

Abstract
Proline is a readily released stress substrate that can be metabolized by proline oxidase (POX) to generate either reactive oxygen species to induce apoptosis or autophagy or ATP during times of nutrient stress. However, the contribution of proline metabolism to tumorigenesis in hypoxic microenvironments has not been explored. In this study, we investigated the different functions of POX under hypoxia and glucose depletion. We found that hypoxia induced POX expression in cancer cells in vitro and that POX upregulation co-localized with hypoxic tissues in vivo. In addition, the combination of hypoxia and low-glucose showed additive effects on POX expression. Similar to conditions of low glucose, hypoxia-mediated POX induction was dependent on AMP-activated protein kinase (AMPK) activation, but was independent of HIF-1α and HIF-2α. Under low-glucose and combined low-glucose and hypoxic conditions, proline catabolized by POX was used preferentially for ATP production, whereas under hypoxia, POX mediated autophagic signaling for survival by generating ROS. Although the specific mechanism was different for hypoxia and glucose deprivation, POX consistently contributed to tumor cell survival under these conditions. Together, our findings offer new insights into the metabolic reprogramming of tumor cells present within a hostile microenvironment and suggest that proline metabolism is a potential target for cancer therapeutics.
Introduction
Research in cancer metabolism has been re-energized by recent advances in the study of pathways controlling cell growth that reveal their close interaction with metabolic pathways (1-3). Tumor cells fuel their metabolism with glucose and glutamine to meet the bioenergetic and biosynthetic demands of proliferation. The Warburg effect, or aerobic glycolysis, has been considered as the central tenet of cancer cell metabolism for more than 80 years (1, 4). Fogal et al. suggested that oxidative phosphorylation also plays a pivotal role in tumorigenesis (5). In addition, aberrant choline phospholipid metabolism is currently emerging as a metabolic hallmark of oncogenesis and tumor progression (2). Recent studies document an important role of glutamine catabolism stimulated by the Myc oncogene in tumor metabolism (3). However, due to the rapid growth of tumors and associated vascular insufficiency, many tumor cells are depleted of oxygen and nutrients, i.e. glucose and glutamine. The hypoxic, low-glucose or combined hypoxic and low-glucose regions in tumors make characterizing tumor metabolism difficult. With these regionally hostile microenvironments, the high bioenergetic demands imposed by transformation require that tumors reprogram their metabolic mode to meet the demands of proliferation and/or survival. Proline as a microenvironmental stress substrate attracted our attention due to its availability in tumors, its unique metabolism and its response to various stresses. With glucose deprivation and upregulation of proline oxidase (POX), proline can be metabolized to provide ATP (6). However, the effect of hypoxia on proline metabolism has not yet been explored.
Proline is one of the most abundant amino acids in the cellular microenvironment. Together with hydroxyproline, proline constitutes more than 25% of residues in collagen, the predominant protein (80%) in extracellular matrix (ECM) (7). With the breakdown of collagen by matrix metalloproteinases (MMP), proline is readily available. Unlike other amino acids, proline has its own metabolic enzymes; it is catabolized to pyrroline-5-carboxylate (P5C) by proline oxidase (POX), a.k.a. proline dehydrogenase (PRODH), a mitochondrial inner membrane enzyme; PRODH was identified as one of a few genes rapidly and robustly induced by p53 (8). Subsequently, its role in cell survival, apoptotic cell death, and autophagy in cancer cells was investigated and characterized (9-11). The conversion of proline to P5C donates electrons, which may directly generate superoxide through flavine adenine dinucleotide (FAD), or enter the electron transport chain to either produce ROS or generate ATP (6, 12, 13). POX was upregulated by p53, PPARγ ligands (a signaling system responding to inflammatory stress) and oxidized low-density lipoproteins to generate superoxide radicals, which initiate apoptotic cell death or prosurvival autophagy depending on the specific stresses (8-11). However, under conditions of nutrient stress, proline could act as an energy source providing ATP (6). In the present study, we investigated the effect of hypoxia on the expression and functions of POX, and explored the differential functions of proline metabolism catalyzed by POX under oxygen and/or glucose deprivation. The evaluation of the importance of proline catabolism in cancer metabolism will provide a better understanding of the tumor metabolic reprogramming in hostile microenvironments.
Materials and Methods
Cell culture
The human colon (HCT116, HCT15, HT29), renal (TK10 and 786-0), breast (MCF7 and Hs-578-T), prostate (PC3), melanoma (M14), lung (A549), and ovarian (IGROV1) cancer cell lines were provided by the NCI cell line repository and were cultured in RPMI-1640 or DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin and 2 mM glutamine as recommended.
The triple-negative metastatic human breast cancer cell line MDA-MB-231 was obtained from the American Type Culture Collection (ATCC) and stably transfected with a construct containing five copies of the hypoxia-response element (HRE) (5′-CCA CAG TGC ATA CGT GGG CTC CAA CAG GTC CTC TT-3′) of the human VEGF-A gene ligated to the complementary DNA of enhanced green fluorescent protein (EGFP), which produced MDA-MB-231 HRE-EGFP as previously described (14, 15). MDA-MB-231 HRE-EGFP cells express EGFP under hypoxic conditions as a hypoxia-inducible factor 1 (HIF-1)-driven hypoxia sensor, which was verified in hypoxic cell cultures by fluorescence microscopy and in corresponding protein lysates by SDS-PAGE and immunoblot analysis with anti-EGFP antibody (BD Biosciences) as previously described (14). Detectable EGFP expression is typically observed within 6 hours of exposure to less than 1% O2, and robust EGFP expression by 20 hours in HRE-EGFP expressing cells in culture (14). All cell lines were authenticated by morphology and growth rate and were Mycoplasma free.
POX promoter activity
Before the assay, the cells were cotransfested with the POX promoter-luciferase (POX-Luc) and pRL-null renilla construct (11). The cells were exposed to normoxia or hypoxia (0.05% O2) at 6-10 h post-transfection. POX promoter activity was assessed with Dual-Luciferase reporter Assay (Promega) in accordance with the manufacturer’s instruction.
Statistical analysis
All data are representative of at least 3 independent experiments, and all error bars are mean ± SEM. All P values were calculated by the Student 2-tailed t test unless otherwise noted. Results were considered significantly different at P < 0.05.
Additional methods
Detailed description of methods for hypoxia exposure, real-time RT-PCR, Western blot, tumor xenograft studies, EGFP and POX immunohistochemical staining and colocolization analysis, siRNA transfections, measurement of ROS and ATP, TUNEL staining, detection of autophagosomes are available in Supplemental Materials and Methods.
Results
Hypoxia upregulates the expression of POX in various cancer cell lines in vitro
Hypoxia is an important feature of the microenvironment of solid tumors and it affects tumor progression by causing genetic instability and driving the changes in tumor metabolism (16). To investigate whether hypoxia affects the expression of POX, we first tested POX promoter activity in a variety of cancer cells including colon (HT-29, HCT-116 and HCT-15), renal (TK10 and 786-0), breast (MCF7 and Hs-578-T), prostate (PC3), melanoma (M14), lung (A549), and ovarian (IGROV1) cancer cell lines under hypoxia (0.05% O2). Most of the cancer cell lines except the TK10 renal cancer cell line showed significant activation of the POX promoter after 24 hrs exposure to hypoxia compared to normoxia (Fig.1A). This transient transfection assay directly measures the promoting effect of a defined fragment of DNA sequence, and its accuracy is influenced by many factors, such as the vector, dose of transfection, and host cell characteristics (17-19). Thus, in parallel, we measured the mRNA levels of POX in the same cell lines, which are expressed as absolute genomic equivalents (17). As shown in Fig. 1B, POX mRNA levels increased in all the tested cell lines with hypoxic exposure (0.05% O2). There was a significant correlation between POX promoter activity and mRNA levels (r = 0.40), which was close to those reported by others (17, 19). HT29 showed a strikingly high expression of POX mRNA, up to 10 fold (Fig. 1B). Using the HT29 cell line, we determined the oxygen concentration- and time-dependent response of POX mRNA and protein levels. As the concentration of oxygen decreased, the increases in POX mRNA expression were observed in a time-dependent fashion. After 24 hrs of treatment, there were 1.8-, 5- and 10-fold increases in POX mRNA at the 5%, 0.5% and 0.05% O2 concentrations, respectively (Fig. 1C). A time-dependent increase in POX protein expression was also observed in the 0.05% O2 environment (Fig. 1D). Densitometry analysis showed that the changes of POX protein corresponded to those of POX mRNA levels.
Figure 1.

Hypoxia upregulates POX expression in vitro. POX promoter activity (A) and mRNA expression (B) were measured in various cancer cells as indicated after 24 hrs of hypoxia (0.05% O2) treatment. The results represent the fold change in relative luciferase activity and POX mRNA levels of cells exposed to hypoxia over those exposed to normoxia. C, oxygen concentration- and time-dependent response of POX mRNA expression in HT29 colorectal cancer cells. D, time-dependent effect of 0.05% O2 on POX protein expression in HT29 cells. E, the effect of hypoxia and/or low-glucose (1mM) on POX mRNA expression in HT29 cells. F, POX protein expression in HT29 cells exposed to hypoxia (Hy) and/or low-glucose (LG) (1mM) for 24 hrs. *, P <0.05, **, P <0.001 compared with normoxic control (Ctr).
The glucose distribution in tumors is thought to follow patterns similar to that of oxygen (20). In addition, tumor cells with an activated aerobic glycolytic (Warburg effect) or anaerobic glycolytic (hypoxic cells) pathway use much more glucose than oxidative cells to generate ATP. They tend to deplete their surroundings of glucose. Thus, we tested the effects of hypoxia with or without glucose deprivation on POX mRNA and protein expression (Fig. 1E and 1F). Consistent with the previous report (6), the low-glucose condition (1mM) increased both POX mRNA and protein expression. The combination of hypoxia and low-glucose had an additive effect on POX expression. The increases of POX protein levels were confirmed in TK10 and Hs-578-T cells under the above conditions (Supplemental Fig. 1A).
POX expression increases in the hypoxic tumor microenvironment in a human breast cancer mouse xenograft model
To confirm the upregulation of POX by hypoxia in the microenviroment of the whole tumor in vivo, we developed a human breast cancer cell line, MDA-MB-231 HRE-EGFP, which expresses the EGFP reporter gene under the control of a promoter containing hypoxia-response elements (HREs) that bind to and are induced by HIF-1. Thus, we can locate hypoxic regions, in which hypoxia stabilized HIF-1 leading to EGFP expression, in MDA-MB-231 HRE-EGFP tumor xenografts grown orthotopically in mice by monitoring EGFP expression. We first characterized the expression of POX mRNA in MDA-MB-231 HRE-EGFP under hypoxic conditions (0.3-0.5% O2) in tissue culture. As expected, POX mRNA showed a time-dependent increase under hypoxia (Supplemental Fig. 2), and HIF-1-controlled EGFP expression was confirmed by fluorescence microscopy. We then performed POX and GFP immunohistochemistry (IHC) using tissue slides from MDA-MB-231 HRE-EGFP tumor xenografts. The left panel of Fig. 2 B shows a diagram of how MDA-MB-231 HRE-EGFP tumor xenografts were sectioned, and what sections that were cut throughout the tumor were analyzed by IHC. The right panel of Fig. 2A shows representative images of EGFP and POX IHC staining of slides from the tumor center, which typically contain most of the hypoxic regions of the tumor. Hypoxic regions identified by EGFP expression also had the highest staining for POX. Although the POX and EGFP staining levels were lower in the top and body portions of the tumor, POX showed the same trend of staining intensity as seen with EGFP. Co-localizaion analysis showed that Pearson’s correlation coefficients of POX and EGFP in most slides were over 0.5 (p<0.001), robustly indicating that POX and EGFP expression co-localized (Fig. 2B).
Figure 2.

POX increases in hypoxic tumor microenvironments in vivo. A, left panel, diagram of a MDA-MB-231 HRE-EGFP tumor xenograft showing the positions of tumor sections cut throughout the tumor for POX and GFP immunohistochemical (IHC) staining. Right panel, representative images of POX and EGFP IHC staining. B, colocalization analysis of POX and EGFP IHC staining using Pearson’s correlation coefficient.
Hypoxia-induced POX expression is dependent on AMPK activation, but not HIF-1α or HIF-2α
The AMP-activated protein kinase (AMPK) is a sensor of cellular energy status; it is found in all eukaryotes and is activated under conditions of high AMP/ATP following stresses such as hypoxia or nutrient deprivation (21). Thus, we determined the potential role of AMPK in hypoxia-induced POX expression by using a specific inhibitor of AMPK activation, (6-[4-(2-piperidin-1-yl-ethoxy)-phenyl)]3-pyridin-4-yl-pyrazolo[1,5-a] pyrimidine, which is known as Compound C. Phospho-AMPKα was assessed by Western blot. As reported, exposure of HT29 cells to hypoxia for 24 and 48 hrs activated AMPK, which could be inhibited by Compound C (50μM) (Fig. 3B). Fig. 3A and B clearly show that the activation of AMPK positively regulated both mRNA and protein expression of POX. Compound C decreased POX expression almost to unstimulated basal levels. These results were confirmed in TK10 and Hs-578-T cells (Supplemental Fig. 1B)
Figure 3.

Hypoxia-induced POX expression is dependent on AMPK activation. HT29 cells were treated with the AMPK inhibitor, Compound C (Com C) (50μM) while cells were treated with 0.05% O2. AMPK activation was estimated by Western blot (B). POX mRNA (A) and protein expression (B) were detected by real-time PCR and Western blot, respectively. *, P <0.001 compared with normoxia. #, P <0.001 compared with hypoxia.
Although hypoxia inducible factor (HIF)-1 and HIF-2 are critical transcription factors that activate transcription of target genes in response to hypoxia in tumor (22), our results suggested that both HIF-1α and HIF-2α are not essential for hypoxia-induced POX expression (Supplemental results and Supplemental Fig. 3).
The above results were consistent with the observation of Pandhare et al. (6), that is, the activation of POX under low-glucose conditions is mediated by activated AMPK, and the AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) increases POX. So we can conclude that hypoxia and glucose depletion upregulate POX expression through the same mechanism that is mediated by the AMPK pathway.
POX contributes to the survival of cancer cells in response to hypoxia and glucose deprivation
As mentioned earlier, POX degrades proline to either generate ROS for apoptosis or pro-survival autophagy, or produce ATP according to different stresses (6, 9-11). We examined the effects of upregulated POX on cell viability and the production of ATP and ROS under hypoxia and/or low-glucose conditions. As shown in Fig. 4A, with HT29 cells treated for 48 hrs, low-glucose (1mM) decreased cell proliferation by 46.5%, whereas hypoxia (0.05% O2) produced only a small decrease (7.3%). The combination of the two, however, was synergistic producing a 72% decrease. The POX inhibitor, dehydroproline (DHP) (10mM) further reduced cell viability under all conditions, but especially with low-glucose and hypoxia (73.2% and 60.4%, respectively), suggesting that proline degradation by POX participated in a compensatory mechanism for survival. The addition of exogenous proline (5mM) had little effect on the proliferation, suggesting basal medium proline or endogenous proline was enough for the adaptive changes.
Figure 4.
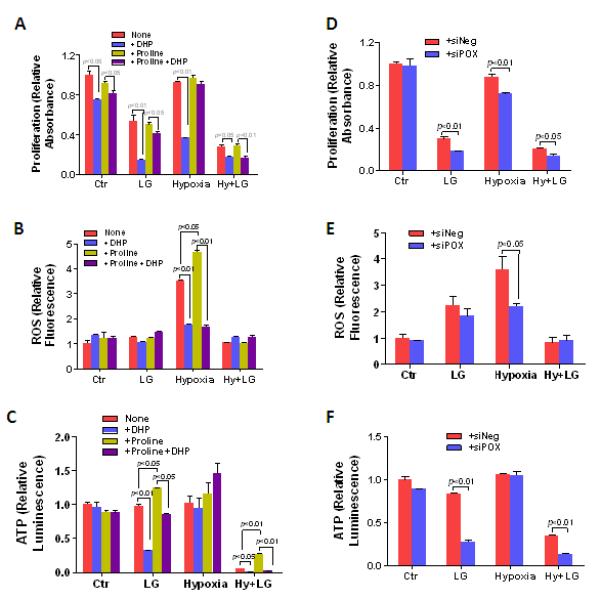
POX contributes to the survival of cancer cells in response to hypoxia and glucose deprivation. HT29 cells were treated with low glucose (1mM) and/or hypoxia (0.05% O2) for 48 hrs. 10mM Dehydroproline (DHP), the POX inhibitor, and/or 5mM proline were given to the cells at the same time as indicated. A, proliferation assay was performed using the WST method. Intracellular ROS (B) and ATP production (C) were performed using the DCF assay and luciferase-based assay, respectively. D to F, POX siRNA (siPOX) was used to knock down POX expression when cells were treated with low glucose and/or hypoxia. Proliferation, ROS, and ATP production were detected as described as above.
Next, we examined the production of ROS and ATP under those conditions to dissect their contributions to cell survival. Hypoxic stress significantly increased ROS production, and this increase could be partially decreased by DHP (Fig. 4B). Exogenous proline addition (5mM) further increased the production of ROS, which was also inhibited by DHP. Interestingly, with hypoxia treatment for 48 hrs, ATP levels were not significantly decreased with or without DHP (Fig. 4C), indicating that upregulated POX with hypoxia had a marked effect on ROS but not on the maintenance of ATP. In contrast, low-glucose, itself, had little effect on either ROS or ATP in HT29 cells. Nevertheless, when DHP was added under low-glucose conditions, intracellular ATP, but not ROS, decreased significantly (Fig. 4B and C), suggesting that with glucose deprivation, proline was a source of energy rather than a source of metabolic signaling. In cells exposed to combined hypoxia and low-glucose, ATP production was dramatically reduced and this was further decreased by DHP. Under this extreme condition, the addition of exogenous proline significantly improved ATP levels (Fig. 4C), but ROS did not show any obvious change (Fig. 4B).
To avoid any potential non-specific side effects of the POX inhibitor DHP, and confirm the above results, we used POX siRNA (siPOX) to knock down POX expression in a series of analogous experiments. The decreased expression of POX was confirmed by western blot and over 80% of POX was knocked down (Supplemental Fig. 4). POX knockdown resulted in decreased cell proliferation with low-glucose, hypoxia, and combined low-glucose and hypoxia (Fig. 4D). POX knockdown significantly decreased ROS production with hypoxia, but only modestly with low-glucose (Fig. 4E). As was seen with DHP inhibition, ATP production was markedly decreased with POX knockdown under low-glucose conditions, whereas it showed no change with hypoxia (Fig. 4C and F). These results corroborated the above findings obtained with POX inhibition by DHP, suggesting that proline degradation by POX led to distinct endpoints depending on the type of metabolic stress. With low-glucose, proline is used as a source of energy, whereas with hypoxia, proline metabolism is a mechanism for ROS signaling. Since POX contributed to cell survival, and not cell death under hypoxia, we hypothesized that ROS induced by POX occurred to achieve protective autophagy, rather than apoptosis. This hypothesis was confirmed by the following experiments.
POX induces protective autophagy, but not apoptosis under hypoxia
It is well-known that hypoxia can increase both apoptotic cell death and autophagy, the latter of which is a survival strategy of cancer cells (23, 24). An important question is whether ROS production from upregulated POX under hypoxia mediates apoptosis or protective autophagy. To this end, we first monitored the apoptotic marker PARP and cleaved-PARP by Western blot in HT29 cells. Hypoxia (0.05% O2) increased the production of cleaved-PARP (Supplemental Fig. 5A). However, POX siRNA (siPOX) slightly increased rather than decreased this hypoxic effect. This small increase was likely due to the transfection procedure itself as the negative control siRNA (siNeg) showed the same effect. TUNEL staining confirmed this result (Supplemental Fig. 5B and C). After HT29 cells were treated by hypoxia with or without siPOX or siNeg for 72 hrs, TUNEL staining was performed and the percentage of TUNEL positive cells (apoptotic cells) was calculated. As compared to normoxia, hypoxia increased the percentage of apoptotic cells from 10% to 62.5%. SiPOX did not show any obviously different effects from siNeg.
Hypoxia-induced autophagy is known to involve the activity of AMPK (24). The fact that AMPK mediated POX upregulation by hypoxia further strengthened our hypothesis that POX may activate autophagy. Thus, we investigated the induction of autophagy by hypoxia and the involvement of AMPK and POX in this induction in HT29 cells. As shown as Fig. 5A, hypoxia (0.05% O2) dramatically induced the formation of LC3II, whose amount correlates well with the number of autophagosomes. The AMPK inhibitor, Compound C, significantly inhibited the induction of autophagy by hypoxia. After we knocked down AMPK-induced POX expression by siRNA, the formation of LC3II was partly blocked while siNeg had no effect. To ensure that this is a dynamic and complete autophagic process, we blocked the fusion of the autophagosome with the lysosome using bafilomycin A1. Bafilomycin A1 led to a dramatic accumulation of the unprocessed LC3-II. In addition, beclin-1, one of the central regulators of autophagy (25), showed a similar pattern of changes as LC3II (Fig. 5B). The production of autophagosomes following GFP-LC3II viral transduction further validated the above results (Fig. 5C and D). The percentage of GFP-LC3II positive cells was much higher under hypoxia than normoxia (35.4% and 2.6%, respectively). Compound C and siPOX decreased the percentage of GFP-LC3II positive cells to 10.8% and 16.05%, respectively. Furthermore, to ascertain that hypoxia-induced autophagy maintained cell survival but did not sensitize cells to cell death, we treated cells with the autophagy inhibitor chloroquine (CQ). As shown in Fig. 6E, two concentrations of CQ (5 and 10 μM) significantly decreased cell proliferation, suggesting that sustained autophagy promoted cell survival, a finding consistent with the effects of POX on cell viability.
Figure 5.

POX induces protective autophagy under hypoxia. A, after HT29 cells were transfected with POX siRNA or scrambled negative siRNA for 18 hrs, cells were exposed to hypoxia (0.05% O2) or treated with Compound C at the same time for different time periods. Autophagic LC3-I and II were measured by Western blot (left panel). Bafilomycin A1 treatment (48 hrs) indicated the presence of the autophagic flux (right panel). B, after cells were exposed to hypoxia for 48 hrs, beclin-1 mRNA expression was examined by real-time RT-PCR. C, autophagosome accumulation was tracked by GFP-LC3II virus transduction. Mutated GFP-LC3 (G120A) was used as the negative control. D, the percentage of GFP-LC3 positive cells was calculated. E, HT29 cells were given the autophagy inhibitor chloroquine (CQ) (5 and 10 μM) while treated with hypoxia (0.05% O2). Proliferation assays at different time points were performed using the WST method. *, P<0.001 compared with normoxia; ##, P<0.05, ##, P<0.01 compared with hypoxia.
Figure 6.

POX-induced ROS is necessary for hypoxia-induced autophagy. HT29 cells were given 5mM of the ROS scavenger, N-acetylcysteine (NAC), while cells were exposed to hypoxia (0.05% O2) for 48 hrs. A, autophagic LC3-I and II were measured by Western blot. B and C, autophagosome accumulation was tracked by GFP-LC3II virus transduction, and the percentage of GFP-LC3 positive cells was calculated. D, proliferation assay was performed using the WST methods. E, POX siRNA or scrambled negative siRNA was transfected into HT29 cells for 18 hrs prior to hypoxic exposure. Then HT29 cells were treated with hypoxia (0.05% O2) for 48 hrs. Compound C was added into the culture medium at the same time. Phospha-mTOR and phospha-S6 levels were determined by Western blot. *, P<0.001 compared with normoxia; #, P<0.001 compared with hypoxia.
POX-induced ROS is necessary for hypoxia-induced autophagy
To confirm that ROS produced by POX is indeed responsible for autophagy, we examined the effect of the ROS scavenger N-acetylcysteine (NAC) on the formation of LC3II and the accumulation of autophagosomes under hypoxia. As shown in Fig. 6A, NAC (5mM) significantly decreased the production of LC3II induced by hypoxia (0.05% O2) in HT29 cells. It also decreased the amount of cells displaying punctuate distribution of GFP-LC3II (Fig. 6B and C). We performed proliferation assays to assess the contribution of hypoxia-induced ROS to cell viability. The defense against ROS-mediated stress provided by NAC significantly improved the cell growth (Fig. 6D), indicating that hypoxia-induced ROS can damage cells and decrease cell viability under our experimental conditions. Because POX is not the only contributor for hypoxia-induced ROS and ROS could serve diverse functions, it is plausible that ROS generated by POX is necessary for protective autophagy.
One of the major downstream signaling pathways regulated by AMPK is the mammalian target-of-rapamycin (mTOR) pathway (26). It is known that AMPK can induce an autophagic response by suppressing mTOR activity (27). Previous work from our lab has shown that mTOR inhibition by rapamycin increased POX and stimulated proline degradation (6). Additionally, work by others demonstrated that the Akt/mTOR pathway was able to induce autophagy mediated by ROS (28). Thus, to assess involvement of the mTOR pathway in the AMPK-POX-ROS-autophagy pathway, we examined the activation of mTOR and its downstream factor S6. As previously reported (26, 27, 29), we showed that phospho-mTOR and phospho-S6 were decreased with hypoxia, and that this effect could be reversed by the AMPK inhibitor, Compound C (Fig. 6E). However, POX siRNA had no effect on the activation of mTOR and S6, which argues against a role for mTOR in POX-induced autophagy with hypoxia.
Autophagy can also be induced under conditions of nutrient-limitation, providing a mechanism for maintaining cell viability (30). Our experiments showed that low-glucose conditions did induce autophagy, but knock-down of POX using siRNA did not change the conversion of LC3I to LC3II (data not shown). This may be because POX was used to produce ATP for direct energy provision rather than for ROS signaling.
Discussion
Hypoxia and nutrient depletion are important characteristics of the tumor microenvironment. One of the hallmarks of tumor cells is their metabolic reprogramming to adapt to these unfavorable conditions (31, 32). Metabolic changes in tumor cells can confer survival advantages and contribute to malignant progression (32, 33). Deprivation of both oxygen and glucose can result in the activation of AMPK, which is a critical factor for tumor cells to mediate those metabolic changes (21, 34). Once stimulated, AMPK inhibits energy-consuming processes and activates energy-producing processes to restore energy homeostasis by direct activation of metabolic enzymes or by inducing specific gene expression, which controls processes relevant to tumor development, including cell cycle progression, protein synthesis, cell growth, and survival (35, 36). Our previous report has shown that POX induction by glucose withdrawal is AMPK dependent (6). Pharmacologic activation of AMPK by AICAR resulted in a dramatic induction of POX (6). Therefore, as demonstrated in this study, it is not surprising that POX upregulation by hypoxia is not related to HIF-1α or HIF-2 α, the master regulators of cell response to hypoxia, but rather due to signaling from the hypoxia-activated AMPK pathway.
Our results show that induction of POX by AMPK signaling contributes to tumor cell survival. POX is a multifunctional enzyme and its structure underlies its multiple functions in a model tested in Thermus thermophilus (12). The pair of electrons transferred from proline to FAD at the active site of POX may directly reduce solvent oxygen to produce superoxide (12, 13). However, an adjacent alpha-helix structure can be conformationally shifted to block the access of FAD to solvent oxygen. Alternatively, the electrons may be donated to the electron transport chain to generate ATP (6, 13). As summarized in Fig. 7, our current work has revealed the different functions of POX under hypoxia and glucose depletion. POX activation produces ATP under low-glucose stress, whereas hypoxia with adequate glucose activates POX-mediated ROS production. As an adaptation to the anaerobic environment, glycolysis is induced and tumors maximize the use of limited quantities of available glucose (37). When glucose supply is limited, cancer cells may be forced to select proline as part of alternative energy source since the ECM/proline source has a marked advantage over fatty acids and glutamine which like glucose also require delivery by the circulation. Although proline is not an efficient substrate for generating ATP, its contribution could be significant in the metabolic adaptation of cancer cells in response to nutrient limitation. Our results suggest that the oxidation of proline by POX may represent a switching point between ATP and ROS production, which results in distinct outcomes according to the specific tumor microenvironmental stresses, e.g. glucose deprivation or hypoxia. The mechanism mediating this switch remains unknown and needs to be further explored. Nevertheless, resulting from the induction of POX, the degradation of proline can augment ATP production under low-glucose conditions and induce ROS generation under hypoxic conditions, both of which are processes that help tumor cells to survive. ATP is used for direct energy provision, while ROS are used for protective autophagy rather than apoptotic cell death.
Figure 7.
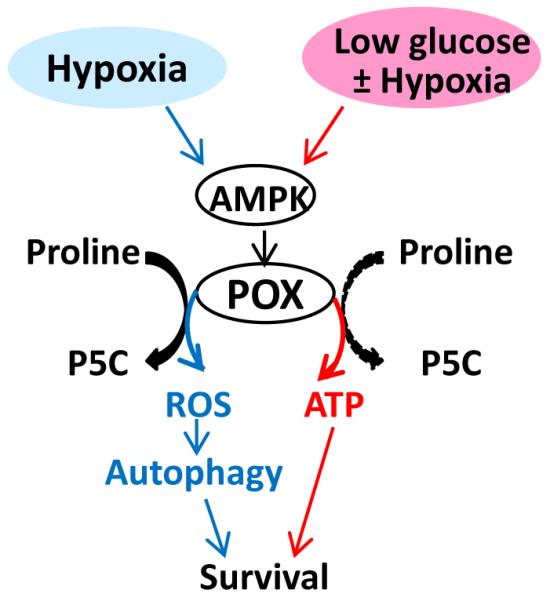
Differential functions of POX under hypoxia and low glucose conditions. Hypoxia, low glucose, and combined low glucose and hypoxia upregulated POX through the same mechanism: the AMPK pathway. Under low glucose condition, POX was used preferentially for ATP production, while under hypoxia with adequate glucose POX mediated ROS production. However, both POX-induced ATP and ROS eventually promoted tumor cell survival through direct energy provision (ATP) or ROS-induced protective autophagy.
Autophagy is a cellular self-catabolic process whereby cytoplasm and cellular organelles are degraded in lysosomes for amino acid recovery and energy recycling (38). It is often induced in cancer cells under the stress of oxygen and/or nutrient limitation, providing a survival mechanism to maintain cell viability (38). Numerous reports have shown that stress-triggered autophagy depends on the AMPK pathway (24, 39). The identified downstream targets of AMPK to regulate autophagy include mTOR (40), p27 (41), and eukaryotic elongation factor-2 kinase (eEF-2 kinase) (42), etc. The present study indicates that POX is an additional downstream target of AMPK to activate autophagy. POX-generated ROS under hypoxia appears to be essential for the induction of autophagy. Although overexpression of beclin-1, the important autophagy inducer and marker, has been shown its association with tumor hypoxia, and its capability of predicting poor prognosis of various cancers (43, 44), the mechanism about its upregulation by hypoxia remains unclear. Protein kinase C (PKC) delta was reported to induce beclin-1 by dissociating the Bcl-2/Beclin 1 complex in the early stage of hypoxic response (45). Chhipa et al. identified Beclin-1 as a potential downstream target of AMPK in turning on the autophagic cascade (46), which was supported by our study showing that POX induced beclin-1 depending on AMPK activation (Fig. 5B).
A number of reports showed that the proline concentration is increased in various tumors (47, 48). An important source of free proline stems from the degradation of collagen I, the predominant protein in the ECM of tumors (7). Kakkad SM et al. reported that hypoxia could induce the degradation of collagen (15). Previous work from our lab showed that glucose depletion activated MMP-2 and -9, which degrade collagen I in the ECM, and that a concomitant increase in intracellular proline levels was observed (6). Intracellular protein that is being degraded by autophagy may also provide an important source of free proline. Ample sources of proline ensure its availability as an alternative stress substrate. Our present study shows that inhibition of POX by DHP or siRNA significantly decreased proliferation, the generation of ATP, or the production of ROS, while exogenous proline addition only slightly influenced those effects, suggesting that endogenous proline is the main source of stress substrate under the experimental conditions used here.
It should be noted that we investigated the function of upregulation of POX by hypoxia and/or glucose deprivation only in HT29 colon cancer cells, although we observed the induction of POX expression in various cancer cells. The downstream effects of POX upregulation may be tissue or context specific and need further study.
In summary, we have demonstrated that hypoxia and/or glucose deprivation induce the expression of POX in various cancer cells. And POX is a critical factor in the AMPK signaling network for adaptive metabolic induction under conditions of hypoxia and glucose deprivation, at least in HT29 colon cancer cells. The induction of proline catabolism contributes an important mechanism by which tumor cells switch to a survival mode. These findings offer new insight into our understanding of metabolic changes in tumor cells beyond the well-recognized Warburg effect of aerobic glycolysis.
Supplementary Material
Acknowledgements
We thank Dr. Ziqiang Zhu for insightful comments. We also thank Donna Butcher for technical help of EGFP and POX immunohistochemistry, and Dr. De Chen and Dr. Stephen Lockett for the colocolization analysis.
Grant Support This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and extramural NIH grants P50 CA103175 and CA154725. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer. 2011;11:835–48. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010;70:859–62. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 5.Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol. 30:1303–18. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandhare J, Donald SP, Cooper SK, Phang JM. Regulation and function of proline oxidase under nutrient stress. J Cell Biochem. 2009;107:759–68. doi: 10.1002/jcb.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ii M, Yamamoto H, Adachi Y, Maruyama Y, Shinomura Y. Role of matrix metalloproteinase-7 (matrilysin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp Biol Med (Maywood) 2006;231:20–7. doi: 10.1177/153537020623100103. [DOI] [PubMed] [Google Scholar]
- 8.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–7. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 10.Zabirnyk O, Liu W, Khalil S, Sharma A, Phang JM. Oxidized low-density lipoproteins upregulate proline oxidase to initiate ROS-dependent autophagy. Carcinogenesis. 2010;31:446–54. doi: 10.1093/carcin/bgp299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem. 2006;281:2044–52. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- 12.White TA, Krishnan N, Becker DF, Tanner JJ. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J Biol Chem. 2007;282:14316–27. doi: 10.1074/jbc.M700912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagedorn CH, Phang JM. Transfer of reducing equivalents into mitochondria by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch Biochem Biophys. 1983;225:95–101. doi: 10.1016/0003-9861(83)90010-3. [DOI] [PubMed] [Google Scholar]
- 14.Raman V, Artemov D, Pathak AP, Winnard PT, Jr., McNutt S, Yudina A, et al. Characterizing vascular parameters in hypoxic regions: a combined magnetic resonance and optical imaging study of a human prostate cancer model. Cancer Res. 2006;66:9929–36. doi: 10.1158/0008-5472.CAN-06-0886. [DOI] [PubMed] [Google Scholar]
- 15.Kakkad SM, Solaiyappan M, O’Rourke B, Stasinopoulos I, Ackerstaff E, Raman V, et al. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia. 2010;12:608–17. doi: 10.1593/neo.10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acker T, Plate KH. A role for hypoxia and hypoxia-inducible transcription factors in tumor physiology. J Mol Med. 2002;80:562–75. doi: 10.1007/s00109-002-0355-1. [DOI] [PubMed] [Google Scholar]
- 17.Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 2006;16:1–10. doi: 10.1101/gr.4222606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue LX, Weng M, Zhang ZY, Tong TJ. Optimization of reporter gene assay: several factors influencing detection of promoter activity. Chin Med J (Engl) 2007;120:965–9. [PubMed] [Google Scholar]
- 19.Landolin JM, Johnson DS, Trinklein ND, Aldred SF, Medina C, Shulha H, et al. Sequence features that drive human promoter function and tissue specificity. Genome Res. 2010;20:890–8. doi: 10.1101/gr.100370.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horsman MR. Nicotinamide and other benzamide analogs as agents for overcoming hypoxic cell radiation resistance in tumours. A review. Acta Oncol. 1995;34:571–87. doi: 10.3109/02841869509094031. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 2009;196:55–63. doi: 10.1111/j.1748-1716.2009.01980.x. [DOI] [PubMed] [Google Scholar]
- 22.Blancher C, Moore JW, Talks KL, Houlbrook S, Harris AL. Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res. 2000;60:7106–13. [PubMed] [Google Scholar]
- 23.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 24.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–81. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 25.Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–49. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- 26.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puissant A, Robert G, Auberger P. Targeting autophagy to fight hematopoietic malignancies. Cell Cycle. 2010;9:3470–8. doi: 10.4161/cc.9.17.13048. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Kong X, Kang J, Su J, Li Y, Zhong J, et al. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol Sci. 2009;110:376–88. doi: 10.1093/toxsci/kfp101. [DOI] [PubMed] [Google Scholar]
- 29.Schneider A, Younis RH, Gutkind JS. Hypoxia-induced energy stress inhibits the mTOR pathway by activating an AMPK/REDD1 signaling axis in head and neck squamous cell carcinoma. Neoplasia. 2008;10:1295–302. doi: 10.1593/neo.08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein promotes autophagy in a survival response to glucose deprivation. Int J Oncol. 2009;34:1691–9. doi: 10.3892/ijo_00000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312:1158–9. doi: 10.1126/science.312.5777.1158. [DOI] [PubMed] [Google Scholar]
- 32.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–7. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardie DG. AMPK and Raptor: matching cell growth to energy supply. Mol Cell. 2008;30:263–5. doi: 10.1016/j.molcel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M, et al. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene. 2002;21:6082–90. doi: 10.1038/sj.onc.1205737. [DOI] [PubMed] [Google Scholar]
- 37.Oliver L, Olivier C, Marhuenda FB, Campone M, Vallette FM. Hypoxia and the malignant glioma microenvironment: regulation and implications for therapy. Curr Mol Pharmacol. 2009;2:263–84. doi: 10.2174/1874467210902030263. [DOI] [PubMed] [Google Scholar]
- 38.Tsuchihara K, Fujii S, Esumi H. Autophagy and cancer: dynamism of the metabolism of tumor cells and tissues. Cancer Lett. 2009;278:130–8. doi: 10.1016/j.canlet.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 39.Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007;3:238–40. doi: 10.4161/auto.3710. [DOI] [PubMed] [Google Scholar]
- 40.Hall MN. mTOR-what does it do? Transplant Proc. 2008;40:S5–8. doi: 10.1016/j.transproceed.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 42.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–31. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 43.Wan XB, Fan XJ, Chen MY, Xiang J, Huang PY, Guo L, et al. Elevated Beclin 1 expression is correlated with HIF-1alpha in predicting poor prognosis of nasopharyngeal carcinoma. Autophagy. 2010;6:395–404. doi: 10.4161/auto.6.3.11303. [DOI] [PubMed] [Google Scholar]
- 44.Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br J Cancer. 2010;103:1209–14. doi: 10.1038/sj.bjc.6605904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen JL, Lin HH, Kim KJ, Lin A, Ou JH, Ann DK. PKC delta signaling: a dual role in regulating hypoxic stress-induced autophagy and apoptosis. Autophagy. 2009;5:244–6. doi: 10.4161/auto.5.2.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011;23:1466–72. doi: 10.1016/j.cellsig.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catchpole G, Platzer A, Weikert C, Kempkensteffen C, Johannsen M, Krause H, et al. Metabolic profiling reveals key metabolic features of renal cell carcinoma. J Cell Mol Med. 2009;15:109–18. doi: 10.1111/j.1582-4934.2009.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–25. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.