

Abstract
Restriction-modification (RM) systems are important for bacteria to limit foreign DNA invasion. The naturally competent bacterium Helicobacter pylori has highly diverse strain-specific type II systems. To evaluate the roles of strain-specific restriction in H. pylori natural transformation, a markerless type II restriction endonuclease-deficient (REd) mutant was constructed. We deleted the genes encoding all four active type II restriction endonucleases in H. pylori strain 26695 using sacB-mediated counterselection. Transformation by donor DNA with exogenous cassettes methylated by Escherichia coli was substantially (1.7 and 2.0 log10 for cat and aphA, respectively) increased in the REd strain. There also was significantly increased transformation of the REd strain by donor DNA from other H. pylori strains, to an extent corresponding to their shared type II R-M system strain specificity with 26695. Comparison of the REd and wild-type strains indicates that restriction did not affect the length of DNA fragment integration during natural transformation. There also were no differentials in cell growth or susceptibility to DNA damage. In total, the data indicate that the type II REd mutant has enhanced competence with no loss of growth or repair facility compared to the wild type, facilitating H. pylori mutant construction and other genetic engineering.
INTRODUCTION
Helicobacter pylori is a bacterium that persistently colonizes the human stomach and dominates gastric microbial populations (5, 12, 22). A striking characteristic of H. pylori is its remarkable genetic diversity (33, 50, 56), including strain-specific genes, mosaicism within some genes, and sequence diversity within conserved genes (36). H. pylori strains are naturally competent (7, 18, 29, 53), and analysis of the population structure provides evidence for extensive recombination, approaching pan-myxis (13, 51). Thus, intergenomic recombination has played an important role in the composition of present-day H. pylori strains (5, 7, 13, 51).
Comparison of the entire genomic sequences of two H. pylori strains 26695 (54) and J99 (1) showed that 6 to 7% of the genes are strain specific and that restriction-modification (R-M) genes are a major source of the differences (1). Recent analysis of the entire genomic sequences of H. pylori strains 98-10 (35), HPAG1 (39), B128 (21), G27 (6), and V225 (32), isolated from subjects from different geographic locales, reveals that a substantial proportion of the strain-specific genes are R-M genes (35). The type II R-M systems, consisting of at least two separate enzymes, a restriction endonuclease (RE) and one (type II) or two (type IIa) DNA methyltransferases (modification), are the major H. pylori R-M systems (3, 4, 31, 52, 59). Although many of the R-M system genes are shared, the enzymatically active R-M proteins in different H. pylori strains vary and are strain specific (27, 31, 52, 59). The REs create barriers against transformation (3, 4, 10) that limit genetic exchange in vivo and on the laboratory bench, which increases the difficulty to genetically manipulate H. pylori.
Strain 26695, the first H. pylori strain for which the whole genome sequence (WGS) was solved, has been extensively studied (43, 44, 54), and, in total, possesses 14 complete or partial type II R-M systems. However, only four are functional: HpyAII (encoded by HP1366), HpyAV (HP0053), HpyAIII (HP0091), and HpyAIV (HP1351) (31, 38). In the present study, we sequentially deleted each of the four active REs from 26695 using sacB-mediated counterselection (9), constructing a markerless strain deficient in type II RE function. We then characterized the effects of these mutations on H. pylori transformation and on growth, DNA repair, and spontaneous mutations in vitro. The constructed restriction endonuclease-deficient (REd) strain has a significantly enhanced natural transformation frequency while retaining similar growth, DNA repair, and spontaneous mutation phenotypes compared to wild-type 26695, indicating its utility for H. pylori mutant construction and other genetic engineering.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
The H. pylori and Escherichia coli strains and plasmids used here are listed in Table S1 in the supplemental material. The H. pylori strains were grown at 37°C in 5% CO2 on Trypticase soy agar (TSA) plates with 5% sheep blood (BBL Microbiology Systems, Cockeysville, MD) or brucella agar (BA) plates (Difco Laboratories, Detroit, MI) supplemented with 10% newborn calf serum (NBCS; Serologicals Corp., Norcross, GA) (30). Antibiotic-resistant H. pylori mutant strains were selected with kanamycin (Km; 10 μg/ml), chloramphenicol (Cm; 10 μg/ml), streptomycin (Str; 25 μg/ml), or rifampin (Rif; 7.5 μg/ml). For counterselection, we modified the Copass method (9), adding 6.5% (wt/vol) sucrose (Fisher Scientific, Gaithersburg, MD) to the BA plates to make sucrose-selective medium (SSM) plates. E. coli strains were grown in Luria-Bertani (LB) medium at 37°C (42). Ampicillin (Ap) (100 μg/ml), Km (50 μg/ml), and Cm (30 μg/ml) were used for selecting vector or constructed plasmids during cloning in E. coli.
Construction of H. pylori markerless restriction endonuclease-deficient mutants.
The restriction endonuclease genes hpyAII (HP1366), hpyAV (HP0053), hpyAIII (HP0091), and hpyAIV (HP1351) of the active type II R-M systems were sequentially deleted from H. pylori 26695 using a modified sacB-mediated counterselection (9). The overall strategy was as follows: a sacB-cat cassette was first constructed in strain 26695 by substituting cat for kan in the described kan-sacB cassette (9), based on our observations in 26695 that mutation construction with a cat cassette as a selective marker is more (>1 log10-fold) efficient than using the kan cassette (data not shown). The first target gene (hpyAII) was then replaced by the sacB-cat cassette by homologous recombination (see Fig. S1 in the supplemental material). After confirming the correct replacement by PCR, using primers IIL-F-SacII and SC-R-XbaI (see Table S2 in the supplemental material), the sacB-cat cassette sequence then was deleted from the locus on SSM, by homologous recombination with a direct fusion of hpyAII upstream and downstream regions, leading to a markerless deletion mutant (KO-1; ΔhpyAII) (see Fig. S1 in the supplemental material). Each of the other target genes was deleted in the same way, leading to a series of multiple markerless mutations (KO-2 [ΔhpyAII ΔhpyAV], KO-3 [ΔhpyAII ΔhpyAV ΔhpyAIII], and KO-4 [ΔhpyAII ΔhpyAV ΔhpyAIII ΔhpyAIV]; REd mutant) in the 26695 background (Fig. 1).
Fig 1.

Construction of H. pylori 26695 mutants deficient in active type II restriction endonucleases. H. pylori wild-type 26695 strain has four active type II restriction endonuclease genes: hpyAII (HP1366), hpyAV (HP0053), hpyAIII (HP0091), and hpyAIV (HP1351) (31). With sacB-mediated counterselection, each of the genes encoding an active restriction endonuclease was sequentially deleted from the genome to finally construct a markerless KO-4 mutant (26695 ΔhpyAII ΔhpyAV ΔhpyAIII ΔhpyAIV [26695-REd]).
These steps were accomplished by first obtaining a 1.7-kb sacB cassette by PCR using pKSF (9) as a template and the primers SC-F-XbaI and S-R-PstI (see Table S2 in the supplemental material) that add XbaI and PstI restriction sites to the ends of the PCR fragment. Then, a 0.8-kb cat cassette was obtained by PCR using pAD1-Cat (25) as a template and primers SC-R-XbaI and C-R-PstI (see Table S2 in the supplemental material) that add XbaI and PstI restriction sites to the ends of the PCR fragment. After digestion with PstI, the two fragments were ligated, and PCR performed with primers SC-F-XbaI and SC-R-XbaI to obtain a 2.5-kb sacB-cat cassette flanked by XbaI restriction sites. A 1.0-kb fragment downstream of hpyAII (IIdown) was obtained by PCR using genomic DNA from the wild-type H. pylori strain 26695 as a template and the primers IIL-F-SacII and IIL-R-SpeI (see Table S2 in the supplemental material) that add SacII and SpeI restriction sites to the ends of the fragment. Similarly, a 1.1-kb fragment upstream of hpyAII (IIup) was obtained by PCR using primers IIR-F-SpeI and IIR-R-PstI (see Table S2 in the supplemental material) that add the SpeI or PstI restriction sites to the ends of the fragment. The fragments IIdown and IIup were digested with SacII/SpeI and SpeI/PstI, respectively, and then ligated together into SacII/PstI-digested pGEM-T Easy (Promega, Madison, WI), creating pXZ017 (see Table S1 in the supplemental material). In the same way, a 1.1-kb fragment downstream of hpyAV (Vdown) was obtained by PCR with primers VL-F-SacII and VL-R-SpeI and a 1.0-kb fragment upstream of hpyAV (Vup) was obtained by PCR with the primers VR-F-SpeI and VR-R-PstI (see Table S2 in the supplemental material). The fragments Vdown and Vup were digested with SacII/SpeI and SpeI/PstI and then ligated together into SacII/PstI-digested pGEM-T Easy, creating pXZ016 (see Table S1 in the supplemental material). A 0.9-kb fragment upstream of hpyAIII (IIIup) was obtained by PCR with the primers IIIL-F-SacII and IIIL-R-SpeI, and a 1.1-kb fragment downstream of hpyAIII (IIIdown) was obtained by PCR with the primers IIIR-F-SpeI and IIIR-R-PstI (see Table S2 in the supplemental material). The fragments IIIup and IIIdown were digested with SacII/SpeI and SpeI/PstI and then ligated together into SacII/PstI-digested pGEM-T Easy, creating pXZ144 (see Table S1 in the supplemental material). A 0.7-kb fragment downstream of hpyAIV (IVdown) was obtained by PCR with the primers IVL-F-SacII and IVL-R-SpeI, and a 1.0-kb fragment upstream of hpyAIV (IVup) was obtained by PCR with the primers IVR-F-SpeI and IVR-R-PstI (see Table S2 in the supplemental material). The fragments IVdown and IVup were digested with SacII/SpeI and SpeI/PstI and then ligated together into SacII/PstI-digested pGEM-T Easy, creating pXZ145 (see Table S1 in the supplemental material). The 2.5-kb sacB-cat cassette was digested with XbaI and then ligated with SpeI-digested pXZ017, pXZ016, pXZ144, or pXZ145, creating pXZ033, pXZ032, pXZ146, and pXZ147, respectively (see Table S1 in the supplemental material).
The wild-type H. pylori strain 26695 was first transformed to Cmr with pXZ033 using Cm-containing plates and then transformed to Cms with pXZ017 using SSM plates to create a markerless single (KO-1) mutant HPXZ285 (26695 ΔhpyAII) (see Table S1 in the supplemental material). In the same way, HPXZ285 was transformed to Cmr with pXZ032 and then transformed to Cms with pXZ016 to create a markerless double (KO-2) mutant HPXZ490 (26695 ΔhpyAII ΔhpyAV) (see Table S1 in the supplemental material). HPXZ490 was then transformed to Cmr with pXZ146 and then transformed to Cms with pXZ144 to create a markerless triple (KO-3) mutant HPXZ560 (26695 ΔhpyAII ΔhpyAV ΔhpyIII) (see Table S1 in the supplemental material). Finally, HPXZ560 was transformed to Cmr with pXZ147 and then transformed to Cms with pXZ145 to create a markerless quadruple (KO-4) mutant HPXZ566 (26695 ΔhpyAII ΔhpyAV ΔhpyIII ΔhpyIV; called the REd [restriction-endonuclease deficient] mutant) (see Table S1 in the supplemental material).
Construction of mutants with Kmr, Cmr, or Strr genetic markers.
An aphA (Km) cassette was introduced into the genomic vacA locus of H. pylori strains 26695, JP26, J99, or J166 by transformation with pCTB8:Km to create Kmr strains (see Table S1 in the supplemental material). PCR, using the primers VacA-F and VacA-R (see Table S2 in the supplemental material), was used to confirm the appropriate constructs in Kmr transformants. A cat cassette was introduced into the genomic ureA locus of 26695, HPXZ566, JP26, J99, or J166 by transformation with pAD1-Cat to create Cmr strains (see Table S1 in the supplemental material), and results confirmed by PCR using Ure-F and Ure-R (see Table S2 in the supplemental material). An rpsL(A128G) point mutation was introduced into the genome of 26695, HPXZ566, JP26, J99, or J166, by transformation with p801R to create Strr strains and similarly introduced into the genome of the Cmr strains to create Cmr/Strr double-marked strains (see Table S1 in the supplemental material).
Natural transformation.
The assay to evaluate natural transformation was performed as described previously (29), with modifications. In brief, recipient H. pylori cells were harvested from 24-h cultures on TSA plate into 1.0 ml of phosphate-buffered saline (PBS; pH 7.4) and centrifuged at 800 × g for 5 min. The cell pellet was resuspended in PBS to a concentration of ∼109 cell/ml. Each transformation mixture with recipient cells (100 μl) and donor DNA (1,000 ng for genomic DNA or plasmids with the aphA [pCTB8:Km] or cat [pAD1-Cat] cassette and 100 ng for the plasmid with the point mutation [p801R] in 10 μl of distilled water) was spotted onto a TSA plate. To detect transformation, we used much higher concentrations of donor DNA since the integration of long cassettes was required to encode selectable phenotypes compared to when point mutations were used for transformation, as described previously (29). Each plate was incubated for 24 h, as we (30) and others (34, 49, 57) have reported, at 37°C in 5% CO2. The transformation mixture then was harvested and placed in 1.0 ml of PBS, and 100-μl 10-fold serial dilutions were plated onto either TSA plates or BA plates containing 10% NBCS and suitable antibiotics. The plates were incubated for 4 days at 37°C in 5% CO2, and the total transformation frequency was determined by the number of colonies on selective plates divided by the total CFU on nonselective media. In each transformation experiment, H. pylori strains with no DNA added also were tested in parallel as negative controls; no colonies were seen in any case. Each experiment was repeated three to five times with independent cultures.
Horizontal DNA transfer during coculture.
An assay was performed to evaluate the ability of H. pylori strains to obtain exogenous DNA during coculture with another strain. After 24 h of growth on TSA plates, cell cultures of a singly marked (Kmr) strain (as donor) and a doubly marked (Strr/Cmr) strain (as recipient) were collected, washed, and resuspended in BB medium. Aliquots of 50 μl containing ∼2.5 × 108 cells of donor and recipient strains were mixed, spotted onto a TSA plate, and incubated for 24 h at 37°C in 5% CO2. The cocultured cells then were collected in 1 ml of PBS and, after a series of suitable dilutions, the cells were plated and incubated onto BA plates containing 10% NBCS and Str/Km/Cm or onto BA/Str/Cm plates for 4 days. Colonies were counted and frequencies of doubly marked Strr/Cmr strains that obtained DNA from a singly marked (Kmr) strain were calculated as the CFU (BA/Strr/Kmr/Cmr)/CFU (BA/Strr/Cmr). As controls, a Kmr Rifr strain (HPXZ660) and a Strr Cmr strain (HPXZ698 or HPXZ641) (see Table S1 in the supplemental material) were cocultured in the same way. No colonies grew on BA/Kmr/Rifr/Strr/Cmr plates in any assay, indicating that no strain was able to obtain both selective markers from a donor strain during coculture. These results suggested that Strr Kmr Cmr colonies obtained during coculture of the Kmr and Strr Cmr strains were Strr Cmr strains (as recipients) that received donor DNA from the Kmr strain. As additional controls, the Kmr, or the Strr Cmr strains alone also were incubated and selected in the same way. No colonies grew on Strr Kmr Cmr plates, which confirmed that under the test conditions, strains obtain the additional antibiotic marker due to DNA invasion during coculture rather than by spontaneous mutation. Coculture of a singly marked (Cmr) strain (as donor) and a doubly marked (Strr Kmr) strain (as recipient) was performed in a similar way, and the frequencies of doubly marked Strr Kmr strains that obtained DNA from a singly marked (Cmr) strain were calculated as the CFU (BA/Strr/Kmr/Cmr)/CFU (BA/Strr/Kmr). All calculations were based on three independent experiments.
Mapping the transforming fragment.
Mapping of the insertion sites was performed as described previously (30). For each of the recipient strains (26695 and 26695-REd), nine sets of transformants, each independently incubated with donor DNA (from strain HPXZ627), were examined. One Cmr transformant from each independent transformation was picked, subcultured on a selective plate (containing Cm), and harvested (30). Chromosomal DNA was prepared using the Wizard genomic DNA purification kit (Promega). To analyze the region of DNA flanking the cat cassette, PCR was performed using isolated chromosomal DNA from each of the 18 (9 + 9) independent transformants, using primers designed to match the sequences in both 26695 and J99 (see Table S2 in the supplemental material). Sequencing was performed at Macrogen (Rockville, MD), and all sequence analysis was performed using Sequencher (Gene Codes, Ann Arbor, MI).
Susceptibility to DNA damage.
To evaluate H. pylori strain recovery from DNA damage induced by UV irradiation, H. pylori cells from 48-h cultures on TSA plates were collected into 1.0 ml of brucella broth, and with suitable dilutions, equal suspensions were inoculated onto TSA plates. When dried, plates were exposed to 254-nm UV generated by USHIO G30T UV lamp (Fisher Scientific) at a distance of 78 cm for 0 to 60 s (resulting in doses of ∼2.0 J/m2/s) and then incubated at 37°C in 5% CO2 for 4 days before the colonies were counted and the survival rates were determined. Each experiment was repeated three times with independent cultures.
Susceptibility to fluoroquinolones was evaluated using levofloxacin Etest strips (bioMérieux, Hazelwood, MO), according to the manufacturer's instructions. Briefly, cells from each H. pylori culture on a TSA plate after 48 h of growth were evenly spread onto a new TSA plate to cover the surface of the plate, and Etest strips were applied. Plates were incubated for 48 h at 37°C in 5% CO2, and the MICs were determined, in at least three independent experiments.
Spontaneous mutation.
The spontaneous mutation rate of H. pylori strains was evaluated as the rate of rifampin resistance, as reported (25), since a point mutation in rpoB converts rifampin-sensitive (Rifs) H. pylori to rifampin-resistant (Rifr) (17). Briefly, single colonies of Rifs H. pylori strains on TSA plates after 48 h of growth were picked, streaked onto new TSA plates, and incubated for 48 h, allowing mutations to occur. The cells were harvested into PBS and serially diluted onto either BA plates containing 10% NBCS and rifampin or TSA alone. The plates were incubated at 37°C in 5% CO2 for 96 h, the colonies were counted, and the spontaneous mutation frequencies were calculated. Each experiment was performed with nine independent single colonies for each strain.
RESULTS
Construction of restriction endonuclease-deficient H. pylori strains.
Most wild-type H. pylori strains are naturally competent for uptake of exogenous DNA (20, 55) and simultaneously have strain-specific active type II R-M systems that limit the effects of natural competence (3, 10, 31, 59). To characterize the roles of R-M systems in H. pylori natural transformation, we sequentially deleted the RE genes within the four sets of 26695 active type II R-M systems (HpyAII, HpyAV, HpyAIII, and HpyAIV) using sacB-mediated counterselection, generating markerless mutants: ΔhpyAII (KO-1), ΔhpyAII ΔhpyAV (KO-2), ΔhpyAII ΔhpyAV ΔhpyIII (KO-3), and ΔhpyAII ΔhpyAV ΔhpyAIII ΔhpyAIV (KO-4; REd) (Fig. 1). Restriction analysis of genomic DNA showed that the REd mutant, as with the 26695 parental strain, is resistant to digestion by each of the four REs (Fig. 2), indicating that the methylases belonging to each of the four affected RMs remained active. Moreover, all of the four RE-deficient mutants had similar growth rates to the wild type (see Fig. S2 in the supplemental material), similar abilities to repair UV-induced DNA damage (see Fig. S3 in the supplemental material) and fluoroquinolone (levofloxacin)-induced DNA damage (see Table S3 in the supplemental material), and similar spontaneous mutation rates (see Fig. S4 in the supplemental material). Thus, the mutant strains resembled the parental in important phenotypes related to growth, specific methylation, and DNA repair.
Fig 2.
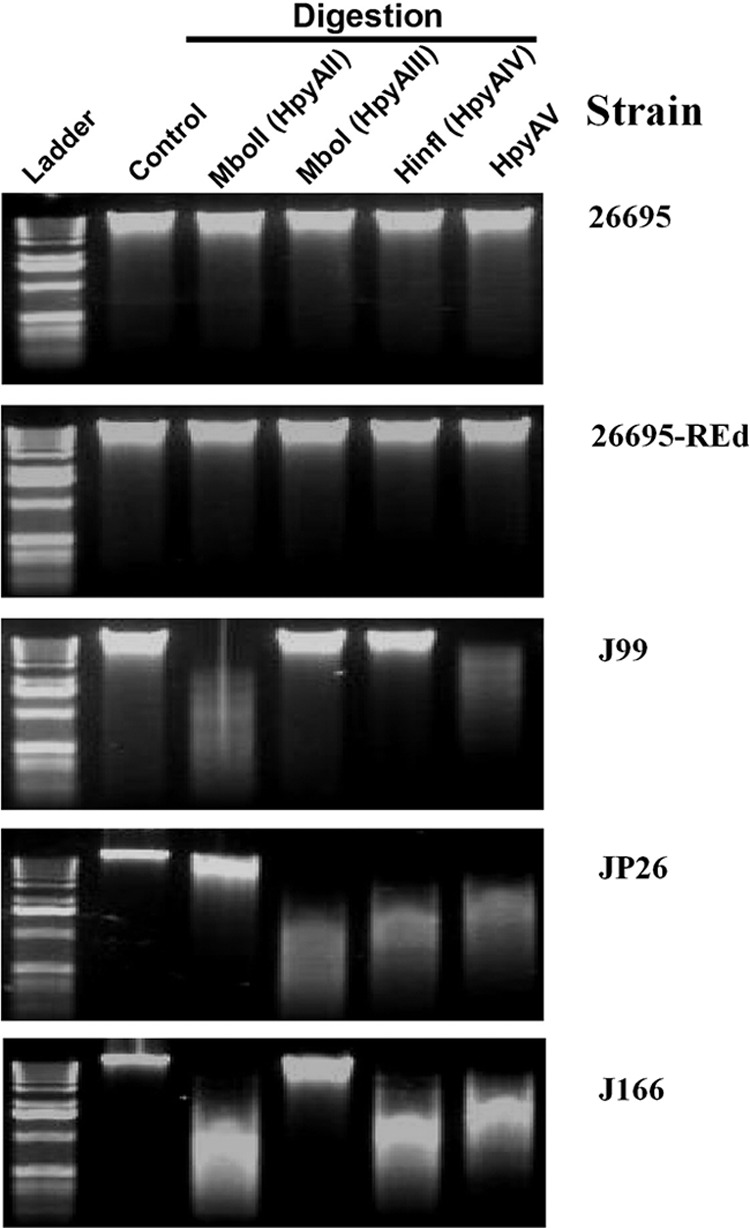
Restriction analysis of genomic DNA of H. pylori strains. Genomic DNA of H. pylori strains 26695, 26695-REd, J99, JP26, and J166 (from top panel to bottom panel) was digested with each of the four active 26695 strain-specific type II restriction endonucleases or their isoenzymes—HpyAII, HpyAIII, HpyAIV, and HpyAV (left to right)—or was not digested (control). The genomic DNA of 26695 and 26695-REd was not digested by any of the four restriction enzymes, indicating that all of the HpyAII, HpyAIII, HpyAIV, and HpyAV restriction sites were protected by methylation. Restriction endonucleases, HpyAII, and HpyAV digested the genomic DNA of strain J99, whereas the HpyAIII and HpyAIV restriction sites in J99 were protected by methylation. REs HpyAIII, HpyAIV, and HpyAV digested the genomic DNA of J99, while the HpyAII restriction sites were protected by methylation; HpyAII, HpyAIV, and HpyAV digested J166 genomic DNA, while the HpyAIII restriction sites were protected by methylation.
Relationship between size of the transforming DNA allele and transformation of RE-deficient strains.
To evaluate the roles of REs in H. pylori natural transformation, we first incubated H. pylori wild-type 26695 and 26695-REd (KO-4) with p801R prepared in E. coli. Transformation by p801R, which carries an 801-bp H. pylori 26695 rpsL fragment with a single central point mutation (A128G), converts Strs H. pylori to Strr (29). A total of six type II RE recognition sites are located within the 801-bp H. pylori sequence that may be subject to cleavage by wild-type 26695 but not by 26695-REd. Under standard test conditions (29), the transformation frequencies (TFs) of the wild-type and 26695-REd strains with p801R were nearly identical (with values of 2.7 ± 0.9 × 10−3 versus 2.0 ± 0.7 × 10−3) (Fig. 3A). These results indicate that, as anticipated by studies involving deletion of a single RE (4), the REs of recipient strains do not affect the transformation of this point mutation.
Fig 3.

Transformation of H. pylori type II restriction endonuclease-deficient mutants with plasmid DNA. H. pylori 26695 and derivative strains were transformed with plasmids with selectable phenotypes cloned into pGEM-T Easy. (A) p801R (an 801-bp H. pylori rpsL fragment from a Strr strain [rpsL A126G]). (B) pAD1-Cat (H. pylori DNA at ureAB locus flanking a 1.2-kb cat cassette conferring Cmr). (C) pCTB8:Km (H. pylori DNA at vacA locus flanking a 1.3-kb aphA cassette conferring Kmr). Transformation frequency was expressed as transformation events/total CFU/μg of donor DNA.
Next, we transformed H. pylori wild-type and RE-deficient mutant strains with pAD1-Cat, produced in E. coli. Transformation by pAD1-Cat, which carries a ureAB fragment from H. pylori strain 60190 with a central exogenous cat cassette (1,127 bp), converts Cms H. pylori to Cmr (30). In pAD1-Cat, the cat cassette is flanked by 570 bp downstream and 339 bp upstream of the ureAB locus sequence. A total of six type II RE recognition sites are located within the 1127-bp cat cassette (two each for HpyAII and HpyAIV, and one each for HpyAIII and HpyAV) (Table 1), and 13 type II RE sites located outside the cat cassette but within H. pylori flanking sequence (six for HpyAII, three for HpyAIII, two each for HpyAIV and HpyAV). Under the standard test conditions, the TF of the wild-type strain was (3.0 ± 0.7) × 10−6. In contrast, the TF of the ΔhpyAII single (KO-1) mutant was (1.0 ± 0.1) × 10−5 (∼0.5 log10-fold significantly increased compared to the wild type [WT], P < 0.05), the TF of the ΔhpyAII ΔhpyAV double (KO-2) mutant was (1.7 ± 0.7) × 10−5 (∼0.8 log10-fold significantly increased compared to the WT, P < 0.05), the TF of the ΔhpyAII ΔhpyAV ΔhpyAIII triple (KO-3) mutant was (4.4 ± 1.1) × 10−5 (∼1.2 log10-fold significantly increased compared to the WT, P < 0.05), and the TF of the 26695-REd mutant (KO-4) was (1.3 ± 0.3) × 10−4 (∼1.7 log10-fold significantly increased compared to the WT, P < 0.05) (Fig. 3B). These results indicated that REs of the recipient strains inhibit transformation by this exogenous 1127-bp cat cassette (synthesized in E. coli), which is consistent with functional restriction barriers in the wild-type strain (4, 48).
Table 1.
Restriction sites in selectable markers recognized by the four active type II restriction endonucleases in H. pylori strain 26695
| Restriction endonuclease | Recognition sequence | No. of restriction sites |
|
|---|---|---|---|
| cat cassette (1,189 bp) | aphA cassette (1,288 bp) | ||
| HpyAII | GAAGA | 2 | 4 |
| HpyAV | CCTTC | 2 | 3 |
| HpyAIII | GATC | 1 | 7 |
| HpyAIV | GANTC | 1 | 7 |
| Total | 6 | 21 | |
To confirm and extend this observation, we also transformed H. pylori wild-type and RE-deficient strains with pCTB8:Km (synthesized in E. coli), which carries an H. pylori 60190 vacA fragment with a central exogenous aphA cassette (1,288 bp) that converts Kms H. pylori to Kmr (30). In pCTB8:Km, the aphA cassette is flanked by 672 bp downstream and 963 bp upstream of the vacA locus. A total of 21 type II RE sites are located within the 1,288-bp aphA cassette (four for HpyAII, seven each for HpyAIII and HpyAIV, and three for HpyAV) (Table 1), and 15 sites located outside the aphA cassette but within the H. pylori flanking sequence (four each for HpyAII and HpyAV, six for HpyAIII, and one for HpyAIV). Under the standard test conditions, the TF of the wild-type strain by pCTB8:Km was (2.5 ± 0.8) × 10−8. The TF by pCTB8:Km of the KO-1 mutant was (1.1 ± 0.2) × 10−7 (∼0.6 log10-fold significantly increased compared to the WT, P < 0.05), the TF of the KO-2 mutant was (3.9 ± 2.5) × 10−7 (∼1.2 log10-fold significantly increased compared to the WT, P < 0.05), the TF of the KO-3 mutant was (1.1 ± 0.4) × 10−6 (∼1.7 log10-fold significantly increased compared to the WT, P < 0.05), and the TF of the 26695-REd mutant (KO-4) was (2.7 ± 1.3) × 10−6 (∼2.0 log10-fold significantly increased compared to the WT, P < 0.05) (Fig. 3C). Consistent with the results for transformation by the cat cassette at the ureAB locus, these results indicate that the active H. pylori REs inhibit transformation of the exogenous (synthesized in E. coli) 1.3-kb aphA cassette within the unrelated vacA locus.
Since H. pylori strain 26695 is not a good recipient for transformation by E. coli-H. pylori shuttle plasmids (3, 47), we also determined whether the 26695-REd strain can be transformed by shuttle plasmids through either natural transformation or electroporation. We found that no transformation of shuttle plasmid pHel3 (see Table S1 in the supplemental material) into 26695 or 26695-REd strains could be detected (data not shown), suggesting that factors other than type II REs affect shuttle plasmid transformation of H. pylori.
Strain-specific REs limit transformation between differing H. pylori source DNAs.
To evaluate the roles of the strain-specific REs in horizontal gene transfer between different H. pylori strains in addition to transfer from E. coli to H. pylori, we transformed wild-type H. pylori strain 26695 and the four RE-deficient mutants with genomic DNA from H. pylori strains J99, JP26, and J166.
Restriction analysis indicates that J99 genomic DNA is digested by HpyAII and HpyAV but protected from digestion by HpyAIII and HpyAIV (Fig. 2), a result consistent with prior observations that J99 possesses the HpyAIII and HpyAIV methylases (3, 27, 31, 59). Genomic DNA from J99 with the cat cassette at the ureAB locus was used to transform 26695 and the RE-deficient mutants (Fig. 4B). Compared to the wild-type 26695, the KO-1 mutant (ΔhpyAII) had a significantly increased TF (∼0.7 log10-fold [P < 0.05]). The KO-2 mutant (ΔhpyAII ΔhpyAV) had a further significantly increased TF (∼0.4 log10-fold [P < 0.05] compared to the ΔhpyAII single mutant and ∼1.2 log10-fold [P < 0.05] compared to the WT). These data indicated that the active HpyAII and HpyAV REs form barriers that inhibit J99 genomic DNA transformation of the wild-type strain 26695. The KO-3 mutant and the REd mutant had similar TF values to the KO-2 mutant (Fig. 4B). Similar results were observed when J99 genomic DNA with the aphA cassette at the vacA locus was used to transform the wild-type and the RE-deficient 26695 mutants (Fig. 5A). Deletion of 26695 HpyAII and HpyAV significantly increased J99 genomic DNA transformation, but there were no further increase with deletion of HpyAIII and HpyAIV. These results indicate that HpyAIII and HpyAIV do not form a barrier inhibiting J99 genomic DNA, which is consistent with the known methylation of HpyAIII and HpyAIV chromosomal sites in strain J99 (31).
Fig 4.

Transformation of H. pylori type II restriction endonuclease-deficient mutants with genomic DNA from Cmr H. pylori strains with different methylation activities. H. pylori 26695 and derivative strains were transformed with genomic DNA from H. pylori strains that had been transformed with pAD1-Cat, yielding chromosomally encoded Cmr (ureA::cat) strains. (A) Strain 26695 specifically methylates to protect at all four restriction sites (HpyAII, HpyAIII, HpyAIV and HpyAV). (B) Strain J99 specifically methylates HpyAIII and HpyAIV restriction sites. (C) Strain JP26 specifically methylates HpyAII restriction sites. (D) Strain J166 specifically methylates at HpyAIII restriction sites.
Fig 5.

Transformation of H. pylori type II restriction endonuclease-deficient mutants with genomic DNA from Kmr H. pylori strains with different methylation activities. H. pylori 26695 and derivative strains were transformed with genomic DNA from H. pylori strains that had been transformed with pCTB8:Km yielding chromosomally encoded Kmr (vacA::aphA) strains. (A) Strain J99 methylates HpyAIII and HpyAIV restriction sites. (B) Strain JP26 methylates HpyAII restriction sites. (C) Strain J166 methylates HpyAIII restriction sites.
The RE analysis showed that JP26 genomic DNA was digested by HpyAIII, HpyAIV, and HpyAV but protected from HpyAII (Fig. 2), indicating that JP26 has methylases that modify HpyAII recognition sites, but not HpyAIII, HpyAIV, or HpyAV sites (3). Genomic DNA from JP26 with the cat cassette at the ureAB locus also was used to transform the wild-type and RE-deficient 26695 mutants (Fig. 4C). Compared to the WT strain, deletion of hpyAII did not significantly increase TF, while double deletion of hpyAII and hpyAV significantly increased TF ∼0.9 log10-fold (P < 0.05). These data indicate that active HpyAV forms a barrier to inhibit JP26 transformation of 26695. However, for JP26 DNA with the cat cassette, the KO-3 and the REd mutants slightly but consistently increased TF (Fig. 4C). Since the aphA cassette contains more sites recognized by the four REs than does the cat cassette, we also transformed the 26695 strains with genomic DNA from JP26 with the aphA cassette at the vacA locus (Fig. 5B). The results showed that hpyAII deletion did not increase JP26 transformation of 26695, whereas deletion of hpyAV, hpyAIII, and hpyAIV did. Compared to the WT, the KO-2, the KO-3, and the REd mutants increased TF ∼0.6, 1.1, and 1.5 log10-fold (P < 0.05), respectively, indicating that HpyAV, HpyAIII, and HpyAIV form barriers to inhibit JP26 transformation of 26695.
J166 genomic DNA was digested by HpyAII, HpyAIV, and HpyAV but protected from HpyAIII (Fig. 2), which indicates that J166 has methylases that modify HpyAIII, but not HpyAII, HpyAIV, or HpyAV recognition sites (3). The wild-type and RE-deficient mutants were transformed by genomic DNA from J166 with the cat cassette at the ureAB locus (Fig. 4D) or with the aphA cassette at the vacA locus (Fig. 5C). Consistent with the digestion analysis, the KO-1, KO-2, and the REd mutants showed significantly increased TF, whereas deletion of hpyAIII did not significantly affect the TF by either J166 donor. Compared to the WT, the REd mutant had increased TF ∼1.3 and ∼1.4 log10-fold (P < 0.05) for the two J166 genomic donors, respectively.
In contrast, when the wild-type and RE-deficient 26695 mutant strains were transformed with 26695 DNA with the cat cassette at the ureAB locus, as expected, the TFs of each strain were similar (Fig. 4A). This served as a control for the above experiments. These results confirm that the RE mutations enhance transformation by reducing donor DNA digestion.
Effect of strain-specific REs on mating.
To further evaluate the effects of the REs on the restriction barriers, we determined the frequency of horizontal DNA transfer into H. pylori wild-type strain 26695 or 26695-REd (KO-4) in mating with H. pylori strains of different methylation patterns (Fig. 6A). The wild-type and 26695-REd strains had similar frequencies of uptake of genetic markers in mating with a Kmr 26695 strain that methylates all four type II RE recognition sites. The TF of 26695-REd was ∼1.2 log10-fold (P < 0.05) significantly higher than the wild type in mating with a Kmr J99 strain, which methylates the recognition sites of only two of the four REs in 26695, and ∼2.0 log10-fold (P < 0.05) significantly higher than the wild type in mating with a Kmr JP26 strain or J166 strain, both of which methylate the recognition sites of only one of the four REs. These results indicate that REd mutation also enhances the 26695 transformation efficiency in cell-to-cell mating. As control experiments, the frequencies of horizontal DNA transfer from H. pylori wild-type strain 26695 and 26695-REd to the same H. pylori strains (26695, J99, JP26, or J166) were evaluated. The lack of significant differences (Fig. 6B), indicates that, as expected, REd mutation does not affect the ability to serve as a DNA donor in cell-to-cell mating.
Fig 6.
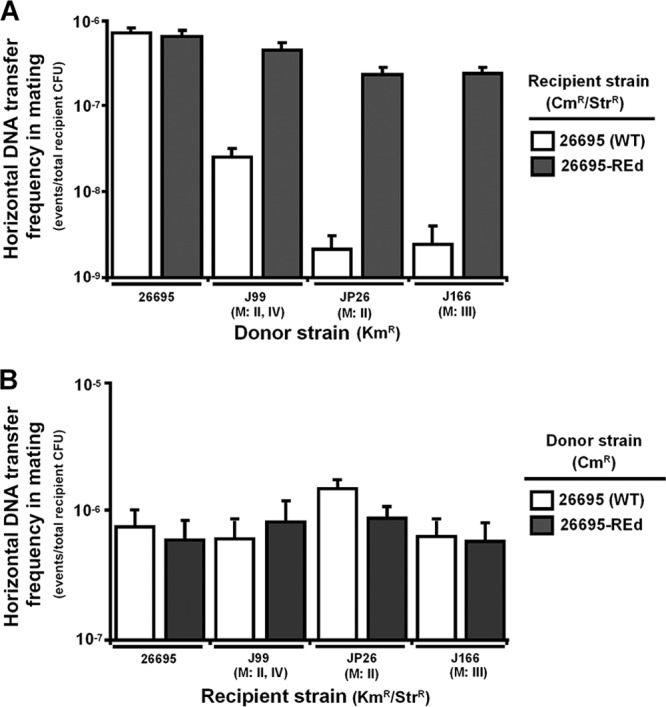
Frequency of horizontal DNA transfer in cell-to-cell mating. (A) Horizontal DNA transfer into H. pylori wild-type strain 26695 and 26695-REd in mating with H. pylori strains with different methylation activities. H. pylori wild-type strain 26695 or 26695-REd with double genetic markers (Cmr Strr) as recipients were mated with H. pylori strains 26695, J99, JP26, or J166, each containing a single genetic marker (Kmr) in the vacA locus, as donors. The numbers of transformants positive for all three phenotypes (Kmr, Cmr, and Strr) were determined, and the frequency of horizontal DNA transfer to the H. pylori wild-type and the REd strain was determined. (B) Horizontal DNA transfer from H. pylori wild-type strain 26695 and 26695-REd in mating with H. pylori strains. H. pylori wild-type strain 26695 or 26695-REd with a single genetic marker (Cmr) in the ureAB locus as donors were mated with H. pylori strains 26695, J99, JP26, or J166, each containing double genetic markers (Kmr Strr) as recipients. The numbers of transformants positive for all three phenotypes (Kmr, Cmr, and Strr) were determined, and the frequency of horizontal DNA transfer from the H. pylori wild type and the REd mutant to each recipient strain was determined.
Strain-specific restriction endonucleases do not affect the import size of exogenous DNA during transformation.
To evaluate whether RE mutations affect the pattern of DNA integration into the recipient genome, we performed independent transformations of wild-type 26695 or 26695-REd using genomic DNA from Cmr strain J99. The length of DNA insert flanking the cat cassette at the ureAB locus was determined (Fig. 7). For nine independent transformants, a mean length of (3,221 ± 1,546)-bp donor (J99) genomic DNA was integrated into the wild-type 26695 chromosome, and similarly, a mean length of (3,202 ± 1,640)-bp donor (J99) genomic DNA into the 26695-REd chromosome (Table 2). These results indicate that restriction does not affect the DNA import size in H. pylori recipients, a finding consistent with previous observations of the transformation of wild-type 26695 by donor J99 genomic DNA with the same cassette, but at a different (recG) recipient locus [integration of (3,393 ± 1,013)-bp donor DNA] (30).
Fig 7.

Length of imported DNA flanking the cat cassette following transformation with chromosomal DNA from donor H. pylori strains. The J99-derived donor HPXZ627 was used to transform H. pylori 26695 (A) or 26695-REd (B). The values above the bars indicate the distance (in bp) from the cat cassette to the recombination endpoints (red arrows), as determined by sequence analysis. The numbers reflect the distance from the nearest cassette boundary; the range reflects the locations of informative single-nucleotide polymorphisms (SNPs). Sequences corresponding to J99 are colored blue, those corresponding to 26695 are colored yellow, and those corresponding to 60190, which are from pAD1-Cat that flank the cat cassette, are colored red. All of the H. pylori 26695-strain specific HpyAII and HpyAV restriction sites on donor DNA are indicated. HpyAII cleavage sites (GAAGA N8/N7↓) are indicated by blue | lines, and HpyAV cleavage sites (CCTTCN6/N5↓) are indicated by red | lines. Within the ∼4-kb flanking region on each side of the cat cassette, 26 restriction sites (17 HpyAII sites and 9 HpyAV sites) are present in the downstream flanking region, and 16 restriction sites (15 HpyAII sites and 1 HpyAV site) are present in the upstream flanking region. A high-density region of restriction sites (with 7 HpyAII sites within 156 bp) is present ca. 1,744 to 1,899 bp upstream of the cat cassette and is indicated by an asterisk (*).
Table 2.
Comparison of lengths of imported flanking DNAs in homologous transformation of H. pylori strains 26695 and 26695-REd with donor DNA from strain J99 ureA::cat (generated by transforming J99 with pAD1-Cat)
| Category | Mean length (bp) ± SD for recipient straina |
|
|---|---|---|
| 26695 | 26695-REd | |
| Length of flanking DNA upstream of cat cassette | 1,537 ± 1,079 | 2,341 ± 1,467 |
| Length of flanking DNA downstream of cat cassette | 1,684 ± 1,132 | 861 ± 451 |
| Total length of imported DNA | 3,221 ± 1,546 | 3,202 ± 1,640 |
Mean of nine independent transformants.
Since restriction analysis indicates that J99 genomic DNA is digested by HpyAII and HpyAV but protected from digestion by HpyAIII and HpyAIV (Fig. 2), we then analyzed the HpyAII and HpyAV restriction of the donor DNA at this locus (the cat cassette ± the 4-kb flanking region) (Fig. 7). In total, 16 restriction sites, with a high-density region (7 HpyAII sites within 156 bp), are present upstream of the cat cassette; somewhat more (6 versus 3) 26695-REd transformants have upstream insertions beyond this high-density region than do the wild-type transformants (Fig. 7). Although for the transformants of both the wild-type and 26695-REd strains, the endpoints in upstream flanking DNA appear random, endpoints of the downstream flanking DNA are not. There are only five patterns of insertion for the nine 26695-REd independent transformants and seven patterns of insertion of the nine 26695-independent transformants (Fig. 7). Similar endpoint clusters of import DNA also were observed in transformations involving the rpsL (30) and rpoB loci (28), raising the possibility that the uneven distribution of strain-specific restriction sites affects donor DNA import into particular loci.
DISCUSSION
In this study, we examined the effects of type II restriction on H. pylori transformation by sequentially deleting the genes of the four active type II REs from H. pylori strain 26695 and then introducing exogenous DNA in different forms to the strains. Our observation that the RE-ablating mutations increased transformation frequencies by donor DNA originating from H. pylori strains corresponding to the R-M strain specificities of the strains confirmed that the restriction barriers function to suppress H. pylori natural competence and to help maintain strain-specific genetic integrity. Our findings also support the previously proposed concept that H. pylori interstrain R-M diversity prevents genome subversion by chromosomal DNA from competing strains (4). For DNA of non-H. pylori origin (e.g., E. coli), the barriers to transformation are substantially higher.
The transformation frequency of the wild-type strain by the 1.3-kb aphA cassette was ∼2.0 log10-fold lower than the 1.1-kb cat cassette (Fig. 3C and B), a finding consistent with the previous observation of transformation of 26695 strains by donor DNA with cat or aphA cassette at different loci (16). This marked difference may reflect that fewer sites recognized by the four active type II REs are present within the cat cassette (6 sites) than within the aphA cassette (21 sites) (Table 1). Another possibility is that other REs (type I and/or III) may differentially affect the exogenous cassettes. Identification of such alternative active endonucleases will further characterize H. pylori restriction. The results indicate that the constructed 26695-REd strain with lessened barriers to transformation will be useful for H. pylori genetic engineering, especially for genomic integration of exogenous DNA cassettes.
DNA import during horizontal gene transfer between different strains was investigated to evaluate the effects of strain-specific restriction, in a manner that may be relevant to in vivo conditions, since multiple H. pylori strains may cocolonize an individual host (15, 24, 37, 50) and, in some cases, different strains may coexist at a single biopsy site (24, 37). Upon comparing independent transformations of the wild-type and 26695-REd strains, restriction did not affect donor DNA import sizes (Table 2). The mean DNA import sizes of the wild-type and REd strains were nearly identical (3.2 ± 1.5 kb and 3.2 ± 1.6 kb, respectively). Another group recently showed that mutation of two type II restriction enzyme genes (Hpy188III and HpyCH4V) in H. pylori strain NSH57 led to statistically significant increases in donor DNA import sizes at the cagH locus (5.7 ± 2.7 kb in the RE double mutant versus 4.1 ± 2.4 kb in the wild-type strain NSH57) (19), although with considerable variation in import size (standard deviations = 2.7 and 2.4) in the two strains. Comparison of those results (19) to ours may suggest that restriction effects on DNA import sizes during natural transformation may be strain dependent or locus dependent, since other undefined factors involved appear to modify restriction effects.
Deletion of these four REs did not appear to enhance the growth characteristics of the H. pylori cells, confirming the utility of these engineered strains for future studies and indicating that the biological cost of carrying these genes is low in the presence of functioning methylases. That the deletions did not impair growth suggests H. pylori does not have alternative uses for DNA restriction other than for controlling incoming DNA. This contrasts with the subversion of methylases in introduced R-M systems that may provide regulatory functions for the host cells (2, 8, 41, 46). Considering that DNA methylation potentially affects bacterial cell cycle, DNA replication, repair, and mutation rates (23, 40, 41), the phenotypes we observed suggest that the targeted deletion of the RE genes most likely does not affect transcription of the adjacent DNA methylases. Although elements in upstream type II restriction genes (such as hpyIR [iceA]) also may affect H. pylori virulence in vivo through transcriptional regulation of the downstream DNA methylase (11, 14, 58), there is no evidence that the methylases of the four active type II R-M system REs affect H. pylori 26695 colonization. Similarly, the lack of effect on UV-induced or fluoroquinolone-induced DNA damage repair, or spontaneous mutation indicates that the endonuclease-induced double strand breaks are completely functionally suppressed by the paired endogenous methylases. These studies provide experimental verification of the hypothesized strong methylation phenotypes to control restriction (26, 45), and provide further rationale for the use of these engineered strains in future H. pylori research.
It should also be noted that all four active type II RE genes have immediate downstream genes with intergenic distances ranging from 86 to 432 bp. Our mRNA-based analysis indicated expression of these immediate downstream genes in the REd strain is not significantly different from that in the wild-type strain (<2-fold) when grown in nutrient-rich medium to mid-log phase (data not shown), which suggests deletion of the four RE genes may have no or only very limited polar effect on their downstream genes.
H. pylori strains (including strain 26695) not only have the active type II restriction-methylation systems but also contain restriction-methylation gene sequence remnants in their genome. Several display characteristics that indicate potential phase variation (31). It is possible that these phase-variable inactive restriction genes are in the process of disappearing, but they also may be reactivated and restore endonuclease activity. Whether abolishing active type II R-M systems would exert a selective pressure on those putative phase-variable inactive restriction genes awaits further investigation, using the REd strain as a tool. As such, our REd mutant strain also will be useful in R-M systems research in the perspective of H. pylori evolution.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grant R01GM63270.
We thank Leah Traube and Michael DiBenedetto for technical assistance and Jessica Stangel for assisting with the manuscript.
Footnotes
Published ahead of print 20 April 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Alm RA, et al. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180 [DOI] [PubMed] [Google Scholar]
- 2. Ando T, et al. 2010. Restriction-modification systems may be associated with Helicobacter pylori virulence. J. Gastroenterol. Hepatol. 25(Suppl 1):S95–S98 [DOI] [PubMed] [Google Scholar]
- 3. Ando T, et al. 2000. Restriction-modification system differences in Helicobacter pylori are a barrier to interstrain plasmid transfer. Mol. Microbiol. 37:1052–1065 [DOI] [PubMed] [Google Scholar]
- 4. Aras RA, Small AJ, Ando T, Blaser MJ. 2002. Helicobacter pylori interstrain restriction-modification diversity prevents genome subversion by chromosomal DNA from competing strains. Nucleic Acids Res. 30:5391–5397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atherton JC, Blaser MJ. 2009. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest. 119:2475–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baltrus DA, et al. 2009. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191:447–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baltrus DA, Guillemin K. 2006. Multiple phases of competence occur during the Helicobacter pylori growth cycle. FEMS Microbiol. Lett. 255:148–155 [DOI] [PubMed] [Google Scholar]
- 8. Collier J. 2009. Epigenetic regulation of the bacterial cell cycle. Curr. Opin. Microbiol. 12:722–729 [DOI] [PubMed] [Google Scholar]
- 9. Copass M, Grandi G, Rappuoli R. 1997. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect. Immun. 65:1949–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donahue JP, Israel DA, Peek RM, Blaser MJ, Miller GG. 2000. Overcoming the restriction barrier to plasmid transformation of Helicobacter pylori. Mol. Microbiol. 37:1066–1074 [DOI] [PubMed] [Google Scholar]
- 11. Donahue JP, et al. 2000. Analysis of iceA1 transcription in Helicobacter pylori. Helicobacter 5:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dorer MS, Talarico S, Salama NR. 2009. Helicobacter pylori's unconventional role in health and disease. PLoS Pathog. 5:e1000544 doi:10.1371/journal.ppat.1000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falush D, et al. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. U. S. A. 98:15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Figueiredo C, et al. 2000. Genetic organization and heterogeneity of the iceA locus of Helicobacter pylori. Gene 246:59–68 [DOI] [PubMed] [Google Scholar]
- 15. Ghose C, Perez-Perez GI, van Doorn LJ, Dominguez-Bello MG, Blaser MJ. 2005. High frequency of gastric colonization with multiple Helicobacter pylori strains in Venezuelan subjects. J. Clin. Microbiol. 43:2635–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorrell RJ, Yang J, Kusters JG, van Vliet AH, Robins-Browne RM. 2005. Restriction of DNA encoding selectable markers decreases the transformation efficiency of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 44:213–219 [DOI] [PubMed] [Google Scholar]
- 17. Heep M, et al. 2000. Mutations at four distinct regions of the rpoB gene can reduce the susceptibility of Helicobacter pylori to rifamycins. Antimicrob. Agents Chemother. 44:1713–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hofreuter D, Odenbreit S, Puls J, Schwan D, Haas R. 2000. Genetic competence in Helicobacter pylori: mechanisms and biological implications. Res. Microbiol. 151:487–491 [DOI] [PubMed] [Google Scholar]
- 19. Humbert O, Dorer MS, Salama NR. 2011. Characterization of Helicobacter pylori factors that control transformation frequency and integration length during inter-strain DNA recombination. Mol. Microbiol. 79:387–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Israel DA, Lou AS, Blaser MJ. 2000. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiol. Lett. 186:275–280 [DOI] [PubMed] [Google Scholar]
- 21. Israel DA, et al. 2001. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc. Natl. Acad. Sci. U. S. A. 98:14625–14630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jakobsson HE, et al. 2010. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 5:e9836 doi:10.1371/journal.pone.0009836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jeltsch A, et al. 2007. Application of DNA methyltransferases in targeted DNA methylation. Appl. Microbiol. Biotechnol. 75:1233–1240 [DOI] [PubMed] [Google Scholar]
- 24. Jorgensen M, Daskalopoulos G, Warburton V, Mitchell HM, Hazell SL. 1996. Multiple strain colonization and metronidazole resistance in Helicobacter pylori-infected patients: identification from sequential and multiple biopsy specimens. J. Infect. Dis. 174:631–635 [DOI] [PubMed] [Google Scholar]
- 25. Kang J, Huang S, Blaser MJ. 2005. Structural and functional divergence of MutS2 from bacterial MutS1 and eukaryotic MSH4-MSH5 homologs. J. Bacteriol. 187:3528–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi I. 2001. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29:3742–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kong H, et al. 2000. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 28:3216–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kulick S, et al. 2008. Mosaic DNA imports with interspersions of recipient sequence after natural transformation of Helicobacter pylori. PLoS One 3:e3797 doi:10.1371/journal.pone.0003797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levine SM, et al. 2007. Plastic cells and populations: DNA substrate characteristics in Helicobacter pylori transformation define a flexible but conservative system for genomic variation. FASEB J. 21:3458–3467 [DOI] [PubMed] [Google Scholar]
- 30. Lin EA, et al. 2009. Natural transformation of Helicobacter pylori involves the integration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 5:e1000337 doi:10.1371/journal.ppat.1000337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin LF, Posfai J, Roberts RJ, Kong H. 2001. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 98:2740–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mane SP, et al. 2010. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J. Bacteriol. 192:3078–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marshall DG, Dundon WG, Beesley SM, Smyth CJ. 1998. Helicobacter pylori: a conundrum of genetic diversity. Microbiology 144:2925–2939 [DOI] [PubMed] [Google Scholar]
- 34. Marsin S, et al. 2010. Genetic dissection of Helicobacter pylori AddAB role in homologous recombination. FEMS Microbiol. Lett. 311:44–50 [DOI] [PubMed] [Google Scholar]
- 35. McClain MS, Shaffer CL, Israel DA, Peek RM, Jr., Cover TL. 2009. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mobley HL, Mendez GL, Hazell SL. 2001. Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC: [PubMed] [Google Scholar]
- 37. Morales-Espinosa R, et al. 1999. Colonization of Mexican patients by multiple Helicobacter pylori strains with different vacA and cagA genotypes. J. Clin. Microbiol. 37:3001–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nobusato A, Uchiyama I, Kobayashi I. 2000. Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene 259:89–98 [DOI] [PubMed] [Google Scholar]
- 39. Oh JD, et al. 2006. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc. Natl. Acad. Sci. U. S. A. 103:9999–10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Radman M, Wagner R. 1984. Effects of DNA methylation on mismatch repair, mutagenesis, and recombination in Escherichia coli. Curr. Top. Microbiol. Immunol. 108:23–28 [DOI] [PubMed] [Google Scholar]
- 41. Reisenauer A, Kahng LS, McCollum S, Shapiro L. 1999. Bacterial DNA methylation: a cell cycle regulator? J. Bacteriol. 181:5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 43. Schilling CH, et al. 2002. Genome-scale metabolic model of Helicobacter pylori 26695. J. Bacteriol. 184:4582–4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma CM, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 45. Simala-Grant JL, Lam E, Keelan M, Taylor DE. 2004. Characterization of the DNA adenine 5′-GATC-3′ methylase HpyIIIM from Helicobacter pylori. Curr. Microbiol. 49:47–54 [DOI] [PubMed] [Google Scholar]
- 46. Skoglund A, et al. 2007. Functional analysis of the M. HpyAIV DNA methyltransferase of Helicobacter pylori. J. Bacteriol. 189:8914–8921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smeets LC, Bijlsma JJ, Boomkens SY, Vandenbroucke-Grauls CM, Kusters JG. 2000. comH, a novel gene essential for natural transformation of Helicobacter pylori. J. Bacteriol. 182:3948–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smeets LC, Kusters JG. 2002. Natural transformation in Helicobacter pylori: DNA transport in an unexpected way. Trends Microbiol. 10:159–162 [DOI] [PubMed] [Google Scholar]
- 49. Stingl K, Muller S, Scheidgen-Kleyboldt G, Clausen M, Maier B. 2010. Composite system mediates two-step DNA uptake into Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suerbaum S, Josenhans C. 2007. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 5:441–452 [DOI] [PubMed] [Google Scholar]
- 51. Suerbaum S, et al. 1998. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 95:12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takata T, et al. 2002. Phenotypic and genotypic variation in methylases involved in type II restriction-modification systems in Helicobacter pylori. Nucleic Acids Res. 30:2444–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taylor DE. 1992. Genetics of Campylobacter and Helicobacter. Annu. Rev. Microbiol. 46:35–64 [DOI] [PubMed] [Google Scholar]
- 54. Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 55. Tsuda M, Karita M, Nakazawa T. 1993. Genetic transformation in Helicobacter pylori. Microbiol. Immunol. 37:85–89 [DOI] [PubMed] [Google Scholar]
- 56. Wang G, Humayun MZ, Taylor DE. 1999. Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol. 7:488–493 [DOI] [PubMed] [Google Scholar]
- 57. Wang G, Maier RJ. 2008. Critical role of RecN in recombinational DNA repair and survival of Helicobacter pylori. Infect. Immun. 76:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu Q, Blaser MJ. 2001. Promoters of the CATG-specific methyltransferase gene hpyIM differ between iceA1 and iceA2 Helicobacter pylori strains. J. Bacteriol. 183:3875–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu Q, Morgan RD, Roberts RJ, Blaser MJ. 2000. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc. Natl. Acad. Sci. U. S. A. 97:9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.