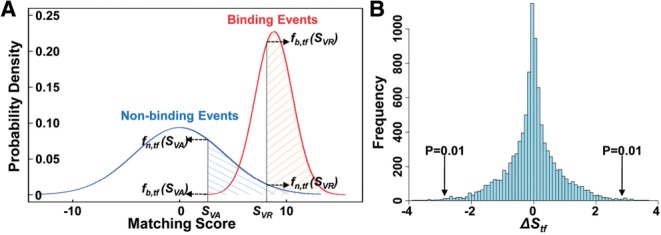
The effect of one SNP on the binding affinity of a specific transcription factor. (A). Binding patterns of TFBS M00803 for E2F1. The red distribution represents matching scores from binding events and the blue distribution represents matching scores from non-binding events. For a candidate SNP, SVR and SVA characterize the binding affinities of the reference and alternative alleles to the transcription factor, respectively. Then, four intersects from these binding patterns corresponds to the candidate SNP, including fn,tf(SVR), fn,tf(SVA), fb,tf(SVR) and fb,tf(SVA). And the SNP's effect on the binding affinity of the transcription factor (ΔSv) can be calculated based on the two binding patterns and four intersects, as characterized in Equation (2). (B) Distribution of ΔStf score from 10 000 random SNPs which may or not take influence on M00761 binding. A two-tail P-value calculation method is used to calculate candidate SNPs' significant P-value
3.1.2 Prioritize transcription factors that are related to a specific disease/phenotype
In this step, we used Endeavour (Aerts et al., 2006), a knowledge-learning tool for gene prioritization, to rank transcription factors by their relevance to the phenotype being investigated. Endeavour prioritizes candidate genes on the basis of how similar a candidate gene was to a profile derived from genes already known to be involved in the specific biological process (training set). Here, human transcription factors selected from the TRANSFAC database (Supplementary Data 1) were treated as candidate genes. Genes known to be involved in certain biological disease/phenotype features were collected from OMIM (Online Mendelian Inheritance in Man) (McKusick, 2007) and/or Ingenuity Pathway Analysis (IPA) software (http://www.ingenuity.com/), and were then used as the training set. The Endeavour program assigned a rank to each transcription factor based upon how likely it was to be involved in the disease/phenotype being studied.
3.1.3 Identify a subset of SNPs that are likely to affect the binding of transcription factors important to the phenotype/disease
In this step, we defined a score for each SNP that weighted the likelihood that the observed SNP affected the binding of a transcription factor as well as the likelihood that the transcription factors were important to the phenotype/disease of interest [PS calculated as Equation (3) in Section 2]. The lower the score is, the higher the likelihood that the SNP in question would affect transcriptional regulation related to the disease/phenotype. To evaluate the statistical significance of candidate SNPs, we performed permutation analysis by randomly selecting HapMap SNPs (Gibbs et al., 2003) within promoter regions of human protein coding genes, and by randomizing the ranking of the transcription factors with respect to phenotypes (Section 2). This allowed calculation of the FDR for each candidate SNP.
3.2 Evaluation of regSNPs using promoter mutations in the HGMD
The HGMD (Stenson et al., 2009) documents known mutations causing human inherited disease, as well as disease-associated and/or functional polymorphisms reported in the literature. It does not include mutations that lack phenotypic consequences. To evaluate the performance of our strategy for prioritizing regulatory SNPs, we tested the ability of regSNPs to select disease-causing regulatory SNPs/mutations (positive set) from an equal number of randomly selected regulatory HapMap SNPs (negative set).
We selected for evaluation 13 different disease states (Table 1), based on having ≥9 disease-causing mutations from promoter regions recorded in HGMD and with at least two disease-associated genes in OMIM. For each disease state, we extracted the disease-causing promoter regulatory SNPs/mutations from HGMD and constructed a negative dataset of an equal number of randomly selected regulatory SNPs from the HapMap database. For example, in the case of diabetes, 40 SNPs/mutations in promoter regions that have been reported (in HGMD) to be associated with diabetes were the positive dataset, while 40 randomly selected regulatory SNPs from HapMap were used as the negative set. It should be appreciated that it is conceivable, albeit rather unlikely, that some of the selected SNPs in the negative set may have exert a functional effect in the development of diabetes. Then, the balanced evaluation set (positive set + negative set) for each respective disease state was assessed using regSNPs. A PS score [Equation (3) in Section 2] was calculated for each variant, where lower values corresponded to a more significant relationship with the disease. Then, a ROC curve was constructed based upon the true positives, true negatives, false positives and false negatives, calculated at different score thresholds. For each disease state, we performed 1000 iterations with a different negative set randomly sampled from the HapMap promoter SNPs (Section 2). The average AUC of ROCs from the 1000 iterations was plotted as shown in Figure 3A. The precision of the proposed methodology varied among the 13 diseases. Breast cancer showed the highest precision (AUC = 77.9%), whereas schizophrenia had the lowest (AUC = 53.7%).
Table 1.
Validated diseases in OMIM and HGMD
| Disease | Related genes in OMIM | Related mutations in HGMD | Promoter region mutations |
|---|---|---|---|
| Diabetes | 57 | 60 | 40 |
| Thalassaemia | 5 | 56 | 43 |
| Obesity | 24 | 30 | 18 |
| Hemophilia | 2 | 28 | 25 |
| Hypertension | 19 | 25 | 16 |
| Alzheimer disease | 14 | 23 | 19 |
| Schizophrenia | 20 | 22 | 17 |
| Hypercholesterolemia | 9 | 20 | 19 |
| Myocardial infarction | 14 | 19 | 12 |
| Lung cancer | 22 | 16 | 13 |
| Prostate cancer | 17 | 14 | 10 |
| Colorectal cancer | 31 | 14 | 10 |
| Breast cancer | 25 | 12 | 9 |
Fig. 3.
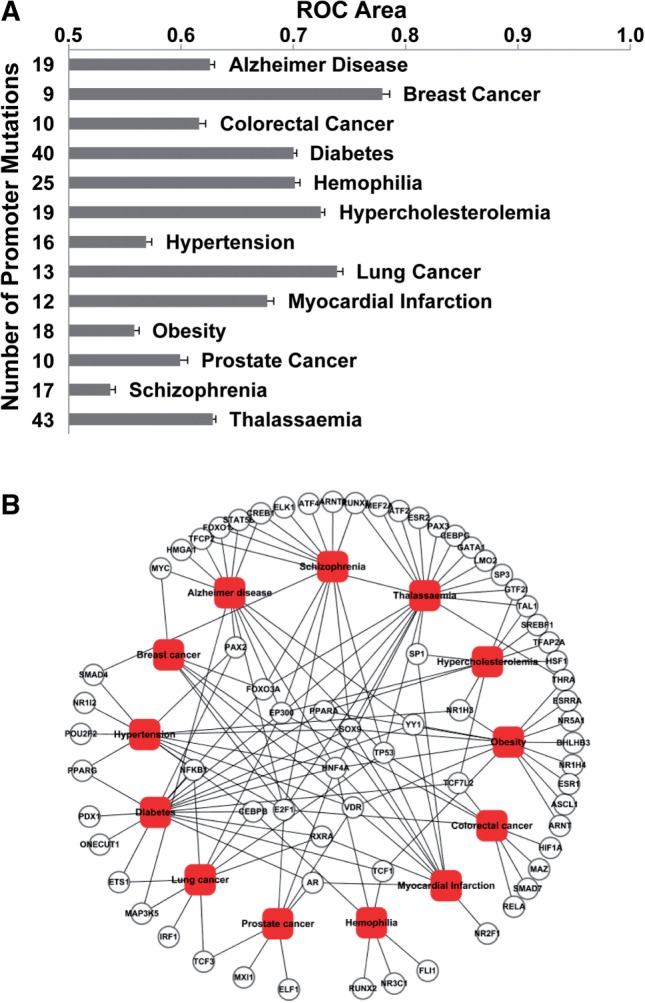
The AUC of the ROC for prioritization of 13 diseases and predicted associations between transcription factors and selected diseases. (A) ROC area (horizontal axis) of different diseases as well as the numbers (vertical axis) of tested promoter mutations in diseases. Thirteen diseases as alphabetical ranked in vertical. (B) Sixty-nine gene symbols of transcription factors, 13 diseases and 147 associations are included in this network, where solid small circles represent TFs and rounded rectangles represent diseases. Transcription factors associated with more than two diseases are inside the circle area of disease rectangles, those with fewer are outside
A major advantage of regSNPs is that it not only selects the DNA variants that are associated with a specific phenotype, but also identifies the transcription factors whose DNA binding affinities are putatively affected by the variant in question. Supplementary Data 2 lists the 69 different transcription factors whose DNA binding affinity was predicted to be affected by the 251 selected HGMD regulatory SNPs/mutations from the 13 different disease states under study. A bipartite graph was created to describe the network of 147 distinct relationships between the 13 disease states and the 69 transcription factors (Fig. 3B), where circles and rectangles represented transcription factors and diseases, respectively. An association (represented by an edge in the network in Fig. 3B) was placed between a transcription factor and a disease if the binding of that transcription factor was potentially affected by one of the mutations held to be responsible (HGMD) for the disease. Interestingly, HNF4α (hepatocyte nuclear factor 4, alpha) was predicted to be important in 9 of the 13 selected disease states, including breast cancer, diabetes, hemophilia, prostate cancer, Alzheimer disease, obesity, thalassaemia, hypertension and myocardial infarction. In addition, a total of 10 transcription factors were found to be associated with more than 3 of the 13 disease states, including E2F1 (7), p300 (7), SOX-9 (6), p53 (6), PPARα (peroxisome proliferator-activated receptor α, 5), VDR (vitamin D receptor, 5), C/EBPβ (4), FoxO3A (Forkhead box O3, 4), RXRα (Retinoic acid receptor α, 4) and YY1 (4).
3.3 Prioritizing SNPs in the genetic regions associated with BMD
We applied regSNPs to prioritize regulatory SNPs within the genetic regions known to be associated with BMD from a list of candidate SNPs in the HapMap database. We previously reported that a linkage disequilibrium block on chromosome 1q is associated with BMD (Ichikawa et al., 2008). This region was initially identified through linkage analysis using spine BMD as the phenotype of interest (Econs et al., 2004). Later, SNPs were genotyped across the linkage region and evidence of association with both spine and femoral neck BMD was identified in a 230 kb LD block (Ichikawa et al., 2008). Due to the extensive LD within the region, we could not determine which of the 11 candidate genes within this region was contributing to the variation in BMD. According to the HapMap CEU population records, 51 SNPs were in gene promoter regions within this block. We then used regSNPs to ascertain which of these SNPs would be most likely to affect transcription factor binding affinities, our tacit assumption being that SNPs which affect transcription factor binding would also have a high likelihood of altering gene expression/regulation.
First, all 51 promoter SNPs within the 230 kb LD block in chromosome 1q were evaluated for their potential to affect the binding affinities of known human transcription factors. In this study, we focused on 323 human transcription factors, whose PWMs, a commonly used representation of the DNA binding motifs, were available in the TRANSFAC database version 9.2 (Wingender et al., 1996). The empirical P-value (Section 2) was calculated for each SNP-transcription factor pair.
Second, existing bioinformatics tools, including the IPA software (http://www.ingenuity.com/) and Endeavour (Aerts et al., 2006) were used to evaluate the potential impact of individual transcription factors based upon their relevance to bone biology. We used IPA software to compile a list of genes whose functions are related to bone biology. A total of 869 genes were retrieved using IPA software by querying bone-related genes in the system (Supplementary Data 3). We then used Endeavour software to rank the 323 transcription factors according to how relevant a transcription factor was to the 869 genes that are related to biology, based upon prior information including functional annotations, protein–protein interactions, expression data and other information in literature (Aerts et al., 2006). Endeavour provided a score that evaluated the relationship between different transcription factors and bone biology. Top ranking transcription factors include p53, STAT1, p65 and E2F1 etc.; the full ranking of these transcription factors is listed in Supplementary Data 4.
Third, the regSNPs scores were calculated by jointly considering the capabilities of the 51 SNPs to affect transcription factor binding and the relevance of the affected transcription factors to bone biology (Section 2). Three SNPs received lowest scores with FDR ≤25%. These three SNPs, rs34612917, rs11265251 and rs6661009, reside in the regulatory regions of the following genes: SLC39A1 (solute carrier family 39, zinc transporter, member 1), TPM3 (tropomyosin 3) and CREB3L4 (cAMP responsive element binding protein 3-like 4) (Table 2). These SNPs are predicted to affect the binding affinities of p53 (tumor protein p53), p65 (v-rel reticuloendotheliosis viral oncogene homolog A) and E2F1 (E2F transcription factor 1), respectively. The sequence logos, a form of graphical representation of nucleic acid multiple sequence alignment (Schneider and Stephens, 1990), for the binding sites of these three transcription factors are shown in Figure 4A. Clearly, all three SNP sites are located at positions that allow less degeneracy (i.e. larger letters), and therefore have the potential to dramatically alter the binding affinities of their cognate transcription factors. The allele frequencies of these three sites vary quite markedly. Thus, with rs34612917, only 0.5% of the Caucasian population contains the T allele, a variant that may cause the loss of binding of p53. The other two SNPs, rs11265251 (60.6% A, 39.4% G) and rs6661009 (58.8% A, 41.2% C) exhibit much higher minor allele frequencies, which may give rise to gain of binding of p65 and E2F1, respectively.
Table 2.
Top SNPs in a bone-related LD block in chromosome 1q
| SNP | PS(v) | Gene | Location | TFBS | TF | FDR |
|---|---|---|---|---|---|---|
| rs34612917 | 0.00062 | SLC39A1 | −1692 | M00761 | p53 | 0.25 |
| rs11265251 | 0.00098 | TPM3 | −1365 | M00054 | p65 | 0.25 |
| rs6661009 | 0.00110 | SLC39A1/CREB3L4 | −551/+342 | M00803 | E2F1 | 0.25 |
Three SNPs are significant at FDR <25%. Score is the final PS score of prioritization; location is relative to transcription start site (TSS), while negative location indicates upstream of TSS and positive location indicates downstream of TSS; TFBS is the identity of the TFBS in TRANSFAC database; TF is the name of the transcription factor; FDR is FDR based on permutation analysis.
Fig. 4.
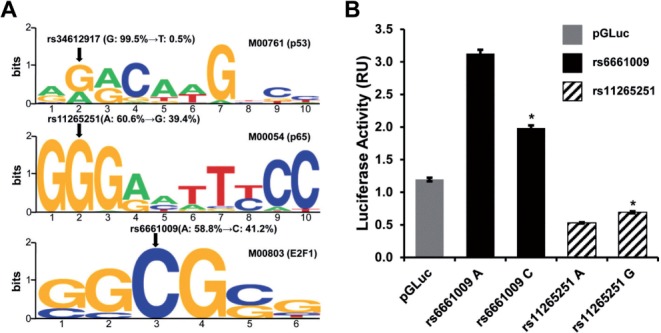
Top prioritized SNPs in BMD and luciferase reporter assay validation. (A) Detailed binding alterations by three top prioritized SNPs in BMD. Three transcription factors, with detailed disrupted positions in their binding sites, are shown in the form of sequence logos. The names of transcription factors, rsID of SNPs and allele frequencies of SNPs are marked. For each SNP, the left allele and right allele with an arrow separated, are the reference and alternative alleles, respectively, based on Caucasian population in HapMap. (B) Relative promoter strengths determined by luciferase reporter assay. RU: relative units. *P!<!0.01 compared with alternative alleles that are predicted to disrupt binding of transcription factors
3.4 Biological validation of selected SNPs
To verify the ability of the predicted SNPs to modulate transcription, SLC39A1 and TPM3 promoters harboring rs6661009 and rs11265251 were cloned upstream of a luciferase reporter gene (Section 2). SNP rs34612917 was excluded from this validation because of its very low minor allele frequency in the general population. The luciferase activities for both alleles of rs11265251 (residing in the TPM3 promoter) were lower than the basic pGLuc vector (background) activity. This may be due to the low activity of the TPM3 promoter in HEK293 cells, the cell system used to test the promoter activity. Consistent with this interpretation, the expression level of the TPM3 gene was found to be low in kidney cells and in all fetal tissues, as documented in the microarray gene expression database at the UCSC Genome Browser website (Fujita et al., 2011). In contrast, luciferase assays demonstrated that SNP rs6661009 significantly altered the activity of the SLC39A1 promoter (Fig. 4B). Our original prediction by regSNPs was that the A allele of rs6661009 would disrupt the binding of E2F1 to the SLC39A1 promoter; however, in vitro the A allele induced 58% higher luciferase activity (P=0.00075; Fig. 4B). It has been reported that E2F1 can serve as both positive and negative regulator on their target genes (Crowe et al., 2001; Croxton et al., 2002; Stanelle et al., 2002).
4 DISCUSSION
In this study, we proposed a three-step bioinformatics approach to identify functional SNPs within the regulatory regions following GWAS. Promoter regions were used as an example of regulatory regions to illustrate the procedures involved. First, we tested whether the identified variant is within the target sequence of a known transcription factor and hence could affect its binding affinity. Second, we prioritized those transcription factors that are related to a specific phenotype. Third, we identified a subset of sequence variants that are likely to affect the binding of a transcription factors important to the disease/phenotype under study. We tested the performance of regSNPs in identifying the promoter mutations in 13 diseases, using experimentally validated SNPs from HGMD. The results of this analysis showed that the prioritization was sufficiently high and could be useful, at least in some disease states (e.g. breast cancer: 77.9%). Our analysis also shows that some transcription factors (e.g. HNF4α) may play a key role in multiple disease states. Finally, our model identified three putative functional promoter SNPs, with a FDR estimated to be 25%, in a region that influences BMD. We demonstrated that one of these SNPs, rs6661009, significantly altered luciferase activity between the A and C alleles, suggesting that regSNPs has identified a causal SNP associated with BMD. The direction of effect, in which the variant predicted to reduce binding of a transcription factor increased promoter activity, could be due to assembly of an alternative promoter complex in the cells used. It is also reported that the E2F1 binding can be associated with both positive and negative regulation on the target genes (Crowe et al., 2001; Croxton et al., 2002; Stanelle et al., 2002).
regSNPs offers several major improvements over other methods used for SNP prioritization. First, it is a phenotype-specific approach to prioritize functional regulatory variants, while most existing methods (Conde et al., 2006; Lee and Shatkay, 2009; Pico et al., 2009; Yuan et al., 2006) only generate putative candidates based on the sequence composition of the TFBSs, irrespective of whether the specific transcription factor is functionally important in the disease or phenotypic status. By integrating different bioinformatics tools and databases (Endeavour and IPA/OMIM), regSNPs has the advantage of narrowing the search to those transcription factors that are likely to influence the phenotype of interest. Second, unlike the previous methods, which only yield the putative SNPs, regSNPs identifies SNPs as well as the transcription factors which are most likely to influence the phenotype. The identified transcription factors associated with specific phenotypes provide candidate proteins for further research. Third, statistical evaluation is included in the regSNPs output that allows quantification of the false positive rate that can be placed in the prioritizations. Thus, at each step, a P-value indicates the likelihood that a given SNP will alter transcription factor-binding affinity, the likelihood of an association between a transcription factor and a specific phenotype, and the likelihood of a particular SNP exerting a regulatory role in the context of a specific phenotype. This statistical information will enable investigators to prioritize particular genes/SNPs for more in depth molecular study.
When we used regSNPs to analyze promoter mutations from HGMD to test the program, we found variable utility across disease states, ranging from 53.7% (schizophrenia) to 77.9% (breast cancer). One reason for this variability may be that some disease genes are more heavily reliant upon genetic regulation, and are therefore likely to be more sensitive to the influence of polymorphic variation within promoter regions. Breast cancer is a widely studied disease, reported to be highly associated with gene regulation (Dunning et al., 1999). Thus, when ranking validated disease-associated promoter mutations (positives) with randomly selected SNPs (negatives), reasonably high precision can be achieved. In contrast, in the case of complex traits, such as hypertension, their pathophysiological mechanisms involve multiple genetic factors together with a potent environmental influence (Kosugi et al., 2009; Rahmouni et al., 2005; Riserus et al., 2009; Sesso et al., 2008); in such cases, the ability of regulatory variant identification may be limited.
Limitations of regSNPs include its reliance on existing knowledge (e.g. disease/phenotype-related genes, and binding models for transcription factors). Despite these, regSNPs provides an important strategy for prioritizing causal regulatory DNA variants with respect to specific disease/phenotype of interest. It is especially valuable for prioritizing candidate SNPs identified by the GWAS, or novel or rare variants discovered from the direct human genome sequencing.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the valuable discussion from Dr Lang Li in the Center for Computational Biology and Bioinformatics at Indiana University School of Medicine.
Funding: National Institutes of Health grants AA017941 and AG041517.
Conflict of Interest: none declared.
Footnotes
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
REFERENCES
- Aerts S., et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- Capon F., et al. A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum. Mol. Genet. 2004;13:2361–2368. doi: 10.1093/hmg/ddh273. [DOI] [PubMed] [Google Scholar]
- Chen R., et al. Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS One. 2010;5:e13574. doi: 10.1371/journal.pone.0013574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde L., et al. PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res. 2006;34:W621–W625. doi: 10.1093/nar/gkl071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D.N., et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum. Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- Crowe D.L., et al. E2F-1 represses transcription of the human telomerase reverse transcriptase gene. Nucleic Acids Res. 2001;29:2789–2794. doi: 10.1093/nar/29.13.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxton R., et al. Direct repression of the Mcl-1 promoter by E2F1. Oncogene. 2002;21:1359–1369. doi: 10.1038/sj.onc.1205157. [DOI] [PubMed] [Google Scholar]
- Dickson S.P., et al. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning A.M., et al. A systematic review of genetic polymorphisms and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 1999;8:843–854. [PubMed] [Google Scholar]
- Econs M.J., et al. Confirmation of linkage to chromosome 1q for peak vertebral bone mineral density in premenopausal white women. Am. J. Hum. Genet. 2004;74:223–228. doi: 10.1086/381401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgar G., Vavouri T. Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 2008;24:344–352. doi: 10.1016/j.tig.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C., et al. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Fujita P.A., et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs R.A., et al. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Glinskii A.B., et al. Identification of intergenic trans-regulatory RNAs containing a disease-linked SNP sequence and targeting cell cycle progression/differentiation pathways in multiple common human disorders. Cell Cycle. 2009;8:3925–3942. doi: 10.4161/cc.8.23.10113. [DOI] [PubMed] [Google Scholar]
- Higuchi R., et al. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff L.A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.N., et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hunt R., et al. Silent (synonymous) SNPs: should we care about them? Methods Mol. Biol. 2009;578:23–39. doi: 10.1007/978-1-60327-411-1_2. [DOI] [PubMed] [Google Scholar]
- Ichikawa S., et al. Identification of a linkage disequilibrium block in chromosome 1q associated with BMD in premenopausal white women. J. Bone Miner. Res. 2008;23:1680–1688. doi: 10.1359/JBMR.080509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegga A.G., et al. PolyDoms: a whole genome database for the identification of non-synonymous coding SNPs with the potential to impact disease. Nucleic Acids Res. 2007;35:D700–D706. doi: 10.1093/nar/gkl826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.D., O'Donnell C.J. An open access database of genome-wide association results. BMC Med. Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchin R., et al. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C., et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- Kosugi T., et al. Uric acid and hypertension: an age-related relationship? J. Hum. Hypertens. 2009;23:75–76. doi: 10.1038/jhh.2008.110. [DOI] [PubMed] [Google Scholar]
- Lee P.H., Shatkay H. An integrative scoring system for ranking SNPs by their potential deleterious effects. Bioinformatics. 2009;25:1048–1055. doi: 10.1093/bioinformatics/btp103. [DOI] [PubMed] [Google Scholar]
- McKusick V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J., et al. Functional impact of endotoxin receptor CD14 polymorphisms on transcriptional activity. J. Mol. Med. 2009;87:815–824. doi: 10.1007/s00109-009-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica A.C., et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloso M.S., et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–2798. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico A.R., et al. SNPLogic: an interactive single nucleotide polymorphism selection, annotation, and prioritization system. Nucleic Acids Res. 2009;37:D803–D809. doi: 10.1093/nar/gkn756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmouni K., et al. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45:9–14. doi: 10.1161/01.HYP.0000151325.83008.b4. [DOI] [PubMed] [Google Scholar]
- Ramensky V., et al. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reumers J., et al. SNPeffect v2.0: a new step in investigating the molecular phenotypic effects of human non-synonymous SNPs. Bioinformatics. 2006;22:2183–2185. doi: 10.1093/bioinformatics/btl348. [DOI] [PubMed] [Google Scholar]
- Riserus U., et al. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009;48:44–51. doi: 10.1016/j.plipres.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T.D., Stephens R.M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesso H.D., et al. Alcohol consumption and the risk of hypertension in women and men. Hypertension. 2008;51:1080–1087. doi: 10.1161/HYPERTENSIONAHA.107.104968. [DOI] [PubMed] [Google Scholar]
- Stanelle J., et al. Gene expression changes in response to E2F1 activation. Nucleic Acids Res. 2002;30:1859–1867. doi: 10.1093/nar/30.8.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson P.D., et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitziel N.O., et al. topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Res. 2004;32:D520–D522. doi: 10.1093/nar/gkh104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., et al. Transcription factor and microRNA regulation in androgen-dependent and -independent prostate cancer cells. BMC Genomics. 2008;9(Suppl. 2):S22. doi: 10.1186/1471-2164-9-S2-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E., et al. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.Y., et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–W641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P., et al. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.