

Abstract
Interindividual variability in activity of uptake transporters is evident in vivo, yet limited data exist in vitro, confounding in vitro-in vivo extrapolation. The uptake kinetics of seven organic anion-transporting polypeptide substrates was investigated over a concentration range in plated cryopreserved human hepatocytes. Active uptake clearance (CLactive, u), bidirectional passive diffusion (Pdiff), intracellular binding, and metabolism were estimated for bosentan, pitavastatin, pravastatin, repaglinide, rosuvastatin, telmisartan, and valsartan in HU4122 donor using a mechanistic two-compartment model in Matlab. Full uptake kinetics of rosuvastatin and repaglinide were also characterized in two additional donors, whereas for the remaining drugs CLactive, u was estimated at a single concentration. The unbound affinity constant (Km, u) and Pdiff values were consistent across donors, whereas Vmax was on average up to 2.8-fold greater in donor HU4122. Consistency in Km, u values allowed extrapolation of single concentration uptake activity data and assessment of interindividual variability in CLactive across donors. The maximal contribution of active transport to total uptake differed among donors, for example, 85 to 96% and 68 to 87% for rosuvastatin and repaglinide, respectively; however, in all cases the active process was the major contributor. In vitro-in vivo extrapolation indicated a general underprediction of hepatic intrinsic clearance, an average empirical scaling factor of 17.1 was estimated on the basis of seven drugs investigated in three hepatocyte donors, and donor-specific differences in empirical factors are discussed. Uptake Km, u and CLactive, u were on average 4.3- and 7.1-fold lower in human hepatocytes compared with our previously published rat data. A strategy for the use of rat uptake data to facilitate the experimental design in human hepatocytes is discussed.
Introduction
Over the past decade human hepatocytes have become the tool of choice to study metabolism of new chemical entities (NCEs) because of the expression of phase I and II metabolizing enzymes alongside transporters (Hewitt et al., 2007; Soars et al., 2007; Hallifax et al., 2010; Ohtsuki et al., 2012). As the quality and characterization of the cells available have improved, investigation of hepatic disposition of new NCEs in donors expressing various levels of enzyme and transport is becoming more plausible (Parker and Houston, 2008; Chiba et al., 2009; Badolo et al., 2011; Ulvestad et al., 2011; Jones et al., 2012). However, interdonor variability in transporter expression and activity has yet to be addressed; experience with metabolic clearance predictions (Hallifax and Houston, 2009) indicates that this may be problematic. Reports have recently been published on characterization of active uptake in reasonably large sets of human hepatocyte donors using typical OATP probes (Badolo et al., 2011; De Bruyn et al., 2011). Badolo et al. (2011) reported variation of 53% in the uptake clearance of estradiol-17β-glucuronide when studied in six lots of cryopreserved human hepatocytes. De Bruyn et al. (2011) observed a 30-fold range in estrone-3-sulfate uptake when measured in 14 human hepatocyte donors. However, to our knowledge, there has been no investigation to date of the uptake of a substantial set of drugs in the same human hepatocyte donors and following a standardized experimental approach.
In the present study, uptake of seven OATP substrates was characterized over a range of concentration and time points in plated cryopreserved human hepatocytes (donor HU4122). Rosuvastatin, pravastatin, pitavastatin, telmisartan, valsartan, repaglinide, and bosentan were selected, because these drugs have been reported to interact with at least one of the major OATPs expressed in the liver (OATP1B1 or OATP1B3), in either in vitro systems and/or in clinical studies involving patients with polymorphic variants of these transporters (Niemi et al., 2005; Ieiri et al., 2009; Watanabe et al., 2009; Yabe et al., 2011). These drugs have also been involved in clinical drug-drug interactions (DDIs) due to inhibition of active transport, either in isolation or in conjunction with a reduction in metabolism (Niemi et al., 2003; Simonson et al., 2004; Kajosaari et al., 2005). In addition, rosuvastatin, pravastatin, and pitavastatin have been recommended by the International Transporter Consortium as probe substrates to assess the potential risk of DDIs involving OATPs of NCE in development (Giacomini et al., 2010). Therefore, a mechanistic characterization of their in vitro uptake kinetics is required. In this study, advantages and limitations associated with each of these substrates have been highlighted.
In the case of rosuvastatin and repaglinide, full kinetic profiles were assessed in three human hepatocyte donors. Rosuvastatin was selected as a representative of a drug undergoing minimal metabolism, whereas repaglinide allowed the simultaneous assessment of uptake and metabolism. For the remaining drugs, uptake was assessed at a single low concentration in two additional hepatocyte donors. In those cases, a priori information obtained from extensive rat hepatocyte studies (Ménochet et al., 2012) and detailed kinetic data from the donor HU4122 were applied in the mechanistic model to estimate uptake kinetic parameters and assess interindividual variability in active uptake across hepatocyte donors.
In a recent study, we have proposed a mechanistic two-compartment model for simultaneous assessment of uptake, passive diffusion, intracellular binding, and metabolism in vitro (Ménochet et al., 2012). Application of the mechanistic model developed to describe the interplay among multiple processes was previously assessed in rat hepatocytes and extended here to human hepatocytes; hence, the interspecies differences in uptake kinetics of seven OATP substrates were investigated. This investigation is of particular relevance because the data generated in rat hepatocytes are valuable to improve our understanding of mechanisms driving the disposition of the compound using physiologically based pharmacokinetic models and subsequent translation to human transporter-mediated pharmacokinetics (Poirier et al., 2009; Watanabe et al., 2009). Although the similarities and differences in metabolizing enzymes between rat and human are acknowledged (Martignoni et al., 2006; Hagenbuch and Gui, 2008; Kotani et al., 2011), a limited number of studies have elucidated the details of species differences in substrate specificities and activity of uptake transporters (Hagenbuch and Meier, 2004; Nakakariya et al., 2008).
The aim of the present study was to apply the mechanistic two-compartment model to estimate uptake kinetics of seven selected substrates in human hepatocytes. The use of single concentration experiments in conjunction with the mechanistic model was explored for characterization of uptake kinetics of these substrates in three human hepatocyte donors; subsequently, the impact of interindividual variability in the uptake activity on the prediction of hepatic clearance in vivo was investigated. Finally, comparison of uptake kinetic parameters obtained in rat and human hepatocytes was performed for the drugs selected.
Materials and Methods
Chemicals.
Bosentan, pitavastatin, pravastatin, rosuvastatin, telmisartan, and valsartan were obtained from Sequoia Research Products (Pangbourne, UK). Repaglinide, mibefradil, verapamil, and indomethacin were purchased from Sigma-Aldrich (Poole, UK). Telmisartan acyl-β-d-glucuronide, 2-despiperidyl-2-amino repaglinide (M1), 2-despiperidyl-2(5-carboxypentylamine) repaglinide (M2), 3′-hydroxy repaglinide (M4), and repaglinide acyl-β-d-glucuronide were obtained from Toronto Research Chemicals (North York, ON, Canada). All other reagents were purchased from Sigma-Aldrich and were of the highest grade available.
Preparation of Human Hepatocytes.
The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents. Cryopreserved human hepatocytes from three donors (lots HU4122, HU4199, and HU8089) were purchased from Invitrogen (Paisley, UK). The hepatocytes were removed from liquid nitrogen storage, immediately thawed at 37°C for 90 s, and transferred in prewarmed Cryopreserved Hepatocyte Recovery Medium (AP Sciences, Columbia, MD). Cells were centrifuged at 100g for 10 min and resuspended in Cryopreserved Hepatocyte Plating Medium at 4°C. Cells were counted under a microscope using a hemocytometer, and the viability was assessed using the trypan blue exclusion method. Only cell suspensions with viability >80% were used. Cell suspension was diluted to a density of 750,000 (donor HU4122) or 800,000 cells/ml (donors HU4199 and HU8089), and 0.5 ml of cell suspension was added to each well of 24-well collagen I-coated plates (BD Biosciences, Oxford, UK). Cells were incubated for 6 h at 37°C in an atmosphere containing 5% CO2 to allow to adhere to the collagen. Incubations were performed with cells from a single donor to avoid issues associated with differential plating efficiency among the three batches, which could result in unknown proportion of cells from individual donor.
Measurement of Uptake in Human Hepatocytes.
Uptake studies were performed as described previously (Ménochet et al., 2012). In brief, for all drugs of interest, uptake was measured over a range of seven concentrations between 0.1 and 300 μM in donor HU4122 (0.1, 1, 10, 20, 30, 100, and 300 μM). The maximum concentration studied for telmisartan was 100 μM due to the limited solubility of this compound in aqueous buffer. The maximum concentration used for repaglinide was 100 μM because formation of the metabolites was linear only up to that concentration. These concentrations were chosen because uptake Km values ranging between 11.5 and 48.5 μM have been reported in human hepatocytes for pravastatin and telmisartan, respectively (Nakai et al., 2001; Ishiguro et al., 2006). No corresponding data were available for bosentan, pitavastatin, and rosuvastatin in this cellular system. In addition, full uptake kinetics of rosuvastatin and repaglinide was measured in donors HU4199 and HU8089. In these donors, uptake of bosentan, pitavastatin, telmisartan, and valsartan was measured at a single concentration of 1 μM. A single concentration of 10 μM was used for pravastatin because of limitations in the analytical method for this drug. Substrates were dissolved in dimethyl sulfoxide and diluted in Dulbecco's phosphate-buffered saline (DPBS) (maximum 1% dimethyl sulfoxide). Plating medium was removed. Cell monolayers were rinsed twice with prewarmed DPBS and preincubated with fresh DPBS for at least 20 min. Incubations were started by the addition of 400 μl of substrate prewarmed at 37°C. After incubation for 30, 60, 90, and 120 s at 37°C, substrate was collected for analysis. The cell monolayers were rinsed three times with 800 μl of DPBS, and 200 μl of water was added in each well to lyse the cells. Substrate samples and cell lysates were stored at −20°C until analysis. To inhibit phase I metabolism of repaglinide and bosentan, a cytochrome P450 pan-inhibitor 1-aminobenzotriazole (ABT) was added at a concentration of 1 mM in the preincubation and incubation buffers used for these two drugs (Mico et al., 1988). ABT has been reported not to affect the activity of uptake transporters in transfected human embryonic kidney 293 cells (Plise et al., 2010). Because of a lack of a nonspecific UGT inhibitor, formation of repaglinide and telmisartan glucuronide was monitored during the uptake experiment. Pitavastatin glucuronidation was not monitored, considering the minor importance of this pathway (Fujino et al., 2003), and inclusion of metabolic clearance was expected to have a minimal impact on the uptake parameter estimates for this drug. In each experiment, uptake of 1 μM rosuvastatin and 1 μM estrone-3-sulfate was measured as a control of the uptake activity. Each time and concentration point was measured in duplicate.
Sample Preparation and LC-MS/MS Analysis.
Cell lysates and substrate samples were thawed and quenched with an equal volume of methanol containing 1 μM internal standard. Samples were placed at −20°C for at least 1 h before being centrifuged for 10 min at 6720g. Then 20 μl of supernatant was analyzed by LC-MS/MS on either a Waters 2795 HPLC coupled with a Micromass Quattro Ultima mass spectrometer (Waters, Milford, MA) or a Waters 2695 HPLC coupled with a Micromass Quattro Micro mass spectrometer (Waters). Analytes were separated through a Luna C18 column (3 μm, 50 × 4.6 mm; Phenomenex, Torrance, CA). The flow rate through the HPLC system was 1 ml/min and was split to 0.25 ml/min before entering the mass spectrometer. With the exception of estrone-3-sulfate, all analytes were ionized by positive electrospray. LC-MS/MS analysis of all the drugs studied was performed as described previously (Ménochet et al., 2012). Telmisartan glucuronide, M1, M2, M4, and repaglinide glucuronide cell concentrations were also monitored for modeling purposes.
Determination of Unbound Fraction in the Incubation Media.
Nonspecific binding of the drugs to the cellular membrane and the experimental support was assessed by measuring the media concentrations over time at each incubation concentrations, as described previously (Ménochet et al., 2012). Media were assumed to be free of protein because the hepatocytes were attached to the support, and the monolayers were rinsed thoroughly before the start of the incubation. Media concentrations at t0 were estimated by extrapolation of the linear regression of the media concentration versus time plot at each incubation concentration. Unbound fraction in the media (fumed) was subsequently expressed as the slope of the linear regression of the measured media concentrations at t0 versus the initial incubation concentrations.
Determination of Uptake Kinetic Parameters Using a Mechanistic Two-Compartment Modeling Approach.
A mechanistic two-compartment model was previously developed by the authors to estimate the uptake kinetics of the same seven OATP substrates in plated rat hepatocytes (Ménochet et al., 2012). In that case, experiments performed at 10 substrate concentrations over 45 to 90 min (eight time points) were suitable to estimate with precision active uptake kinetics (Km, u and Vmax), bidirectional passive diffusion clearance (Pdiff, u), and intracellular binding (fucell). Considering the cost of human hepatocytes, experiments in the present study were limited to seven concentrations over 2 min (four time points), restricting the number of data points available for modeling in comparison with those for rat hepatocytes. In the initial analysis, fucell estimates obtained from the human hepatocyte data were associated with large S.E. (CV >100%; data not shown), analogous to the trends seen in rat data when reduced numbers of time and concentration points were used (Ménochet et al., 2012). To maintain similar degrees of freedom and level of precision in the parameter estimates, fucell obtained in the rat hepatocytes after incubations over 45 to 90 min were set as constant in the mechanistic two-compartment model used for the analysis of the human hepatocyte data; fucell values used are listed in Table 1. Use of extended incubation times in the rat study resulted in CV <21% for the fucell for all drugs studied with the exception of rosuvastatin.
TABLE 1.
Uptake kinetic parameters of seven OATP substrates estimated in human hepatocyte using a mechanistic two-compartment model
Uptake kinetics was measured in cryopreserved human hepatocytes (donor HU4122) plated for 6 h over 2 min at seven concentrations (0.1–300 μM).
| Drug | Km, u | Vmax | Pdiff,u | fucella | fumed | CLactive, u |
|---|---|---|---|---|---|---|
| μM | pmol · min−1 · 106 cells−1 | μl · min−1 · 106 cells−1 | μl · min−1 · 106 cells−1 | |||
| Bosentan | 22.5 | 402 | 9.74 | 0.096 | 0.85 | 17.9 |
| Pitavastatin | 1.59 | 65 | 13.2 | 0.053 | 0.63 | 40.7 |
| Pravastatin | 2.25 | 6.24 | 0.208 | 0.963 | 0.91 | 2.77 |
| Repaglinide | 15.6 | 1236 | 10.0 | 0.074 | 0.95 | 79.0 |
| Rosuvastatin | 11.2 | 104 | 0.391 | 0.477 | 0.93 | 9.21 |
| Telmisartanb | 2.03 | 193 | 14.1 | 0.024 | 0.71 | 95.2 |
| Valsartan | 10.4 | 30 | 0.064 | 1 | 1 | 2.88 |
Data from uptake experiments performed in rat hepatocytes (Ménochet et al., 2012).
Kinetics parameters obtained when telmisartan metabolism was incorporated in the modeling (CLmet = 4.3 μl · min−1 · 106 cells−1).
The model, implemented in Matlab (version 7.10, 2010; Mathworks, Natick, MA), allows simultaneous fitting of all concentration and time points. In addition to the kinetics of the active uptake, this model describes the bidirectional passive diffusion of the drug through the cellular membrane and the intracellular binding. After isolation, hepatocytes lose their polarization and efflux transporters are internalized (Hewitt et al., 2007; Bow et al., 2008). Therefore, active efflux was assumed to be negligible, consistent with other studies focusing on estimation of uptake in isolated or short-term cultured hepatocytes. Equations 1 and 2 describe the changes in cell and media concentrations over time, respectively.
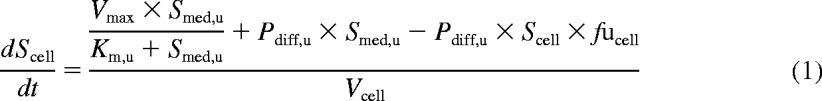 |
 |
where Scell and Smed, u are the intracellular and unbound media concentrations, respectively. Km, u, Vmax, Pdiff, u, and fucell are the unbound affinity constant, the maximum uptake rate, the unbound passive diffusion clearance, and the unbound intracellular fraction, respectively. An intracellular volume, Vcell, of 3.9 μl/106 cells is used in this study (Reinoso et al., 2001). Media volume, Vmed, is 400 μl. CLactive, u, the unbound active uptake clearance, was expressed as the ratio of Vmax over Km, u. The total unbound uptake clearance (CLuptake, u) included both the active component (CLactive, u) and clearance via passive diffusion. The contribution of the active transport to the total uptake was expressed as the ratio of CLactive, u over CLuptake, u and was calculated over the range of concentrations studied.
Initial media concentrations were obtained by correcting the nominal substrate concentrations by fumed. Initial cell concentrations were obtained from the linear regression of the first four time points for each of the concentrations at time 0. The rationale was that not all the drug could be washed from the cell membranes or experimental plates with DPBS during the washing steps. However, because of the differences in volumes between the cell and media compartments, the largest amount of drug found in the cells at time 0 represented <1% of the total amount of compound present in the incubation. Cell concentrations were used as input data in the model. At least 48 data points were available to model the uptake kinetics of each drug. In the first instance, kinetic parameters were optimized with open boundaries for Km, u, Vmax, and Pdiff, u. The greatest uncertainty was associated with the estimation of Pdiff, u. To improve the precision of this estimate, a second round of optimization was performed using the results of the preliminary optimization as a priori information. For that purpose, the following boundaries were set: Km, u ± 20%, Vmax ± 50%, and Pdiff, u ± 100%.
Simultaneous Modeling of Telmisartan Uptake and Metabolism.
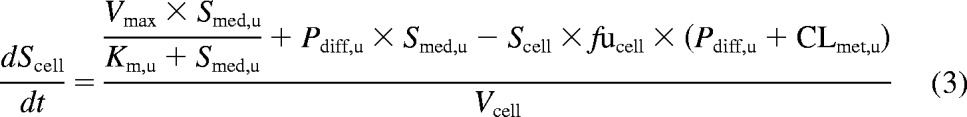
In humans, telmisartan is transformed to a single glucuronide metabolite by UGT1A3 (Stangier et al., 2000; Yamada et al., 2011). Because no specific inhibitor of UGTs is available, modeling of telmisartan uptake and metabolism was performed simultaneously. The mechanistic two-compartment model was extended to account for the formation of telmisartan glucuronide over time, as illustrated in Fig. 1. Equation 3 describes the changes in telmisartan cell concentrations during the uptake experiment, whereas changes in telmisartan glucuronide cell concentrations are defined by eq. 4. Changes in media concentrations over time were defined as shown above in eq. 2.
 |
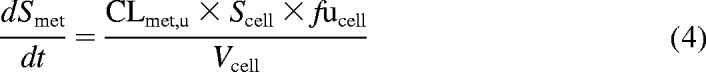 |
where Smet and CLmet, u are telmisartan glucuronide cell concentrations and the unbound metabolic clearance, respectively. CLmet, u obtained previously from the metabolite formation assay in donor HU4122 was used as a constant in the model in addition to fucell to estimate Km, u, Vmax, and Pdiff, u accurately, because telmisartan glucuronide was not monitored in this initial uptake assay. In donors HU4199 and HU8089, formation of telmisartan glucuronide was monitored in the uptake assay performed at 1 μM.
Fig. 1.

Extended two-compartment model (eqs. 5–8) describing the uptake and metabolism of repaglinide in human hepatocytes over time. SGluc, cell, SM2, cell, and SM4, cell represent concentrations of major repaglinide metabolites in the cell. CLmet,gluc, u is the glucuronidation clearance; Km, M2, u, Vmax, M2, Km, M4, u, and Vmax, M4 are the Michaelis-Menten parameters describing the concentration-dependent saturation of the formation of M2 and M4. In the case of telmisartan, the model was limited to the parent compound and the glucuronide metabolite.
Determination of Uptake Kinetic Parameters and Metabolic Clearance of Repaglinide Using a Mechanistic Modeling Approach.
In humans, repaglinide is metabolized by CYP3A4 and CYP2C8 to five metabolites, including M1, M2, and M4. A glucuronide metabolite formed by UGT1A1 has also been reported recently (Bidstrup et al., 2003; Yamada et al., 2011; Säll et al., 2012). In the first instance, uptake of repaglinide was investigated in isolation (in the presence of ABT) in donor HU4122, and no metabolism was taken into account, because repaglinide glucuronide was not commercially available at the time. The mechanistic two-compartment model described by eqs. 1 and 2 was used to analyze the data. Subsequently, uptake and metabolism of repaglinide were studied simultaneously in donors HU4199 and HU8089. Incubations were performed in the absence of ABT and extended to 15 min (six time points). M1, M2, M4, and repaglinide glucuronide cell concentrations were monitored in addition to repaglinide and the linearity of the metabolite formation was confirmed over the incubation period. Preliminary analysis indicated that intrinsic clearance (CLint) for M1 was 16- and 28-fold lower than that for M2 and M4 in donor HU8089. Therefore, subsequent analysis focused on monitoring the formation of M2, M4, and repaglinide glucuronide in human hepatocytes during the uptake experiment (Supplemental Fig. S1), and these metabolites were taken into account for modeling purposes. In a manner similar to that for telmisartan, cell subcompartments were added to the mechanistic two-compartment model to describe the formation of each metabolite over time (Fig. 1). Equation 5 describes the changes in repaglinide cell concentrations over time, whereas eqs. 6 to 8 describe the formation of M2, M4, and repaglinide glucuronide, respectively. Changes in media concentrations are described by eq. 2.
 |
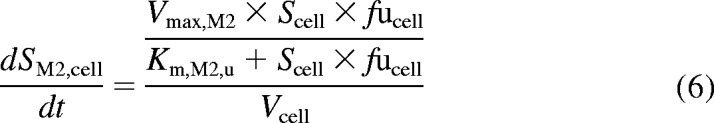 |
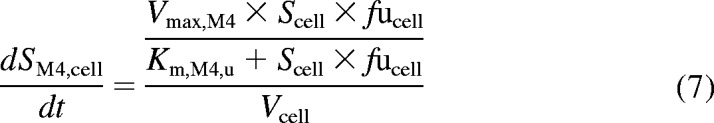 |
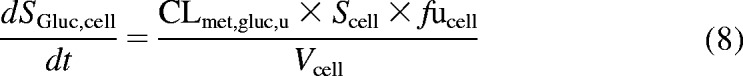 |
where SM2, cell, SM4, cell, and SGluc, cell are M2, M4, and repaglinide glucuronide cell concentrations, respectively. Km,M2, u, Vmax,M2, Km,M4, u, Vmax,M4, and CLmet,gluc, u are the kinetic parameters describing the formation of M2, M4, and repaglinide glucuronide, respectively. Cell and media initial concentrations were estimated as described previously (Ménochet et al., 2012). Initial metabolite cell concentrations were obtained from the linear regression of the first four time points. Initial metabolite cell concentrations were never greater than 0.1% of the total amount of drug present in the incubation. To optimize the amount of information obtained from each experiment and to describe the potential saturation of the formation of each metabolite, metabolic clearances were replaced by Michaelis-Menten kinetic parameters (Km, u and Vmax) in eqs. 6 to 8. The impact of the use of the various modeling approaches and associated error on the uptake and metabolism kinetic parameter estimates was investigated.
Interindividual Variability in Uptake among Three Human Hepatocyte Donors.
To investigate variability in uptake activity among human hepatocyte donors, uptake of rosuvastatin and repaglinide was measured in three human hepatocyte donors over a range of concentrations, following mechanistic modeling data analysis, as described in sections above. For the remaining drugs in the dataset (pravastatin, valsartan, bosentan, and pitavastatin) uptake was measured over 2 min in donors HU4199 and HU8089 using a single substrate concentration. On the basis of full kinetic characterization data obtained for rosuvastatin and repaglinide in three donors, Km, u and Pdiff, u values were assumed to be constant among donors (Tables 2 and 3). The mechanistic model defined in eq. 1 was therefore used to fit the Vmax using the data obtained over 2 min at a single substrate concentration, keeping Km, u, Pdiff, u, and fucell constant (as obtained in donor HU4122). The fitting routine was performed in Matlab using the lsqnonlin function; the differential equations were solved using the ODE45 solver. An analogous method was applied for telmisartan, but in this case metabolic clearance was also taken into account in the fitting routine as shown in eq. 3. Subsequently, CLactive, u for the remaining five drugs could be calculated as the Vmax/Km, u ratio. To verify the validity of the assumptions stated above, CLactive, u was calculated for each drug in donor HU4122 using the single concentration (1 μM) method and compared with the CLactive, u values obtained from the full kinetic characterization.
TABLE 2.
Uptake and metabolism kinetic parameters of repaglinide estimated in three human hepatocyte donors using an extended mechanistic two-compartment model
Uptake kinetics was measured in cryopreserved human hepatocytes plated for 6 h at seven concentrations (0.1–300 μM) in the presence (HU4122) or absence (HU4199 and HU8089) of 1 mM ABT.
| Donor | HU4122 | HU4199 | HU8089 | Mean |
|---|---|---|---|---|
| Km, u, μM | 15.6 | 8.97 | 13.8 | 12.8 ± 3.4 |
| Vmax, pmol · min−1 · 106 cells−1 | 1236 | 310 | 464 | 670 ± 496 |
| Pdiff, u, μl · min−1 · 106 cells−1 | 10.0 | 16.0 | 12.0 | 12.7 ± 3.1 |
| fucella | 0.074 | 0.074 | 0.074 | 0.074 |
| fumed | 0.95 | 0.94 | 0.96 | 0.95 ± 0.01 |
| CLactive, u, μl · min−1 · 106 cells−1 | 79.0 | 34.6 | 33.6 | 49.1 ± 25.9 |
| CLmet, gluc, u, μl · min−1 · 106 cells−1 | 0.412 | 0.242 | 0.327 | |
| Km, M2, u, μM | 41.5 | 7.40 | 24.4 | |
| CLmet, M2, u, μl · min−1 · 106 cells−1 | 0.950 | 2.42 | 1.68 | |
| Km, M4, u, μM | 5.32 | 8.83 | 7.07 | |
| CLmet, M4, u, μl · min−1 · 106 cells−1 | 0.619 | 1.29 | 0.957 | |
| CLmet,t otal, u, μl · min−1 · 106 cells−1 | 1.98 | 3.96 | 2.97 |
Data from uptake experiments performed in rat hepatocytes over an extended incubation time (Ménochet et al., 2012).
TABLE 3.
Uptake kinetic parameters of rosuvastatin estimated in three human hepatocyte donors using a mechanistic two-compartment model
Uptake kinetics was measured in cryopreserved human hepatocytes plated for 6 h over 2 min at seven concentrations (0.1–300 μM).
| Donor | Km, u | Vmax | Pdiff, u | fumed | CLactive, u |
|---|---|---|---|---|---|
| μM | pmol · min−1 · 106 cells−1 | μl · min−1 · 106 cells−1 | μl · min−1 · 106 cells−1 | ||
| HU4122 | 11.2 | 104 | 0.391 | 0.80 | 9.21 |
| HU4199 | 9.80 | 16.6 | 0.148 | 1 | 1.69 |
| HU8089 | 12.0 | 16.3 | 0.243 | 0.96 | 1.35 |
| Mean ± S.D. | 11.0 ± 1.1 | 45.5 ± 50.3 | 0.261 ± 0.123 | 0.93 ± 0.13 | 4.09 ± 4.44 |
Statistical Analysis.
A Jacobian approach was used in Matlab to estimate S.E. associated with each parameter estimate generated from the mechanistic two-compartment model (Landaw and DiStefano, 1984). CVs were calculated and are expressed as percentages to assess the quality of each parameter estimate. Goodness of fit was assessed by visual inspection of the data and minimum objective function value. Arithmetic means, S.E.s, and CVs associated with each kinetic parameter were calculated for each drug when results in the three human hepatocyte donors were available. p values were calculated using a Student's t test, and results were deemed significant for p < 0.05.
Collation of Uptake Measurement Data for Estrone-3-Sulfate in Human Hepatocytes from Commercial Supplier.
Uptake rate values of estrone-3-sulfate in human cryopreserved hepatocytes were collated from the characterization spreadsheets available from BD Gentest (Oxford, UK) between September 2008 and September 2011 (http://www.bdbiosciences.com/nvCategory.jsp?action=SELECT&form=formTree_catBean&item=774729). Uptake was measured at 2 μM over 3 min using 200,000 human hepatocytes in suspension per incubation. By using the uptake rates provided, uptake clearances were calculated from the ratio of the uptake rate over the substrate concentration to allow comparison with data obtained in the three human hepatocyte donors studied here. Demographic information (age and gender) for each donor available from BD Gentest was also collated, and data were assessed for any trends. Collated data are summarized in Supplemental Table S1.
Prediction of Hepatic Clearance in Human.
In vivo clearance was predicted using the unbound intrinsic clearance (CLint, u) term derived previously (Shitara and Sugiyama, 2006), as described in eq. 9:
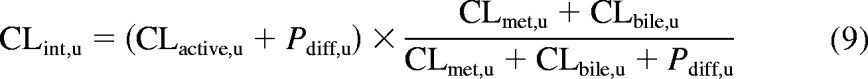 |
CLactive, u, CLmet, u, and Pdiff, u represent the total unbound uptake clearance, the unbound metabolic clearance, and the passive diffusion clearance; all parameters were obtained experimentally and scaled up to a whole liver using physiological scaling factors and are expressed in milliliters per minute per kilogram. Hepatocellularity and liver weight values of 120 × 106 cells/g liver and 21.4 g of liver/kg b.wt., respectively, were used (Brown et al., 2007). CLbile, u is the unbound biliary clearance and was obtained by multiplying the fraction of the dose recovered unchanged in the feces after intravenous administration by the unbound total plasma clearance. In the case of bosentan, CLmet, u was obtained from the literature (Lave et al., 1996). Details for the individual drugs are provided in Supplemental Table S2.
Values obtained from in vitro results were compared with the in vivo intrinsic clearances estimated from the literature data and are expressed as described in eq. 10:
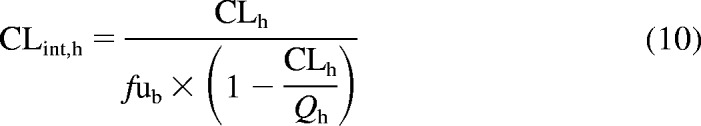 |
where CLh is the hepatic blood clearance after intravenous administration corrected for renal clearance, Qh is hepatic blood flow, and fub is the unbound fraction in the blood. No CLh was available for pitavastatin; therefore, this drug was omitted from the comparison between predicted and observed hepatic clearances.
Preliminary in vitro-in vivo extrapolation has shown underprediction of the intrinsic hepatic clearance. Therefore, empirical scaling factors required for CLactive, u were calculated for each drug in all three donors. In the first instance, a mean scaling factor per individual donor was used, and the three individual empirical scaling factors were then applied to correct corresponding CLactive, u in that particular donor. In addition, the average empirical scaling factor obtained for all drugs in all three donors (16 data points in total) was calculated and applied to correct CLactive, u. The accuracy and bias associated with each predictive approach were compared by calculation of the root mean square error (rmse) and the geometric mean fold error (gmfe) (Gertz et al., 2010), as described in eqs. 11 and 12, respectively.
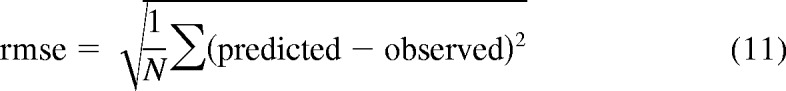 |
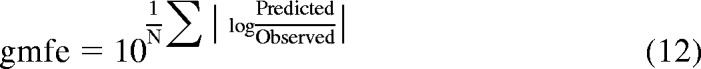 |
Comparison of Uptake Data Obtained in Rat and Human Hepatocytes.
Uptake data obtained in three human hepatocyte donors were compared with the results generated previously in rat hepatocytes for the same seven drugs using a similar experimental and modeling approach (Ménochet et al., 2012); data for the distribution coefficient between octanol and water at pH 7.4 (LogD7.4) were also taken from previous publications. Fold difference in Km, u, CLactive, u, and Pdiff, u were calculated and interspecies differences were considered.
Results
Mechanistic Modeling of Uptake in Donor HU4122.
Full uptake kinetics of seven drugs was investigated in human hepatocyte donor HU4122 using a mechanistic modeling approach defined previously. Km, u, Vmax, and Pdiff, u were estimated from the data generated over 2-min incubations and are summarized in Table 1. Corresponding kinetic profiles and goodness of fit plots obtained with this model are shown in Supplemental Figs. S2 and S3, respectively. Km, u values ranged 14-fold, from 1.59 to 22.5 μM for pitavastatin and bosentan, respectively. A greater difference in Vmax among the drugs investigated resulted in a 34-fold range in CLactive, u. Pravastatin showed the lowest active uptake clearance (2.77 μl · min−1 · 106 cells−1), whereas telmisartan was the drug most readily taken up by the cell with CLactive, u of 95.2 μl · min−1 · 106 cells−1. Pdiff, u was the kinetic parameter that differed the most among the seven drugs, with a range of 222-fold, from 0.064 to 14.1 μl · min−1 · 106 cells−1 for valsartan and telmisartan, respectively.
All media concentrations were corrected for nonspecific binding to the experimental support; fumed was >0.6 for all drugs investigated in the current dataset. Nonspecific binding had a minimal influence on the uptake kinetics of all drugs of interest and most specifically on rosuvastatin, repaglinide, and pravastatin, which showed fumed values >0.9. Use of fucell values derived from rat studies allowed modeling of human hepatocyte uptake data and accurate estimation of all kinetic parameters for the drugs in the current dataset (CV <30%), with the exception of the Km, u and Vmax values for pravastatin (CVs 194 and 36%, respectively). fucell values ranged from 0.024 to 1 for telmisartan and valsartan, respectively. Interexperiment variability of rosuvastatin and estrone-3-sulfate uptake used as controls was 39 and 40%, respectively (n = 8, donor HU4122).
Differing extents of active uptake and passive diffusion were observed among the seven drugs investigated, as shown in Fig. 2. Only in the case of rosuvastatin, repaglinide, and valsartan was the active process responsible for ≥50% of the total uptake over the full range of concentrations investigated. However, at the lowest concentrations studied, the maximal proportion of total uptake due to the active process was >90% for all the drugs investigated. Thus, at low, therapeutically relevant concentrations, the passive permeation is expected to have a limited impact on total uptake into the hepatocytes of the drugs investigated. Analogous to rat hepatocytes, a positive linear correlation was observed between the logarithm of the passive diffusion clearance and LogD7.4 (R2 = 0.886) (Fig. 8C).
Fig. 2.

Proportion of total uptake due to active transport and passive diffusion in human hepatocytes over a range of concentrations. Active transport and passive diffusion contributions were estimated using either a generic or extended two-compartment mechanistic model and on the basis of uptake kinetic estimates obtained in human hepatocyte donors HU4122 (black line), HU4199 (red line), and HU8089 (green line). ——, proportion of active transport; – – –, proportion of passive diffusion. A, rosuvastatin; B, repaglinide; C, bosentan; D, pitavastatin; E, pravastatin; F, telmisartan; G, valsartan.
Fig. 8.

Comparison of the uptake affinity (A), active uptake clearance (B), and passive diffusion clearance (C) of seven OATP substrates in rat and human hepatocytes. Km, u, CLactive, u, and Pdiff, u estimates in rat hepatocytes were obtained from full kinetic experiments for all drugs. Data represent the mean ± S.D. of three experiments. Km, u and Pdiff, u in human hepatocytes were obtained from full kinetic experiments in donor HU4122, with the exception of rosuvastatin and repaglinide, for which full kinetic experiments were conducted in all three human hepatocyte donors. CLactive, u in humans is the mean ± S.D. of estimates obtained from the full kinetic experiment in donor HU4122 and extrapolated from experiments conducted at 1 μM in donors HU4199 and HU8089. In A and B, ——, line of unity. – – –, 2-fold difference. In C, ——, line of best fit between LogD7.4 and the passive diffusion clearances generated in human hepatocytes. ●, data generated in human hepatocytes; ○, data obtained in rat hepatocytes. 1, pitavastatin; 2, pravastatin; 3, telmisartan; 4, repaglinide; 5, rosuvastatin; 6, valsartan; 7, bosentan.
Simultaneous Modeling of Uptake and Metabolism of Repaglinide.
Uptake and metabolism of repaglinide were studied in three human hepatocyte donors. The kinetic estimates are summarized in Table 2. In donor HU4122, uptake of repaglinide was investigated in the presence of 1 mM ABT to inhibit phase I metabolism and allow only uptake to be characterized in the modeling process. The repaglinide fucell value of 0.074 was held constant in the mechanistic model and is based on values obtained previously in rat hepatocytes. In donors HU4199 and HU8089, the uptake of repaglinide was studied in the absence of ABT and the formation of three metabolites was incorporated in the modeling. Repaglinide cell concentrations were fitted with good precision in all cases, as illustrated in the diagnostic goodness-of-fit plots shown in the Supplemental Fig. S3B. Uptake Km, u of repaglinide was moderate (12.8 ± 3.4 μM) and consistent among the three hepatocyte donors. Pdiff, u was among the highest values measured in the current dataset (12.7 ± 3.1 μl · min−1 · 106 cells−1), and no significant difference was observed in this parameter across the donors investigated. Variation in uptake activity resulted in up to 2.4-fold difference between CLactive, u values measured in HU4122 and the additional donors (HU4199 and HU8089) (Fig. 3A). Differences in uptake were reflected in the proportion of total uptake due to active transport over the range of concentrations. In HU4122, active uptake was the predominant process responsible for total uptake over the range of concentrations studied. In HU4199 and HU8089, contribution of the active process of >70% was observed only for concentrations ≤10 μM (Fig. 2).
Fig. 3.

Total uptake kinetic profiles of repaglinide (A) and rosuvastatin (B) measured in three batches of cryopreserved human hepatocyte plated for 6 h. Data represent the mean of uptake rates measured in duplicate at 2 min in donor HU4122 (●), HU4199 (○) and HU8089 ( ). Six concentrations were used for repaglinide (0.1–100 μM) and seven for rosuvastatin (0.1–300 μM). Lines represent the simulated total uptake based on uptake kinetic estimates obtained from the two-compartment mechanistic model, including metabolism in the case of repaglinide.
). Six concentrations were used for repaglinide (0.1–100 μM) and seven for rosuvastatin (0.1–300 μM). Lines represent the simulated total uptake based on uptake kinetic estimates obtained from the two-compartment mechanistic model, including metabolism in the case of repaglinide.
The kinetics of repaglinide metabolite formation was also investigated in donors HU4199 and HU8089. Because the uptake experiment was performed without ABT, M2, M4, and repaglinide glucuronide were monitored (Supplemental Fig. S1). Kinetics of repaglinide glucuronide formation was best described by a single clearance parameter, whereas formation of M2 and M4 was defined by Michaelis-Menten parameters used to describe potential saturation of metabolism over the range of concentrations studied. Clearance due to the formation of repaglinide glucuronide was low, contributing 21 and 6% to total metabolism in donors HU4199 and HU8089, respectively. Km, u value for the M2 pathway was 6-fold greater in HU4199 than in HU8089. However, this resulted in only 2.5-fold greater CLmet,M2, u, due to differences in metabolic capacity between the two donors. Overall, metabolic clearances in HU4199 and HU8089 were 1.98 and 3.96 μl · min−1 · 106 cells−1, respectively. This was lower than the value obtained in donor HU4122 when measured in a separate depletion assay (6.61 μl · min−1 · 106 cells−1; data not shown). Uptake clearances were 17- and 8.5-fold greater than metabolism for donor HU8089 and HU4199, respectively. Km,M4, u was consistently lower than the uptake Km, u by on average 2-fold, but the difference in Km,M2, u was less clear because of donor variation. CLmet, M2 was greater in comparison with the metabolic clearances of other metabolites formed, consistent with recent studies in pooled cryopreserved hepatocytes, in which repaglinide metabolism was monitored in isolation (Säll et al., 2012).
Interindividual Variability in Uptake and Metabolism Activity between Human Hepatocyte Donors.
Analysis of uptake kinetic data showed Km to be the most consistent parameter among the three donors with CVs of 10 and 27% for rosuvastatin and repaglinide, respectively (Tables 2 and 3). Pdiff, u was also similar among the three donors (CVs 47 and 24% for rosuvastatin and repaglinide, respectively), consistent with the reliance of passive permeation on lipophilicity of the drugs studied. However, for both drugs, uptake activity varied among the donors, because HU4122 uptake activity was up to 2.4- and 6.8-fold greater than activity observed in the two other donors for repaglinide and rosuvastatin, respectively (Fig. 3).
To explore the use of uptake estimates obtained at a single low concentration in donors HU4199 and HU8089, CLactive, u for pitavastatin, valsartan, bosentan, pravastatin, and telmisartan was estimated using the uptake rate data generated at 1 μM (10 μM for pravastatin) over 2 min only. Uptake Km and Pdiff were assumed to be consistent among donors on the basis of full kinetic profiles for repaglinide and rosuvastatin. Keeping these parameters constant in the mechanistic model allowed fitting of the Vmax and subsequent estimation of the CLactive, u. This approach allowed the delineation of active and passive uptake from a single concentration experiment in a mechanistic manner, using only a fraction of the number of cells required to investigate the full uptake kinetics of a drug. For validation purposes, the same analysis was performed in HU4122, and estimates based on this approach were compared with the CLactive, u obtained from the full kinetic profile in the same donor; the analysis was also performed for rosuvastatin in all three human hepatocyte donors. As illustrated in Fig. 4, the CLactive, u values obtained using single concentration data were within 2-fold of the value generated from the full kinetic profile for all drugs. In the case of pravastatin, because the concentration used (10 μM) was greater than uptake Km, u, CLactive, u could not be estimated from single concentration uptake data. Therefore, results presented for this drug are solely based on the full kinetic experiment performed in donor HU4122. Precision of the estimated uptake clearance obtained at the single concentration varied from 22% lower for rosuvastatin in donor HU8089 to 47% higher for bosentan in donor HU4122, but overall no significant bias was observed (gmfe of 1.2). This approach was therefore considered suitable to estimate CLactive, u in donors HU4199 and HU8089 from single low concentration data.
Fig. 4.

Comparison between uptake clearances obtained from full kinetic experiments in human hepatocytes and the value estimated on the basis of data generated at 1 μM only using mechanistic model.
Donor HU4122 showed higher uptake activity than HU4199 and HU8089 for all drugs investigated, with the exception of bosentan and pitavastatin (Fig. 5). On average, uptake clearance in this donor was 2.3- and 2.8-fold greater than that in HU4199 and HU8089, respectively. The greatest difference between donors was observed for rosuvastatin. For this drug, uptake clearance in HU4122 was 5.4- and 6.8-fold greater than that in HU4199 and HU8089. In contrast, uptake of telmisartan was consistent among the three donors, with a CV of only 7.8%. Overall, HU4199 and HU8089 showed similar levels of uptake activity. Uptake clearances in these donors were within 30% of each other for all drugs, with the exception of pitavastatin and bosentan where uptake clearances in HU4199 were 1.9-fold greater than those in HU8089. This difference among the three donors was reflected in the uptake of rosuvastatin when this drug was used as a positive control at 1 μM (Fig. 6A). However, the use of estrone-3-sulfate at 1 μM did not distinguish the three donors in their uptake activity, because the differences seen were not statistically significant (p = 0.35) (Fig. 6B). In addition to current data, uptake clearance values for estrone-3-sulfate were collated from BD Gentest characterization spreadsheets for 28 individual human hepatocyte donors (details shown in Supplemental Table S1). Clearances ranged from 4 to 35.5 μl · min−1 · 106 cells−1 with a mean clearance of 21.4 ± 8.5 μl · min−1 · 106 cells−1 (CV 40%). No correlation was observed between uptake activity and either gender or age for this dataset (Supplemental Fig. S4). Mean CLuptake of estrone-3-sulfate in the current study (19.5 ± 7.7 μl · min−1 · 106 cells−1) was comparable to the mean value obtained from this larger dataset (p = 0.48).
Fig. 5.

Active uptake clearance of six OATP substrates in human hepatocyte donors generated using a full kinetic approach (●, HU4122) or extrapolated from measurements at 1 μM (○, HU4199; , HU8089). Line represents the mean of the three clearance estimates.
Fig. 6.

Uptake clearances of rosuvastatin (A) and estrone-3-sulfate (B) measured as a control in human hepatocyte donors HU4122 (n = 8), HU4199 (n = 3), and HU8089 (n = 4) at 1 μM. ——, represents the median; □, represents the mean. The box represents the 25th and 75th percentiles; whiskers are the 5th and 95th percentiles.
Metabolism of telmisartan was studied in all three donors. Unbound metabolic clearance of telmisartan in HU4122 was 22-fold lower than the active uptake clearance (Table 1). However, metabolic clearance obtained in this donor was 6-fold greater than the values measured in the other two donors (4.3 versus 0.7 μl · min−1 · 106 cells−1, respectively). Although this difference could be due to variability in metabolic capacity between the donors, it is noteworthy that telmisartan glucuronide was monitored in the entire incubation (cell and media) and in the metabolite formation assay performed in donor HU4122, whereas it was only measured in the cell in the uptake assay in donors HU4199 and HU8089, because this metabolite could not be detected in the media.
Prediction of Hepatic Clearance in Human.
Comparison of the observed and predicted hepatic intrinsic clearances for six of the drugs investigated is shown in Fig. 7. The importance of active uptake on the prediction of CLint, h was drug-dependent. In the case of pravastatin, rosuvastatin, telmisartan, and valsartan, hepatic clearance was mainly driven by CLactive, u, because the combined CLmet, u and CLbile, u were much greater than Pdiff, u. For bosentan and repaglinide, CLint, u was a composite of a number of processes, because passive diffusion was greater than metabolic and biliary clearances combined. In vitro-in vivo extrapolation of these data resulted in systematic underprediction of hepatic intrinsic clearance for all drugs in the dataset, with the exception of valsartan, which was predicted within 2-fold of the observed data. The extent of the underprediction was compound- and donor-dependent, ranging from 5-fold for pravastatin in donor HU4122 to 66-fold for rosuvastatin in donor HU8089. Hence, the empirical scaling factors for CLactive per individual donor were necessary to recover the observed clearance in vivo and also resulted in a large variation (Table 4). For example, clearances in donor HU4122 were corrected by a factor of 6.9 on average, whereas values of 25.0 and 25.6 were required in donors HU4199 and HU8089, respectively. Nine of 16 CLint, u values were predicted within 2-fold of the observed data (Fig. 7A). The approach involving empirical scaling factors resulted in low overall bias (2.3), but poor precision (rmse of 801).
Fig. 7.

Comparison of predicted and observed hepatic clearances for the drugs investigated. Scaling factors were based either on the donor-specific (A) or empirical scaling factors obtained using the whole dataset (B). Hepatic clearance was predicted from uptake, metabolic and passive diffusion clearances obtained in three human hepatocyte donors, and biliary clearance values reported in the literature. ●, bosentan; ○, pravastatin; ■, repaglinide; □, rosuvastatin; ▴, telmisartan; ▵, valsartan. ——, line of unity. – – –, 2-fold difference; - - - - -, 5-fold difference.
TABLE 4.
Empirical scaling factors applied to the active uptake clearances to recover the intrinsic uptake clearance in human livers
| Drug | Scaling Factor |
Mean | ||
|---|---|---|---|---|
| HU4122 | HU4199 | HU8089 | ||
| Bosentan | 16.1 | 13.0 | 24.8 | 17.9 ± 6.1 |
| Pravastatin | 6.67 | ND | ND | 6.67 |
| Repaglinide | 4.70 | 39.2 | 17.7 | 20.5 ± 17.4 |
| Rosuvastatin | 9.75 | 51.9 | 65.3 | 42.3 ± 29.0 |
| Telmisartan | 3.23 | 18.7 | 17.9 | 13.3 ± 8.7 |
| Valsartan | 0.740 | 2.00 | 2.20 | 1.64 ± 0.79 |
| Mean | 6.87 ± 5.46 | 25.0 ± 20.2 | 25.6 ± 23.7 | 17.1 ± 18.8 |
N.D., not determined, because CLuptake, u could not be measured accurately at a single concentration.
In addition, the whole dataset was considered to accommodate both drug and donor variability, resulting in an average empirical scaling factor of 17.1. Use of this approach resulted in 8 of 16 values predicted within 2-fold, improved precision (rmse of 471), and bias comparable to that for the previous scenario (gmfe of 2.52) (Fig. 7B). In all analyses, predicted valsartan CLint, u was more than 5-fold greater than the observed CLint, h value. In general, use of the average dataset scaling factor resulted in an overprediction of clearance when data from donor HU4122 were used (with the exception of bosentan) and underprediction when clearances from donors HU4199 and HU8089 were used.
Comparison between Uptake in Rat and Human Hepatocytes.
Uptake kinetic parameters of the seven drugs investigated in the present study were estimated previously in rat hepatocytes using a mechanistic modeling approach (Ménochet et al., 2012). Km, u values were on average 4.3-fold greater in rat than in human hepatocytes, mainly because of the higher affinity of pitavastatin and pravastatin toward human transporters (labeled as 1 and 2 in Fig. 8A). Valsartan and bosentan were the only drugs in the dataset showing the opposite trend with higher affinity in rat hepatocytes. Pitavastatin had the lowest Km, u in human hepatocytes (1.59 μM), whereas telmisartan showed the highest affinity in rat hepatocytes (3.4 μM). Pravastatin and bosentan showed the lowest affinity toward rat and human transporters with Km, u of 37.0 and 22.5 μM in rat and human hepatocytes, respectively. Less than a 2-fold difference was observed between Km, u in rat and human hepatocytes for telmisartan, rosuvastatin, valsartan, and bosentan. Overall, the drugs studied covered a similar range of affinity in both species: 11-fold versus 14-fold in rat and human, respectively. Despite the greater affinity observed in human hepatocytes, uptake clearances were on average 7.1-fold greater in rat hepatocytes than in human hepatocytes, due to a large difference in activity between the two species (Fig. 8B). The greatest difference was observed for valsartan and rosuvastatin for which CLactive, u values were 16- and 21-fold greater in rat than in human hepatocytes, respectively. All other drugs had CLactive, u values within 5-fold of each other in the two species. The lowest uptake clearance was observed for valsartan in human hepatocytes (1.64 μl · min−1 · 106 cells−1 in HU8089) and for pravastatin in rat hepatocytes (12.5 μl · min−1 · 106 cells−1). Telmisartan showed the greatest extent of active uptake in human hepatocytes with CLactive, u of 95.2 μl · min−1 · 106, whereas active uptake of repaglinide was the greatest in rat hepatocytes (119 μl · min−1 · 106 cells−1). Therefore, individual CLactive, u covered a wider range in human than in rat hepatocytes: 58-fold versus 8-fold, respectively. As anticipated, interindividual variability in uptake clearance was greater in human hepatocytes than in rat hepatocytes for all drugs excluding telmisartan.
Unlike active uptake, passive diffusion clearance was consistent between the two species, with Pdiff, u in rat being only 1.7-fold greater on average than in human hepatocytes (Fig. 8C). However, Pdiff, u values obtained for the poorly permeable compounds were much lower in human than in rat cells, whereas the Pdiff, u values at the higher end were comparable between the two species (21.4 and 14.1 μl · min−1 · 106 cells−1 for telmisartan in rat and human hepatocytes, respectively); Pdiff, u values in human span 222-fold, whereas they only covered a 60-fold range in rat hepatocytes. Pdiff, u in human hepatocytes was positively correlated with LogD7.4, consistent with the previous analyses in rat hepatocytes. At low concentrations, active transport was the major process driving total uptake for all drugs in both species.
A large difference in telmisartan metabolism was observed between rat and human hepatocytes; glucuronidation clearance was 32-fold greater in rat hepatocytes than in human hepatocytes. Metabolic clearance was only 4-fold lower than active uptake in rat, whereas the difference between these two processes was up to 71-fold in human hepatocytes, depending on the individual donor. In contrast, repaglinide glucuronidation and overall metabolism data were more consistent between the two species in comparison to telmisartan. However, species differences were evident, because the formation of M2 was increased by 5-fold between rat and human hepatocytes. In addition, M4 metabolite was not identified as a major metabolite in rat hepatocytes, but a clearance of 1.26 μl · min−1 · 106 cells−1 was measured in human. These differences resulted in a repaglinide total metabolic clearance in humans 4-fold greater than that in rat. Total metabolic clearance was lower than the active uptake in either species; however, this difference was 13-fold on average in human hepatocytes in contrast to 112-fold reported for the rat data (Ménochet et al., 2012), probably reflecting the differences in uptake activity between the two species.
Discussion
A mechanistic two-compartment model, previously developed in rat hepatocytes was applied here for the analysis of human hepatocyte data, allowing description of the interplay between active uptake, passive diffusion, intracellular binding, and metabolism. Uptake kinetics of seven OATP substrates was investigated in three human hepatocyte donors using a standardized experimental method and modeling approach. The data generated were applied for the prediction of in vivo hepatic clearance and comparison with previously obtained parameters in rat hepatocytes.
The seven drugs investigated showed a 14-fold range in Km, u values (Fig. 8A), but because of greater variation in their uptake capacity, mean CLactive, u ranged 54-fold (Fig. 8B). The CLactive, u values presented here are generally consistent with literature data. However, higher uptake clearances have been reported for pitavastatin and valsartan (Hirano et al., 2004; Poirier et al., 2009), whereas the opposite was observed for pravastatin and telmisartan (Nakai et al., 2001; Ishiguro et al., 2006). Discrepancies observed may be associated with inconsistencies in the uptake experimental design (use of single concentration versus full kinetic profile) and data analysis among the studies published to date. Uptake activity and passive diffusion were both correlated with lipophilicity, as was the extent of intracellular binding; however, no correlation could be established between CLuptake, u and Km, u, LogD7.4, or fucell. Unlike the approach traditionally used to analyze in vitro uptake data, the mechanistic two-compartment model provides insight into the effect of the bidirectional passive diffusion over a range of concentrations. At low concentrations, the passive diffusion had limited contribution to the overall uptake for all drugs investigated (Fig. 2), which is of relevance because the unbound Cmax (after a therapeutic dose) of the drugs studied ranges from 0.80 nM to 0.52 μM for pitavastatin and valsartan, respectively (Sunkara et al., 2007; Deng et al., 2008). However, because these drugs are administered orally, concentrations in the hepatic portal vein could be higher and the contribution of passive processes may differ.
The fucell term in the mechanistic uptake model was based on studies in rat hepatocytes after an extended incubation time (45–90 min) to allow a steady state between intracellular concentration and media to be reached. Use of these fucell values allowed a reasonably precise estimation of kinetic parameters for all drugs (CV <30%, with the exception of pravastatin), despite the reduced number of data points available for analysis compared with that for rat hepatocytes. This insight into the extent of intracellular binding also permitted the simultaneous assessment of uptake and metabolism, giving a further understanding of the interplay between these two processes compared with previous studies (Paine et al., 2008; Poirier et al., 2008; Watanabe et al., 2009). In the case of repaglinide, active uptake was on average 4.3- and 13-fold greater than passive diffusion and metabolic clearances, respectively. Consistent with the rat data, active uptake has been identified as the major contributor to repaglinide overall clearance. However, a significant contribution of the passive component to the total uptake should not be neglected, as supported by only a 2.4-fold increase in repaglinide area under the curve when coadministered with cyclosporine (potent OATP1B1 inhibitor) (Kajosaari et al., 2005; Amundsen et al., 2010). Considering that metabolism represents the rate-limiting process for repaglinide hepatic clearance, one may expect that the DDIs observed for this type of drug would be metabolism driven. However, such interpretation of the current findings is inappropriate for inhibitors showing dual effects on uptake transporters and metabolism, as illustrated in the repaglinide-gemfibrozil/gemfibrozil glucuronide interaction, leading to a marked increase in repaglinide exposure (Niemi et al., 2003). The data analysis presented here provides a mechanistic description of the processes occurring in the hepatocytes that should improve our ability to quantitatively predict the risk of complex DDIs involving both uptake and metabolism.
Analogous to Km, u, passive diffusion clearances showed minimal donor dependence, although no correlation was evident between Km, u and Pdiff. Consistency in these parameters across donors allowed the use of the mechanistic two-compartment model to successfully estimate CLactive, u from experiments performed at a single concentration. Although this approach requires prior knowledge of Km, u and Pdiff, u, it permits the investigation of interindividual variability from a reduced number of human cells, while allowing the mechanistic nature of the data analysis. Although donor HU4122 exhibited the greatest uptake capacity overall, drugs investigated showed different extent of uptake, depending on the donor used. Rosuvastatin demonstrated the greatest variability in uptake clearance between donors with a CV of 108% (Table 3), reflecting the variability in uptake Vmax (CV 111%). A similar trend was more evident for drugs with predominant active uptake in the current dataset. A comparable degree of interindividual variability has been reported previously (Ishiguro et al., 2006; Yamada et al., 2007). Whereas Yamada et al. (2007) found a variation of 10% in olmesartan uptake clearance among three donors, Ishiguro et al. (2006) found CVs as high as 68% when they investigated the clearance of estradiol-17β-glucuronide in three donors. Emerging absolute quantification data of uptake transporter expression in a number of human livers support this interindividual variability (Ohtsuki et al., 2012). However, the extent to which this variability in transporter expression in the actual tissue relates to the interindividual variability in uptake activity in isolated human hepatocytes remains to be established.
In vitro-in vivo extrapolation resulted in systematic underprediction of hepatic intrinsic clearances, because in some instances predicted values represented <2% of the observed data (rosuvastatin in donor HU8089). Interindividual variability observed in uptake activity resulted in drug and donor-specific empirical scaling factors, ranging from 6.9 to 25.6 (Table 4). Overall, scaling factors required here were greater than the values reported previously (3.7–6.5) for rat plated hepatocytes and estimated for individual drugs or small datasets (Poirier et al., 2009; Watanabe et al., 2009; Gardiner and Paine, 2011). This species difference in prediction success is also evident for metabolic clearance based on large datasets (Ito and Houston, 2005). These results also emphasize the difficulty one faces when trying to identify suitable human hepatocyte donors to investigate the uptake of NCEs. Considering the large range in individual empirical scaling factors and compound differences, it is not surprising that the use of an average factor of 17.1 was not satisfactory for some drugs and in particular for data obtained in HU4122 donor. These findings emphasize the need to characterize human hepatocytes against a set of uptake markers to allow prediction of the impact of active uptake on NCE pharmacokinetics in human.
Giacomini et al. (2010) recommended the use of rosuvastatin, pravastatin, or pitavastatin as probes to study DDIs involving OATP1B1 in vivo. In the present study, we have investigated whether these drugs could be used as markers of variability in uptake activity across different hepatocyte donors. Pravastatin exhibited limited ionization when analyzed by LC-MS/MS, and reliable results could not be generated in all hepatocyte donors. Unlike the general trend observed in the dataset, pitavastatin showed greater active uptake in donor HU4199 than in HU4122 (Fig. 5). This could be due to the fact that pitavastatin is almost exclusively transported by OATP1B1 (Hirano et al., 2004), and uptake of this compound therefore reflects only variability in a single transporter. Rosuvastatin uptake was a good marker of observed interindividual variability in uptake activity regardless of whether full kinetics or a single concentration was used. A significant difference was observed between donors HU4122 and HU8089 when uptake was measured at 1 μM as a control (p < 0.05) but not between HU4122 and HU4199. Rosuvastatin is transported not only by OATP1B1 and OATP1B3, but also by Na+-taurocholate cotransporting polypeptide (Ho et al., 2006; Kitamura et al., 2008). Therefore, uptake clearance of rosuvastatin might reflect better the overall uptake capabilities of cells, despite its limited application as a specific marker of OATP1B1 activity.
The final aim of this study was to investigate differences between uptake in rat and human hepatocytes. Km, u values in both species were within 2-fold for all drugs studied (Fig. 8A), with the exception of pravastatin and pitavastatin, despite the differences in OATPs expressed (Hagenbuch and Gui, 2008). Likewise, Pdiff was consistent between the two species, showing on average less than a 2-fold difference. The greatest interspecies differences were observed for CLactive, u, because this parameter was on average 7-fold greater in rat than in human hepatocytes, with rosuvastatin and valsartan exhibiting differences of up to 21-fold (Fig. 8B). Although this difference could be explained by variation in transporter expression and specificity between the two species, it is noteworthy that while rat hepatocytes were freshly isolated, human cryopreserved hepatocytes were used. Uptake activity has been shown to decrease after cryopreservation and over time in culture (Badolo et al., 2011; Ulvestad et al., 2011). Therefore, although rat hepatocytes can be a useful surrogate for human cells in the development of the experimental method, the discrepancy in transporter expression and activity occurring naturally or as a result of the isolation/storage techniques confounds direct scaling of clearances obtained in rat hepatocytes to human.
In conclusion, a mechanistic two-compartment model was applied to uptake experiments performed in human hepatocytes to simultaneously estimate active uptake, passive diffusion, intracellular binding, and metabolism. Although the affinity of the drugs toward the transporter proteins remained constant, large variability was observed in uptake activity between the three human hepatocyte donors investigated. Uptake activity differed between rat and human hepatocytes; however, information obtained in rat hepatocytes on the extent of intracellular binding was essential for the modeling of human hepatocytes data and for more a informative experimental design of uptake studies in human hepatocytes with a reduced number of concentration and time points. Underprediction of hepatic intrinsic clearances emphasizes the need to account for differences in both transporter expression and activity between hepatocytes and liver tissue and the associated variability.
Supplementary Material
Acknowledgments
We acknowledge Dr. David Hallifax and Sue Murby for their assistance with the LC-MS/MS analysis and Dr. Michael Gertz (all University of Manchester) for help with the modeling of uptake data.
This work was supported by GlaxoSmithKline, Ware, UK (Ph.D. studentship to K.M.) and the Biotechnology and Biological Sciences Research Council, UK.
Parts of this work were previously presented at the following conference: Ménochet K, Kenworthy KE, Houston JB, and Galetin A (2010) Contribution of active uptake to the hepatic clearance of seven OATP substrates in rat and human plated hepatocytes. Proceedings of the 9th International ISSX Meeting; 2010 Sept 4–8; Istanbul, Turkey. International Society for the Study of Xenobiotics, http://www.issx.org/.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
- NCE
- new chemical entity
- ABT
- 1-aminobenzotriazole
- OATP
- organic anion-transporting polypeptide
- DDI
- drug-drug interaction
- DPBS
- Dulbecco's phosphate-buffered saline
- UGT
- UDP-glucuronosyltransferase
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- CV
- coefficients of variation
- rmse
- root mean square error
- gmfe
- geometric mean fold error
- HPLC
- high-performance liquid chromatography.
Authorship Contributions
Participated in research design: Ménochet, Kenworthy, Houston, and Galetin.
Conducted experiments: Ménochet.
Performed data analysis: Ménochet and Galetin.
Wrote or contributed to the writing of the manuscript: Ménochet, Houston, and Galetin.
References
- Amundsen R, Christensen H, Zabihyan B, Asberg A. (2010) Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion-transporting protein 1B1-mediated transport of atorvastatin. Drug Metab Dispos 38:1499–1504 [DOI] [PubMed] [Google Scholar]
- Badolo L, Trancart MM, Gustavsson L, Chesné C. (2011) Effect of cryopreservation on the activity of OATP1B1/3 and OCT1 in isolated human hepatocytes. Chem Biol Interact 190:165–170 [DOI] [PubMed] [Google Scholar]
- Bidstrup TB, Bjørnsdottir I, Sidelmann UG, Thomsen MS, Hansen KT. (2003) CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br J Clin Pharmacol 56:305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bow DA, Perry JL, Miller DS, Pritchard JB, Brouwer KL. (2008) Localization of P-gp (Abcb1) and Mrp2 (Abcc2) in freshly isolated rat hepatocytes. Drug Metab Dispos 36:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HS, Griffin M, Houston JB. (2007) Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos 35:293–301 [DOI] [PubMed] [Google Scholar]
- Chiba M, Ishii Y, Sugiyama Y. (2009) Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J 11:262–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruyn T, Ye ZW, Peeters A, Sahi J, Baes M, Augustijns PF, Annaert PP. (2011) Determination of OATP-, NTCP- and OCT-mediated substrate uptake activities in individual and pooled batches of cryopreserved human hepatocytes. Eur J Pharm Sci 43:297–307 [DOI] [PubMed] [Google Scholar]
- Deng JW, Song IS, Shin HJ, Yeo CW, Cho DY, Shon JH, Shin JG. (2008) The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet Genomics 18:424–433 [DOI] [PubMed] [Google Scholar]
- Fujino H, Yamada I, Shimada S, Yoneda M, Kojima J. (2003) Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: human UDP-glucuronosyltransferase enzymes involved in lactonization. Xenobiotica 33:27–41 [DOI] [PubMed] [Google Scholar]
- Gardiner P, Paine SW. (2011) The impact of hepatic uptake on the pharmacokinetics of organic anions. Drug Metab Dispos 39:1930–1938 [DOI] [PubMed] [Google Scholar]
- Gertz M, Harrison A, Houston JB, Galetin A. (2010) Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos 38:1147–1158 [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuch B, Gui C. (2008) Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica 38:778–801 [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. (2004) Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch 447:653–665 [DOI] [PubMed] [Google Scholar]
- Hallifax D, Foster JA, Houston JB. (2010) Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm Res 27:2150–2161 [DOI] [PubMed] [Google Scholar]
- Hallifax D, Houston JB. (2009) Methodological uncertainty in quantitative prediction of human hepatic clearance from in vitro experimental systems. Curr Drug Metab 10:307–321 [DOI] [PubMed] [Google Scholar]
- Hewitt NJ, Lechón MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, et al. (2007) Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev 39:159–234 [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, Sugiyama Y. (2004) Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther 311:139–146 [DOI] [PubMed] [Google Scholar]
- Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, Wang Y, Kim RB. (2006) Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology 130:1793–1806 [DOI] [PubMed] [Google Scholar]
- Ieiri I, Higuchi S, Sugiyama Y. (2009) Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: implications for inter-individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs. Expert Opin Drug Metab Toxicol 5:703–729 [DOI] [PubMed] [Google Scholar]
- Ishiguro N, Maeda K, Kishimoto W, Saito A, Harada A, Ebner T, Roth W, Igarashi T, Sugiyama Y. (2006) Predominant contribution of OATP1B3 to the hepatic uptake of telmisartan, an angiotensin II receptor antagonist, in humans. Drug Metab Dispos 34:1109–1115 [DOI] [PubMed] [Google Scholar]
- Ito K, Houston JB. (2005) Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res 22:103–112 [DOI] [PubMed] [Google Scholar]
- Jones HM, Barton HA, Lai Y, Bi YA, Kimoto E, Kempshall S, Tate SC, El-Kattan A, Houston JB, Galetin A, et al. (2012) Mechanistic pharmacokinetic modelling for the prediction of transporter-mediated disposition in human from sandwich culture human hepatocyte data. Drug Metab Dispos 40:1007–1017 [DOI] [PubMed] [Google Scholar]
- Kajosaari LI, Niemi M, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. (2005) Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin Pharmacol Ther 78:388–399 [DOI] [PubMed] [Google Scholar]
- Kitamura S, Maeda K, Wang Y, Sugiyama Y. (2008) Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos 36:2014–2023 [DOI] [PubMed] [Google Scholar]
- Kotani N, Maeda K, Watanabe T, Hiramatsu M, Gong LK, Bi YA, Takezawa T, Kusuhara H, Sugiyama Y. (2011) Culture period-dependent changes in the uptake of transporter substrates in sandwich-cultured rat and human hepatocytes. Drug Metab Dispos 39:1503–1510 [DOI] [PubMed] [Google Scholar]
- Landaw EM, DiStefano JJ., 3rd (1984) Multiexponential, multicompartmental, and noncompartmental modeling. II. Data analysis and statistical considerations. Am J Physiol 246:R665–R677 [DOI] [PubMed] [Google Scholar]
- Lave T, Coassolo P, Ubeaud G, Brandt R, Schmitt C, Dupin S, Jaeck D, Chou RC. (1996) Interspecies scaling of bosentan, a new endothelin receptor antagonist and integration of in vitro data into allometric scaling. Pharm Res 13:97–101 [DOI] [PubMed] [Google Scholar]
- Martignoni M, Groothuis GM, de Kanter R. (2006) Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2:875–894 [DOI] [PubMed] [Google Scholar]
- Ménochet K, Kenworthy KE, Houston JB, Galetin A. (2012) Simultaneous assessment of uptake and metabolism in rat hepatocytes: a comprehensive mechanistic model. J Pharmacol Exp Ther 341:2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mico BA, Federowicz DA, Ripple MG, Kerns W. (1988) In vivo inhibition of oxidative drug metabolism by, and acute toxicity of, 1-aminobenzotriazole (ABT). A tool for biochemical toxicology. Biochem Pharmacol 37:2515–2519 [DOI] [PubMed] [Google Scholar]
- Nakai D, Nakagomi R, Furuta Y, Tokui T, Abe T, Ikeda T, Nishimura K. (2001) Human liver-specific organic anion transporter, LST-1, mediates uptake of pravastatin by human hepatocytes. J Pharmacol Exp Ther 297:861–867 [PubMed] [Google Scholar]
- Nakakariya M, Shimada T, Irokawa M, Maeda T, Tamai I. (2008) Identification and species similarity of OATP transporters responsible for hepatic uptake of beta-lactam antibiotics. Drug Metab Pharmacokinet 23:347–355 [DOI] [PubMed] [Google Scholar]
- Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, Eichelbaum M, Kivistö KT, Neuvonen PJ. (2005) Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther 77:468–478 [DOI] [PubMed] [Google Scholar]
- Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. (2003) Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 46:347–351 [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A, Ishiguro N, Kishimoto W, Ludwig-Schwellinger E, Ebner T, et al. (2012) Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos 40:83–92 [DOI] [PubMed] [Google Scholar]
- Paine SW, Parker AJ, Gardiner P, Webborn PJ, Riley RJ. (2008) Prediction of the pharmacokinetics of atorvastatin, cerivastatin, and indomethacin using kinetic models applied to isolated rat hepatocytes. Drug Metab Dispos 36:1365–1374 [DOI] [PubMed] [Google Scholar]
- Parker AJ, Houston JB. (2008) Rate-limiting steps in hepatic drug clearance: comparison of hepatocellular uptake and metabolism with microsomal metabolism of saquinavir, nelfinavir, and ritonavir. Drug Metab Dispos 36:1375–1384 [DOI] [PubMed] [Google Scholar]
- Plise EG, Halladay JS, Cheong J, Sodhi J, Salphati L. (2010) Commonly used inhibitors of drug metabolizing enzymes: do they also inhibit drug transporters? Drug Metab Rev 42:139 [Google Scholar]
- Poirier A, Cascais AC, Funk C, Lavé T. (2009) Prediction of pharmacokinetic profile of valsartan in humans based on in vitro uptake-transport data. Chem Biodivers 6:1975–1987 [DOI] [PubMed] [Google Scholar]
- Poirier A, Lavé T, Portmann R, Brun ME, Senner F, Kansy M, Grimm HP, Funk C. (2008) Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab Dispos 36:2434–2444 [DOI] [PubMed] [Google Scholar]
- Reinoso RF, Telfer BA, Brennan BS, Rowland M. (2001) Uptake of teicoplanin by isolated rat hepatocytes: comparison with in vivo hepatic distribution. Drug Metab Dispos 29:453–459 [PubMed] [Google Scholar]
- Säll C, Houston JB, Galetin A. (2012) A comprehensive assessment of repaglinide metabolic pathways: impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab Dispos 40:1279–1289 [DOI] [PubMed] [Google Scholar]
- Shitara Y, Sugiyama Y. (2006) Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther 112:71–105 [DOI] [PubMed] [Google Scholar]
- Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, Windass AS, Schneck DW. (2004) Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther 76:167–177 [DOI] [PubMed] [Google Scholar]
- Soars MG, Grime K, Sproston JL, Webborn PJ, Riley RJ. (2007) Use of hepatocytes to assess the contribution of hepatic uptake to clearance in vivo. Drug Metab Dispos 35:859–865 [DOI] [PubMed] [Google Scholar]
- Stangier J, Schmid J, Türck D, Switek H, Verhagen A, Peeters PA, van Marle SP, Tamminga WJ, Sollie FA, Jonkman JH. (2000) Absorption, metabolism, and excretion of intravenously and orally administered [14C]telmisartan in healthy volunteers. J Clin Pharmacol 40:1312–1322 [PubMed] [Google Scholar]
- Sunkara G, Reynolds CV, Pommier F, Humbert H, Yeh C, Prasad P. (2007) Evaluation of a pharmacokinetic interaction between valsartan and simvastatin in healthy subjects. Curr Med Res Opin 23:631–640 [DOI] [PubMed] [Google Scholar]
- Ulvestad M, Björquist P, Molden E, Asberg A, Andersson TB. (2011) OATP1B1/1B3 activity in plated primary human hepatocytes over time in culture. Biochem Pharmacol 82:1219–1226 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. (2009) Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328:652–662 [DOI] [PubMed] [Google Scholar]
- Yabe Y, Galetin A, Houston JB. (2011) Kinetic characterization of rat hepatic uptake of 16 actively transported drugs. Drug Metab Dispos 39:1808–1814 [DOI] [PubMed] [Google Scholar]
- Yamada A, Maeda K, Ishiguro N, Tsuda Y, Igarashi T, Ebner T, Roth W, Ikushiro S, Sugiyama Y. (2011) The impact of pharmacogenetics of metabolic enzymes and transporters on the pharmacokinetics of telmisartan in healthy volunteers. Pharmacogenet Genomics 21:523–530 [DOI] [PubMed] [Google Scholar]
- Yamada A, Maeda K, Kamiyama E, Sugiyama D, Kondo T, Shiroyanagi Y, Nakazawa H, Okano T, Adachi M, Schuetz JD, et al. (2007) Multiple human isoforms of drug transporters contribute to the hepatic and renal transport of olmesartan, a selective antagonist of the angiotensin II AT1-receptor. Drug Metab Dispos 35:2166–2176 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.