

Summary
Despite aggressive multi-modality therapy including surgery, radiation, and chemotherapy, the prognosis for patients with malignant primary brain tumors remains very poor. Moreover, the non-specific nature of conventional therapy for brain tumors often results in incapacitating damage to surrounding normal brain and systemic tissues. Thus, there is an urgent need for the development of therapeutic strategies that precisely target tumor cells while minimizing collateral damage to neighboring eloquent cerebral cortex. The rationale for using the immune system to target brain tumors is based on the premise that the inherent specificity of immunologic reactivity could meet the clear need for more specific and precise therapy. The success of this modality is dependent on our ability to understand the mechanisms of immune regulation within the central nervous system (CNS), as well as counter the broad defects in host cell-mediated immunity that malignant gliomas are known to elicit. Recent advances in our understanding of tumor-induced and host-mediated immunosuppressive mechanisms, the development of effective strategies to combat these suppressive effects, and a better understanding of how to deliver immunologic effector molecules more efficiently to CNS tumors have all facilitated significant progress toward the realization of true clinical benefit from immunotherapeutic treatment of malignant gliomas.
Keywords: glioma, immunotherapy, brain tumor, cancer vaccines
Immunotherapy of malignant brain tumors
Immunotherapy holds the promise of targeting tumor cells for destruction with an exquisite specificity and efficiency, while at the same time almost completely sparing normal cells from harm. The sensitivity and specificity of the immune system is refined to the point at which the body is capable of recognizing foreign pathogens such as viruses within minutes of infection, responding with an array of innate, humoral, and cellular effector mechanisms that can control a rapidly expansive viral infection, and eliminating almost every infected cell from the body. For more than 100 years, tumor immunologists have hoped to harness this amazing cytotoxic power for use against malignant cancer cells, which, although following entirely different physiologic mechanisms for invasion than viruses, also spread through the body with deadly consequences.
The immunologic treatment of high-grade malignant brain tumors has discriminating considerations compared with other malignant diseases with regard to its central nervous system (CNS) immune privilege and concerns of organ-specific autoimmunity. The development of effective immunotherapy against brain tumors, therefore, represents a unique challenge in the field of tumor immunotherapy. Despite aggressive multimodality therapy, including surgery, radiation therapy, and chemotherapy, the prognosis for patients diagnosed with high-grade brain tumors remains very poor. Patients diagnosed with glioblastoma multiforme (GBM), the most aggressive and unfortunately most common type of adult brain tumor, have a median survival of only 15 months (1). In addition, the standard treatments for malignant brain tumors often result in debilitating motor and neurological deficits in treated patients. Therefore, there is a paramount need for the development of more effective and specific therapies, such as immunotherapy for the treatment of malignant brain tumors.
The present day thinking within the field of tumor immunology has moved beyond a debate as to whether tumors express antigens that can be recognized and targeted by the immune system. Experimental cancer models have undoubtedly demonstrated that the immune system is highly capable of effectively eradicating malignant tumor cells. Classical transplantation models were used in the first experiments examining the immunogenicity of tumors to demonstrate that chemically-induced tumors contain antigens that can lead to the specific recognition and rejection of tumors in immunocompetent mice (2). Although chemically-induced tumor cell lines demonstrated the capacity of the immune system to mediate tumor rejection, their strong immunogenic properties did not closely parallel the presumably non-immunogenic nature of most human tumors. Subsequently, using more relevant tumor models, it was demonstrated that even less immunogenic or non-immunogenic tumor cell lines expressed antigens that could be recognized by the immune system (3). These experiments demonstrated the existence of tumor antigens in rodent tumor lines, but the first human tumor antigens were discovered in malignant melanoma through the notable efforts of van der Bruggen et al. (4) and Boon et al. (5). Since that time, an explosion has occurred in the identification of tumor antigens and the development of approaches toward the immunologic treatment of cancer. Current research focuses on continuing to identify new antigens in human tumors, understanding how tumors effectively evade the physiologic anti-tumor immune response, and translating the preclinical successes in experimental tumor immunotherapy models into a clinical reality in human patients.
Early efforts in tumor immunotherapy focused on the use of non-specific immune stimulators to expand an anti-tumor immune response in the host. Injection of adjuvants, such as heat-killed bacille Calmette-Guerin or Corynebacterium, directly into peripheral tumors was attempted to treat malignant melanoma and other cancers but was generally unsuccessful. However, there have been a few notable successes, such as the treatment of bladder cancers with locally injected, non-specific adjuvants, that supports the validity of non-specific adjuvant approaches to treating malignancy (6).
In recent years, more potent immunostimulatory agents that act directly on antigen-presenting cells (APCs) and effector cells of the immune system, such as Toll-like receptor agonists, have gained interest in immunotherapy (7, 8). These agonists lead to signaling through a family of Toll-like receptors on APCs of the immune system that results in the upregulation of costimulatory molecules and cytokine production such as interferon γ (IFNγ) and interleukin-12 (IL-12) (9). Toll-like receptor agonists such as lipopolysaccharide (LPS) (10), double-stranded RNA (11), heat shock protein 70 (12), imiquimod (13), and CpG oligonucleotides (8) have all demonstrated the capacity to enhance immunologic responses against malignant gliomas (and other tumors). Although these agonists alone can enhance physiologic anti-tumor immune responses, most experimental protocols with demonstrated anti-tumor efficacy use the agonists in combination with some form of specific anti-tumor immunotherapy.
The majority of current efforts in immunotherapy are directed primarily at the induction of specific immune responses against tumor antigens using either active immunization strategies, called ‘cancer vaccines,’ or adoptive transfer of tumor-specific effector cells or antibodies. While it was initially believed that effective immune responses against tumors within the CNS would be prevented by the ‘immunoprivileged’ status of the brain, studies have demonstrated that immune effector cells and antibodies can gain access to the CNS and leverage potent effector mechanisms against recognized target cells within the brain (reviewed in 14).
CNS immunity
The wide variety of aberrantly expressed and mutated proteins present in tumor cells should, in theory, permit them to be identified as foreign and ultimately rejected by the immune system. This concept of tumor surveillance as a normal function of the immune system has supporting evidence in that physiologic anti-tumor immune responses can be detected in patients with cancer, although obviously at an insufficient level of function to prevent the progression of malignancy. Circulating tumor-specific antibodies and cytotoxic T lymphocytes (CTL) have been isolated from the peripheral blood of patients with malignant glioma (15). It is also evident, however, that normal immune mechanisms are not sufficient, as tumors indeed continue to grow in the face of an apparently intact immune response. The potential conclusions are that in patients who present with cancer, the immune system either mounts a response that is incapable of eradicating tumor cells, or that by the time malignant tumor growth reaches clinical detection, the resulting tumor has been selected for immunologic escape under the pressure of what was once an effective physiologic anti-tumor immune response (16).
Malignant gliomas and the surrounding CNS each supply additional challenges to an already inadequate cancer response. A purported immune privilege of the brain and a glioma-induced local and systemic immunosuppression may further limit the efficacy of any existing natural or therapeutically manipulated immune responses against malignant brain tumor cells.
Immunologic privilege
It has long been contended that the immune system has limited access to the brain and thus would have negligible contact with neoplastic cells that are harbored within the CNS. The concept of CNS immune privilege has its origins in studies by Medawar in 1948 (17), who demonstrated the failure to reject allogeneic tissue grafts placed within the brains of experimental animals. The capacity for allografts to survive in the CNS was attributed to the presence of the blood–brain barrier, the absence of a lymphatic drainage system within the brain, and a void of resident specialized APC within the CNS (18, 19).
This model of immune privilege has been challenged, particularly as it has become evident that connections do exist between cerebrospinal fluid (CSF) compartments and cervical lymphatics (20), that microglia can fill the role of resident APC within the CNS (21), and that professional APCs (i.e. dendritic cells) are present in both the choroid plexus (22) and meninges (23). Furthermore, careful studies have demonstrated that an active pattern of T-cell trafficking to and from the brain indeed does occur (24). In fact, up to 30–60% of primary human glial tumors contain mononuclear infiltrates at the time of pathologic examination (25, 26). Thus, the question of whether immune responses are mounted within the CNS may be more refined to the degree of intensity of these responses compared with the periphery, rather than an anergic state of the CNS with respect to immunity. Nevertheless, while it is now clear that the CNS is not isolated from immune surveillance, access is also clearly not liberal, and if a major mechanism of tumor escape from immunologic destruction is inadequate access to the CNS by effector immune cells, then the potential exists to develop ways to increase the delivery of these cells to CNS lesions and produce an effective immunologic response (16).
Immune responses to antigen in the brain
Animal studies have demonstrated that antigen presentation in cervical lymph nodes occurs due to drainage via non-classical lymphatic pathways along cranial nerves and that activated lymphocytes enter the brain despite the presence of the blood–brain barrier (27). While it is clear that immunologic responses to CNS antigens are normally induced, the presence of the blood–brain barrier creates a carefully regulated environment with distinct composition of immunoregulatory molecules such as neuronal growth factors, cytokines, chemokines, and neuropeptides. Thus, response to antigens within the CNS occurs with a distinct hierarchy in terms of the types of responses induced (humoral, cellular, and innate) and the character of these responses (28). Antigen draining the CNS induces responses initially in the periphery within the context of the cervical lymph nodes. This response is characterized by a strong antibody response, the priming of cytotoxic T-cell responses, but an absence of the induction of delayed-type hypersensitivity (DTH) responses (29). Furthermore, the microenvironment of the brain permits the full development of effector function of B lymphoblasts (antibody secretion) but inhibits the full development of cell-mediated immunity(CMI) either entirely (DTH) or partially (cytotoxic T-cell responses) (28). Thus, the net effect of antigens introduced into the CNS is a T helper-2 (Th2) skewing of T-cell responses and the induction of strong humoral responses to antigenic challenge (27-32). While there is a bias toward Th2 responses to antigens derived from the CNS, this skewing can be affected by changes in the inflammatory microenvironment within the brain, alteration in the composition of immune cells accumulating at sites of antigenic challenge, or a shift in the production of cytokines or other immunoregulatory molecules within the CNS. Therefore, the microenvironment of the CNS is entirely capable of supporting the full spectrum of cellular and humoral immune responses observed during immune responses in the periphery, although with a significant skewing toward Th2 lymphocyte and humoral responses.
Antigen presentation and APCs/microglia
A consideration of the flow of cerebral extracellular fluids, cerebral interstitial fluid (CIF) and CSF, is essential to understanding antigen presentation within the CNS. CIF is secreted at the blood–brain barrier (BBB) and flows within the spaces between cells of the brain parenchyma. CSF is formed by the choroid plexus within the ventricles and subarachnoid membrane and circulates in a rostral to caudal direction through the ventricles to the basal cisterns. CSF then moves into the subarachnoid space, which is contiguous with the spinal cord (27, 33). Although the brain is absent of specialized lymphatic vessels, there is efficient drainage of CIF to the deep cervical lymph nodes via the nasal submucosa, along cranial nerve tracks, and around perivascular sheaths. Convective flow of CSF allows for antigens to gain access to outlets within the arachnoid membrane and cribiform plate. These fluids exit the subarachnoid space through flow across the arachnoid granulations and through drainage along the olfactory nerve across the cribriform plate into the blood and cervical lymph pathways (29, 34, 35). Thus, the initial site of immune activation for CNS-derived antigens occurs at extracranial sites within the deep cervical lymph nodes. Antigen draining to cervical lymph nodes from the CNS can encounter cognate B lymphocytes and also be processed and presented to circulating naive T cells by professional APC such as dendritic cells (DCs) present within these lymph nodes and lead to the activation of immune effector mechanisms. While naive lymphocytes do not cross the BBB, activated lymphocytes patrol the CNS freely and are actively recruited to sites of inflammation. It is less clear what immune mechanisms are utilized within the parenchyma of the brain for the reactivation of effector lymphocytes, due to the fact that immunologic responses in the CNS are the result of a complex interaction between resident immune cells such as microglia and astrocytes, as well as recruited macrophages, lymphocytes, and DCs from the periphery (36-39).
Professional APCs such as DCs have not formally been demonstrated in the CNS parenchyma (40). It has been suggested that microglia might be the major or exclusive APC in the CNS, and human microglial cells have been shown to have phenotypic and functional characteristics of both macrophages and DCs (41, 42). Strong evidence has been provided that these cells, predominately located in the perivascular spaces and the leptomeninges, are bone marrow-derived cells capable of presenting antigen to helper T cells in vivo (43). Although resting microglial cells appear quiescent with regard to endocytic and secretory function, they constitutively express class II antigens in situ (42, 44) express T-cell costimulatory molecules such as leukocyte function-associated antigen-1 (LFA-1), LFA-3, intercellular adhesion molecule-1 (ICAM-1), and B7 (45), can cluster CD4+ T cells (42), and can induce T-cell responses in a mixed lymphocyte-type reaction in vitro (42, 45). Incidentally, astrocytes also express ICAM-1 and LFA-3, although to a lesser degree than microglia (45). Astrocytes, while capable of processing and presenting antigens to lymphocytes and being activated to produce immunoregulatory cytokines, are thought to be relatively poor APCs and unlikely to efficiently lead to activation of T lymphocytes (46).
T-cell trafficking
Naive T cells are not found within the CNS due to their inability to pass through the BBB. When T cells have been activated against neurotrophic pathogens or CNS autoantigens, they cross the BBB and are restimulated upon encounter with their cognate antigen on target cells and local APCs. Murine models of experimental autoimmune encephalitis (EAE) have demonstrated that activated T cells infiltrating the CNS secrete effector cytokines but do not proliferate and undergo apoptosis (46, 47). Recent studies, however, employing brain tumor models have shown conflicting data, with the brain microenvironment promoting the proliferation of tumor-specific T cells within brain parenchyma and differentiation into cells with enhanced effector function (48). While activated T cells are thought to patrol the CNS in an antigen-independent manner, cells that encounter their cognate antigen are retained for longer periods within the CNS than those that do not encounter target antigen within the brain (48). Studies examining the exit of T cells from the brain have shown these cells to uniformly pass the cribroid plate and reach the naval mucosa and eventually cervical lymph nodes (49).
Antibody penetration
It is a generally held notion that antibodies do not effectively penetrate the CNS except in cases of disruption of the BBB due to inflammatory processes. This assumption is based largely on the low prevalence of globulin proteins within the CSF compared with peripheral blood of normal individuals. However, careful experimental studies have demonstrated rapid accumulation of antibodies within the CSF and brain parenchyma after passive or active peripheral immunization in experimental animals (50). These studies confirmed that antibodies distribute throughout the CNS with similar kinetics to other peripheral organs, albeit at a ratio of approximately 0.1-1% the titer of antibody found in the serum. While it is apparent that antibody penetrates the CNS in the absence of BBB disruption, it is unclear whether the levels of antibody achieved within the CNS in the absence of BBB disruption are sufficient to mediate effector functions in the brain. Evidence supportive of the physiologic relevance of CNS antibody titers come from studies of active or passive immunization in which antibodies seem to play a significant role.
The interesting case of Alzheimer’s vaccines
The apparent clinical efficacy and the rather dramatic toxicity recently attributed to vaccines targeting amyloid-β (AB) protein in patients with Alzheimer’s disease (AD) has recently provided a number of novel insights into the potential role of immunotherapy against diseases within the CNS. It has also raised a number of questions, which mostly remain unanswered.
AB protein has long been suspected to play a role in AD. This role is supported by significant genetic and pathologic data. Mice transgenic for amyloid precursor protein (APP) mutants also develop early onset forms of AD that mirrors the human condition. Monitoring AB plaque burden and behavioral endpoints in these models provided the initial evidence that vaccinations with AB peptide may have efficacy in this disease (51, 52). The hypothesis driving these initial studies was that AB peptide vaccinations would produce antibodies that would bind AB and enhance clearance or prevent deposition.
In human trials, antibodies to AB were induced, but titers failed to correlate with clinical responses and a dramatic meningoencephalitis developed in a small but significant subset (approximately 6%) of patients who received the vaccine, resulting in the trials being halted prematurely. The bulk of evidence currently supports a CD4+CD45RO+ T-cell-mediated mechanism for the meningoencephalitis but still supports an antibody-mediated mechanism for the efficacious effects (53, 54).
Three distinct, not mutually exclusive, and possibly overlapping mechanisms have been proposed for the ability of even passive antibody-based immunotherapy to reverse or prevent the pathologic and clinical effects of AB-peptide plaque accumulation. The first two require that antibodies enter the CNS and once there either directly dissolve the plaques (55) or mediate opsonization and clearance of the AB peptide in a microglial dependent manner (56, 57). The third hypothesis postulates that antibody restricted to the periphery sequesters AB-peptide and establishes a concentration gradient that drives efflux from the CNS (58).
A successful humoral immunotherapy for CNS diseases, especially within the context of an intact BBB, was counterintuitive, given the known limited penetration of large molecules like antibodies across the BBB. As a result, the early reports in animals and humans demonstrating the effectiveness of an active immunotherapy against AD were reviewed with surprise and some skepticism.
Further evidence that physiologically relevant levels of antibody can accumulate in non-inflamed CNS is supported by the observation that passive administration of AB-specific antibodies is sufficient to produce most if not all of the beneficial effects of the vaccination in murine models (56, 57). Intraperitoneally administered antibody can be detected clearly within the CNS, and where strong antibody staining was found, it correlated with efficacy of the antibodies (56). Further support for the theory that antibodies must penetrate the CNS to be efficacious in this context is provided by the finding that microglia were capable of removing amyloid plaque material from mouse and human brain sections only in the presence of intact, AB-specific antibodies (56). AB-specific antibodies were also detected in the brains of vaccinated humans postmortem. Although the antibody levels were not higher than in unimmunized controls (53), this may have been due to the time elapse between the vaccination and autopsy. In vaccinated humans, AB was also found to be associated with microglia, and microglia Fc receptor staining was increased, consistent with Fc-mediated phagocytosis (53) as seen in mouse models (56).
The best evidence to support a physiologic role for vaccine-induced antibodies within the CNS thus comes from the above-described trials targeting AB plaques in patients with AD. While it remains under debate whether the antibodies induced by these vaccines are responsible for any clinical responses and whether or not the antibodies entered the CNS to mediate such effects (58), much of the data supports this conclusion and provides a rationale for using systemically derived antibody therapies against intracranial tumors (53, 56, 57, 59). A recent Phase I clinical trial in eight patients using a systemically administered chimeric monoclonal antibody (ch806) that recognizes mutant forms and overexpressed epidermal growth factor receptor (EGFR) but not normal EGFR demonstrated surprising efficient localization of antibody to tumor in a patient with malignant glioma (anaplastic astrocytoma) (60). Specific accumulation of indium-111-labeled antibody was visualized by single photon emission computed tomography (SPECT) imaging by day 3 after injection, and antibody deposition at the tumor site accumulated over the 7-day observation period. This study showed that human CNS tumors can serve as a ‘sink’ for the extravasation of passively administered antibody across the BBB from the periphery, if competing target antigen is absent elsewhere in the body, and thus, further supports the notion that the BBB may not be as significant a barrier to immunity as once thought.
Immunosuppression
Patients with malignant gliomas typically exhibit a comprehensive suppression of their cell-CMI, which serves not only to disrupt their physiologic immune responses but to hinder the success of immune-based therapies as well (reviewed in 61). Cutaneous anergy, lymphopenia, impaired antibody production, reduced lymphocyte protein synthesis, and diminished lymphocyte responsiveness have all been reported (62-75). Defects in the T-cell compartment are especially well documented, as peripheral blood lymphocytes (PBLs) from glioma patients proliferate poorly in response to T-cell mitogens, anti-CD3, and T-dependent B-cell mitogens (76-79). Although invariably present, T-cell lymphopenia does not sufficiently explain the CMI impairment, since purified T-cell populations also demonstrate a limited ability to respond to mitogenic stimulation (73, 76, 77, 80). Interestingly, many of the proliferative defects appear to be concentrated in the CD4+ subset of T cells (77), and these fail to provide helper activity in allogeneic pokeweed mitogen cultures (81). Thus, defects intrinsic to the CD4+ T-cell pool appear to play a major role in the impairment of CMI.
Immunosuppressive cytokines: transforming growth factor-β, vascular endothelial growth factor
One possible explanation for the impairment of CMI suffered by patients with primary brain tumors is the secretion of transforming growth factor-β (TGF-β) by their tumors (82-84). TGF-β is a homodimer of two disulfide-linked subunits, each with a molecular mass of 12.5 kDa. It has been isolated from malignant glioma cell supernatants, and subsequently, the gene for TGF-β2 was cloned from a malignant glioma cell line (85). TGF-β has been shown to suppress the generation of CTLs from PBLs and tumor-infiltrating lymphocytes by IL-2, to inhibit IL-2 receptor expression on T cells (86), to reduce IL-1 (87) and IL-2 (86, 88), to be involved in proliferation of T and B lymphocytes, to depress the cytotoxicity of natural killer (NK) cells and their activation by IFN-γ (89), to inhibit the development of CTLs (90), to reduce IFN-γ production (91), and to downregulate major histocompatibility complex (MHC) class II-dependent antigen expression (92, 93). The potential of immunosuppressive factors, such as TGF-β, to abolish a cell-mediated anti-tumor immune response has been demonstrated experimentally. Torre-Amione et al. (94) transfected a highly immunogenic and easily rejected tumor cell line with TGF-β and demonstrated that, after transfection, it was able to completely escape immune rejection. Thus, immunosuppressive factors, such as TGF-β, which are commonly secreted by primary brain tumors, may have a tremendous negative impact on the efficacy of active, specific immunotherapies. In addition, the dependence of many patients with malignant gliomas on corticosteroids may also have a significant impact. Corticosteroids, including hydrocortisone (t1/2 = 8–12 h), prednisone (t1/2 = 18–36 h), or dexamethasone (t1/2 = 36–54 h), are known to decrease migration of leukocytes into inflamed tissues, reduce peripheral blood counts of all leukocyte subsets but especially T cells, decrease immunoglobulin G (IgG) levels, and impair cutaneous DTH responses (95, 96).
Another soluble factor secreted by tumor cells that may play a significant role in immunosuppression is vascular endothelial growth factor (VEGF). VEGF, produced by most solid cancers, plays a key role in tumor angiogenesis but also has been found to be directly responsible for inhibition of the maturation of DCs from progenitor cells from the bone marrow (97, 98). The molecular mechanism by which VEGF inhibits DC maturation has been shown to be a VEGF-induced inhibition of NF-κB signaling in hematopoietic progenitor cells. Furthermore, inhibition of VEGF production using an anti-VEGF monoclonal antibody was shown in murine tumor models to enhance the efficacy of cancer immunotherapy when given in combination with DC vaccination (99), highlighting that VEGF-mediated immunosuppression may represent a reversible axis of immunologic inhibition in patients with malignancy. Malignant gliomas are notoriously vascularized tumors that produce abundant quantities of VEGF and TGF-β, which very likely contributes significantly to the immunosuppressed phenotype observed in these patients. The development of pharmacological agents that inhibit the production of these factors, however, offers a tremendous opportunity to evaluate the capacity to enhance immunotherapy against malignant gliomas through synergistic use of agents that block the VEGF and TGF-β pathways. Anti-VEGF monoclonal antibody treatment (bevacizumab) has already been shown to have efficacy in the treatment of a variety of tumors, including malignant gliomas (100, 101). Besides the known anti-angiogenic effects of VEGF-specific antibodies (102, 103), it is possible that inhibition of VEGF-mediated immunosuppression may play a significant role in the efficacy of bevacizumab against human tumors.
Tumor defenses: B7-homolog 1, Fas/Fas ligand
In addition to the production of soluble ligands that inhibit immunologic function, malignant gliomas also produce surface ligands that can directly downregulate immunologic effector mechanisms. B7-homolog 1 (B7-H1) is a recently identified homolog of B7.1/2 (CD80/86) that has been shown to exert immune regulatory function on T lymphocytes. B7-H1 has been shown to mediate attenuation of T-cell receptor (TCR) function through engagement of the programmed death-1 (PD-1) receptor on the surface of T cells. This engagement leads to rapid phosphorylation of the Src homology region 2-containing protein tyrosine phosphatase-2, which downregulates TCR signaling (104). In addition, B7-H1 engagement of PD-1 on T cells promotes apoptosis and renders immunologic tumor cells resistant to immune-mediated rejection in murine tumor models (105). The finding of ubiquitous expression of B7-H1 in all malignant glioma lines and the increased expression of this molecule in response to TGF-γ, suggests that B7-H1 expression may be a potent mechanism for immunologic escape in malignant gliomas. The loss of phosphatase and tensin homolog (PTEN), a genetic alteration frequented in malignant gliomas, results in increases in B7-H1 expression in gliomas and correlates with immunoresistance (106). Several experimental model systems have demonstrated that blockade of the B7-H1/PD-1 pathway can lead to increased cytokine production, improved DC function, reactivation of anergic T-cell lymphocytes, and improved anti-viral and anti-tumor immune responses (107-115).
Fas ligand (FasL, CD95L) is a membrane protein belonging to the tumor necrosis factor (TNF) family that is responsible for induction of apoptosis in Fas-bearing cells (116). It is utilized by CTLs as an effector molecule for cytolytic killing, and the Fas/FasL interaction has been suggested to mediate the relative immune privilege of cells within the eye and testis, which express FasL on the surface of restricted cell types at these organ sites (117-119). Malignant gliomas have been shown to frequently express both Fas and FasL on the cell surface, and FasL positive gliomas can induce cell-contact-dependent apoptosis in Fas-expressing lymphocytes and other target cells (120, 121). However, the co-expression of Fas/FasL on gliomas also offers the opportunity to mediate apoptosis of tumor cells through engagement of Fas signaling within the malignant tumor cells. While it is unclear what mechanisms prevent the autolytic killing of Fas/FasL co-expressing tumor cells, engagement of Fas on the surface of glial tumors using soluble FasL (122) or agonistic antibodies to CD95 induces apoptosis in human malignant glioma cells in vitro (123). The co-expression of Fas/FasL by human gliomas in vivo, however, and the finding of soluble FasL in the cyst fluids of malignant gliomas (124) calls into question whether these tumors will be susceptible to Fas-mediated apoptotic mechanisms in a therapeutic setting.
Regulatory T cells
Although immunotherapy has emerged as a means of designing more tumor-specific treatment, the success of this modality is linked to our ability to understand and counter the broad defects in host CMI that malignant gliomas are known to elicit. Decades of patient studies have revealed lymphocyte proliferative defects that are concentrated in the T-cell IL-2/IL-2R system (76, 125), but their underlying cause is yet to be well elucidated.
In the late 1990s, however, the concept of T-cell-induced tolerance re-emerged with the identification of a population of T cells designated as regulatory T cells (Tregs). Tregs represent a physiologic subset of CD4+ T cells that constitutively express high levels of CD25, the IL-2Rα chain (126). They potently inhibit T-cell activation and proliferative responses by downregulating IL-2 production in their target responder cells (127-129), a penchant, which recapitulates exactly those defects observed in the peripheral blood of patients with malignant glioma. Tregs require stimulation through their TCR for activity, and though anergic in vitro, may be induced to proliferate in vivo in an antigen-specific fashion (130, 131). As they recognize self-antigens (132-134), Tregs are now known to elicit tolerance to tumor antigens, and supra-physiologic levels of functional Tregs have been found in the peripheral blood and/or tumors of patients with a variety of cancers (135-140). Tumor models employing in vivo depletion or inhibition of Tregs have demonstrated prolonged survival when used as a single modality treatment as well as enhanced efficacy of tumor cell- and DC-vaccines when used in conjunction with immunotherapy (141, 142).
Upon even the initial discovery that purified T-cell populations from patients with glioma were not capable of normal responsiveness and that defects intrinsic to the T-cell compartment were at least partially responsible for impaired patient CMI, it was suggested that the ‘results might indicate the expansion of an otherwise normally present subpopulation of cells which … are capable of modulating the responsiveness of other lymphocytes’ (73). The existence of suppressor cells had first been proposed by Gershon in 1971 (143), and others had shown them to be maximally responsive at suboptimal concentrations of mitogen, a notion which remains today, and which was also consistent with the observed pattern of mitogenic responsiveness for patient lymphocytes at the time. As a result, an entire paper was dedicated to the assessment of the presence of ‘Concanavalin-A-activated, glass-adherent, and spontaneous, nonspecific suppressor cells’ (71). The authors concluded that although suppressor cell activity was inducible in patients with primary brain tumors, it did not differ from normal activity.
Another paper, however, found that of six patients studied before surgical exploration, all six exhibited significantly depressed levels of PHA responsiveness in association with notably increased levels of regulatory cell function, where regulatory activity was defined as being indomethacin-sensitive, glass-adherent, and/or preculture-sensitive. Furthermore, two of four patients with recurrent disease demonstrated reduced T-cell function in association with enhanced immunoregulatory cell activity. This study also examined the existence of a lymphocyte-mediated immunoregulation and found that two of nine patients demonstrated significantly increased lymphocyte-mediated suppressor activity. This activity was determined simply by depleting peripheral blood mononuclear cells (PBMCs) of latex-ingesting cells and evaluating the ability of the remaining patient lymphocytes to regulate lymphocyte DNA synthesis when co-cultured with normal allogeneic cells (68). Similar co-culture experiments by Roszman et al. (81) using polkweed mitogen as the stimulator instead did not reproduce these results, and it was asserted in the literature that increased suppressor cell activity was not at the root of lymphocyte proliferative defects in patients with glioma.
It is now established that the T-cell repertoire of normal animals is indeed able to thwart pathological responses toward self-antigens. For years, the model of central tolerance had been one in which the thymus clonally deleted self-reactive T cells with high avidity TCRs specific for self-antigens that are thymically presented (144, 145). These mechanisms alone, however, prove insufficient, as self-reactive T cells with pathogenic potential (including those directed against normal brain) are present in the periphery of normal individuals (146, 147). T-cell-mediated autoimmune diseases can be produced in rodents by elimination of a sub-population of peripheral CD4+ T cells, suggesting that a model of central tolerance is incomplete and that the thymus may also produce a population of immunoregulatory T cells capable of inhibiting self-reactive T cells in the periphery (126, 148, 149). It is now known that this ‘immunoregulatory’ cell activity is concentrated neither within the monocyte sub-fraction nor the CD8+ T-cell subset, but instead, is focused in the CD25+ (IL-2Rα+) fraction of CD4+ T cells (Tregs) (126, 148, 150-156).
Studies into their generation and antigen specificity requirements have yielded that Tregs are generated in the thymus by means of a moderate avidity interaction with self-peptide presented by the thymic epithelium (132-134). Furthermore, the presence of their relevant autoantigen in the periphery is requisite for their generation, as Tregs preventing organ-specific autoimmunity fail to be generated in vivo if the relevant organ is removed early in development (157). The requirement for thymic presentation of antigen to select Tregs of a given specificity is perhaps most elegantly demonstrated by the fact that mice transgenic for a TCR specific for an antigen not expressed in the thymus will develop Tregs only in the event that endogenous α-chain rearrangement allows co-expression of a second TCR on the Treg surface that is permissive of thymic selection (132, 158). Conversely, the same TCR transgenic mice bred onto a recombinase activating gene knockout (RAG KO) background, in which no α-chain rearrangement is permitted, fail to develop Tregs.
Tregs constitute 5–10% of CD4+ T cells in both mice and humans (126, 128, 149, 159, 160). In vitro studies of their functional properties have revealed that they potently inhibit polyclonal CD4+ T-cell activation and proliferative responses (127-129). They accomplish this through downregulating IL-2 production and transcription in their target responder cells (127, 161), a mechanism, which recapitulates precisely those defects observed in malignant glioma patient lymphocytes (76, 125). Tregs have also proved capable of inhibiting CD8+ T-cell proliferation and IFN-γ production (162-166). Their suppressive capacities are dependent upon signaling via their own TCR, and, at least in vitro, upon cell–cell contact with target T cells (127, 167), a mechanism that may involve surface-bound TGF-β (168, 169). Human Tregs appear to ‘transfer’ tolerizing function to their targets, eliciting in these cells a long-lasting anergy and prompting them to secrete the cytokines IL-10 (170) and TGF-β (171, 172). These two cytokines, which are also the primary cytokines secreted by malignant gliomas and the T cells isolated from these patients (82, 84, 88, 173-175), propagate the transferred tolerance by further inhibiting surrounding T cells and by even conferring on them a regulatory phenotype (176-178).
Tregs and cancer
Tregs are capable of not only preventing autoimmunity but also of decreasing immune responses to non-self-antigens, including those involved in the rejection of viral infection (166) and allogeneic tissue grafts (126, 162). It is now accepted that Tregs play a role in hindering immunity to tumor antigens (179, 180), and increased levels of functional Tregs have been found in the tumors and/or peripheral blood of patients with pancreatic (135, 136), breast (135, 136), gastrointestinal/colorectal (136-138), esophageal (136, 137), ovarian (139), lung cancer (136, 139, 140), and malignant glioma (181).
In tumor models, in vivo depletion of Tregs resulted in prolonged survival without concomitant autoimmunity when depleted mice were subsequently challenged subcutaneously with tumor (182-184) and similarly augmented the efficacy of a tumor cell-based vaccine in both tumor challenge and therapeutic models (184). We have shown in a murine astrocytoma model that blockade of Treg function using an anti-CD25 monoclonal antibody (PC61) leads to an enhancement of the physiologic immunity against astrocytic tumors, restores dysregulated lymphocyte function to normal, and prolongs survival in mice with intracranial tumors (142). We have shown that patients with malignant glioma exhibit profound lymphopenia but an elevated proportion of Tregs compared with normal individuals (181). We observed that the well-characterized CMI defects in patients with malignant glioma had significant overlap with know Treg functions (Table 1), and we demonstrated that these defects could be reversed by removal of Tregs in vitro, such that Treg-depleted lymphocytes from patients with GBM functioned equivalently to normal individuals (181). These studies demonstrated that Tregs represent a major and, importantly, potentially reversible source of immunosuppression in patients with malignant gliomas. The removal of Tregs has therefore gained justification in the context of tumor immunotherapy and is a rational approach to investigate toward optimizing anti-tumor immune responses.
Table 1.
Overlap Between Cell Mediated Immune Defects and Regulatory T cell Functions in Patients with Malignant Glioma
| CMI Deficits in Patients with Malignant Glioma | Regulatory T cell functions |
|---|---|
| Concentrated in CD4+ T-cell subset | Concentrated in CD4+ T-cell subset |
| T-cell IL-2R defects observed | Characterized by CD25 (IL-2Rα) expression and induce IL-2R defects in target cells |
| T-cells anergic: fail to proliferate and produce IL-2 | Anergic. Inhibit T-cell proliferation and IL-2 production |
| Tumors produce TGF-β and IL-10 | TGF-β and IL-10 induce regulatory phenotype in T lymphocytes |
| Lymphocytes produce TGF-β and IL-10 in response to stimulation, and fail to produce IFN-γ | Induce TGF-β and IL-10 production in surrounding T-cells, and inhibit IFN-γ production. |
The well-described CMI defects found in patients with malignant glioma (left column) exhibit significant overlap in phenotype with the more recently described inhibitory properties of CD4+CD25+FOXP3+ Tregs (right column). We found that patients with malignant glioma have elevated proportion of Tregs amidst an overall diminished CD4+T cell compartment and proliferative and cytokine production defects observed in lymphocytes from these patients could be completely restored by removal of Tregs in vitro (373). These studies suggest that Tregs are a major source of potentially reversible immunosuppression in patients with malignant glioma.
Clinical targeting of Tregs
The most immediately available clinical strategy to eliminate Tregs from patients is the employment of an immunotoxin to the IL-2 receptor. Currently, there are two available clinical grade reagents that have proven capable of successfully targeting CD25+ cells. The first, anti-Tac(Fv)-PE38 (LMB-2) is an immunotoxin that contains the variable heavy domain (VH) of anti-Tac (anti-CD25) fused via a 15 amino acid linker to the variable light domain (VL), which in turn is fused to the amino terminus of a 38 kDa truncated form (amino acids 253–364 and 381–613) of the bacterial toxin Pseudomonas exotoxin that is devoid of its binding domain. LMB-2 has demonstrated cytotoxicity in phase I trials against hematologic malignancies that express CD25, including cutaneous T-cell lymphoma (CTCL), hairy cell leukemia (HCL), Hodgkin’s lymphoma, and chronic lymphocytic leukemia (CLL) (185). The mode of cytotoxicity appears to include binding to CD25, internalization and processing of the toxin within its translocation domain, binding of the 35 kDa carboxyl-terminus of the toxin intracellularly to the KDEL receptor that carries it to the endoplasmic reticulum (ER), translocation of the toxin into the cytoplasm, and catalytic adenosine diphosphate (ADP)-ribosylation of elongation factor 2 (EF-2), leading to apoptosis and cell death. LMB-2 has demonstrated a degree of non-specific liver toxicity that appears reducible in mice by site-specific modification of the molecule with polyethylene glycol (186). In vitro LMB-2 has been shown to selectively eliminate CD25+ Tregs and showed partial reduction of Tregs in vivo in patients with malignant melanoma (187, 188).
The second available reagent, denileukin diftitox (ONTAK®), is a recombinant DNA-derived, 58-kDa cytotoxic protein composed of the amino acid sequences for diphtheria toxin fragments A and B (Met1-Thr387)-His followed by the sequences for IL-2 (Ala1-Thr133). Denileukin diftitox has demonstrated efficacy in a phase III trial for the treatment of CTCL (189) and was shown to reduce the levels of peripheral blood Tregs and enhance immunologic responses to DC vaccination when administered to patients with renal carcinoma (190) and to peptide vaccination in patients with melanoma (191). However, a conflicted report from the Attia et al. (192) at the National Cancer Institute demonstrated an inability to modulate Treg levels in patients using denileukin diftitox and lack of improvement in immunologic responses. It is unclear whether differences in the dose administration of denileukin diftitox account for these differences with the group reporting a depletive effect using successive treatments with 5 μm/kg body-weight and the group reporting no effect on Treg levels using a single 9 or 18 μm/kg dose.
Targeting Tregs and risks of autoimmunity
In light of the documented expression of normal and fetal brain antigens on human glioma cell lines (193) and brain tumor tissue (194-197), attempts to remove barriers to immunity against tumor antigens that are shared with the normal CNS and to actively immunize against the same antigens risks inducing an uncontrolled autoimmune response against the normal CNS, similar to that of EAE. In models of EAE, myelin basic protein (MBP) is the most common known antigenic trigger, but myelin proteolipid protein (MPP) (198, 199), myelin oligodendrocyte glycoprotein (MOG) (200), glial fibrillary acidic protein (GFAP), and S-100β (201) are also antigens whose targeting is sufficient, and many other antigens remain unidentified. The susceptibility of humans to EAE was discovered accidentally when patients were vaccinated against rabies using spinal cord homogenate derived from rabbits that were infected with the rabies virus (202-206). EAE has also been induced in monkeys after repeated injections of homogenized normal CNS tissue (207) and can be readily induced in the various species of rats, guinea pigs, mice, sheep, and monkeys with a single injection of a potent adjuvant and either autologous or heterologous CNS tissue homogenate or tumor (208). Given the role of Tregs in preventing autoimmunity, the risks typically associated with immunotherapy may be compounded by employing Treg depletion as an immunotherapeutic adjuvant. In mice, removal of Tregs alone has not proven sufficient for eliciting EAE (209). However, the adoptive transfer of CNS antigen-specific Tregs into EAE-susceptible mice proved capable of preventing or alleviating the autoimmunity (158, 210), asserting an EAE-protective role for Tregs possessing TCRs with CNS-specificity. Furthermore, mice transgenic for an anti-MBP TCR spontaneously develop EAE when deficient for the RAG-1 gene (a situation in which no Tregs develop), while RAG-1 competent transgenic animals remain protected by Tregs that co-express MBP-specific TCRs and TCRs with endogenously rearranged α-chains (158). This finding leaves open the hypothesis that the removal of Tregs sharing specificity for tumor and CNS antigens could constitute an intervention predisposing to EAE, particularly if combined with immunotherapy directed against CNS targets. The potential for such autoimmune responses must be afforded its due attention when constructing an immunotherapy platform directed against tumors harbored within the CNS, where the consequences of such autoimmunity are exceedingly dire.
The induction of lethal EAE has been described in primates and guinea pigs after vaccination with human glioblastoma tissue (208). This situation has raised concerns that vaccination with DCs pulsed with unfractionated tumor-derived antigens may similarly elicit autoimmunity. These concerns have gathered support for the notion of vaccination with DCs pulsed with defined antigens or total tumor RNA, the latter of which could allow the use of subtractive hybridization to reduce the number of shared antigens between tumor and normal CNS (211). However, clinical studies to date using DCs pulsed with unfractionated glioma antigens have failed to induce significant toxicity in human patients (212, 213).
Immunotherapeutic treatment of malignant glioma
The development of more effective and specific therapies, such as passive immunotherapy, for the treatment of brain tumors is a high priority for clinicians and patients who deal with this fatal disease. Immunologic treatments for cancer can take the form of passive immunotherapy (involving the administration of antibodies or toxins to patients without specifically inducing or expanding a host antitumor response), active immunotherapy (immunization of the tumor-bearing host to expand an antitumor immune response in vivo), or adoptive immunotherapy (the ex vivo expansion of effector cells and return of these effectors to the tumor bearing host).
Passive antibody treatment
Passive immunotherapy may be defined as the transfer of immunity to a patient in the form of either antibodies (not developed by the patient) or targeted toxins. The rationale is that therapeutic use of antibodies, for example, should be feasible if tumor-specific or operationally specific tumor-associated antigens can be identified, and if monoclonal antibodies (mAbs) can recognize them without being significantly retained in normal tissue (214).
It is important in discussing some of the stipulations for antibody therapy to clarify the distinction between tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs). The term ‘tumor-specific’ may truly only be applied when referring to an antigen that is expressed solely by tumor and never anywhere in the normal tissues of the body. In the case of brain tumors, a truncated variant of the EGFR, referred to as EGFRvIII, is the lone consistently expressed TSA discovered to date (215). TAAs, instead, are those that are only relatively overexpressed in tumors as compared with expression in normal tissue. The archetype TAA is the MAGE-1 antigen, discovered on malignant melanoma (3). Its concomitant expression in normal testis, making it part of the cancer-testes antigen group, designates it as a TAA. With the emergence of powerful tools such as serial analysis of gene expression (SAGE) and tissue microarray analysis and the subsequent availability of invaluable gene expression information for brain tumors (216), a number of TAAs have indeed been, and continue to be, identified for gliomas, thereby providing potentially useful targets for immunotherapy. A major limitation of this type of antigen discovery, however, is that genetic mutations in normal proteins that are truly TSAs and more likely to serve as potent rejection antigens are not identified by these techniques, which focus on relative expression levels for identifying candidate genes of interest. Once target antigens are identified, they can be used in vaccine preparations for active immunotherapeutic applications or antibodies raised against these targets can be used in passive immunotherapeutic strategies. Antibodies raised against TAAs or TSAs have been used as biologic response modifiers (217) and as delivery systems for a range of other effector agents such as chemotherapeutic agents, plant or bacterial toxins, or a host of radionucleotides (218).
mAbs as biologic response modifiers
EGFR is expressed on the plasma membranes of up to 100% of malignant glioma cells, but it is essentially absent from normal brain (219, 220). Unpublished observations in our laboratory have demonstrated the presence of EGFR on every one of more than 100 consecutive malignant glioma samples. It has long been established that growth factors and their receptors, including EGFR, play a role in oncogenesis and tumor progression (221). The logical theory that evolves is that blockade of such an overexpressed receptor should inhibit proliferation of the tumor cells (222). In 1996, Faillot et al. (223) established the ability of EMD55900 (an anti-EGFR mAb) to bind specifically and without toxicity to in vivo malignant gliomas, when administered to human patients in a single intravenous dose. In the same year, Stragliotto et al. (214) published results of a phase I/II trial involving repeated intravenous administration of EMD55900 to 16 recurrent glioma patients. Unfortunately, therapeutic response was poor, as no measurable tumor regression was observed (214). Despite evidence of accumulation of antibody in malignant gliomas using a systemic route of administration, this may have limited the levels of effective antibody at the tumor site as a possible confounding factor (224). Targeting of EGFR still exhibits potential, however, and imaging studies have demonstrated that appreciable antibodies can reach intracranial tumors through systemic route of administration (60). Partial responses have been obtained by directing the anti-EGFR mAb C225 against a variety of non-CNS tumors (225, 226), and recently published early phase trials have demonstrated objective responses to humanized forms of anti-EGFR antibodies (227-229). Improvements in survival over standard therapy alone, however, have not been demonstrated, although phase II and III trials are ongoing.
Although studies targeting EGFR in brain tumors have generally met with disappointment, a still promising use of antibodies as response modifiers involves targeting a mutant variant of EGFR, known as EGFRvIII. Through use of reverse transcriptase-polymerase chain reaction (RT-PCR) and fluorescent in situ hybridization (FISH), it has been estimated that the wildtype EGFR gene is amplified in approximately 45–62% of grade III/IV gliomas (230-233). Often, the amplified gene has undergone a rearrangement, resulting in one of a number of deletion-induced truncations (221, 234, 235). The most common of these is EGFRvIII, which RT-PCR and FISH have detected on 21–66.7% of the grade III/IV gliomas that have amplified EGFR (231, 236, 237). EGFRvIII exhibits a deletion of exons 2–7, producing a truncated protein with constitutively active tyrosine kinase activity (235, 238). In addition, a novel glycine is inserted at the fusion junction of normally distant parts of the extracellular domain, resulting in a tumor-specific epitope. By very sensitive, quantitative fluorescent activated cell sorting (FACS) analysis, 50% of GBMs were found to be positive for EGFRvIII (239).
In preclinical studies, targeting of EGFRvIII with a single intratumoral injection of Y10 (anti-EGFRvIII) increased the median survival of mice bearing EGFRvIII-expressing tumors in the brain by an average of 286% (218). In vitro, Y10 was found to inhibit DNA synthesis and cellular proliferation, and to induce autonomous, complement-mediated, and antibody-dependent cell-mediated cytotoxicity. Treatment with Y10 in Fc receptor-knockout mice demonstrated the activity of Y10 to be Fc receptor dependent (218). These data on efficacy and mechanism support further study into the use of unarmed tumor-specific antibodies as biologic response modifiers.
Perhaps the most exciting and promising advance in the use of mAbs as biologic response modifiers has been the use of the anti-VEGF mAb bevacizumab (Avastin®) in combination with standard chemotherapy in the treatment of patients with recurrent glioma. Patients with recurrent GBM have an extremely poor prognosis with a reported median survival of 3–6 months. Recent studies examining the capacity to inhibit the growth of recurrent disease by combination of blockade of the VEGF axis with bevacizumab and chemotherapy with irinotecan have shown promising response rates and evidence of prolongation of survival in patients with recurrent GBM (101, 240). It is unclear what mechanisms allow for the synergy between the anti-tumor effects of bevacizumab and chemotherapy, but increased permeability of tumor blood vessels to chemotherapy, disruption of critical cancer cell ‘niches’ within the perivascular space, and attenuation of the development of angiogenic blood vessels and resultant slowing of tumor growth have been advanced as possible explanations for the efficacy seen with anti-VEGF mAb treatment (102, 241, 242). Understanding the mechanisms of the anti-tumor effects of VEGF mAb therapy and its synergistic effects with chemotherapy are areas of intense study at our institution and others.
mAbs as delivery systems: delivery of radionucleotides
Unconjugated antibodies may not be sufficient to mediate anti-tumor effects, particularly if the recognized target does not play a key role in tumorigenesis. However, the specificity of an antibody for a TAA maintains its use as a delivery system for a variety of effectors that may be conjugated artificially to the antibody. These effector molecules are guided specifically to their tumor targets by the antibody specificity. Radionucleotides have been the most commonly utilized conjugate in mAb therapy, and although antibodies to EGFR have been used for delivery (243, 244), tenascin has been the most widely evaluated antigenic target. Tenascin is a distinct extracellular matrix protein that is prominent in mesenchymal tumors and carcinomas, including gliomas (217). Furthermore, its prominence among gliomas increases with tumor malignancy, such that it is present in up to 99% of GBMs (245). It has therefore frequently been the target of trials using radioimmunotherapy.
At our institution, we have administered the radiolabeled antibody 81C6 in a number of clinical studies (217, 246-249). 81C6 reacts with an alternatively spliced segment of the tenascin molecule at the fibronectin type III domain (250), and its immunoreactivity and tumor-localizing capacity have been reported as superior to other anti-glioma mAbs (251). It has demonstrated both safety and the ability to increase survival in patients with leptomeningeal neoplasms (246), recurrent glioma (217, 248), and newly diagnosed glioma (247, 249). This latter application, in particular, has demonstrated promising results (247, 252). Reardon et al. (252) treated 33 patients with newly diagnosed malignant glioma (GBM, n = 27; AA, n = 4; and AO, n = 2) with 120mCi of (131) I-labeled murine 81C6 into the surgically created resection cavity and observed a median survival of 86.7 weeks for all patients and 79.4 weeks for patients with GBM. These results were favorable in comparison with prognostic factor-matched historical controls and an acceptable toxicity profile was observed during this trial (252). A phase III study of the clinical efficacy of 81C6 mAb treatment is planned based on these encouraging data.
Our group also reported a long term response in a patient with a neoplastic meningitis that developed secondary to malignant melanoma when the patient was treated with intrathecal 131I-labeled Mel-14 F(ab′)2 (253). Neoplastic meningitis typically represents a terminal stage of malignant melanoma. Yet, despite having an expected survival of only 2–6 months following diagnosis (254), the patient in this report demonstrated no abnormal contrast enhancement on cranial magnetic resonance imaging (MRI) even 3 years pursuant to treatment (253).
Focus in recent research has been directed toward the use of alternative radioisotopes. At present, the α-emitter astatine-211 (211At) is generally considered the most promising radionuclide for radiotherapeutic applications. Because it possesses an electron capture branch, 211At also emits polonium K X-rays of 77–92 keV. These X-rays are of sufficient energy and quantity as to permit both γ-counting of tissue samples and external imaging, including SPECT (255). mAbs labeled with α-particle emitting radionuclides such as 211At (as opposed to β-emitters like 131I) may be valuable in the treatment of CNS malignancies (256) for several reasons. The range of 211At α-particles in tissue is only 55–70 μm, so their toxic effects are confined to a region equivalent to only a few cell diameters. Toxicity is thus limited to those normal cells that are in immediate proximity to tumor cells. Their high energy and short range combine to give 211At α-particles a linear energy transfer (LET) that is about 500 times higher than the LET for the β-particles of 131I (257). The LET of 211At α-emissions is nearly ideal for maximizing biologic efficacy, as the distance between ionizing events approximates the distance between DNA strands. Thus, the probability of inducing irreparable double-strand DNA breaks is high, thereby increasing the potential for cytotoxicity (258). Indeed, α-particles have been shown to be exquisitely cytotoxic, with a D0 equivalent to < 10 211At atoms bound per cell (259).
Preclinical studies have demonstrated decreased thyroid uptake, increased tumor retention, and increased therapeutic efficacy in an athymic mouse/human glioma xenograft model (260). In a phase I safety study at our institution, 18 patients were treated with 211At-labeled antitenascin mAb administered into a surgically created resection cavity and then treated with salvage chemotherapy (261). There were no dose-limiting toxicities observed, and promising median survival in patients with recurrent GBM (52 weeks) were observed. This study demonstrated that regional administration of 211At-81C6 is feasible, safe, and associated with promising antitumor benefit in patients with malignant gliomas.
Immunotoxins and convection enhanced delivery
Plant and bacterial toxins represent alternative biologic effectors that may be conjugated to either antibodies or peptide ligands in order to produce either immunotoxins or fusion toxins, respectively (262). These, in turn, are designed to selectively deliver toxins into tumors. Once arrived at their target, the toxins exhibit cytotoxicity via a mode characteristic of their natural mechanisms of action. Such mechanisms may hold important advantages for targeted toxins over modalities employing simply radiation or chemotherapeutic agents. Toxins are effective against hypoxic cells (the main instigators of radiation resistance in gliomas), and they are far more potent as well: in some cases, even a single molecule can react enzymatically within the cytosol to cause cell death (263). It is therefore conceivable that cancer cells exhibiting resistance to both radiotherapy and chemotherapy may nonetheless be susceptible to the cytotoxic effects of a targeted toxin. Furthermore, selective targeting of toxins with, for instance, targeted antibody delivery systems, limits the toxins’ otherwise non-specific side effects.
Secondary to increased iron requirements, transferrin receptors (Tfr) are often highly expressed both in vitro and in vivo on rapidly dividing cells, including those of GBM (264). Thus, the natural ligand (transferrin) may be used to deliver therapeutic molecules via Tfr. Laske and colleagues conjugated human transferrin (tf) to a mutated diphtheria toxin (CRM107) that lacked native toxin binding ability and adopted a high flow microinfusion technique to administer the recombinant toxin intratumorally. This represented the first clinical use of what has been called convection-enhanced delivery (CED) or intratumoral microinfusion. Clinical response rate was significant but was hampered by dose-limiting toxicity (265). Tfrs are expressed at substantial levels on the endothelial cells of normal brain capillaries (266), and it was thought that the local brain injury observed in this study may be due to the targeting of endothelial cells by the diphtheria conjugate.
The rationale for delivery of to the brain using CED is as follows: diffusion of most therapeutic agents within the brain is severely limited, as the rate of diffusion is (i) dependent on a concentration gradient, (ii) inversely related to the size of the agent, and (iii) usually slow relative to tissue clearance. Thus, diffusion results in inhomogeneous distribution of most agents, with steep concentration gradients evolving between the point of delivery and the advancing tumor border. Consequently, lethal tumor cell populations that exist only a few millimeters beyond the tumor border may escape exposure to the therapeutic agent. In contrast to diffusion, however, CED has been predicted to produce a bulk flow current that possesses the potential to homogeneously distribute even large molecules over much greater distances in the extracellular spaces of the brain (267). Such enhancement of drug distribution has already been demonstrated in animal models (268, 269) and has been confirmed in our clinical studies using SPECT imaging to track distribution of the large molecules delivered via CED. Therefore, CED should allow delivery of large therapeutic constructs to a greater portion of the tumor and should saturate invasive neoplastic cells far from the site of infusion.
Imaging studies of the biodistribution of macromolecules delivered by CED into the brain of large animals and humans have shown the potential for this modality to significantly enhance drug delivery to the tumor bed lining resection cavities using CED but also have demonstrated that target-tissue anatomy and patient-specific physiology play significant roles in actual drug distribution (270-275). These studies highlighted the need to improve prospective selection of catheter trajectories to achieve adequate drug distribution in all the areas at risk for tumor recurrence lining the resection cavity.
The truncated form of the Pseudomonas exotoxin has also been used as a targeted toxin, which was conjugated to a circularly permuted IL-4 (276) as a ligand-toxin moiety. The use of IL-4 as the delivery system made avail of a proposed enrichment of IL-4 receptor (IL-4R) on the surface of malignant glioma cells (277). The IL-4-toxin conjugate is internalized after binding IL-4R, and subsequently, the exotoxin ADP-ribosylates EF-2, arresting tumor cell protein synthesis (278). One patient remained disease-free more than 18 months after the procedure, and no systemic toxicities were observed. Yet, seven of nine patients treated required craniotomy during the protocol for the relief of increased intracranial pressure (276). Further studies are underway, although there has been some debate as to the true existence of IL-4R on glioma cells in vivo.
A similar line of preclinical work has recently been translated into clinical trials and entails targeting of the IL-13 receptor 2 α chain (IL-13R2α). IL-13Rs are known to be expressed on a significant proportion of gliomas, although the relevance of this expression is yet to be determined (279, 280). However, a wide array of human glioblastoma cell lines expressing the IL-13R were killed by a chimeric protein (IL-13PE38QQR) composed of human IL-13 and a mutated form of Pseudomonas exotoxin (279). Furthermore, a variety of intratumoral dosing schedules proved effective against subcutaneous human U251 glioblastoma tumors in nude mice (280). Recently published results of clinical trials using intracerebral CED of IL-13PE38QQR showed capacity to induce tumor necrosis but no clinical benefit in over 50 treated patients with malignant glioma (271). Also, a recently completed phase III study of treatment of first recurrence of GBM with CED of IL-13PE38QQR compared with carmustine wafers showed that IL-13PE38QQR was comparable but not statistically superior to Gliadel® (authors’ unpublished data). The conclusions from these trials demonstrate the need for better catheter placement, accurate modeling of in vivo drug distribution, or novel delivery mechanisms to improve outcomes using this promising modality of localized drug delivery. Also, a reported significant heterogeneity in the expression of the intended target of IL-13PE38QQR, IL-13R2α, among malignant glioma specimens, underscores the challenges but potential benefits of identifying patients prospectively who are likely to respond to targeted therapies (281).
Our group recently reported progress on a phase I/II trial using TP-38 in patients with malignant brain tumors. TP-38 is a recombinant chimeric protein composed of TGF-α (which binds wildtype EGFR with high affinity) and a genetically engineered form of the Pseudomonas exotoxin, PE-38. Patients enrolled on the trial (n = 20) had recurrent malignant glioma and < 5 cm lesion and received microinfusion of TP-38 in a dose-escalation trial to define the MTD. Therapy was well tolerated, and although the median survival for all patients was not significantly longer than historical controls (23 weeks), three patients had demonstrated radiographic responses. One patient with a bifrontal tumor has experienced a durable near complete radiographic response, with no further therapy administered (282).
Advantages and disadvantages of passive antibody therapy
Results of passive immunotherapy trials have left reason for optimism. Indeed, they offer much improvement over current modalities in the arena of specificity. However, a number of limitations still exist on the clinical efficacy of targeted treatment with antibodies or operationally tumor-specific ligands. The main unresolved problem stems from the question whether treatments administered systemically can achieve clinically significant levels of the targeting molecule at the site of intracranial tumors. Systemic therapy with antibodies, as in the phase I/II trial with EMD55900 discussed above (214), may not have been successful for a number of reasons. Sufficient levels of antibody to mediate an antitumor response may not have been achieved with the dosage and systemic route of administration. Our group has demonstrated in human imaging studies using an antigen-specific radiolabeled mAb that 0.0006–0.0043% of the total injected dose localized to the intracerebral tumor following systemic intravenous administration (224); thus, sufficient targeting of intracranial tumors may require higher or more sustained doses of antibody in the peripheral blood or a lack of any competing antigen source for binding antibody in the periphery to facilitate tumor accumulation. Clinical attempts to ‘open up’ the BBB to drug delivery with mannitol or RMP-7, a bradykinin agonist, have met with both failure and toxicity (283-286).
The BBB aside, other problems remain for systemic delivery. High interstitial pressures in the tumor and surrounding tissue are prohibitive for perfusion (287). Furthermore, systemic administration would require clearance by the liver and/or kidney. This would be particularly problematic during delivery of conjugated radioisotopes or toxins, for it would tend to concentrate the toxic substances in these organs.
As a result, antibodies have been administered in an almost exclusively loco-regional fashion, providing them operational specificity and enhanced targeting. Because gliomas demonstrate a very low propensity for spread outside the CNS (0.1–0.5% of cases) (288, 289), this regional mode of delivery is perhaps more apropos for gliomas than for other tumors, and its effectiveness has been further enhanced by the emergence of CED.
Other challenges exist for the passive immunotherapy of gliomas. Even afforded a TSA, the heterogeneity of these tumors makes it extraordinarily unlikely that targeting a single antigen would produce a curative therapeutic effect. In addition, the use of antibodies introduces a problem unique unto itself, as the antibodies themselves are antigenic. The development of human anti-mouse antibodies (HAMA) and human anti-human antibodies (HAHA) against the therapeutic Igs is a not infrequent event. In one of Riva’s RAIT trials, HAMA generation evolved in 59% of the patients, although no clinical correlate was identified in this case (290). This outcome may impose strict limitations regarding repeated use of the therapy, however.
Active immunotherapy strategies
The term active immunotherapy defines a strategy wherein antitumor immunity is initiated in vivo following immunization with tumor antigen. With evidence mounting that such peripherally administered immunizations are capable of inducing responses against tumors located within the CNS (291-293), it seems likely that peripheral immunization may successfully bypass the immune privilege of the brain. This rationale supports a number of trials in active immunotherapy.
In 1983, Mahaley et al. (294) administered monthly subcutaneous inoculations consisting of one of two human glioma cell lines to 20 postoperative patients with malignant glioma. Each patient also received levamisole. Patients inoculated with one of the cell lines (U-251MG) exhibited prolonged survival when compared with historic controls (294). This represented one of the earliest trials to adopt an active strategy. Since its publication, numerous immunotherapy studies have followed suit.
DC therapy
DC are the specialized APCs of the immune system that have established a foundation for therapeutic immunizations against cancers such as lymphoma, multiple myeloma, melanoma, prostate cancer, renal cell carcinoma, non-small cell lung carcinoma, colon cancer, and malignant gliomas (295-297). The discovery of DCs has redirected the approach to cancer immunotherapy (298). The capacity to generate large numbers of DCs in vitro from monocytes or myeloid bone marrow precursor cells (107, 112, 299-304) led to the emergence of ex vivo loading of DCs with tumor antigens and administration of ‘DC vaccines’ as a prominent strategy for induction of antitumor immunity.
Preclinical studies have shown DCs to be the most potent activators of de novo and recall responses in B and T lymphocytes and therefore are regarded as one of the most promising entities for the realization of successful tumor immunotherapy. The safety and efficacy of DCs pulsed with tumor antigens and administered as vaccines for the treatment of malignant glioma has been evaluated in a number of reported and ongoing clinical trials (305-312). In the initial clinical study, conducted by Yu et al. (293), a demonstrable increase in tumor-specific cytotoxicity was successfully developed in four out of seven testable patients who received DCs pulsed with MHC class I peptides eluted from the surface of autologous glioma cells. Furthermore, two out of four that underwent re-operation demonstrated robust CD8+ and memory (CD45RO+) T-cell infiltrates in areas of tumor. Based on the small sample size, no reliable data on survival could be generated, but the treatment proved safe (293).
A phase I clinical trial at Duke University has been completed in which 16 patients with malignant gliomas received intradermal immunizations with autologous DCs pulsed with a keyhole limpet hemocyanin (KLH) conjugate of a peptide spanning the mutated region of EGFRvIII (PEPvIII). The intradermal route of administration is supported by evidence that DCs delivered in this manner will migrate to lymph nodes (313, 314) and subsequently present antigen to T lymphocytes, as well as by prior studies comparing the ability of various routes of administration to elicit strong T-cell-mediated immunity (315). The enrolled patient population consists of adults with malignant gliomas who have undergone gross total tumor resection and radiotherapy. Patients undergo leukapheresis to remove autologous PBMCs, which are cultured in granulocyte-macrophage (GM)-CSF and IL-4 to generate DCs. These are pulsed with PEPvIII/KLH and matured in a cocktail of TNF-α, IL-1β, and IL-6 [but not prostaglandin E2, due to some concern over a counterproductive effect on DC IL-12 production (316)] before being delivered back to the patient in three biweekly intradermal injections.
Sixteen patients have completed vaccination with no adverse events. No patient showed a positive DTH reaction to KLH or PEPvIII before vaccination, and of the evaluable patients after vaccination, 13/13 (100%) patients reacted to KLH and 5/13 (38.5%) reacted to PEPvIII. In vitro proliferation in response to PEPvIII was seen in 10/13 (76.9%) and to KLH in 12/13 (92.3%) patients tested. Two patients, one with anaplastic astrocytoma and one with GBM with residual radiographic disease after resection and radiation, have had a nearly complete response following completion of vaccination. These patients have remained stable for > 66.7 and 56.9 months, respectively. Of the 14 patients without radiographically evident disease, 2/14 (14.3%) have not progressed at 70.2 and 65.9 months with a median overall time to progression of 13.2 months. For patients with GBM, the median survival time was 25.6 months, which compares favorably with recently published trials. These findings suggest that autologous mature DCs loaded with the tumor-specific antigen PEPvIII are safe and may induce a beneficial immunologic response in patients with malignant gliomas.
We have also targeted the EGFRvIII mutation in patients with malignant glioma using the same PEPvIII/KLH conjugate administered as a peptide-based vaccine in combination with GM-CSF in two consecutive and one multi-institutional phase II immunotherapy trials. These trials have demonstrated the capacity to induce potent EGFRvIII-specific immunity in treated patients and have shown promising survival times (317). A multi-institutional randomized phase III trial is underway for formal evaluation of the efficacy of this treatment.
Liau et al. (305) reported the results of a phase I trial of DCs pulsed with peptides acid-eluted from the surface of resected autologous tumor and administered to 12 GBM patients in three bi-weekly intradermal injections. There were no adverse effects of treatment, and evidence of increased immunologic responses against autologous tumor was observed in half of the treated patients. Promising prolongation of survival (median survival 23.4 months) compared with historical controls was observed and a multi-center randomized clinical trial has been initiated based on these results.
Sloan et al. (318) have reported the results of an in vitro human DC study that examined the ability of different DC-based strategies to induce effective T-cell responses against malignant astrocytomas. DCs were generated from patient PBMCs and were fused with autologous tumor cells or pulsed with total tumor RNA or tumor lysate. They were then assayed for their respective abilities to stimulate tumor-specific T-cell proliferation and CTL responses in vitro. No significant differences were found between the various DC arms in their T-cell stimulatory capacity; all showed enhanced cytotoxicity that was further augmented by addition of CD40 ligand during T-cell stimulation (318). The data should be helpful in designing protocols for DC-based immunotherapy of malignant astrocytomas.
Immunization with DCs is an attractive route to pursue in the search for more specific and more effective treatments for malignant brain tumors. Currently, no consensus exists on the optimal DC subtype, generation, loading method, maturation, dose, or route of delivery. An understanding of the roles that each of these factors plays in influencing clinical outcomes is essential.
Adoptive immunotherapy strategies
Adoptive immunotherapy encompasses treatments that involve transfer of immune cells manipulated in some fashion ex vivo to patients. Treatment approaches have differed in the types of cells administered, the route of administration, and the activation status of the cells. Cell types that have been used in adoptive immunotherapy include (i) PBMCs (319, 320), (ii) lymphokine-activated killer cells (LAKs) (321-323), (iii) mitogen-activated killer cells (MAKs) (324, 325), (iv) tumor-infiltrating lymphocytes (TILs) (326), and (v) antigen-specific and unselected CTLs (327, 328). Routes of administration have generally been either systemic or into the tumor cavity (intralesional or loco-regional).
PBLs/PBMCs
One of the earlier trials to establish the feasibility of adoptive immunotherapy for tumors of the CNS was performed in 1984 by Steinbok et al. (319). In the years before the study, it was observed that approximately 30% of GBM patients demonstrated lymphocytic infiltrates within their tumors at autopsy (319). Mounting clinical opinion at the time contended that these infiltrates marked an attempted immune response by the patient and that they might be beneficial (329, 330). With this opinion and with some knowledge of the immune deficits that glioma patients suffered, Steinbok et al. (319) obtained PBMCs from four patients and simply reinfused them through a reservoir into the resection cavity. The theory adopted was that such direct infusion of cells into the brain would circumvent whatever afferent and efferent limb defects were operating and would likewise avoid any ‘blocking factors’ present in the serum that had allegedly been elaborated by the tumor. The study established the safety of such an experimental design but provided no measurable benefit to the patients enlisted (319).
LAK cells
The emphasis shifted toward the use of PBMCs activated in vitro by IL-2 and cultured to become LAK cells. Rosenberg et al. (331) demonstrated that LAK cells had the capacity to lyse a wide variety of autologous and allogeneic tumors but had little cytotoxicity against normal tissues. Although the antigenic recognition mechanisms used by LAKs were and still remain largely not understood, efforts to treat tumors with the use of LAK cells were initiated in the early 1980s. High-dose IL-2 was also infused directly into patients to expand tumor-specific lymphocytes in vivo, but these efforts were limited drastically by dose-dependent toxicity, particularly in patients with brain tumors (332).
In 1986, Jacobs et al. (333) locally infused LAK cells to patients with brain tumors and were able to avoid the toxicity of systemic IL-2 while providing the advantages of preactivation of the infused lymphocytes. It was hoped that IL-2 culture ex vivo would bypass the in vivo ability of gliomas to depress lymphocyte activation and induction of cytolytic function. This trial, along with others using LAK cell therapy, demonstrated minimal benefit to the treated patients but did show the overall safety of the approach (334-336). One trial of note, conducted by Hayes et al. (321), was able to demonstrate a median survival of 53 weeks in patients with recurrent GBM or anaplastic astrocytoma treated by LAK therapy and IL-2, compared with a chemotherapy-treated control group, which had a median survival of 26 weeks.
In 2004, Dillman et al. (337) performed a clinical trial using 40 patients with GBM who all received a placement of autologous LAK cells into the tumor cavity. The median interval from the original diagnosis of glioma to LAK treatment was 10.9 months. Patients received an average of 2.0 ± 1.0 × 109 LAK cells, with a viability of 91 ± 6.8%. The treatment was well tolerated by patients. At the median follow-up of 2.5 years, the median survival post-LAK was 9 months, and 34% of the patients sustained 1 year of survival. This clinical trial supported the safety and feasibility of the treatment but was inconclusive for a survival benefit.
Ishikawa et al. (338) presented the efficiency of NK cells in the cellular immune response against gliomas. They used PBMCs prepared from patients with malignant gliomas, and co-cultured them with an irradiated human feeder cell line, HFWT, in RHAM-α medium supplemented with 5% autologous plasma and IL-2. These enriched NK effector cells were then infused into nine patients (six grade III gliomas and three cases of GBM) over 16 different courses. The mean frequency of NK cells among lymphocytes was 82.2 ± 10.5%. There was no severe neurological toxicity observed in any of the cases. NK cells were successfully expanded ex vivo from PBMCs in all of the cases and this approach was safe and partially effective in these patients.
The not well-understood mechanism by which LAK cells recognize and kill various tumor cells has limited application of LAK therapy. Also these cells do not home specifically to tumor sites (339). This lack in homing capacity requires loco-regional administration, which may not be a severe limitation with brain tumors because of restriction to CNS. However, the demonstration that tumor-specific T-cell lymphocytes are more effective in killing tumor targets on a cell-by-cell basis than LAK cells has shifted the major emphasis of adoptive cellular therapy of glioma and other tumors away from the use of LAKs in adoptive immunotherapy protocols to the use of T lymphocytes in this modality.
Tumor-draining lymph node T cells and TILs
Additional trials with adoptive therapy were fashioned to solve some of the above problems by increasing the specificity of the cells delivered. In 1987, Kitahara et al. (328) isolated PBLs from patients and sensitized them in vitro with autologous tumor cells in the presence of IL-2 to generate CTLs, which were then re-administered intracranially. This strategy offered the advantage of tumor specificity but required isolation and maintenance of tumor cells, whose availability is frequently limited. Newer approaches proposed to avoid this prerequisite for tumor cell isolation by stimulating instead with anti-CD3, an antibody against a costimulatory molecule of the TCR complex (327). Engagement of CD3 by the antibody was sufficient to elicit activation of the T cells. However, none of the approaches using this strategy are necessarily antigen specific.
Another means to select for the most tumor-specific cells has been to isolate the lymphocytes that have already infiltrated the tumor (TILs) at the time of resection (326). These cells, although limited in number, presumably already possess specificity for tumor and, in initial trials, were simply redelivered into the tumor cavity with IL-2. In a study by Quattrocchi et al. (326) involving six patients, there were no serious complications, although all developed transient cerebral swelling. One patient, with a diagnosis of AA, demonstrated no radiologic evidence of disease at 45-month follow-up (326).
One of the most interesting trials in adoptive therapy, however, has exhibited some success by taking advantage of purported differences in the level of MHC antigen expressed by gliomas and normal CNS. Normal brain expresses exceedingly low amounts of MHC, with expression generally limited to non-neuronal cells such as those of the vascular endothelium (340, 341). By contrast, in vitro cell lines derived from human gliomas have demonstrated the capacity for moderate levels of expression of MHC class I (342, 343). Although the results of studies looking at in vivo tumor expression levels have varied (340, 341, 344, 345), this may potentially afford the right to consider any MHC class I expression by glioma cells to be that of a TAA. With this idea, and with the understanding that antigenic responses to allogeneic antigens are stronger than those to syngeneic antigens, Kruse et al. (346) removed CTLs from healthy non-immunosuppressed donors, sensitized them in vitro to the patients’ MHC, and administered them into the resection cavity of five patients with recurrent glioma. Two patients with diagnoses of anaplastic oligodendroglioma had no evidence of disease 80 months following the initiation of therapy (347).
Toward a more specific cellular therapy
Infusing T-cell populations previously activated against specific tumor antigens that have been identified in a specific tumor is one strategy of improvement for adoptive T-cell therapy. However, the lack of identified glioma-specific antigens poses as an obstacle for this method of treatment with brain tumors. EGFRvIII once represented the only identified glioma antigen that could potentially be targeted using an antigen-specific based approach in malignant glioma (348-350). While vaccine-induced responses against EGFRvIII in both rodents and humans have been reported, no reports targeting this antigen using adoptive T-cell therapy have been published (350, 351). Recently, glioma antigens that may be suitable for targeted immunotherapy have been identified such as IL-13R2α (with caveat that reported to be expressed also in the kidney) (281, 352-354), survivin (355), and telomerase (356). Another identified glioma antigen that is recognized by T cells is the UDP-Gal:βG1cNAc β1, 3-galactosyltransferase, polypeptide 3, in which several peptides have been identified that can elicit tumor-reactive CTL induction (357). Other T-cell antigens have been identified in brain tumors, including SART3, ADP-ribosylation factor 4-like protein, and tyrosinase-related protein-2 (358-360). Interestingly, we and others (361, 362) have found viral antigens unique to human cytomegalovirus to be expressed within the vast majority of high-grade gliomas but not within surrounding normal brain, lending the possibility of leveraging these highly immunogenic and well-characterized viral antigens as tumor-associated targets. Each of these antigens lends themselves to inducing antigen-specific T cells for use in cellular immunotherapy through either in vivo vaccination or in vitro expansion of antigen-specific T cells by potent APCs such as DCs. Antigen-specific T-cell populations can be purified with the use of newly developed methodologies, such as tetramer-based sorting or cytokine secretion-based sorting protocols, making it feasible to generate highly pure populations of T cells that recognize these specific antigens (363-365).
While it is believed that targeting such antigens and selecting for enriched populations of T cells with amplified specificity will lead to more effective treatment strategies, such approaches will also have to contend with the reality that not all tumor cells within a given tumor will likely express a single antigen, as demonstrated in the case of EGFRvIII (366). Consequently, a highly effective T-cell therapy that targets a single antigen will likely select for escape variants, as has been observed in adoptive T-cell therapy trials targeted against melanoma-specific antigens (367, 368). The most effective strategies in using highly enriched populations of tumor-specific T cells for adoptive immunotherapy are likely to be those that target multiple specific antigens or the total antigenic content of the tumor cells and allow for the isolation and expansion of this broad repertoire of tumor-reactive lymphocytes.
Future developments in cellular immunotherapy
Although earlier trials in adoptive T-cell therapy for gliomas have demonstrated the potential for the immune system to mediate regression of high-grade tumors in the CNS, current research realizes that these early trials involved the infusion of inefficiently activated T-cell populations. Such populations contained a small portion of potentially tumor-reactive cells, and the small number of clinical responses in these trials represents the expectation of being able to significantly improve adoptive immunotherapeutic protocols. There has been a considerable gain in experience with the maintenance and expansion of T cells ex vivo, but this experience is only the beginning of understanding how to make a ‘better T cell’ for therapy. Hence, the future of adoptive T-cell therapy for the treatment of malignant brain tumors involves exciting new developments at all phases of T-cell activation, expansion, and in vivo trafficking, maintenance, and effector function.
CTLA-4 inhibition and Treg blockade
Specific mAbs that block immunosuppressive pathways in tumor-bearing hosts have demonstrated considerable benefit in preclinical and clinical immunotherapy settings. CTLA-4 is a receptor constitutively expressed on Tregs and upregulated during activation of effector T cells, whose engagement with its ligands mediate downregulation of the immunologic response in activated lymphocytes (369-371). The use of blocking antibodies to CTLA-4 has shown considerable efficacy in enhancing antitumor immunity in preclinical tumor models as well as efficacy in patients with advanced malignancy (372). We have shown CTLA-4 blockade to be a potent modality for mediating the immunologic rejection of advanced intracranial astrocytomas in our murine models and reversing glioma-induced immunosuppression (373) and are currently pursuing evaluation of the clinical safety and efficacy of this modality in patients with malignant gliomas. CTLA-4 blockade led to resistance to Treg-mediated suppression in effector cells in treated mice without affecting the suppressive capacity of Tregs from CTLA-4 treated animals, indicating that despite constitutive expression of CTLA-4 on Tregs, the major impact of CTLA-4 blockade is on activated effector cells (Fig. 1).
Fig. 1. CD25 and CTLA-4 blockade: complementary strategies to overcome regulatory T-cell immunosuppression.
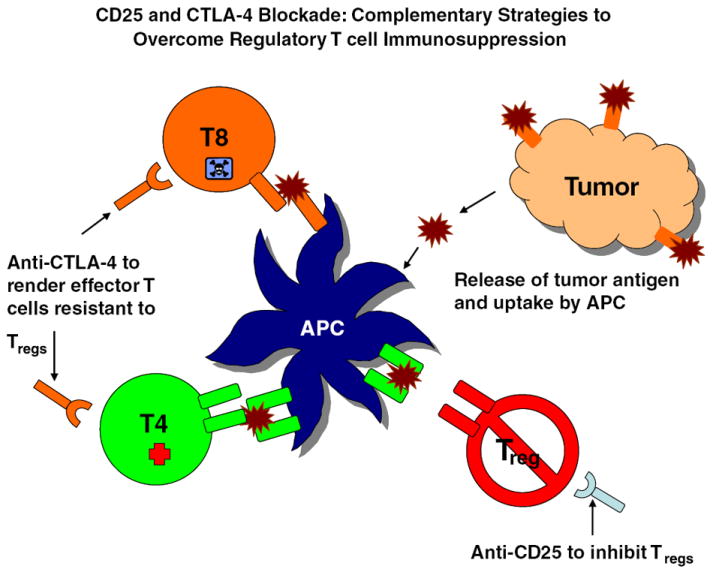
Systemic CTLA-4 blockade in experimental mice enhances CD4+CD25− T-cell proliferation and makes them resistant to Treg-mediated suppression but does not alter Treg function (373). Systemic anti-CD25 administration only partially depletes Tregs but renders remaining Tregs incapable of mediating T-cell suppression (142). Thus, CD25 and CTLA-4 blockade represent potentially complementary strategies for overcoming Treg-mediated immunosuppression in patients with malignant glioma. The diagram shows potential synergistic effects of anti-CTLA-4 and anti-CD25 mAb treatment in enhancing activation of tumor-specific lymphocytes. Antigen released by dying tumor cells is taken up and processed by resident or infiltrating APCs and presented as peptides to CD4+ and CD8+ T cells. Tregs, which are elevated in proportion in patients with malignant glioma, attenuate these responses through interaction with APCs and T cells, and these suppressive effects may be counteracted through anti-CD25 treatment to partially deplete Tregs and functionally inactivate remaining Tregs as well as through CTLA-4 blockade, which renders activated effector cells resistant to Treg-mediated immunosuppression.
Strategies to block the function of Tregs have shown promise in other clinical settings, and recent studies from our laboratory and others have demonstrated the potential benefits of Treg blockade using anti-CD25 mAbs in enhancing immunotherapeutic treatment of malignant gliomas in preclinical settings (142, 181, 374). We have shown that anti-CD25 mAb treatment only partially depletes Tregs but renders remaining Tregs incapable of mediating suppression in in vitro assays (142). While the mechanisms of this functional inhibition are under study, these studies suggest that CTLA-4 and anti-CD25 mAb treatments represent potentially complementary strategies for overcoming Treg immunosuppression in patients with malignant glioma. Studies in human patients with malignant glioma employing Treg blockade with mAbs have not yet been reported.
Lymphodepletion and homeostatic proliferation
Dudley et al. (375) demonstrated a breakthrough in achieving high and sustainable numbers of tumor-specific T cells in human patients by using a lymphodepleting chemotherapy conditioning regimen that was given to patients before the transfer of melanoma-specific T-cells. Six of 13 patients with widespread malignant melanoma had objective clinical responses, and another four had mixed responses with significant shrinkage of one or more metastatic deposits. The advantage of lymphodepletion T-cell infusion for the maintenance and function of adoptively transferred T cells was noted in early animal studies and is now being born out as being of paramount importance in clinical treatment involving adoptive T-cell transfer (376). An understanding of the factors that promote the survival of the transferred T cells in this setting may lead to the ability to substitute lymphodepletive chemotherapy with more specific forms of supplementation or depletion of specific cell subsets. Vaccination against tumor-specific antigens during recovery from periods of lymphopenia has also shown promise as a method to enhance cellular therapy against malignant tumor cells (377-379).
TILs were effective in mediating regression of malignant disease in lymphodepleted hosts, while similar attempts using melanoma-specific clones expanded from PBLs failed to persist and did not initiate any responses in treated patients (375, 380). It is unknown whether intrinsic differences between TILs and PBLs or differences in the methods used in the generation of these cells account for the marked difference in the efficacy observed in these trials. TILs, theoretically, are enriched for tumor-specific T cells that have migrated into the tumor site and therefore have been proposed to be an advantageous source of T cells for use in adoptive transfer (381, 382). However, few studies have examined whether TILs in fact possess any intrinsic advantages over T cells expanded from PBLs, a much more readily available source of lymphocytes (381).
Although it is clear that the major effector cell in adoptive T-cell therapy protocols is the CD8+ CTL, the importance of the presence of CD4+ T cells for the survival and function of transferred CTLs has been highlighted in adoptive T-cell therapy trials against viral diseases, such as cytomegalovirus infection and Epstein-Barr virus (383, 384). Just as the understanding of the mechanisms of T-cell activation has evolved significantly since the earliest attempts at adoptive T-cell therapy, it is now apparent that the role of the CD4+ T cell cannot simply be substituted by administration of IL-2 (385-387). In fact, adoptive transfer of pure populations of tumor-specific CD4+ T cells has been shown to have efficacy against experimental brain tumors, highlighting the many functions of these cells (386). A major area of future development in cellular immunotherapy will be the identification of brain tumor antigens recognized by CD4+ T cells and the development of methods of enriching and expanding tumor-specific CD4+ T cells in addition to CTLs in vitro and in vivo.
The modification of T cells through genetic engineering for enhanced T-cell function continues to be an area of great interest. Genetic engineering has been used to enhance the longevity and efficacy of administered T cells by augmenting the secretion of cytokines such as IL-2 and TNF-α (388). The specificity of T cells has been redirected toward tumor recognition through the transfer of genes encoding for tumor-specific TCRs or T-cell chimeric receptors composed of an intracellular T-cell signaling domain coupled to an extracellular antibody recognition domain (389, 390). The use of TCR or chimeric TCR gene transfer allows for the desired high frequency of tumor-specific T cells to be achieved without complex and often unpredictable antigen presentation platforms.
Tumor blood vessels
A stimulating new approach to immunotherapy for tumors involves the immunologic targeting of tumor endothelial cells for destruction instead of the targeting of the tumors themselves. Tumor endothelial cells express distinct antigenic markers that are largely absent from the normal vasculature and can be exploited for recognition by the immune system in potent vaccine strategies (391-393). We are currently investigating whether such strategies involving the adoptive transfer of tumor endothelium-specific T cells can effectively inhibit tumor growth through the cutting off of the blood supply to the growing tumor. There are several theoretical advantages to this approach: (i) the potential for killing many more tumor cells per effector lymphocyte generated through the disruption of blood vessels that supply many tumor cells; (ii) the ability to avoid escape mutations because the tumor vasculature represents a non-malignant, genetically stable target; and (iii) the lack of concern of T-cell trafficking across the BBB because the endothelial targets are in constant contact with the blood. Such a cytotoxic approach toward targeting angiogenic vessels may also overcome the observed limitations of cytostatic angiogenic inhibitor therapy, in which escape mechanisms can frequently emerge (394).
‘Brain tumor stem cells’
A small subset of tumor cells in GBM have been identified that are uniquely capable of unlimited self-renewal and of recapitulating the heterogeneity of the original tumor when orthotopically transplanted into immunoincompetent mice (395-397). These brain tumor stem cells (BTSCs), which have been isolated by the neural stem cell surface marker CD133, are widely believed to be responsible for tumor propagation and resistance to conventional radiation (398, 399) and chemotherapy (400), although not definitively established. Still, indiscriminate anti-tumor approaches may spare BTSCs because of their idiosyncratic properties and divergent gene expression (398-400), and therapeutic approaches that target the molecular properties that define these cells may have a greater biologic impact and even reduce the need for intensive conventional therapies.
Although BTSCs appear resistant to radiation (398, 399) and chemotherapy (400), their susceptibility to immunotherapeutic attack has not been formally tested. A recent study in a murine glioma model has shown that the immunologic targeting of brain tumors using DCs pulsed with lysates from tumor cells grown in stem-cell culture media was more efficacious than DCs pulsed with tumor lysates from cells grown in standard culture, suggesting that the targeting of BTSCs may be of therapeutic benefit (401). Thus, the identification of antigens expressed in BTSCs and development of immunotherapy that effectively targets these cells is a promising approach toward improving the outcome of immunotherapy for malignant gliomas.
These exhilarating advances contain great promise for the development of cellular immunotherapies that will ultimately lead to significant and reliable anti-tumor responses. They also hold significant promise for having a favorable impact on patient survival. Although immunotherapy is not likely to emerge as a single-modality treatment for brain tumors that will replace surgery, radiation, and chemotherapy, it will almost certainly take its place as a very effective adjuvant therapy for the treatment of patients with malignant brain tumors.
References
- 1.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Ehrlich P. Collected Studies on Immunity. New York: John Wiley & Sons; 1906. [Google Scholar]
- 3.van der Bruggen P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 4.van der Bruggen P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 5.Boon T, et al. Identification of tumour rejection antigens recognized by T lymphocytes. Cancer Surv. 1992;13:23–37. [PubMed] [Google Scholar]
- 6.Abubakr YA, Redman BG. The role of immunotherapy in urologic malignancies. Cancer Treat Res. 1996;88:235–248. doi: 10.1007/978-1-4615-6343-3_15. [DOI] [PubMed] [Google Scholar]
- 7.Hussain SF, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 8.Wu A, et al. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J Immunother. 2007;30:789–797. doi: 10.1097/CJI.0b013e318155a0f6. [DOI] [PubMed] [Google Scholar]
- 9.Paulos CM, et al. Toll-like receptors in tumor immunotherapy. Clin Cancer Res. 2007;13:5280–5289. doi: 10.1158/1078-0432.CCR-07-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirsch M, Fischer H, Schackert G. Activated monocytes kill malignant brain tumor cells in vitro. J Neurooncol. 1994;20:35–45. doi: 10.1007/BF01057959. [DOI] [PubMed] [Google Scholar]
- 11.Zhu X, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito A, et al. Heat shock protein 70 expression induces antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol Immunother. 2003;52:80–88. doi: 10.1007/s00262-002-0335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prins RM, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 14.Sehgal A, Berger MS. Basic concepts of immunology and neuroimmunology. Neurosurg Focus. 2000;9:e1. doi: 10.3171/foc.2000.9.6.2. [DOI] [PubMed] [Google Scholar]
- 15.Miescher S, Whiteside TL, de Tribolet N, von Fliedner V. In situ characterization, clonogenic potential, and antitumor cytolytic activity of T lymphocytes infiltrating human brain cancers. J Neurosurg. 1988;68:438–448. doi: 10.3171/jns.1988.68.3.0438. [DOI] [PubMed] [Google Scholar]
- 16.Fine HA. Novel biologic therapies for malignant gliomas. Antiangiogenesis, immunotherapy, and gene therapy. Neurologic Clinics. 1995;13:827–846. [PubMed] [Google Scholar]
- 17.Medawar PB. Immunity to homologous grafted skin: III – The fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 18.Parney IF, Hao C, Petruk KC. Glioma immunology and immunotherapy. Neurosurgery. 2000;46:778–791. doi: 10.1097/00006123-200004000-00002. discussion 791–792. [DOI] [PubMed] [Google Scholar]
- 19.Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol Today. 1994;15:218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- 20.Cserr HF, Knopf PM. Cervical lymphatics, the blood–brain barrier and the immunoreactivity of the brain: a new view. Immunol Today. 1992;13:507–512. doi: 10.1016/0167-5699(92)90027-5. [DOI] [PubMed] [Google Scholar]
- 21.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 22.Serot JM, Foliguet B, Bene MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport. 1997;8:1995–1998. doi: 10.1097/00001756-199705260-00039. [DOI] [PubMed] [Google Scholar]
- 23.McMenamin PG, Forrester JV. Dendritic cells in the central nervous system and eye and their associated supporting tissues. In: Loetze MT, Thomson AW, editors. Dendritic Cells: Biology and Clinical Applications. New York: Academic; 1999. pp. 205–248. [Google Scholar]
- 24.Owens T, Renno T, Taupin V, Krakowski M. Inflammatory cytokines in the brain: does the CNS shape immune responses? Immunol Today. 1994;15:566–571. doi: 10.1016/0167-5699(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 25.Stevens A, Kloter I, Roggendorf W. Inflammatory infiltrates and natural killer cell presence in human brain tumors. Cancer. 1988;61:738–743. doi: 10.1002/1097-0142(19880215)61:4<738::aid-cncr2820610417>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 26.Bullard DE, Gillespie GY, Mahaley MS, Bigner DD. Immunobiology of human gliomas. Semin Oncol. 1986;13:94–109. [PubMed] [Google Scholar]
- 27.Harling-Berg C, Knopf PM, Merriam J, Cserr HF. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol. 1989;25:185–193. doi: 10.1016/0165-5728(89)90136-7. [DOI] [PubMed] [Google Scholar]
- 28.Harling-Berg CJ, Hallett JJ, Park JT, Knopf PM. Hierarchy of immune responses to antigen in the normal brain. Curr Top Microbiol Immunol. 2002;265:1–22. doi: 10.1007/978-3-662-09525-6_1. [DOI] [PubMed] [Google Scholar]
- 29.Harling-Berg CJ, Park TJ, Knopf PM. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J Neuroimmunol. 1999;101:111–127. doi: 10.1016/s0165-5728(99)00130-7. [DOI] [PubMed] [Google Scholar]
- 30.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 31.Street NE, Mosmann TR. Functional diversity of T lymphocytes due to secretion of different cytokine patterns. FSDRB J. 1991;5:171–177. doi: 10.1096/fasebj.5.2.1825981. [DOI] [PubMed] [Google Scholar]
- 32.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 33.Cserr HF, DePasquale M, Harling-Berg CJ, Park JT, Knopf PM. Afferent and efferent arms of the humoral immune response to CSF-administered albumins in a rat model with normal blood–brain barrier permeability. J Neuroimmunol. 1992;41:195–202. doi: 10.1016/0165-5728(92)90070-2. [DOI] [PubMed] [Google Scholar]
- 34.Weller RO, Engelhardt B, Phillips MJ. Lymphocyte targeting of the central nervous system: a review of afferent and efferent CNS-immune pathways. Brain Pathol. 1996;6:275–288. doi: 10.1111/j.1750-3639.1996.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 35.Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992;2:269–276. doi: 10.1111/j.1750-3639.1992.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 36.Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med. 1996;184:1737–1745. doi: 10.1084/jem.184.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sedgwick JD, Ford AL, Foulcher E, Airriess R. Central nervous system microglial cell activation and proliferation follows direct interaction with tissue-infiltrating T cell blasts. J Immunol. 1998;160:5320–5330. [PubMed] [Google Scholar]
- 38.Hatalski CG, Hickey WF, Lipkin WI. Evolution of the immune response in the central nervous system following infection with Borna disease virus. J Neuroimmunol. 1998;90:137–142. doi: 10.1016/s0165-5728(98)00076-9. [DOI] [PubMed] [Google Scholar]
- 39.Stohlman SA, Bergmann CC, Lin MT, Cua DJ, Hinton DR. CTL effector function within the central nervous system requires CD4+ T cells. J Immunol. 1998;160:2896–2904. [PubMed] [Google Scholar]
- 40.Hart DN, Fabre JW. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J Exp Med. 1981;154:347–361. doi: 10.1084/jem.154.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe J, MacLennan KA, Powe DG, Pound JD, Palmer JB. Microglial cells in human brain have phenotypic characteristics related to possible function as dendritic antigen presenting cells. J Pathol. 1989;159:143–149. doi: 10.1002/path.1711590209. [DOI] [PubMed] [Google Scholar]
- 42.Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- 43.Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 44.Gehrmann J, Banati RB, Kreutzberg GW. Microglia in the immune surveillance of the brain: human microglia constitutively express HLA-DR molecules. J Neuroimmunol. 1993;48:189–198. doi: 10.1016/0165-5728(93)90191-z. [DOI] [PubMed] [Google Scholar]
- 45.Williams K, Jr, Ulvestad E, Cragg L, Blain M, Antel JP. Induction of primary T cell responses by human glial cells. J Neurosci Res. 1993;36:382–390. doi: 10.1002/jnr.490360404. [DOI] [PubMed] [Google Scholar]
- 46.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141–147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 47.Irani DN, Lin KI, Griffin DE. Regulation of brain-derived T cells during acute central nervous system inflammation. J Immunol. 1997;158:2318–2326. [PubMed] [Google Scholar]
- 48.Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol. 2007;179:845–853. doi: 10.4049/jimmunol.179.2.845. [DOI] [PubMed] [Google Scholar]
- 49.Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol. 2006;80:797–801. doi: 10.1189/jlb.0306176. [DOI] [PubMed] [Google Scholar]
- 50.Freund J. Accumulation of antibodies in the central nervous system. J Exp Med. 1930;51:889–902. doi: 10.1084/jem.51.6.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koller MF, et al. Active immunization of mice with an Abeta-Hsp70 vaccine. Neurodegener Dis. 2004;1:20–28. doi: 10.1159/000076666. [DOI] [PubMed] [Google Scholar]
- 52.Brendza RP, Holtzman DM. Amyloid-beta immunotherapies in mice and men. Alzheimer Dis Assoc Disord. 2006;20:118–123. doi: 10.1097/01.wad.0000213810.89681.a5. [DOI] [PubMed] [Google Scholar]
- 53.Nicoll JA, Kinrade E, Love S. PCR-mediated search for herpes simplex virus DNA in sections of brain from patients with multiple sclerosis and other neurological disorders. J Neurol Sci. 1992;113:144–151. doi: 10.1016/0022-510x(92)90242-d. [DOI] [PubMed] [Google Scholar]
- 54.Orgogozo JM, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 55.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bard F, et al. Peripherally administered anti-bodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 57.Bacskai BJ, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 58.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 60.Scott AM, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA. 2007;104:4071–4076. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100:216–232. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 62.Brooks WH, Netsky MG, Normansell DE, Horwitz DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med. 1972;136:1631–1647. doi: 10.1084/jem.136.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brooks WH, Caldwell HD, Mortara RH. Immune responses in patients with gliomas. Surg Neurol. 1974;2:419–423. [PubMed] [Google Scholar]
- 64.Mahaley MS, Jr, Brooks WH, Roszman TL, Bigner DD, Dudka L, Richardson S. Immunobiology of primary intracranial tumors. Part 1: studies of the cellular and humoral general immune competence of brain-tumor patients. J Neurosurg. 1977;46:467–476. doi: 10.3171/jns.1977.46.4.0467. [DOI] [PubMed] [Google Scholar]
- 65.Menzies CB, Gunar M, Thomas DG, Behan PO. Impaired thymus-derived lymphocyte function in patients with malignant brain tumour. Clin Neurol Neurosurg. 1980;82:157–168. doi: 10.1016/0303-8467(80)90033-5. [DOI] [PubMed] [Google Scholar]
- 66.Brooks WH, Roszman TL, Rogers AS. Impairment of rosette-forming T lymphoctyes in patients with primary intracranial tumors. Cancer. 1976;37:1869–1873. doi: 10.1002/1097-0142(197604)37:4<1869::aid-cncr2820370435>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 67.Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin Exp Immunol. 1977;29:61–66. [PMC free article] [PubMed] [Google Scholar]
- 68.Braun DP, Penn RD, Flannery AM, Harris JE. Immunoregulatory cell function in peripheral blood leukocytes of patients with intracranial gliomas. Neurosurg. 1982;10:203–209. [PubMed] [Google Scholar]
- 69.Elliott LH, Brooks WH, Roszman TL. Inability of mitogen-activated lymphocytes obtained from patients with malignant primary intracranial tumors to express high affinity interleukin 2 receptors. J Clin Invest. 1990;86:80–86. doi: 10.1172/JCI114719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miescher S, Whiteside TL, Carrel S, von Fliedner V. Functional properties of tumor-infiltrating and blood lymphocytes in patients with solid tumors: effects of tumor cells and their supernatants on proliferative responses of lymphocytes. J Immunol. 1986;136:1899–1907. [PubMed] [Google Scholar]
- 71.Roszman TL, Brooks WH, Elliott LH. Immunobiology of primary intracranial tumors. VI. Suppressor cell function and lectin-binding lymphocyte subpopulations in patients with cerebral tumors. Cancer. 1982;50:1273–1279. doi: 10.1002/1097-0142(19821001)50:7<1273::aid-cncr2820500709>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 72.Roszman TL, Elliott LH, Brooks WH. Proliferative potential of T-cell lymphocytes from gliomas. J Neurosurg. 1992;77:820–821. doi: 10.3171/jns.1992.77.5.0820. [DOI] [PubMed] [Google Scholar]
- 73.Roszman TL, Brooks WH. Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin Exp Immunol. 1980;39:395–402. [PMC free article] [PubMed] [Google Scholar]
- 74.Thomas DG, Lannigan CB, Behan PO. Letter: impaired cell-mediated immunity in human brain tumours. Lancet. 1975;1:1389–1390. doi: 10.1016/s0140-6736(75)92308-9. [DOI] [PubMed] [Google Scholar]
- 75.Young HF, Sakalas R, Kaplan AM. Immunologic depression in cerebral gliomas. Adv Neurol. 1976;15:327–335. [PubMed] [Google Scholar]
- 76.Elliott LH, Brooks WH, Roszman TL. Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. J Immunol. 1984;132:1208–1215. [PubMed] [Google Scholar]
- 77.Elliott LH, Brooks WH, Roszman TL. Activation of immunoregulatory lymphocytes obtained from patients with malignant gliomas. J Neurosurg. 1987;67:231–236. doi: 10.3171/jns.1987.67.2.0231. [DOI] [PubMed] [Google Scholar]
- 78.McVicar DW, Davis DF, Merchant RE. In vitro analysis of the proliferative potential of T cells from patients with brain tumor: glioma-associated immunosuppression unrelated to intrinsic cellular defect. J Neurosug. 1992;76:251–260. doi: 10.3171/jns.1992.76.2.0251. [DOI] [PubMed] [Google Scholar]
- 79.Ashkenazi E, Deutsch M, Tirosh R, Weinreb A, Tsukerman A, Brodie C. A selective impairment of the IL-2 system in lymphocytes of patients with glioblastomas: increased level of soluble IL-2R and reduced protein tyrosine phosphorylation. Neuroimmunomodulation. 1997;4:49–56. doi: 10.1159/000097315. [DOI] [PubMed] [Google Scholar]
- 80.Morford LA, Elliott LH, Carlson SL, Brooks WH, Roszman TL. T cell receptor-mediated signaling is defective in T cells obtained from patients with primary intracranial tumors. J Immunol. 1997;159:4415–4425. [PubMed] [Google Scholar]
- 81.Roszman TL, Brooks WH, Steele C, Elliott LH. Pokeweed mitogen-induced immunoglobulin secretion by peripheral blood lymphocytes from patients with primary intracranial tumors. Characterization of T helper and B cell function. J Immunol. 1985;134:1545–1550. [PubMed] [Google Scholar]
- 82.Bodmer S, et al. Title: Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol. 1989;143:3222–3229. [PubMed] [Google Scholar]
- 83.Fontana A, Constam DB, Frei K, Malipiero U, Pfister HW. Modulation of the immune response by transforming growth factor beta. Int Arch Allergy Immunol. 1992;99:1–7. doi: 10.1159/000236328. [DOI] [PubMed] [Google Scholar]
- 84.Wrann M, et al. T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-beta. EMBO J. 1987;6:1633–1636. doi: 10.1002/j.1460-2075.1987.tb02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Martin R, et al. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-beta gene family. EMBO J. 1987;6:3673–3677. doi: 10.1002/j.1460-2075.1987.tb02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kehrl JH, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wahl SM, et al. Transforming growth factor-beta is a potent immunosuppressive agent that inhibits IL-1-dependent lymphocyte proliferation. J Immunol. 1988;140:3026–3032. [PubMed] [Google Scholar]
- 88.Fontana A, Hengartner H, de Tribolet N, Weber E. Glioblastoma cells release interleukin 1 and factors inhibiting interleukin 2-mediated effects. J Immunol. 1984;132:1837–1844. [PubMed] [Google Scholar]
- 89.Rook AH, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. 1986;136:3916–3920. [PubMed] [Google Scholar]
- 90.Ranges GE, Figari IS, Espevik T, Palladino MA., Jr Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med. 1987;166:991–998. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Espevik T, Figari IS, Ranges GE, Palladino MA., Jr Transforming growth factor-beta 1 (TGF-beta 1) and recombinant human tumor necrosis factor-alpha reciprocally regulate the generation of lymphokine-activated killer cell activity. Comparison between natural porcine platelet-derived TGF-beta 1 and TGF beta 2, and recombinant human TGF-beta 1. J Immunol. 1988;140:2312–2316. [PubMed] [Google Scholar]
- 92.Czarniecki CW, Chiu HH, Wong GH, McCabe SM, Palladino MA. Transforming growth factor-beta 1 modulates the expression of class II histocompatibility antigens on human cells. J Immunol. 1988;140:4217–4223. [PubMed] [Google Scholar]
- 93.Zuber P, Kuppner MC, de Tribolet N. Transforming growth factor-beta 2 down-regulates HLA-DR antigen expression on human malignant glioma cells. Eur J Immunol. 1988;18:1623–1626. doi: 10.1002/eji.1830181023. [DOI] [PubMed] [Google Scholar]
- 94.Torre-Amione G, et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci USA. 1990;87:1486–1490. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parrillo JE, Fauci AS. Mechanisms of corticosteroid action on lymphocyte subpopulations. III. Differential effects of dexamethasone administration on subpopulations of effector cells mediating cellular cytotoxicity in man. Clin Exp Immunol. 1978;31:116–125. [PMC free article] [PubMed] [Google Scholar]
- 96.Fauci AS. Mechanisms of corticosteroid action on lymphocyte subpopulations. II. Differential effects of in vivo hydrocortisone, prednisone and dexamethasone on in vitro expression of lymphocyte function. Clin Exp Immunol. 1976;24:54–62. [PMC free article] [PubMed] [Google Scholar]
- 97.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23:263–272. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 98.Ohm JE, Shurin MR, Esche C, Lotze MT, Carbone DP, Gabrilovich DI. Effect of vascular endothelial growth factor and FLT3 ligand on dendritic cell generation in vivo. J Immunol. 1999;163:3260–3268. [PubMed] [Google Scholar]
- 99.Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. 1999;5:2963–2970. [PubMed] [Google Scholar]
- 100.Lien S, Lowman HB. Therapeutic anti-VEGF antibodies. Handb Exp Pharmacol. 2008:131–150. doi: 10.1007/978-3-540-73259-4_6. [DOI] [PubMed] [Google Scholar]
- 101.Vredenburgh JJ, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 102.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 103.Tonra JR, Hicklin DJ. Targeting the vascular endothelial growth factor pathway in the treatment of human malignancy. Immunol Invest. 2007;36:3–23. doi: 10.1080/08820130600991794. [DOI] [PubMed] [Google Scholar]
- 104.Wintterle S, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7467. [PubMed] [Google Scholar]
- 105.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 106.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 107.Kugler A, et al. Regression of human meta-static renal cell carcinoma after vaccination with tumor cell-dendritic cell hybrids. Nat Med. 2000;6:332–336. doi: 10.1038/73193. [DOI] [PubMed] [Google Scholar]
- 108.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–745. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yao S, Chen L. Reviving exhausted T lymphocytes during chronic virus infection by B7-H1 blockade. Trends Mol Med. 2006;12:244–246. doi: 10.1016/j.molmed.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 110.Thompson RH, et al. B7-H1 glycoprotein blockade: a novel strategy to enhance immunotherapy in patients with renal cell carcinoma. Urology. 2005;66:10–14. doi: 10.1016/j.urology.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 111.Thompson RH, et al. B7-H1 glycoprotein blockade: a novel strategy to enhance immunotherapy in patients with renal cell carcinoma. Urology. 2005;66:10–14. doi: 10.1016/j.urology.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 112.Peng JC, Thomas R, Nielsen LK. Generation and maturation of dendritic cells for clinical application under serum-free conditions. J Immunother. 2005;28:599–609. doi: 10.1097/01.cji.0000175491.21099.04. [DOI] [PubMed] [Google Scholar]
- 113.Hirano F, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 114.Strome SE, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. [PubMed] [Google Scholar]
- 115.Curiel TJ, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 116.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 117.Ferguson TA, Griffith TS. The role of Fas ligand and TNF-related apoptosis-inducing ligand (TRAIL) in the ocular immune response. Chem Immunol Allergy. 2007;92:140–154. doi: 10.1159/000099265. [DOI] [PubMed] [Google Scholar]
- 118.Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev. 2004;44:65–81. doi: 10.1016/j.brainresrev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 119.Bart J, et al. An oncological view on the blood-testis barrier. Lancet Oncol. 2002;3:357–363. doi: 10.1016/s1470-2045(02)00776-3. [DOI] [PubMed] [Google Scholar]
- 120.Ichinose M, Masuoka J, Shiraishi T, Mineta T, Tabuchi K. Fas ligand expression and depletion of T-cell infiltration in astrocytic tumors. Brain Tumor Pathol. 2001;18:37–42. doi: 10.1007/BF02478923. [DOI] [PubMed] [Google Scholar]
- 121.Gratas C, et al. Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors. Brain Pathol. 1997;7:863–869. doi: 10.1111/j.1750-3639.1997.tb00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weller M, Kleihues P, Dichgans J, Ohgaki H. CD95 ligand: lethal weapon against malignant glioma? Brain Pathol. 1998;8:285–293. doi: 10.1111/j.1750-3639.1998.tb00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Krause MF, Lienhart HG, Haberstroh J, Hoehn T, Schulte-Monting J, Leititis JU. Effect of inhaled nitric oxide on intrapulmonary right-to-left-shunting in two rabbit models of saline lavage induced surfactant deficiency and meconium instillation. Eur J Pediatr. 1998;157:410–415. doi: 10.1007/s004310050841. [DOI] [PubMed] [Google Scholar]
- 124.Frankel B, Longo SL, Canute GW. Soluble Fas-ligand (sFasL) in human astrocytoma cyst fluid is cytotoxic to T-cells: another potential means of immune evasion. J Neurooncol. 2000;48:21–26. doi: 10.1023/a:1006473800589. [DOI] [PubMed] [Google Scholar]
- 125.Ausiello CM, et al. Cell mediated cytotoxicity and cytokine production in peripheral blood mononuclear cells of glioma patients. Eur J Cancer. 1991;27:646–650. doi: 10.1016/0277-5379(91)90235-6. [DOI] [PubMed] [Google Scholar]
- 126.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25) J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 127.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med. 2001;193:1285–1294. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Klein L, Khazaie K, von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci USA. 2003;100:8886–8891. doi: 10.1073/pnas.1533365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Itoh M, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. 1999;162:5317–5326. [PubMed] [Google Scholar]
- 133.Bruder CE, et al. High resolution deletion analysis of constitutional DNA from neurofibromatosis type 2 (NF2) patients using microarray-CGH. HumMol Genet. 2001;10:271–282. doi: 10.1093/hmg/10.3.271. [DOI] [PubMed] [Google Scholar]
- 134.Pacholczyk R, Kraj P, Ignatowicz L. Peptide specificity of thymic selection of CD4+CD25+ T cells. J Immunol. 2002;168:613–620. doi: 10.4049/jimmunol.168.2.613. [DOI] [PubMed] [Google Scholar]
- 135.Liyanage UK, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 136.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. [PubMed] [Google Scholar]
- 137.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–4408. [PubMed] [Google Scholar]
- 138.Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. 2003;98:1089–1099. doi: 10.1002/cncr.11618. [DOI] [PubMed] [Google Scholar]
- 139.Woo EY, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- 140.Woo EY, et al. Cutting edge: regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 141.Yu P, et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fecci PE, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006;12:4294–4305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- 143.Gershon RK, Kondo K. Infectious immunological tolerance. Immunology. 1971;21:903–914. [PMC free article] [PubMed] [Google Scholar]
- 144.Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 145.Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 146.Schmitz M, et al. Identification of a naturally processed T cell epitope derived from the glioma-associated protein SOX11. Cancer Lett. 2007;245:331–336. doi: 10.1016/j.canlet.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 147.Wekerle H, Bradl M, Linington C, Kaab G, Kojima K. The shaping of the brain-specific T lymphocyte repertoire in the thymus. Immunol Rev. 1996;149:231–243. doi: 10.1111/j.1600-065x.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 148.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Suri-Payer E, Amar AZ, Thornton AM, Shevach EM. CD4+CD25+ T cells inhibit both the induction and effector function of auto-reactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol. 1998;160:1212–1218. [PubMed] [Google Scholar]
- 150.Green DR, Webb DR. Saying the ‘S’ word in public. Immunol Today. 1993;14:523–525. doi: 10.1016/0167-5699(93)90180-S. [DOI] [PubMed] [Google Scholar]
- 151.Salomon B, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 152.Stephens LA, Mason D. CD25 is a marker for CD4+ thymocytes that prevent autoimmune diabetes in rats, but peripheral T cells with this function are found in both CD25+ and CD25 – subpopulations. J Immunol. 2000;165:3105–3110. doi: 10.4049/jimmunol.165.6.3105. [DOI] [PubMed] [Google Scholar]
- 153.Taguchi O, Nishizuka Y. Self tolerance and localized autoimmunity. Mouse models of autoimmune disease that suggest tissue-specific suppressor T cells are involved in self tolerance. J Exp Med. 1987;165:146–156. doi: 10.1084/jem.165.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taguchi O, Kontani K, Ikeda H, Kezuka T, Takeuchi M, Takahashi T. Tissue-specific suppressor T cells involved in self-tolerance are activated extrathymically by self-antigens. Immunology. 1994;82:365–369. [PMC free article] [PubMed] [Google Scholar]
- 155.Seddon B, Mason D. Regulatory T cells in the control of autoimmunity: the essential role of transforming growth factor beta and interleukin 4 in the prevention of autoimmune thyroiditis in rats by peripheral CD4(+)CD45RC-cells and CD4(+)CD8(–) thymocytes. J Exp Med. 1999;189:279–288. doi: 10.1084/jem.189.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bagavant H, Thompson C, Ohno K, Setiady Y, Tung K. Differential effect of neonatal thymectomy on systemic and organ-specific autoimmune disease. Int Immunol. 2002;14:1397–1406. doi: 10.1093/intimm/dxf105. [DOI] [PubMed] [Google Scholar]
- 157.Seddon B, Mason D. Peripheral autoantigen induces regulatory T cells that prevent auto-immunity. J Exp Med. 1999;189:877–882. doi: 10.1084/jem.189.5.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc Natl Acad Sci USA. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 160.Ng WF, et al. Human CD4(+)CD25(+) cells: a naturally occurring population of regulatory T cells. Blood. 2001;98:2736–2744. doi: 10.1182/blood.v98.9.2736. [DOI] [PubMed] [Google Scholar]
- 161.Ermann J, Szanya V, Ford GS, Paragas V, Fathman CG, Lejon K. CD4(+)CD25(+) T cells facilitate the induction of T cell anergy. J Immunol. 2001;167:4271–4275. doi: 10.4049/jimmunol.167.8.4271. [DOI] [PubMed] [Google Scholar]
- 162.Dai Z, et al. CD4+CD25+ regulatory T cells suppress allograft rejection mediated by memory CD8+ T cells via a CD30-dependent mechanism. J Clin Invest. 2004;113:310–317. doi: 10.1172/JCI19727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Camara NO, Sebille F, Lechler RI. Human CD4+CD25+ regulatory cells have marked and sustained effects on CD8+ T cell activation. Eur J Immunol. 2003;33:3473–3483. doi: 10.1002/eji.200323966. [DOI] [PubMed] [Google Scholar]
- 164.Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Piccirillo CA, Shevach EM. Cutting edge: control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J Immunol. 2001;167:1137–1140. doi: 10.4049/jimmunol.167.3.1137. [DOI] [PubMed] [Google Scholar]
- 166.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 168.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen W, Wahl SM. TGF-beta: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine Growth Factor Rev. 2003;14:85–89. doi: 10.1016/s1359-6101(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 170.Dieckmann D, Bruett CH, Ploettner H, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory, contact-dependent T cells induce interleukin 10-producing, contact-independent type 1-like regulatory T cells [corrected] J Exp Med. 2002;196:247–253. doi: 10.1084/jem.20020642. erratum appears in J Exp Med 2002; 196:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med. 2002;196:255–260. doi: 10.1084/jem.20020394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Huber S, et al. Cutting Edge: TGF-β signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol. 2004;173:6526–6531. doi: 10.4049/jimmunol.173.11.6526. [DOI] [PubMed] [Google Scholar]
- 173.de Martin R, et al. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-beta gene family. EMBO J. 1987;6:3673–3677. doi: 10.1002/j.1460-2075.1987.tb02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Roussel E, Gingras MC, Grimm EA, Bruner JM, Moser RP. Predominance of a type 2 intratumoural immune response in fresh tumour-infiltrating lymphocytes from human gliomas. Clin Exp Immunol. 1996;105:344–352. doi: 10.1046/j.1365-2249.1996.d01-753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zou JP, et al. Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J Immunol. 1999;162:4882–4892. [PubMed] [Google Scholar]
- 176.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25 – cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 177.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25 – T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 178.Chen ZM, et al. IL-10 and TGF-beta induce alloreactive CD4+CD25 – T cells to acquire regulatory cell function. Blood. 2003;101:5076–5083. doi: 10.1182/blood-2002-09-2798. [DOI] [PubMed] [Google Scholar]
- 179.Somasundaram R, et al. Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-beta. Cancer Res. 2002;62:5267–5272. [PubMed] [Google Scholar]
- 180.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 181.Fecci PE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 182.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 183.Steitz J, Bruck J, Lenz J, Knop J, Tuting T. Depletion of CD25(+) CD4(+) T cells and treatment with tyrosinase-related protein 2-transduced dendritic cells enhance the interferon alpha-induced, CD8(+) T-cell-dependent immune defense of B16 melanoma. Cancer Res. 2001;61:8643–8646. [PubMed] [Google Scholar]
- 184.Sutmuller RP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18:1622–1636. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 186.Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc Natl Acad Sci USA. 2000;97:8548–8553. doi: 10.1073/pnas.140210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Attia P, Powell DJ, Jr, Maker AV, Kreitman RJ, Pastan I, Rosenberg SA. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J Immunother. 2006;29:208–214. doi: 10.1097/01.cji.0000187959.45803.0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Powell DJ, Jr, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–4928. doi: 10.4049/jimmunol.179.7.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Olsen E, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol. 2001;19:376–388. doi: 10.1200/JCO.2001.19.2.376. [DOI] [PubMed] [Google Scholar]
- 190.Dannull J, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mahnke K, et al. Depletion of CD4+CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int J Cancer. 2007;120:2723–2733. doi: 10.1002/ijc.22617. [DOI] [PubMed] [Google Scholar]
- 192.Attia P, Maker AV, Haworth LR, Rogers-Freeze L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–592. doi: 10.1097/01.cji.0000175468.19742.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wahlstrom T, Linder E, Saksela E. Glia-specific antigens in cell cultures from rabbit brain, human foetal and adult brain, and gliomas. Acta Pathol Microbiol Scand [B] Microbiol Immunol. 1973;81:768–774. doi: 10.1111/j.1699-0463.1973.tb02274.x. [DOI] [PubMed] [Google Scholar]
- 194.Siris JH. Concerning the immunological specificity of glioblastoma multiforme. Bull Neurol NY. 1936;4:597–601. [Google Scholar]
- 195.Slagel DE, Wilson CB, Simmons PB. Polyacrylamide electrophoresis and immunodiffusion studies of brain tumor proteins. Ann NY Acad Sci. 1969;159:490–496. [Google Scholar]
- 196.Wickremesinghe HR, Yates PO. Immunological properties of neoplastic neural tissues. Bri J Cancer. 1971;25:711–720. doi: 10.1038/bjc.1971.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wikstrand CJ, Mahaley MS, Bigner DD. Surface antigenic characteristics of human glial brain tumor cells. Cancer Res. 1977;37:4267–4275. [PubMed] [Google Scholar]
- 198.Waksman BH, Porter H, Lees MD, Adams RD, Folch J. A study of the chemical nature of components of bovine white matter effective in producing allergic encephalomyelitis in the rabbit. J Exp Med. 1954;100:451–471. doi: 10.1084/jem.100.5.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tuohy VK, Lu ZJ, Sobel RA, Laursen RA, Lees MB. A synthetic peptide from myelin proteolipid protein induces experimental allergic encephalomyelitis. J Immunol. 1988;141:1126–1130. [PubMed] [Google Scholar]
- 200.Linington C, et al. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. European J Immunol. 1993;23:1364–1372. doi: 10.1002/eji.1830230627. [DOI] [PubMed] [Google Scholar]
- 201.Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C. Animal models. Ann Neurol. 1994;36(Suppl):S47–53. doi: 10.1002/ana.410360714. [DOI] [PubMed] [Google Scholar]
- 202.Pasteur L. Methode pour prevenir la rage apres morsure. Comp Acad Sci (Paris) 1885;101:765–774. [Google Scholar]
- 203.Remlinger P. Accidents paralytiques au cours du traitement antirabique. Ann Inst Pasteur. 1905;19:625–646. [PubMed] [Google Scholar]
- 204.Remlinger P. Contribution a l’etude de la toxine rabique (faits experimentaux et clinique) Comp seances Soc Biol. 1904;56:348–350. [Google Scholar]
- 205.Stuart G, Krikorian K. The neuro-paralytic accidents of anti-rabies treatment. Ann Trop Med. 1928;22:327–377. [Google Scholar]
- 206.Stuart G, Krikorian K. A fatal neuro-paralytic accident of anti-rabies treatment. Lancet. 1930;1:1123–1125. [Google Scholar]
- 207.Rivers TM, Schwentker FF. Encephalomyelitis accompanied by myelin destruction experimentally produced in monkeys. J Exp Med. 1935;61:689–702. doi: 10.1084/jem.61.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bigner DD, Pitts OM, Wikstrand CJ. Induction of lethal experimental allergic encephalomyelitis in nonhuman primates and guinea pigs with human glioblastoma multiforme tissue. J Neurosurg. 1981;55:32–42. doi: 10.3171/jns.1981.55.1.0032. [DOI] [PubMed] [Google Scholar]
- 209.Kohm AP, Williams JS, Miller SD. Cutting edge: ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4+ T cell activation and experimental autoimmune encephalomyelitis. J Immunol. 2004;172:4686–4690. doi: 10.4049/jimmunol.172.8.4686. [DOI] [PubMed] [Google Scholar]
- 210.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 211.Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186:1177–1182. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fecci PE, et al. The history, evolution, and clinical use of dendritic cell-based immunization strategies in the therapy of brain tumors. J Neuro-Oncol. 2003;64:161–176. doi: 10.1007/BF02700031. [DOI] [PubMed] [Google Scholar]
- 213.Liau LM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 214.Stragliotto G, Vega F, Stasiecki P, Gropp P, Poisson M, Delattre JY. Multiple infusions of anti-epidermal growth factor receptor (EGFR) monoclonal antibody (EMD 55,900) in patients with recurrent malignant gliomas. Eur J Cancer. 1996;32A:636–640. doi: 10.1016/0959-8049(95)00592-7. [DOI] [PubMed] [Google Scholar]
- 215.Wong AJ, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lal A, Sui I, Riggins GJ. Serial analysis of gene expression: probing transcriptomes for molecular targets. Curr Opin Mol Ther. 1999;1:720–726. [PubMed] [Google Scholar]
- 217.Bigner DD, et al. Iodine-131-labeled anti-tenascin monoclonal antibody 81C6 treatment of patients with recurrent malignant gliomas: phase I trial results. J Clin Oncol. 1998;16:2202–2212. doi: 10.1200/JCO.1998.16.6.2202. [DOI] [PubMed] [Google Scholar]
- 218.Sampson JH, et al. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc Natl Acad Sci USA. 2000;97:7503–7508. doi: 10.1073/pnas.130166597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Humphrey PA, et al. Anti-synthetic peptide antibody reachingn at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA. 1990;87:4207–42011. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wong H, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor gene in malignant gliomas is invariably related with gene amplification. Proc Natl Acad Sci USA. 1987;84:6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Libermann TA, et al. Amplification, enhanced expression and possible rearrangement of the EGF receptor gene in primary human brain tumors of glial origin. Nature. 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 222.Kawamoto T, Sato JD, Le A, Polikoff J, Sato GH, Mendelsohn J. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci USA. 1983;80:1337–1341. doi: 10.1073/pnas.80.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Faillot T, et al. A phase I study of an anti-epidermal growth factor receptor monoclonal antibody for the treatment of malignant gliomas. Neurosurgery. 1996;39:478–483. doi: 10.1097/00006123-199609000-00009. [DOI] [PubMed] [Google Scholar]
- 224.Zalutsky MR, Moseley RP, Coakham HB, Coleman RE, Bigner DD. Pharmacokinetics and tumor localization of 131I-labeled anti-tenascin monoclonal antibody 81C6 in patients with gliomas and other intracranial malignancies. Cancer Res. 1989;49:2807–2813. [PubMed] [Google Scholar]
- 225.Baselga J. Clinical trials of single-agent trastuzumab (Herceptin) Semin Oncol. 2000;27:20–26. [PubMed] [Google Scholar]
- 226.Shin DM, et al. Epidermal growth factor receptor-targeted therapy with C225 and cisplatin in patients with head and neck cancer. Clin Cancer Res. 2001;7:1204–1213. [PubMed] [Google Scholar]
- 227.Crombet T, et al. Use of the humanized anti-epidermal growth factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J Clin Oncol. 2004;22:1646–1654. doi: 10.1200/JCO.2004.03.089. [DOI] [PubMed] [Google Scholar]
- 228.Ramos TC, et al. Treatment of high-grade glioma patients with the humanized anti-epidermal growth factor receptor (EGFR) antibody h-R3: report from a phase I/II trial. Cancer Biol Ther. 2006;5:375–379. doi: 10.4161/cbt.5.4.2522. [DOI] [PubMed] [Google Scholar]
- 229.Torres LA, et al. Phase I/II clinical trial of the humanized anti-EGF-r monoclonal antibody h-R3 labelled with 99mTc in patients with tumour of epithelial origin. Nucl Med Commun. 2005;26:1049–1057. doi: 10.1097/00006231-200512000-00002. [DOI] [PubMed] [Google Scholar]
- 230.Schober R, et al. The epidermal growth factor receptor in glioblastoma: genomic amplification, protein expression, and patient survival data in a therapeutic trial. Clin Neuropathol. 1995;14:169–174. [PubMed] [Google Scholar]
- 231.Schlegel J, et al. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behaviour in human glioblastoma. Int J Cancer. 1994;56:72–77. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- 232.Schwechheimer K, Huang S, Cavenee WK. EGFR gene amplification – rearrangement in human glioblastomas. Int J Cancer. 1995;62:145–148. doi: 10.1002/ijc.2910620206. [DOI] [PubMed] [Google Scholar]
- 233.Sauter G, Maeda T, Waldman FM, Davis RL, Feuerstein BG. Patterns of epidermal growth factor receptor amplification in malignant gliomas. Am J Pathol. 1996;148:1047–1053. [PMC free article] [PubMed] [Google Scholar]
- 234.Bigner SH, Vogelstein B, Bigner DD. Chromosomal abnormalities and gene amplification in malignant gliomas. ISI Atlas of Sci: Biochem. 1988;1:333–336. [Google Scholar]
- 235.Bigner SH, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–8022. [PubMed] [Google Scholar]
- 236.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 237.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII) Biochem J. 1997;324:855–861. doi: 10.1042/bj3240855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–4140. [PubMed] [Google Scholar]
- 240.Vredenburgh JJ, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 241.Brandsma D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant gliomas. Curr Opin Oncol. 2007;19:598–605. doi: 10.1097/CCO.0b013e3282f0313b. [DOI] [PubMed] [Google Scholar]
- 242.Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96:1788–1795. doi: 10.1038/sj.bjc.6603813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Brady LW, et al. Malignant astrocytomas treated with iodine-125 labeled monoclonal antibody 425 against epidermal growth factor receptor: a phase II trial. Int J Rad Oncol Biol Phys. 1992;22:225–230. doi: 10.1016/0360-3016(92)91009-c. [DOI] [PubMed] [Google Scholar]
- 244.Brady LW. A new treatment for high grade gliomas of the brain. Bull Mem Academ Roy Med Belg. 1998;153:255–261. discussion 261–252. [PubMed] [Google Scholar]
- 245.Bourdon MA, et al. Human gliomamesenchymal extracellular matrix antigen defined by monoclonal antibody. Cancer Res. 1983;43:2796–2805. [PubMed] [Google Scholar]
- 246.Brown MT, et al. Intrathecal 131I-labeled antitenascin monoclonal antibody 81C6 treatment of patients with leptomeningeal neoplasms or primary brain tumor resection cavities with subarachnoid communication: phase I trial results. Clin Cancer Res. 1996;2:963–972. [PubMed] [Google Scholar]
- 247.Cokgor I, et al. Phase I trial results of iodine-131-labeled antitenascin monoclonal antibody 81C6 treatment of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2000;18:3862–3872. doi: 10.1200/JCO.2000.18.22.3862. [DOI] [PubMed] [Google Scholar]
- 248.Reardon DA, et al. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: phase II study results. J Clin Oncol. 2006;24:115–122. doi: 10.1200/JCO.2005.03.4082. [DOI] [PubMed] [Google Scholar]
- 249.Reardon DA, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20:1389–1397. doi: 10.1200/JCO.2002.20.5.1389. [DOI] [PubMed] [Google Scholar]
- 250.Murphy-Ullrich JE, Lightner VA, Aukhil I, Yan YZ, Erickson HP, Hook M. Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol. 1991;115:1127–1136. doi: 10.1083/jcb.115.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wikstrand CJ, et al. Comparative localization of glioma-reactive monoclonal antibodies in vivo in an athymic mouse human glioma xenograft model. J Neuroimmunol. 1987;15:37–56. doi: 10.1016/0165-5728(87)90005-1. [DOI] [PubMed] [Google Scholar]
- 252.Reardon DA, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20:1389–1397. doi: 10.1200/JCO.2002.20.5.1389. [DOI] [PubMed] [Google Scholar]
- 253.Cokgor I, et al. Long term response in a patient with neoplastic meningitis secondary to melanoma treated with (131)I-radiolabeled antichondroitin proteoglycan sulfate Mel-14 F(ab′)(2): a case study. Cancer. 2001;91:1809–1813. [PubMed] [Google Scholar]
- 254.Chamberlain MC. Current concepts in leptomeningeal metastasis. Curr Opin Oncol. 1992;4:533–539. doi: 10.1097/00001622-199206000-00018. [DOI] [PubMed] [Google Scholar]
- 255.Turkington TG, Zalutsky MR, Jaszczak RJ, Garg PK, Vaidyanathan G, Coleman RE. Measuring astatine-211 distributions with SPECT. Phys Med Biol. 1993;38:1121–1130. doi: 10.1088/0031-9155/38/8/010. [DOI] [PubMed] [Google Scholar]
- 256.Zalutsky MR, et al. Phase I Trial of alpha-particle-emitting astatine-211 chimeric anti-tenascin antibody in recurrent malignant glioma patients. Proc Am Assoc Cancer Res; Ninety-First Meeting of the American Association of Cancer Research; San Francisco, CA. 2000. p. 544. [Google Scholar]
- 257.Hall EJ. Radiobiology for the Radiologist. Philadelphia: Lippincott; 1988. [Google Scholar]
- 258.Kampf G. Induction of DNA double-strand breaks by ionizing radiation of different quality and their relevance for cell inactivation. Radiobiol Radiother (Berl) 1988;29:631–658. [PubMed] [Google Scholar]
- 259.Strickland DK, Vaidyanathan G, Zalutsky MR. Cytotoxicity of alpha-particle-emitting m- [211At]astatobenzylguanidine on human neuroblastoma cells. Cancer Res. 1994;54:5414–5419. [PubMed] [Google Scholar]
- 260.Zalutsky MR, Garg PK, Friedman HS, Bigner DD. Labeling monoclonal antibodies and F(ab′)2 fragments with the alpha-particle-emitting nuclide astatine-211: preservation of immunoreactivity and in vivo localizing capacity. Proc Natl Acad Sci USA. 1989;86:7149–7153. doi: 10.1073/pnas.86.18.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zalutsky MR, et al. Clinical Experience with {alpha}-Particle Emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49:30–38. doi: 10.2967/jnumed.107.046938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Frankel AE, Tagge EP, Willingham MC. Clinical trials of targeted toxins. Sem Cancer Biol. 1995;6:307–317. doi: 10.1006/scbi.1995.0039. [DOI] [PubMed] [Google Scholar]
- 263.Hall WA, Fodstad O. Immunotoxins and central nervous system neoplasia. J Neurosurg. 1992;76:1–12. doi: 10.3171/jns.1992.76.1.0001. [DOI] [PubMed] [Google Scholar]
- 264.Recht L, Torres CO, Smith TW, Raso V, Griffin TW. Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J Neurosurg. 1990;72:941–945. doi: 10.3171/jns.1990.72.6.0941. [DOI] [PubMed] [Google Scholar]
- 265.Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med. 1997;3:1362–1368. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- 266.Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY. Transferrin receptor on endothelium of brain capillaries. Nature. 1984;312:162–163. doi: 10.1038/312162a0. [DOI] [PubMed] [Google Scholar]
- 267.Morrison PF, Laske DW, Bobo H, Oldfield EH, Dedrick RL. High-flow microinfusion: tissue penetration and pharmacodynamics. Am J Physiol. 1994;266:R292–305. doi: 10.1152/ajpregu.1994.266.1.R292. [DOI] [PubMed] [Google Scholar]
- 268.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lieberman DM, Laske DW, Morrison PF, Bankiewicz KS, Oldfield EH. Convection-enhanced distribution of large molecules in gray matter during interstitial drug infusion. J Neurosurg. 1995;82:1021–1029. doi: 10.3171/jns.1995.82.6.1021. [DOI] [PubMed] [Google Scholar]
- 270.Sampson JH, et al. Intracerebral infusate distribution by convection-enhanced delivery in humans with malignant gliomas: descriptive effects of target anatomy and catheter positioning. Neurosurg. 2007;60:ONS89–98. doi: 10.1227/01.NEU.0000249256.09289.5F. discussion ONS98–89. [DOI] [PubMed] [Google Scholar]
- 271.Kunwar S, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the cintredekin besudotox intraparenchymal study group. J Clin Oncol. 2007;25:837–844. doi: 10.1200/JCO.2006.08.1117. [DOI] [PubMed] [Google Scholar]
- 272.Sampson JH, et al. Clinical utility of a patient-specific algorithm for simulating intracerebral drug infusions. Neuro Oncol. 2007;9:343–353. doi: 10.1215/15228517-2007-007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sampson JH, et al. Induction of hyperintense signal on T2-weighted MR images correlates with infusion distribution from intracerebral convection-enhanced delivery of a tumor-targeted cytotoxin. AJR Am J Roentgenol. 2007;188:703–709. doi: 10.2214/AJR.06.0428. [DOI] [PubMed] [Google Scholar]
- 274.Sampson JH, Akabani G, Friedman AH, Bigner D, Kunwar S, Berger MS, Bankiewicz KS. Comparison of intratumoral bolus injection and convection-enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus. 2006;20:E14. doi: 10.3171/foc.2006.20.4.9. [DOI] [PubMed] [Google Scholar]
- 275.Raghavan R, Brady ML, Rodriguez-Ponce MI, Hartlep A, Pedain C, Sampson JH. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20:E12. doi: 10.3171/foc.2006.20.4.7. [DOI] [PubMed] [Google Scholar]
- 276.Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin Cancer Res. 2000;6:2157–2165. [PubMed] [Google Scholar]
- 277.Puri RK, Leland P, Kreitman RJ, Pastan I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. Int J Cancer. 1994;58:574–581. doi: 10.1002/ijc.2910580421. [DOI] [PubMed] [Google Scholar]
- 278.Pastan I, Chaudhary V, FitzGerald DJ. Recombinant toxins as novel therapeutic agents. Annu Rev Biochem. 1992;61:331–354. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- 279.Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin Cancer Res. 1995;1:1253–1258. [PubMed] [Google Scholar]
- 280.Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999;5:985–990. [PubMed] [Google Scholar]
- 281.Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor {alpha}2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–7986. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 282.Sampson JH, et al. Sustained radiographic and clinical response in patient with bifrontal recurrent glioblastoma multiforme with intracerebral infusion of the recombinant targeted toxin TP-38: case study. Neuro-Oncology. 2005;7:90–96. doi: 10.1215/S1152851703000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.LeMay DR, et al. Intravenous RMP-7 increases delivery of ganciclovir into rat brain tumors and enhances the effects of herpes simplex virus thymidine kinase gene therapy. Hum Gene Ther. 1998;9:989–995. doi: 10.1089/hum.1998.9.7-989. [DOI] [PubMed] [Google Scholar]
- 284.Nilaver G, et al. Delivery of herpesvirus and adenovirus to nude rat intracerebral tumors after osmotic blood–brain barrier disruption. Proc Natl Acad Sci USA. 1995;92:9829–9833. doi: 10.1073/pnas.92.21.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ford J, Osborn C, Barton T, Bleehen NM. A phase I study of intravenous RMP-7 with carboplatin in patients with progression of malignant glioma. Eur J Cancer. 1998;34:1807–1811. doi: 10.1016/s0959-8049(98)00155-5. [DOI] [PubMed] [Google Scholar]
- 286.Elliott PJ, Hayward NJ, Huff MR, Nagle TL, Black KL, Bartus RT. Unlocking the blood–brain barrier: a role for RMP-7 in brain tumor therapy. Exp Neurol. 1996;141:214–224. doi: 10.1006/exnr.1996.0156. [DOI] [PubMed] [Google Scholar]
- 287.Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50:814s–819s. [PubMed] [Google Scholar]
- 288.Choucair AK, Scott C, Urtasun R, Nelson D, Mousas B, Curran W. Quality of life and neuropsychological evaluation for patients with malignant astrocytomas: RTOG 91-14. Radiation therapy oncology group. Int J Rad Oncol Biol Phys. 1997;38:9–20. doi: 10.1016/s0360-3016(97)00223-x. [DOI] [PubMed] [Google Scholar]
- 289.Pasquier B, Pasquier D, N’Golet A, Panh MH, Couderc P. Extraneural metastases of astrocytomas and glioblastomas: clinicopathological study of two cases and review of literature. Cancer. 1980;45:112–125. doi: 10.1002/1097-0142(19800101)45:1<112::aid-cncr2820450121>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 290.Riva P, et al. 131I radioconjugated antibodies for the locoregional radioimmunotherapy of high-grade malignant glioma—phase I and II study. Acta Oncologica. 1999;38:351–359. doi: 10.1080/028418699431438. [DOI] [PubMed] [Google Scholar]
- 291.Sampson JH, et al. Subcutaneous vaccination with irradiated, cytokine-producing tumor cells stimulates CD8+ cell-mediated immunity against tumors located in the “immunologically privileged” central nervous system. Proc Natl Acad Sci USA. 1996;93:10399–10404. doi: 10.1073/pnas.93.19.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wood GW, Turner T, Wang YY, Holladay FP. Immune rejection of intracerebral gliomas using lymphocytes from glioma-bearing rats. J Immunother. 1999;22:497–505. doi: 10.1097/00002371-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 293.Yu JS, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–847. [PubMed] [Google Scholar]
- 294.Mahaley MS, Jr, et al. Immunobiology of primary intracranial tumors. Part 7: Active immunization of patients with anaplastic human glioma cells: a pilot study. J Neurosurg. 1983;59:201–207. doi: 10.3171/jns.1983.59.2.0201. [DOI] [PubMed] [Google Scholar]
- 295.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 296.Mosca PJ, Lyerly HK, Clay TM, Morse MA, Lyerly HK. Dendritic cell vaccines. Front Biosci. 2007;12:4050–4060. doi: 10.2741/2371. [DOI] [PubMed] [Google Scholar]
- 297.Parajuli P, Mathupala S, Mittal S, Sloan AE. Dendritic cell-based active specific immunotherapy for malignant glioma. Expert Opin Biol Ther. 2007;7:439–448. doi: 10.1517/14712598.7.4.439. [DOI] [PubMed] [Google Scholar]
- 298.Fecci PE, et al. The history, evolution, and clinical use of dendritic cell-based immunization strategies in the therapy of brain tumors. J Neurooncol. 2003;64:161–176. doi: 10.1007/BF02700031. [DOI] [PubMed] [Google Scholar]
- 299.Inaba K, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Campbell JD, et al. Isolation and generation of clinical-grade dendritic cells using the CliniMACS system. Methods Mol Med. 2005;109:55–70. doi: 10.1385/1-59259-862-5:055. [DOI] [PubMed] [Google Scholar]
- 301.Mu LJ, Gaudernack G, Saeboe-Larssen S, Hammerstad H, Tierens A, Kvalheim G. A protocol for generation of clinical grade mRNA-transfected monocyte-derived dendritic cells for cancer vaccines. Scand J Immunol. 2003;58:578–586. doi: 10.1046/j.1365-3083.2003.01333.x. [DOI] [PubMed] [Google Scholar]
- 302.Sorg RV, et al. Clinical-scale generation of dendritic cells in a closed system. J Immunother. 2003;26:374–383. doi: 10.1097/00002371-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 303.Tuyaerts S, et al. Generation of large numbers of dendritic cells in a closed system using Cell Factories. J Immunol Methods. 2002;264:135–151. doi: 10.1016/s0022-1759(02)00099-6. [DOI] [PubMed] [Google Scholar]
- 304.Goxe B, Latour N, Chokri M, Abastado JP, Salcedo M. Simplified method to generate large quantities of dendritic cells suitable for clinical applications. Immunol Invest. 2000;29:319–336. doi: 10.3109/08820130009060870. [DOI] [PubMed] [Google Scholar]
- 305.Liau LM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 306.Yamanaka R, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11:4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 307.Kikuchi T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother Emphas Tumor Immunol. 2004;27:452–459. doi: 10.1097/00002371-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 308.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–4979. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 309.Yamanaka R, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172–1179. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Sampson JH. Neuro Oncol. Vol. 1. La Jolla, Calif: 1999. The Preuss Foundation Seminar on vaccine therapy for malignant primary brain tumors. February 15–17, 1998; pp. 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Yu JS, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–847. [PubMed] [Google Scholar]
- 312.Okada H, et al. Gene therapy of malignant gliomas: a pilot study of vaccination with irradiated autologous glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune response. Hum Gene Ther. 2001;12:575–595. doi: 10.1089/104303401300042528. [DOI] [PubMed] [Google Scholar]
- 313.Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56–58. [PubMed] [Google Scholar]
- 314.Barratt-Boyes SM, et al. Maturation and trafficking of monocyte-derived dendritic cells in monkeys: implications for dendritic cell-based vaccines. J Immunol. 2000;164:2487–2495. doi: 10.4049/jimmunol.164.5.2487. [DOI] [PubMed] [Google Scholar]
- 315.Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166:4254–4259. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 316.Kalinski P, Vieira PL, Schuitemaker JH, de Jong EC, Kapsenberg ML. Prostaglandin E(2) is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood. 2001;97:3466–3469. doi: 10.1182/blood.v97.11.3466. [DOI] [PubMed] [Google Scholar]
- 317.Heimberger AB, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol. 2008;10:98–103. doi: 10.1215/15228517-2007-046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Sloan AE, Parajuli P, Mathupala S. DC-tumor cell fusion for induction of tumor-specific T-cell response against malignant brain tumors: comparison with DC pulsed with total tumor RNA or tumor lysate; Proceedings of the American Association of Cancer Research; San Francisco, CA. 2002. [Google Scholar]
- 319.Steinbok P, Thomas JP, Grossman L, Dolman CL. Intratumoral autologous mononuclear cells in the treatment of recurrent glioblastoma multiforme. A phase 1 (toxicity) study. J Neurooncol. 1984;2:147–151. doi: 10.1007/BF00177901. [DOI] [PubMed] [Google Scholar]
- 320.Young H, Kaplan A, Regelson W. Immunotherapy with autologous white cell infusions (“lymphocytes”) in the treatment of recurrrent glioblastoma multiforme: a preliminary report. Cancer. 1977;40:1037–1044. doi: 10.1002/1097-0142(197709)40:3<1037::aid-cncr2820400311>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 321.Hayes RL, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76:840–852. doi: 10.1002/1097-0142(19950901)76:5<840::aid-cncr2820760519>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 322.Blancher A, et al. Local immunotherapy of recurrent glioblastoma multiforme by intracerebral perfusion of interleukin-2 and LAK cells. Eur Cytokine Netw. 1993;4:331–341. [PubMed] [Google Scholar]
- 323.Jacobs SK, Wilson DJ, Kornblith PL, Grimm EA. Interleukin-2 or autologous lymphokine-activated killer cell treatment of malignant glioma: phase I trial. Cancer Res. 1986;46:2101–2104. [PubMed] [Google Scholar]
- 324.Jeffes EW, III, et al. Therapy of recurrent high grade gliomas with surgery, and autologous mitogen activated IL-2 stimulated killer (MAK) lymphocytes: I. Enhancement of MAK lytic activity and cytokine production by PHA and clinical use of PHA. Neuro-Oncology. 1993;15:141–155. doi: 10.1007/BF01053935. [DOI] [PubMed] [Google Scholar]
- 325.Ingram M, et al. Immunotherapy for recurrent malignant glioma: an interim report on survival. Neurol Res. 1990;12:265–273. doi: 10.1080/01616412.1990.11739955. [DOI] [PubMed] [Google Scholar]
- 326.Quattrocchi KB, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neuro-Oncol. 1999;45:141–157. doi: 10.1023/a:1006293606710. [DOI] [PubMed] [Google Scholar]
- 327.Tsurushima H, et al. Reduction of end-stage malignant glioma by injection with autologous cytotoxic T lymphocytes. Japanese J Cancer Res. 1999;90:536–545. doi: 10.1111/j.1349-7006.1999.tb00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Kitahara T, et al. Establishment of interleukin 2 dependent cytotoxic T lymphocyte cell line specific for autologous brain tumor and its intracranial administration for therapy of the tumor. J Neurooncol. 1987;4:329–336. doi: 10.1007/BF00195603. [DOI] [PubMed] [Google Scholar]
- 329.Brooks WH, Markesberry MD, Gupta GF, Roszman TL. Relationship of lymphocyte invasion and survival of brain tumor patients. Ann Neurol. 1978;4:219–224. doi: 10.1002/ana.410040305. [DOI] [PubMed] [Google Scholar]
- 330.Palma L, DiLorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivious gliomas. J Neurosurg. 1978;49:845–861. doi: 10.3171/jns.1978.49.6.0854. [DOI] [PubMed] [Google Scholar]
- 331.Rosenberg SA. The development of new immunotherapies for the treatment of cancer using interleukin-2. A review. Ann Surg. 1988;208:121–135. doi: 10.1097/00000658-198808000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Merchant RE, Ellison MD, Young HF. Immunotherapy for malignant glioma using human recombinant interleukin-2 and activated autologous lymphocytes. A review of pre-clinical and clinical investigations. J Neuro-Oncol. 1990;8:173–188. doi: 10.1007/BF00177842. [DOI] [PubMed] [Google Scholar]
- 333.Jacobs SK, Wilson DJ, Kornblith PL, Grimm EA. Interleukin-2 and autologous lymphokine-activated killer cells in the treatment of malignant glioma. J Neurosurg. 1986;64:743–749. doi: 10.3171/jns.1986.64.5.0743. [DOI] [PubMed] [Google Scholar]
- 334.Merchant RE, Merchant LH, Cook SH, McVicar DW, Young HF. Intralesional infusion of lymphokine-activated killer (LAK) cells and recombinant interleukin-2 (rIL-2) for the treatment of patients with malignant brain tumor. Neurosurgery. 1988;23:725–732. doi: 10.1227/00006123-198812000-00007. [DOI] [PubMed] [Google Scholar]
- 335.Blacklock JB, Grimm EA. Lymphokine-activated killer lymphocytes: LAK and interleukin-2 in the treatment of malignancies of the central nervous system. Immunol Series. 1989;48:93–99. [PubMed] [Google Scholar]
- 336.Lillehei KO, Mitchell DH, Johnson SD, McCleary EL, Kruse CA. Long-term follow-up of patients with recurrent malignant gliomas treated with adjuvant adoptive immunotherapy. Neurosurgery. 1991;28:16–23. doi: 10.1097/00006123-199101000-00003. [DOI] [PubMed] [Google Scholar]
- 337.Dillman RO, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. 2004;27:398–404. doi: 10.1097/00002371-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 338.Ishikawa E, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004;24:1861–1871. [PubMed] [Google Scholar]
- 339.Ames IH, et al. Preferential homing of tumor-infiltrating lymphocytes in tumor-bearing mice. Cancer Immunol Immunother. 1989;29:93–100. doi: 10.1007/BF00199283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lampson LA, Hickey WF. Monoclonal antibody analysis of MHC expression in human brain biopsies: tissue ranging from “histologically normal” to that showing different levels of glial tumor involvement. J Immunol. 1986;136:4054–4062. [PubMed] [Google Scholar]
- 341.Lampson LA. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: reconciling divergent findings. Microsc Res Tech. 1995;32:267–285. doi: 10.1002/jemt.1070320402. [DOI] [PubMed] [Google Scholar]
- 342.Bigner DD, et al. Heterogeneity of Genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. J Neuropathol Exp Neurol. 1981;40:201–229. doi: 10.1097/00005072-198105000-00001. [DOI] [PubMed] [Google Scholar]
- 343.Kruse CA, et al. Characterization of a continuous human glioma cell line DBTRG-05MG: growth kinetics, karyotype, receptor expression, and tumor suppressor gene analyses. In Vitro Cell Dev Biol. 1992;28A:609–614. doi: 10.1007/BF02631035. [DOI] [PubMed] [Google Scholar]
- 344.Whelan SA, Hightower LE. Differential induction of glucose-regulated and heat shock proteins: effects of pH and sulfhydryl-reducing agents on chicken embryo cells. J Cell Physiol. 1985;125:251–258. doi: 10.1002/jcp.1041250212. [DOI] [PubMed] [Google Scholar]
- 345.Natali PG, Bigotti A, Nicotra MR, Viora M, Manfredi D, Ferrone S. Distribution of human Class I (HLA-A,B,C) histocompatibility antigens in normal and malignant tissues of nonlymphoid origin. Cancer Res. 1984;44:4679–4687. [PubMed] [Google Scholar]
- 346.Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, Lillehei KO. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77–87. doi: 10.1007/s002620050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Virasch N, Kruse CA. Strategies using the immune system for therapy of brain tumors. Hematol Oncol Clin North Am. 2001;15:1053–1071. doi: 10.1016/s0889-8588(05)70267-7. [DOI] [PubMed] [Google Scholar]
- 348.Bigner SH, et al. Gene amplification in malignant human gliomas: clinical and histopathologic aspects. J Neuropathol Exp Neurol. 1988;47:191–205. doi: 10.1097/00005072-198805000-00001. [DOI] [PubMed] [Google Scholar]
- 349.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–4140. [PubMed] [Google Scholar]
- 350.Heimberger AB, et al. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery. 2002;50:158–164. doi: 10.1097/00006123-200201000-00024. discussion 164–156. [DOI] [PubMed] [Google Scholar]
- 351.Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000;60:3081–3087. [PubMed] [Google Scholar]
- 352.Han J, Yang L, Puri RK. Analysis of target genes induced by IL-13 cytotoxin in human glioblastoma cells. J Neuro-Oncol. 2005;72:35–46. doi: 10.1007/s11060-004-3119-7. [DOI] [PubMed] [Google Scholar]
- 353.Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60:1168–1172. [PubMed] [Google Scholar]
- 354.Parry RG, Gillespie KM, Mathieson PW. Effects of type 2 cytokines on glomerular epithelial cells. Exp Nephrol. 2001;9:275–283. doi: 10.1159/000052622. [DOI] [PubMed] [Google Scholar]
- 355.Kawada M, et al. Vaccination of fusion cells of rat dendritic and carcinoma cells prevents tumor growth in vivo. Int J Cancer. 2003;105:520–526. doi: 10.1002/ijc.11120. [DOI] [PubMed] [Google Scholar]
- 356.Komata T, Kanzawa T, Kondo Y, Kondo S. Telomerase as a therapeutic target for malignant gliomas. Oncogene. 2002;21:656–663. doi: 10.1038/sj.onc.1205072. [DOI] [PubMed] [Google Scholar]
- 357.Tsuda N, et al. UDP-Gal: betaGlcNAc beta1, 3-galactosyltransferase, polypeptide 3 (GALT3) is a tumour antigen recognised by HLA-A2-restricted cytotoxic T lymphocytes from patients with brain tumour. Br J Cancer. 2002;87:1006–1012. doi: 10.1038/sj.bjc.6600593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Hayashi T, Murayama S, Sakurai M, Kanazawa I. Jugular foramen syndrome caused by varicella zoster virus infection in a patient with ipsilateral hypoplasia of the jugular foramen. J Neurol Sci. 2000;172:70–72. doi: 10.1016/s0022-510x(99)00263-4. [DOI] [PubMed] [Google Scholar]
- 359.Nonaka Y, et al. Recognition of ADP-ribosylation factor 4-like by HLA-A2-restricted and tumor-reactive cytotoxic T lymphocytes from patients with brain tumors. Tiss Antigens. 2002;60:319–327. doi: 10.1034/j.1399-0039.2002.600406.x. [DOI] [PubMed] [Google Scholar]
- 360.Chi DD, et al. Molecular detection of tumor-associated antigens shared by human cutaneous melanomas and gliomas. Am J Pathol. 1997;150:2143–2152. [PMC free article] [PubMed] [Google Scholar]
- 361.Mitchell DA, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with malignant astrocytoma. Neuro Oncol. 2008;10:10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Cobbs CS, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–3350. [PubMed] [Google Scholar]
- 363.Szmania S, et al. Isolation and expansion of cytomegalovirus-specific cytotoxic T lymphocytes to clinical scale from a single blood draw using dendritic cells and HLA-tetramers. Blood. 2001;98:505–512. doi: 10.1182/blood.v98.3.505. [DOI] [PubMed] [Google Scholar]
- 364.Eberl G, Kessler B, Eberl LP, Brunda MJ, Valmori D, Corradin G. Immunodominance of cytotoxic T lymphocyte epitopes co-injected in vivo and modulation by interleukin-12. Eur J Immunol. 1996;26:2709–2716. doi: 10.1002/eji.1830261124. [DOI] [PubMed] [Google Scholar]
- 365.Bissinger AL, Rauser G, Hebart H, Frank F, Jahn G, Einsele H. Isolation and expansion of human cytomegalovirus- specific cytotoxic T lymphocytes using interferon-gamma secretion assay. Exp Hematol. 2002;30:1178–1184. doi: 10.1016/s0301-472x(02)00897-4. [DOI] [PubMed] [Google Scholar]
- 366.Wikstrand CJ, Reist CJ, Archer GE, Zalutsky MR, Bigner DD. The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J Neurovirol. 1998;4:148–158. doi: 10.3109/13550289809114515. [DOI] [PubMed] [Google Scholar]
- 367.Lozupone F, et al. Adoptive transfer of an anti-MART-1(27–35)-specific CD8+ T cell clone leads to immunoselection of human melanoma antigen-loss variants in SCID mice. Eur J Immunol. 2003;33:556–566. doi: 10.1002/immu.200310032. [DOI] [PubMed] [Google Scholar]
- 368.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Chambers CA, et al. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol Rev. 1996;153:27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 370.Brunet JF, et al. A new member of the immunoglobulin superfamily – CTLA-4. Nature. 1987;328:267–270. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 371.Langer LF, Clay TM, Morse MA. Update on anti-CTLA-4 antibodies in clinical trials. Expert Opin Biol Ther. 2007;7:1245–1256. doi: 10.1517/14712598.7.8.1245. [DOI] [PubMed] [Google Scholar]
- 372.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 373.Fecci PE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD41T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 374.Velicu S, et al. Cross-priming of T cells to intracranial tumor antigens elicits an immune response that fails in the effector phase but can be augmented with local immunotherapy. J Neuroimmunol. 2006;174:74–81. doi: 10.1016/j.jneuroim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 375.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Plautz GE, Inoue M, Shu S. Defining the synergistic effects of irradiation and T-cell immunotherapy for murine intracranial tumors. Cell Immunol. 1996;171:277–284. doi: 10.1006/cimm.1996.0204. [DOI] [PubMed] [Google Scholar]
- 377.Maine GN, Mule JJ. Making room for T cells. J Clin Invest. 2002;110:157–159. doi: 10.1172/JCI16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Rapoport AP, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 379.Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]
- 380.Dudley ME, et al. A phase I study of non-myeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Skornick Y, Topalian S, Rosenberg SA. Comparative studies of the long-term growth of lymphocytes from tumor infiltrates, tumor-draining lymph nodes, and peripheral blood by repeated in vitro stimulation with autologous tumor. J Biol Response Mod. 1990;9:431–438. [PubMed] [Google Scholar]
- 382.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Greenberg P, Goodrich J, Riddell S. Adoptive immunotherapy of human cytomegalovirus infection: potential role in protection from disease progression. Transplant Proc. 1991;23:97–101. [PubMed] [Google Scholar]
- 384.Rooney CM, Aguilar LK, Huls MH, Brenner MK, Heslop HE. Adoptive immunotherapy of EBV-associated malignancies with EBV-specific cytotoxic T-cell lines. Curr Top Microbiol Immunol. 2001;258:221–229. doi: 10.1007/978-3-642-56515-1_14. [DOI] [PubMed] [Google Scholar]
- 385.Riddell SR, Greenberg PD. Principles for adoptive T cell therapy of human viral diseases. Annu Rev Immunol. 1995;13:545–586. doi: 10.1146/annurev.iy.13.040195.002553. [DOI] [PubMed] [Google Scholar]
- 386.Cohen PA, Peng L, Plautz GE, Kim JA, Weng DE, Shu S. CD4+ T cells in adoptive immunotherapy and the indirect mechanism of tumor rejection. Crit Rev Immunol. 2000;20:17–56. [PubMed] [Google Scholar]
- 387.Nyika A, Mahan SM, Burridge MJ, McGuire TC, Rurangirwa F, Barbet AF. A DNA vaccine protects mice against the rickettsial agent Cowdria ruminantium. Parasite Immunol. 1998;20:111–119. doi: 10.1046/j.1365-3024.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 388.Greenberg PD, et al. Genetic modification of T-cell clones for therapy of human viral and malignant diseases. Cancer J Sci Am. 1998;4(Suppl):S100–105. [PubMed] [Google Scholar]
- 389.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–2016. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 390.Willemsen RA, Debets R, Chames P, Bolhuis RL. Genetic engineering of T cell specificity for immunotherapy of cancer. Hum Immunol. 2003;64:56–68. doi: 10.1016/s0198-8859(02)00730-9. [DOI] [PubMed] [Google Scholar]
- 391.Niethammer AG, et al. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med. 2002;8:1369–1375. doi: 10.1038/nm1202-794. [DOI] [PubMed] [Google Scholar]
- 392.Wei YQ, et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med. 2002;6:1160–1166. doi: 10.1038/80506. [DOI] [PubMed] [Google Scholar]
- 393.Li Y, et al. Active immunization against the vascular endothelial growth factor receptor flk1 inhibits tumor angiogenesis and metastasis. J Exp Med. 2002;195:1575–1584. doi: 10.1084/jem.20020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Satchi-Fainaro R. Targeting tumor vasculature: reality or a dream? J Drug Target. 2002;10:529–533. doi: 10.1080/1061186021000051062. [DOI] [PubMed] [Google Scholar]
- 395.Singh SK, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 396.Hemmati HD, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Taylor MD, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 398.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 399.Bao S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 400.Liu G, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Pellegatta S, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]