

Abstract
Retroviral replicating vectors (RRVs) are a nonlytic alternative to oncolytic replicating viruses as anticancer agents, being selective both for dividing cells and for cells that have defects in innate immunity and interferon responsiveness. Tumor cells fit both these descriptions. Previous publications have described a prototype based on an amphotropic murine leukemia virus (MLV), encoding yeast cytosine deaminase (CD) that converts the prodrug 5-fluorocytosine (5-FC) to the potent anticancer drug, 5-fluorouracil (5-FU) in an infected tumor. We report here the selection of one lead clinical candidate based on a general design goal to optimize the genetic stability of the virus and the CD activity produced by the delivered transgene. Vectors were tested for titer, genetic stability, CD protein and enzyme activity, ability to confer susceptibility to 5-FC, and preliminary in vivo antitumor activity and stability. One vector, Toca 511, (aka T5.0002) encoding an optimized CD, shows a threefold increased specific activity in infected cells over infection with the prototype RRV and shows markedly higher genetic stability. Animal testing demonstrated that Toca 511 replicates stably in human tumor xenografts and, after 5-FC administration, causes complete regression of such xenografts. Toca 511 (vocimagene amiretrorepvec) has been taken forward to preclinical and clinical trials.
Introduction
Replicating oncolytic viruses proposed mode of action as therapeutic agents for cancer therapy1,2 is to lyse tumor cells as viral infection proceeds. At least three significant issues have limited the clinical success of these approaches: the adaptive immune response to vectors; the interaction and balance with the innate immune system; and infection of off-target (nontumor) tissues, with resulting toxicity.3,4,5 There have been several approaches to these challenges6,7,8 and some potential clinical benefit when infection of the entire tumor is not required.9 An alternative approach is the use of nonlytic replicating γ-retroviruses (retroviral replicating vectors, RRVs), based on murine leukemia virus (MLV),10,11,12,13 engineered to carry an exogenous, prodrug-activating transgene.14,15 These viruses infect dividing, malignant cells much more efficiently than resting or dividing nontransformed cells.15,16,17 In addition, MLV does not normally cause cell lysis or cellular pathology and hence is not inflammatory in nature, which, in turn, allows viral spread throughout a tumor. We recently demonstrated that, after intracranial injection, a prototype of this virus carrying the yeast cytosine deaminase (CD) gene infects and replicates through a brain cancer tumor in an orthotopic mouse glioblastoma xenograft model.15 Subsequent phased administration (1 week of 4) of the prodrug, 5-fluorocytosine (5-FC) which is converted, in the presence of CD, to 5-fluorouracil (5-FU), led to survival of 90% of treated mice at about 100 days, compared with controls that were moribund by about day 40.15 Further experiments with the same prototype virus in a syngeneic immune competent rat brain tumor model also showed efficacy.16 Such RRVs have a number of properties that may fit well with treatment of glioblastoma multiforme (GBM),18 a cancer with a 13-25% 2-year survival and a median life expectancy from diagnoses of 14.5 months (http://www.cbtrus.org/2007-2008/2007-2008/html),19 with current maximal therapies. The properties of RRV include the potential for a viral reservoir that can spontaneously reinfect growing tumor cells after initial infection, and the potential to replicate into and subsequently destroy tumor infiltrates in normal tissue that cannot be surgically removed. 5-FC is an antifungal drug that is orally bioavailable and efficiently crosses the blood brain barrier20 before conversion to 5-FU by the CD enzyme in infected cells. Locally injected 5-FU has been demonstrated to kill GBM tumors in human patients,21 and GBM cell lines are sensitive to 5-FU in vitro.22 However, systemic 5-FU is minimally effective against recurrent primary brain tumors and has marked toxicity,23 so 5-FU is not used as a treatment for GBM. The prototype RRV encoding CD reported by Tai et al.15 had two potential weaknesses for clinical use. First, although genomic stability of the virus was enough to show impressive efficacy in rodent models, increased stability is preferred for the treatment of the larger human tumors. Second, the CD activity appeared suboptimal as the native yeast CD protein is not very efficient at 37 °C24 and the use of yeast codons, rather than human, could limit protein synthesis rates. To address these issues, we built and tested various modifications of the original vector backbone of Logg et al.25 and inserted various modifications of the CD gene into the vector. New RRV therapeutic candidates have been tested for: stability over multiple rounds of replication; CD expression and activity in vitro; and facilitating cell killing with 5-FC in tissue culture. The lead vector, Toca 511 (T5.0002), showed surprisingly improved genomic stability in cell culture, confirmed by in vivo stability in a subcutaneous mouse xenograft model. Compelling efficacy was also observed in this model. This vector was taken forward into preclinical safety, biodistribution and further efficacy experiments26 in two syngeneic immune competent mouse tumor models. Toca 511 (vocimagene amiretrorepvec) is being investigated in clinical trials in the United States in subjects with recurrent high-grade glioma (see NCT01156584 and NCT01470794 at http://www.clinicaltrials.gov).
Results
Modification of vector backbone and insertion of CD gene in place of green fluorescent protein (GFP)
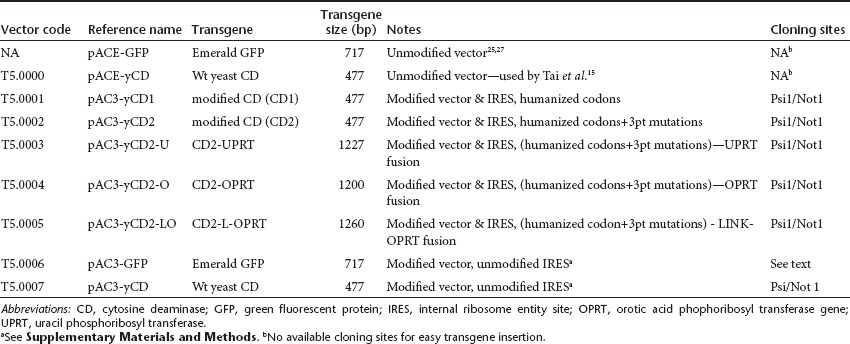
The backbone of the pACE-GFP and pACE-CD (T5.0000) plasmids25,27 was modified at six different sites (one through six, Figure 1) in the GFP plasmid to remove some repeats and unnecessary sequences and to facilitate transgene insertion. These sites were as follows. (i) The DNA sequence downstream of the Cla1 site in the pACE-yCD vector is originally derived from an ecotropic envelope and was changed to the amphotropic (4070A) envelope sequence. This does not change the amino acid sequence in the envelope, only the nucleic acid sequence. (ii) Small repeats on either side of the internal ribosome entity site (IRES)-CD cassette were eliminated to attempt to minimize instability because of homologous recombination. (iii) Four noncoding bases from the 3′ end of the MoMLV env gene region and just 5′ to the Not1 site in this vector were removed.(iv) Two base differences were incorporated in the U3 region of the pAC3 vector, which return the pAC3 vectors to the wild-type MoMLV sequence at these positions. The pACE vector carries two mutations previously inserted to create a unique Pme1 site. Through changes #3 and #4 the 3′ U3 and 3′ long terminal repeats (LTR) region of the pAC3 vector backbone is 100% identical to the 3′U3/LTR of MoMLV (NCBI Reference Sequence: NC 001501.1). (v) Seventy-four bases of the entire primer binding site (PBS) (here primer binding site) and additional 34 bases downstream of the PBS were removed from the 3′end of the 3′ LTR in the plasmid encoding the vector. This does not affect the structure of the retroviral genome and removes the possibility of recombination with the authentic PBS downstream of the 5′ LTR or packaging of 3′ LTR transcripts during the transient transfection to produce infectious vector. (vi) A unique restriction enzyme site Psi I was introduced at the 3′ end of the IRES for convenient insertion of transgene(s), as shown in Figure 1, and described in Materials and Methods and Supplementary Materials and Methods. Some of these backbone modifications have been previously briefly described in other vectors28 but not tested for stability. The resultant plasmid is pAC3-GFP (T5.0006, Table 1, Figure 1), and this plasmid was used as a basis for the vectors encoding CD and variants. We optimized the yeast codons in the CD gene for preferred human codon usage (designated yCD1) without changing the amino acid sequence and inserted it into the AC3 backbone to yield pAC3-yCD1 (T5.0001, see Table 1 and Supplementary Figure S1). The yeast enzyme is known to be optimally active at around 26 °C, but a yeast enzyme modified by alteration of three amino acids has previously been shown to be more active than wild type in bacteria at 37 °C.24 Therefore, we asked if these mutations in the humanized yeast gene (designated yCD2) would show a similar stabilization in human and mammalian cells, and constructed pAC3-yCD2 (T5.0002, Table 1, Figure 1). It has been reported that the fusion of the yeast gene for uracil phosphoribosyl transferase (UPRT; EC 2.4.2.9, the next enzyme in the yeast pyrimidine anabolic salvage pathway) to the CD gene leads to increased sensitivity to 5-FC in mammalian cells carrying the hybrid, compared with cells carrying the CD gene alone.29,30 We synthesized three additional CD hybrid salvage genes and inserted them into the same AC3 backbone (Table 1, Supplementary Figure S1). The first, yCD2-U, is a fusion of the CD2 gene with the yeast UPRT gene to generate pAC3-yCD2-U (T5.0003). The second, yCD-O, used the human orotic acid phophoribosyl transferase gene (OPRT; orotidine-5′- phosphoribosyl transferase; EC 2.4.2.10) fused to the CD2 gene to generate pAC3-yCD2-O (T5.0004). The human OPRT, normally a domain of a multifunctional protein, UMP synthase31 leads to production of UMP. OPRT normally converts 5-FU to 5-fluoro-uridine monophosphate during chemotherapy. Downregulation of this endogenous enzyme can cause 5-FU resistance in tumor cells.32 A second CD-OPRT hybrid gene was constructed with a 60-bp linker encoding an amino acid linker (SGGGASGGGASGGGASGGGA) between the CD and OPRT genes, giving the plasmid pAC3-yCD2-LO (T5.0005). Table 1 summarizes the descriptions of the various CD genes that were inserted into the pAC3 plasmid backbone, and Figure 1 shows the vector configurations of pACE-GFP, pAC3-GFP (T5.0006), and pAC3-yCD2 (T5.0002). The other vector configurations are shown in Supplementary Figure S1. Preparations of infectious vector particles, corresponding to all the vectors shown in Table 1, were obtained by transient transfection. Such preparations are stable for at least 2 months at −65 °C, and all virus preparations had titers of ~5 ×106 TU/ml.
Figure 1.

Alterations in the RVV backbone starting from pACE-GFP to pAC3-GFP (T5.0006) and introduction of the CD gene into pAC3-yCD2 (T5.0002). (a) The original GFP vector plasmid pACE-GFP with five different sites (numbered 1 through 5, and described in the text and Supplementary Materials and Methods) that are modified to produce pAC3-GFP; (b) pAC3-GFP with the improved vector backbone and the introduction of a Psi 1 site (numbered “6”) at the 3′ end of the IRES to allow easier insertion of new genes.; (c) pACE3-yCD2 (T5.0002) with the GFP gene swapped out for the codon optimized heat-stabilized CD gene (yCD2, see Table 2 and the text) using the Not 1 and Psi 1 sites. “CMV-R-U5” is the hybrid CMV-MLV LTR that allows good titers from transient transfection then disappears after a single round of replication; “PBS” is the tRNA (primer) binding site; “5′SS” and “3′SS” are splice donors and acceptors, respectively; “PPT” is the PolyPurine Tract; and “U3-R-U5” are the conventional regions of the retroviral LTR, that end up at the 5′end of the provirus after one round of replication. CD, cytosine deaminase; GFP, green fluorescent protein; IRES, internal ribosome entity site; LTR, long terminal repeat; MLV, murine leukemia virus; PBS, primer binding site.
Table 1. Properties of vectors built and tested in this investigation.
Genome stability over multiple cycles of viral replication
A key consideration in choice of vector was the stability of the genome over multiple cycles of replication. To test genomic stability, infectious virus was serially passaged on U87 cells as described in Materials and Methods for up to 12 serial passages. At the end of each passage, the genomic DNA of the cells was isolated and stored, and at the completion of the serial passages, PCR was performed on all the collected DNA samples using primers spanning the transgene in the provirus. The products were run on a gel to assess the integrity of the transgene. The results for four vectors are shown in Figure 2, and for four more in Supplementary Figure S2. The original prototype vector pACE-yCD (T5.0000), shown to be therapeutic in a mouse xenograft model, is stable out to about passage 5 or 6. The new vector construct, T5.0007 (pAC3-yCD), that has the same transgene (yCD) but an altered viral backbone is stable out to passage 7 or 8. T5.0001 (pAC3-yCD1) is markedly more stable than T5.0000 and T5.0007 and shows stability to passage 11 to 12. This shows that altering the coding sequence of CD while maintaining the same amino acids has, surprisingly, markedly stabilized the genome of the virus. In two such experiments carried out to 12 passages with T5.0001, a small amount of material appeared to run above the major amplified band and appeared at passages 9–12. It seems unlikely that the virus has acquired extra sequence, and the meaning of this observation is unclear. T5.0002 was at least as stable as T5.0001. The vectors with the hybrid gene inserts were less stable (see Supplementary Figure S2), so the majority of additional testing was performed on T5.0000 (pACE-yCD), T5.0007 (pAC3-yCD), T5.0001 (pAC3-yCD1), and T5.0002 (pAC3-yCD2). As T5.0004 and T5.0005 (CD-OPRT hybrids) appeared about as stable as T5.0000 (the original prototype therapeutic construct, see Supplementary Figure S2), some further testing was performed with these vectors. However, T5.0003 (CD-UPRT) was less stable than T5.0000 and was not further evaluated. The serial passage experiments were initially carried out to eight passages in two separate experiments and then to 12 passages in two further separate experiments. Results were consistent for relative stability. However, the amplified PCR band patterns observed in these repeat serial passage experiments were usually not the same as in preceding stability experiments (data not shown) suggesting that the instability is a stochastic process, in agreement with previous data showing that the instabilities can arise from repeats of 4 base pairs or smaller.33
Figure 2.

Stability comparisons of T5.0000, T5.0001, T5.0002, and T5.0007 (see Table 1) upon serial passage on human U87 glioblastoma cells. Passage conditions are described in the text. The figure shows the relative stability of the integrated proviruses at 12 serial passages (P1–P12) by displaying at each passage the PCR amplification product across the IRES transgene from DNA from infected cells after gel electrophoresis. The positive control amplified directly from the plasmid is shown on the far left of each panel, and a size marker is in the next lane, with sizes as shown. Bands with mobility greater than that of the positive control indicate the appearance of replicating viruses with deletions in the IRES-transgene region. The sizes of the amplified bands from the original plasmids are T5.0000: 1,040 bp; T5.0002: 1,038 bp; T5.0001: 1,041 bp; T5.0007: 1,039 bp. Corresponding stability tests for T5.0003, T5.0004, T5.0005, and T5.0006 are shown in the Supplementary Materials and Methods. IRES, internal ribosome entity site.
Expression of CD genes after infection of target cells in tissue culture
To assess the relative kinetics of infection, RNA production, and CD expression, U87 cells were infected at a multiplicity of infection (MOI) of 0.1, cells harvested every day for 6–7 days, RNA or cell extracts prepared, and RT-PCR and western blots performed to assess the relative levels of RNA and protein produced in the tissue culture cells as infection proceeded. The RT-PCR experiments confirmed the presence of increasing amounts of CD RNA in the infected cell cultures over time (data not shown), but as the primers for the humanized and wild-type genes were different, relative quantization was not attempted. Results for Western blots for CD protein production over time in cells infected with T5.0000, T5.0007, T50001, and T5.0002 are shown in Figure 3a. The CD protein levels increase over time for all vectors, but it is not possible to determine if there are quantitative differences between vectors from this experiment. To determine if there were enhancements of enzymatic activity with the modified vectors and in particular with the heat-stabilized CD encoded by T5.0002, we measured the specific activity of the CD in cell extracts of fully infected U87 cells (after 5 days of infection at a MOI of 0.1). The results are shown in Figure 3b and demonstrate that T5.0002 infected cells have a specific activity roughly three times the activity of the original therapeutic vector of Tai et al., T5.0000. The advantage of the humanized codon optimization is also shown in the increased specific activity of T5.0001 versus T5.0007 (which, in turn, were both higher than the original vector with wild-type CD). Therefore, the combination of vector backbone improvements, thermostabilizing point mutations, and human codon optimization of the CD gene (T5.0002) resulted in enhanced specific enzymatic activity relative to prototype vector encoding the wild-type yeast CD (T5.0000).
Figure 3.

CD protein in infected cells. (a) Western blot analyses of the appearance of CD protein from the vectors shown over time (days as indicated on the blots) after virus infection of U87cells at an MOI of 0.1 on day 0. The control lane (C) shows 0.3 µg of purified yeast CD Recombinant Yeast Cytosine Deaminase (Calzyme, Lot no. 6-1-53); marker molecular weights in kiloDaltons are as shown; (b) Specific enzyme activity of CD in U87cell extracts at 37 °C, at 5 days post-infection at an MOI of 1. CD, cytosine deaminase; MOI, multiplicity of infection.
Efficiency of cell killing of infected cells by 5-FC
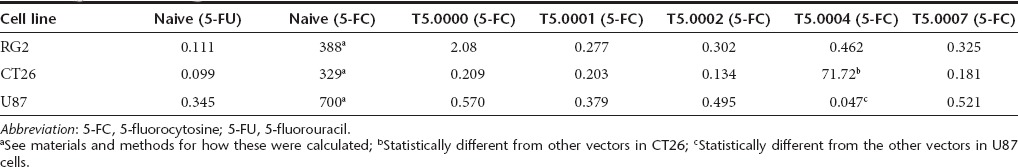
In order to compare the cell killing activity of different vectors in the presence of 5-FC, we examined three cell types: the rat glioblastoma cell line RG2, the mouse colorectal tumor cell line CT26, and the human glioblastoma cell line U87. Cells were fully infected with the relevant vector (minimum 14 days growth after infection with a MOI of 1) and stored as a frozen bank. Cells were plated on day 0 and 5-FC added at different concentrations after 24 hours. The number of live cells was monitored by cell harvest and MTS assay every 2 days. At 5-FC concentrations where cell death occurred, it took about 4 days for the toxicity to be observed by MTS assay. The largest differences in cell numbers between vectors were seen at day 8. Killing curves for naive cells and cells infected with T5.0000, T5.0001, T5.0002 T5.0004, and T5.0007 vectors in the three cell lines at day 8 are shown in Figure 4a–c. Experiments were performed in triplicate, and the three points were, in almost all cases, within the size of the data point on the graphs. The growth kinetics of infected CT26 cells were not affected by infection compared to the parent cell line, but this was not true for RG2 and U87 cells. This could be seen when we plotted our data from 2–8 days for individual cell lines without 5-FC (data not shown). This can also be seen in the differing 8-day cell numbers at 5-FC concentrations that were not toxic, in Figure 4. The extent of these growth differences did not appear to be linked to the vector identity at least across the RG2 and U87 cell lines and likely represents a nonspecific effect, which may also be observed in vivo (see Figure 6 and associated results). A four-parameter curve fit was performed (shown as the lines in the graphs for Figure 4a–c) and IC50 values calculated. The values for these five vectors are shown in Table 2. As expected, all the uninfected cells are highly resistant to 5-FC, and highly susceptible to 5-FU. For the RG2 line, it appears that cells infected with T5.0000 are less susceptible to 5-FC mediated killing (IC50 = 2.08 µg/ml) than RG2 cells infected with T5.0002 (IC50 = 0.302 µg/ml) or T5.0001 (IC50 = 0.277 µg/ml). Vectors T5.0007 and T5.0004 also appear more active (as measured by the IC50 for 5-FC-mediated cell killing) than T5.0000 in RG2 cells. These differences are not statistically significant, but it has previously been suggested that the CD expression levels with T5.0000 (pACE-CD) in the RG2 line are considerably lower than in U87cells.16 This may allow the difference in efficiency of protein expression and stability to be seen in RG2 cells, but not in U87 cells. Nevertheless, this difference in vector activity between U87 and RG2 cells is not obvious for the other vectors. Relative expression levels of the CD protein in RG2 and CT26 lines have not been investigated further, unlike for the U87 line (Figure 3). In the vector infected cells, only two vector differences in the same cell line are statistically significant. First, the CD-OPRT hybrid vector (T5.0004) is clearly inferior to other vectors in 5-FC-mediated killing of CT26 cells but this is not so of RG2 or U87cells. This effect may be due to inferior vector stability in mouse cells,11,34 or to differences in pyrimidine metabolism. Second, T5.0004 made U87cells more sensitive to 5-FC than the other vectors, and this could reflect the reduced sensitivity of U87 cells to 5-FU as shown in Table 2, which could be related to decreased 5-FU salvage, which is then compensated by the OPRT function in T5.0004. However, the relative instability of T5.0004, compared with T5.0002 as shown in Supplementary Figure S2, the large therapeutic window already observed for the CD vectors and the potential for reduction in metabolic cooperation suggest that this vector is not the optimal choice for preclinical development. The reduction in metabolic cooperation refers to the issue that the protein made by T5.0004 should lead to direct formation of 5-5-fluoro-uridine monophosphate, without significant release of 5-FU. 5-5-fluoro-uridine monophosphate cannot readily diffuse across cell membranes, whereas 5-FU can and then can kill neighboring uninfected cells. Therefore, there is a potential for less overall cell killing with partial infection, using a vector that converts 5-FC directly to 5-UMP compared with one that makes only 5-FU.35 The most important observation is that the therapeutic 5-FC serum concentrations (40–80 µg/ml) normally achieved with the top antifungal label dosing of 5-FC in humans (150 mg/kg/day) allows for cerebral fluid (CSF) concentrations (assuming 80% blood brain barrier penetration of 5-FC, per the drug package insert—Ancobon, http://www.accessdata.fda.gov/scripts/cder/drugsatfda/) that are 65- to 130-fold above the IC50 values observed in U87 cells using the clinical candidate vector, T5.0002. The experiment with U87 cells and these vectors, excluding T5.0004, was repeated with the same results. The CT26 and RG2 cell line experiments were not directly repeated. We used a similar experimental system to determine the necessary duration of exposure to 5-FC to achieve 100% cell killing as a function of 5-FC concentration, for U87 cells infected with T5.0002 (Figure 4d). These cells were seeded in 96 well dishes and the same day treated with fresh media with various concentrations of 5-FC (day 0). The 5-FC was withdrawn at days 1, 3, and 5, and the cells allowed to grow without 5-FC to day 8. One further set of cells were kept under 5-FC selection for the full 8 days. On day 8, the cells were assayed for viability by MTS (Figure 4d). As can be seen, the single day 5-FC exposure is already quite efficient in killing the cells, and a four-parameter curve fit shows that 3, 5, and 8 days exposure are statistically indistinguishable (data not shown), although qualitatively 5 and 8 days exposure appear the most effective. Therefore, in tissue culture, 5-FC exposures as short as 5 days are as efficient as longer exposure periods.
Figure 4.

5-FC-mediated killing of infected tissue culture cells. (a) rat RG2 glioma cells transduced with the different vectors indicated and grown for 8 days in different 5-FC concentrations, as shown; (b) mouse CT26 colorectal tumor cells transduced with the different vectors indicated and grown for 8 days in different 5-FC concentrations, as shown; (c) human U87glioma cells transduced with the different vectors indicated and grown for 8 days in different 5-FC concentrations as shown; (d) T5.0002-transduced U87glioma cells treated with 5-FC at different concentrations for different periods of time (1–8 days), and all experiments grown out to 8 days before assessing overall cell growth using the MTS assay. Curves shown in a–c were generated using a four-parameter nonlinear fit for the 5-FC titration points (see Materials and Methods). The lines in d are drawn between the experimental points to allow easy differentiation of the data sets. The 1 mmol/l 5-FU points in all these figures is derived from a titration of the cell lines against 5-FU performed in the same manner as the 5-FC titration, and variation for the different vectors was within the size of the 1 mmol/l 5-FU data point for the different vectors. 5-FC, 5-fluorocytosine; 5-FU, 5-fluorouracil.
Figure 6.

Stability of T5.0002 in vivo in a human xenograft in nude mice. Tumors from five surviving mice that received CD vector (T5.0002) but no 5-FC in the experiment in Figure 5a were excised during killing (day 38), DNA prepared and the DNA examined using the PCR stability assay used in Figure 2 for retention of full-length IRES CD insert. The marker is shown on the left side and the positive control is U87 cells infected with the CD vector T5.0002 in the next lane. Numbers at the top are the numbers of the individual mice. CD, cytosine deaminase; IRES, internal ribosome entity site; 5-FC, 5-fluorocytosine.
Table 2. IC50 values in micrograms/ml for 5-FU and 5-FC in different cell lines infected with different vectors.
In vivo activity of T5.0002
The T5.0002 vector appeared to be at least as stable as T5.0001, and more stable than other vectors on serial passage; to provide the highest specific CD activity in infected human U87 glioma cells; and to efficiently kill cells in culture after short-term 5-FC exposure. In order to confirm the utility of this vector, we tested its antitumor activity in a U87 subcutaneous model in athymic mice. Uninfected U87 cells were mixed with U87 cells infected with T5.0002 in the ratio: uninfected/infected = 98/2, and injected subcutaneously in athymic mice; 100% uninfected U87 cells were also injected in additional athymic mice as controls. Groups were targeted at 10 mice/group, and a few groups had less, with the minimum number being eight. The tumors were allowed to grow until easily palpable and 5-FC therapy initiated continuously for up to 28 days. Figure 5 shows the outcome of an experiment with 5-FC dosing at 500 mg/kg once per day (SID), 200 mg/kg SID, and 50 mg/kg SID. Strikingly, all three 5-FC dosing regimens in conjunction with the vector show complete tumor regression, and the tumors with no vector treatment do not respond to 5-FC. Tumors with vector and no 5-FC do grow more slowly, but eventually expand. A GFP vector control (T5.0006) in Figure 5a shows that this vector also exhibits a growth inhibitory effect, so this effect is not associated with CD expression. This “vector only” slowing of tumor growth is not seen in all models, for example not in models using the CT26 cells.26 Therefore, this phenomenon may reflect the in vitro differences in growth of vector-transduced cells seen for U87 and RG2 cell lines and noted above. It is possible that blockage of the amphotropic receptor (the phosphate transporter PIT-2) by the infecting viruses causes this slowing,36 and this could be cell line dependent.
Figure 5.

T5.0002 spreads through subcutaneous tumors and is therapeutic in conjunction with 5-FC. U87 cells were implanted subcutaneously in nude mice with or without 2% of total cells infected with T5.0002, and tumor growth measured in the presence and absence of 5-FC administration: (a) 500 mg/kg/day 5-FC, T5.0006 (encoding GFP) and T5.0002 + PBS are the vector-only controls; (b) 200 mg/kg/day, T5.0004 + PBS is the vector only control; (c) 50 mg/kg /day 5-FC, T5.0007 + PBS is the vector only control. Error bars are standard deviations; each group had 8–10 mice. 5-FC, 5-fluorocytosine; GFP, green fluorescent protein; PBS, primer binding site.
In vivo stability of the T5.0002 vector
To check the stability of this candidate RRV in vivo, we performed PCR experiments on DNA from U87 human tumor cells implanted in mice that received T5.0002 but not 5-FC in the experiment shown in Figure 5a and harvested at day 38. We performed PCR across the CD gene and IRES as in the in vitro passage experiment (Figure 2). An intact CD gene gives a gel band of about 1.2 kb. The results of this experiment are shown in Figure 6. In all the tumor tissues (N = 5), the vector appears to be stable with the majority of the staining the same size as the control which was amplified from the original plasmid. Some faint bands can be seen at lower positions, in particular for mouse 774. It should be remembered that smaller sequences will be preferentially amplified. We conclude that T5.0002 is largely stable during infection and amplification in vivo in this mouse xenograft model.
Discussion
These experiments describe the optimization and testing of a number of RRV vector constructs encoding CD enzymes derived from the yeast enzyme. A large change in stability of the vectors was observed with coding sequence variations for the same protein. Stability studies with vectors such as the ones used in this report have shown that stability can be highly gene dependent and, for example, the native hygromycin resistance gene is extremely unstable.33 This instability is unlikely to be associated simply with gene size as the coding sequence for the hygromycin resistance gene is only 1,023 bp, comparable in size to genes that were much more stable. One tantalizing concept is that the instability of the foreign transgene results from some as yet uncharacterized mechanism for host cell resistance to foreign genes. So a general strategy would be to vary the codon usage to find highly stable versions of a gene.
The specific activity of the protein from the yCD2 gene was better than those from the other modified and unmodified CD genes, but this improvement was not easily seen in the 5-FC tissue culture cell-killing experiments. We confirmed previous observations that the T5.0000 vector was less efficient in RG2 than in U87 cells,16 but interestingly this seems to be an effect that is specific to this vector. As this vector seems inferior in other ways, such as stability, it is not a candidate for clinical use. The reasons for the marked difference in CD-mediated killing of the RG2 and U87 cell lines with T5.0000 have not been further investigated.
Given the expected use of RRV is for experimental therapy for primary brain cancer in human clinical trials, it seemed advantageous to use a vector which produces more CD activity in human glioma cells. Mouse model tumors may rapidly become nearly 100% infected;15,26 however, it may be more difficult to obtain 100% infection of a human tumor for a patient with GBM. In the context of incomplete intratumoral viral spread, efficacy may be more dependent on the rate of 5-FC conversion to 5-FU in infected tumor cells, and the 5-FU diffusing to, and killing, nearby noninfected dividing tumor cells—the so-called “bystander effect”.37 It is unclear whether, even if there were demonstrably better individual cell killing with the CD-UPRT and CD-OPRT hybrid genes, this would be helpful therapeutically.35 The planned therapeutic strategy relies on efficiently infecting tumors, allowing the virus to replicate, dosing with 5-FC to kill dividing infected tumor cells, then allowing a “rest” period where the virus can replicate and infect any residual dividing, and as yet uninfected tumor cells.15 Highly efficient 5-FC salvage directly to phosphorylated nucleotides may hinder 5-FU diffusion and the by-stander effect or could initially kill off most infected cells, hindering efficient infection of dividing uninfected tumor cells during the 5-FC “rest” period. Such reduced antitumor activity in vivo for hybrid CD-UPRT genes that are more efficient in vitro has been observed by others.35
The 5-FC IC50 experiments show that the vectors examined here would be able to efficiently convert 5-FC to 5-FU in infected human glioblastomas in vivo, as the IC50 values of U87 cells in vitro are approximately two orders of magnitude (estimated 65- to 130-fold) less than the expected 5-FC concentration in the brain of subjects taking therapeutic antifungal doses of 5-FC. Vector T5.0002 is much more stable than the original prototype RRV vector, pACE-yCD (T5.0000), in human U87 cells in culture, and T5.0002 has the best specific activity of CD protein in fully infected U87 cells. In order to further test this vector, we performed in vivo experiments with subcutaneous human U87 xenografts, and the vector appears very capable of rapid spread and therapeutic effect (cell killing) in the presence of a wide range of 5-FC doses. In addition, the IRES-CD insert in the T5.0002 vector is stable in vivo in human tumors grown in athymic mice. Therefore, this vector was chosen as the candidate therapeutic agent to go forward into further preclinical and clinical trials and was renamed Toca 511 (USAN name vocimagene amiretrorepvec).
Materials and Methods
Plasmid constructs. The various constructs used in this study are described in the text—Results, Table 1, Figure 1 and Supplementary Figure S1. Additional details are given in the Supplementary Materials and Methods.
Cells and tissue culture. The U87 cell line (ATCC no.HTB-14) is a human glioblastoma line; the PC3 cell line (ATCC no. CRL-1435) is a human prostate cancer line; the CT26 cell line (ATCC no. 2639) is a mouse colon adenocarcinoma; and the RG2 cell line (ATCC no. CRL-2433) is a rat glioblastoma line. Except where otherwise stated, cell lines were obtained from ATCC, Manassas, VA. RG2 and CT26 cells were originally obtained from the laboratory of Carol Kruse (Sidney Kimmel Cancer Institute, La Jolla, CA); 293T cells were licensed from Stanford University. Tissue culture cells were grown in Dulbecco's Modified Eagle's Medium (DMEM), high glucose supplemented with added 1 mmol/l NaPyruvate (Hyclone), 10% fetal bovine serum (Hyclone), and 200 mmol/l Glutamax (Invitrogen 35050-061). Cells were normally carried without antibiotics but for experiments where there were extensive manipulations, antibiotics (Penicillin:Streptomycin at 1 U/ml:1 µg/ml) were added. 5-FC was USP grade and was contract manufactured for Tocagen by Nantong Jinghua Pharmaceutical Company. 5-FU (Cat no. F6627) was purchased from Sigma Chemicals.
Infectious vector production and titer determination. Infectious RVVs were prepared by standard transient transfections of 293T cells with calcium phosphate precipitates of the relevant plasmid DNA preparations. The resultant vector preparations were tittered on human PC3 cells by single cycle infection of a dilution series of the vector. The single cycle was guaranteed by AZT treatment, followed by qPCR of target cell DNA specific for integrated viral vector to count the number of integrated proviruses. The titer of the original stock was then calculated from the linear portion of the dilution curve. Further details are provided in Supplementary Material and Methods.
Serial passage stability studies. Approximately 105 naive U87 cells were initially infected with the viral vectors at a MOI of 0.1 and grown for 1 week to complete a single cycle of infection. 15 µl of the 1 ml of supernatant was then repassaged onto uninfected cells and the cycle repeated. Genomic stability of the yCD2 sequence was assessed by PCR amplification of the integrated provirus from the infected cells using MLV-specific primers flanking the transgene insertion site. DNA was prepared from each set of infected cells as for the qPCR titer assay. PCR was performed using the following primers: UCLA 5-127 5′-CTGATCTTACTCTTTGGACCTTG-3′ UCLA 3-37 5′-CCCCTTTTTCTGGAGACTAAATAA-3′ which amplify an ~1.2-kb fragment in T5.0000 and related plasmids that span the CD transgene. PCR products were visualized by running the reaction products on a 1% agarose gel. The appearance of any bands, smaller or larger than full-length amplicon, could be an indicator of vector instability.
RT-PCR and western blot measurements on RNA and cell lysates from infected U87cells. U87 cells were harvested from T25 flasks after infection at about 20% confluence and at a MOI of 0.1 on day 0. At days 1, 2, 3, 4, 5, 6, and 7, cells from T25 flasks containing U87 cells transduced with the different vectors (T5.0000, T5.0001, T5.0002, and T5.0007), and noninfected U87cells were harvested. Cell extracts for western blots and total RNA were prepared for the samples as described in the Supplementary Materials and Methods. Western blots were developed using a commercially available sheep polyclonal antibody against yeast CD (ABCAM, Cat. no. ab35251) as the primary antibody.
CD activity. CD activity was measured by conversion of 5-FC to 5-FU by infected cell extracts. Cells were resuspended at 106 cell/ml in PBS after 5 days infection at a MOI of 0.1 and lysed by freeze/thaw (3×) in PBS plus protease inhibitor. Debris was removed by centrifugation and aliquots of soluble cell extract stored at 4 °C. Protein concentrations were measured using the Pierce BCA Protein Assay Kit, Pierce (Cat. no. PI23227); 1 ml CD enzyme reaction volumes had 200 µl of cell extract and 0.1 mg/ml 5-FC in PBS. The reaction was typically allowed to run for 60' to 180', and 5 µl samples were removed at various time points and injected onto an high-performance liquid chromatography (HPLC) column. 5-FU was detected by HPLC using a solid-phase Hypersil BDS C18 4.0 × 250 mm HPLC column (5 µ sphere size) and a mobile phase that was 95% 50 mmol/l ammonium phosphate, pH 2.1 containing 0.01% tert-butylammonium perchlorate; 5% methanol. HPLC runs were at 1 ml/minute mobile phase for 5′, and the Photo Detector Array (PDA) was set to scan from 190 to 350 nm with the chromatogram displaying 264 and 285 nm, Λmax for 5-FU and 5-FC, respectively. The conversion rates of 5-FC to 5-FU were calculated by comparing the peak areas with known amounts on a previously generated standard curve of 5-FU. The rate of 5-FC conversion to 5-FU was derived by plotting the amount of 5-FU generated against its corresponding time interval.
The specific activity of the cell lysate was calculated by dividing the conversion rate (nmol/min) by the amount of protein used in the assay in mg.
5-FC sensitivity of vector-transduced cell lines in tissue culture. RG2, CT26, and U87 cell lines were fully infected with relevant vectors by 14 days of infection and growth, expanded, and then were stored as a bank of frozen cells. Cells were thawed and seeded at 1,000 cells/well in 96-well plates. They were monitored over an 8-day period following treatment with various concentrations of 5-FC, which were first added 1 day after plating and then replenished with whole medium plus 5-FC every 2 days. Cell growth kinetics was assessed every 2 days utilizing Promega's CellTiter 96 AQueous One Solution reagent (MTS). Following the addition of the MTS, OD490 readings were made on the Spectra Max 190 reader at 60-minute post-MTS addition. All time points for each cell line and each concentration were performed in triplicate and the average calculated and plotted to give 5-FC killing profiles. The spread of the data points were smaller than the size of the data points in Figure 4. IC50 values and 95% confidence limits were calculated using a Graphpad (San Diego) Prizm program for nonlinear four-parameter fit of the data points. Statistical significance difference was defined as when the 95% confidence limits of the IC50 values did not overlap. The nontransduced lines were very resistant to 5-FC killing, so in order to obtain an approximate value for the IC50 from the curve fit for these lines only, the 1 mmol/l 5-FU OD value was assumed to represent the baseline with 100% killing and used as a data point equivalent to 100 mmol/l 5-FC. This fit is not shown in Figure 4a–c. Similar experiments, with varying 5-FU, were performed to determine the IC50 of the nontransduced cell lines for 5-FU, but only the single 1 mm 5-FU concentration is shown in Figure 4.
In vivo tumor experiments in nude mice
Mice. Four- to eight-week-old female nude mice (Hsd:Athymic Nude-Foxn1nu, Harlan, IN) were bred and maintained under specific pathogen-free conditions, and all studies conducted under protocols approved by the University of California at Los Angeles Animal Research Committee (UCLA) or the Explora Scientific Animal Use Committee (Tocagen).
Cells and vector. Uninfected parental U87cells (98%) were mixed with pretransduced U87 cells (2%), and these cell mixtures (2 × 106 cells in 100 µl DMEM or PBS) were subcutaneously injected into the flank of each mouse. Intraperitoneal administration of 5-FC (500, 200, or 50 mg/kg, daily, corresponding to 800, 320, or 80 µl of a 12.5 g/ml aliquoted stock preparation of 5-FC stored at room temperature) or PBS (800, 320, or 80 µl, daily) was started when the tumors reached ~5 mm in diameter and continued for up to 28 days. Groups of mice were targeted to be 10 individuals, but some groups had nine or eight depending on experimental variation such as tumor take. Tumor sizes were measured every few days, and tumor volumes were calculated by this formula: volume = length × width2/2.
Statistical analyses. Statistical analyses were carried out with Student's t test or one-way ANOVA to determine significance. P values of <0.05 were considered statistically significant in all analyses, which were performed using the GraphPad Prism 5 software package (GraphPad Software, La Jolla, CA).
PCR analyses of tumor tissue. Tumor tissue was harvested from animals that were killed when the tumor reached a size of 2–5 cm in the T5.0002/no 5-FC groups shown in Figure 5a at day 38. DNA was prepared using the Promega Maxwell 16 Instrument and associated cartridges for tissue preparation, and concentration determined from OD260 measurement on the Nanodrop spectrophotometer. PCR was performed to test for retention of full-length CD gene using the same primers and procedures as used for the in vitro vector passage and stability studies. PCR products were visualized by running the reaction products on a 1% agarose gel.
SUPPLEMENTARY MATERIAL Figure S1. Configuration of plasmid constructs encoding RRV's constructed from Moloney MLV with an amphotropic envelope and various CD and Hybrid CD genes. Figure S2. Stability profile of vectors by serial passage. Supplementary Materials and Methods.
Acknowledgments
We thank Debra Gessner, Dan Pertschuk for discussions on clinical feasibility for the use of Toca 511 and Tocagen Inc. for providing financial and general support. O.D.P., O.D., R.B., D.J., A.L., C.B., C.I., H.E.G., and D.J.J. are full-time employees of Tocagen Inc. K.A., D.J., T.B., and D.L. are former employees of Tocagen. C.R.L. and N.K. are paid consultants to Tocagen and Tocagen provided contract support to UCLA for some experiments described here. This work was supported in part by grants to Tocagen from Accelerate Brain Cancer Cure (ABC2), the National Brain Tumor Association and the American Brain Tumor Association. Support was also previously provided by U.S. National Institutes of Health Grants R01-CA105171 (to N.K.) and P01-CA59318 (to N.K.)
Supplementary Materials
Configuration of plasmid constructs encoding RRV's constructed from Moloney MLV with an amphotropic envelope and various CD and Hybrid CD genes.
Stability profile of vectors by serial passage.
References
- Kelly E., and, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- Stanford MM, Bell JC., and, Vähä-Koskela MJ. Novel oncolytic viruses: riding high on the next wave. Cytokine Growth Factor Rev. 2010;21:177–183. doi: 10.1016/j.cytogfr.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Barral PM, Sarkar D, Su ZZ, Barber GN, DeSalle R, Racaniello VR.et al. (2009Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity Pharmacol Ther 124219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parato KA, Lichty BD., and, Bell JC. Diplomatic immunity: turning a foe into an ally. Curr Opin Mol Ther. 2009;11:13–21. [PubMed] [Google Scholar]
- Naik S., and, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9:1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E.et al. (2010Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production Proc Natl Acad Sci USA 1071576–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo JS, Le Boeuf F, Lai F, Cox J, Vaha-Koskela M, Abdelbary H.et al. (2010A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers Mol Ther 181123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Boeuf F, Diallo JS, McCart JA, Thorne S, Falls T, Stanford M.et al. (2010Synergistic interaction between oncolytic viruses augments tumor killing Mol Ther 18888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G.et al. (2009Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma J Clin Oncol 275763–5771. [DOI] [PubMed] [Google Scholar]
- Logg CR, Tai CK, Logg A, Anderson WF., and, Kasahara N. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 2001;12:921–932. doi: 10.1089/104303401750195881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paar M, Klein D, Salmons B, Günzburg WH, Renner M., and, Portsmouth D. Influence of vector design and host cell on the mechanism of recombination and emergence of mutant subpopulations of replicating retroviral vectors. BMC Mol Biol. 2009;10:8. doi: 10.1186/1471-2199-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerner LJ, Schwantes A, Schneider IC, Cichutek K., and, Buchholz CJ. Cell entry targeting restricts biodistribution of replication-competent retroviruses to tumour tissue. Gene Ther. 2008;15:1500–1510. doi: 10.1038/gt.2008.92. [DOI] [PubMed] [Google Scholar]
- Solly SK, Trajcevski S, Frisén C, Holzer GW, Nelson E, Clerc B.et al. (2003Replicative retroviral vectors for cancer gene therapy Cancer Gene Ther 1030–39. [DOI] [PubMed] [Google Scholar]
- Dalba C, Klatzmann D, Logg CR., and, Kasahara N. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 2005;5:655–667. doi: 10.2174/156652305774964659. [DOI] [PubMed] [Google Scholar]
- Tai CK, Wang WJ, Chen TC., and, Kasahara N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005;12:842–851. doi: 10.1016/j.ymthe.2005.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Tai CK, Kershaw AD, Solly SK, Klatzmann D, Kasahara N.et al. (2006Use of replication-competent retroviral vectors in an immunocompetent intracranial glioma model Neurosurg Focus 20E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavaty J, Jandl G, Liszt M, Petznek H, König-Schuster M, Sedlak J.et al. (2011Comparative evaluation of preclinical in vivo models for the assessment of replicating retroviral vectors for the treatment of glioblastoma J Neurooncol 10259–69. [DOI] [PubMed] [Google Scholar]
- Brandes AA, Tosoni A, Franceschi E, Reni M, Gatta G., and, Vecht C. Glioblastoma in adults. Crit Rev Oncol Hematol. 2008;67:139–152. doi: 10.1016/j.critrevonc.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Block ER., and, Bennett JE. Pharmacological studies with 5-fluorocytosine. Antimicrob Agents Chemother. 1972;1:476–482. doi: 10.1128/aac.1.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menei P, Capelle L, Guyotat J, Fuentes S, Assaker R, Bataille B.et al. (2005Local and sustained delivery of 5-fluorouracil from biodegradable microspheres for the radiosensitization of malignant glioma: a randomized phase II trial Neurosurgery 56242–8; discussion 242. [DOI] [PubMed] [Google Scholar]
- Miller CR, Williams CR, Buchsbaum DJ., and, Gillespie GY. Intratumoral 5-fluorouracil produced by cytosine deaminase/5-fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res. 2002;62:773–780. [PubMed] [Google Scholar]
- Cascino TL, Veeder MH, Buckner JC, O'Fallon JR, Wiesenfeld M, Levitt R.et al. (1996Phase II study of 5-fluorouracil and leucovorin in recurrent primary brain tumor J Neurooncol 30243–246. [DOI] [PubMed] [Google Scholar]
- Korkegian A, Black ME, Baker D., and, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logg CR, Logg A, Matusik RJ, Bochner BH., and, Kasahara N. Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J Virol. 2002;76:12783–12791. doi: 10.1128/JVI.76.24.12783-12791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag D, Amundson KK, Lopez Espinoza F, Martin B, Buckley T, Galvão da Silva AP.et al. (2012Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector Neuro-oncology 14145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WJ, Tai CK, Kasahara N., and, Chen TC. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 2003;14:117–127. doi: 10.1089/104303403321070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Hiraoka K, Kasahara N., and, Logg CR. Optimization of enzyme-substrate pairing for bioluminescence imaging of gene transfer using Renilla and Gaussia luciferases. J Gene Med. 2010;12:528–537. doi: 10.1002/jgm.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F.et al. (2000In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene Cancer Res 603813–3822. [PubMed] [Google Scholar]
- Bourbeau D, Lavoie G, Nalbantoglu J., and, Massie B. Suicide gene therapy with an adenovirus expressing the fusion gene CD::UPRT in human glioblastomas: different sensitivities correlate with p53 status. J Gene Med. 2004;6:1320–1332. doi: 10.1002/jgm.611. [DOI] [PubMed] [Google Scholar]
- Suttle DP, Bugg BY, Winkler JK., and, Kanalas JJ. Molecular cloning and nucleotide sequence for the complete coding region of human UMP synthase. Proc Natl Acad Sci USA. 1988;85:1754–1758. doi: 10.1073/pnas.85.6.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto E, Nagase H, Kobunai T, Oie S, Oka T, Fukushima M.et al. (2007Orotate phosphoribosyltransferase expression level in tumors is a potential determinant of the efficacy of 5-fluorouracil Biochem Biophys Res Commun 363216–222. [DOI] [PubMed] [Google Scholar]
- Logg CR, Logg A, Tai CK, Cannon PM., and, Kasahara N. Genomic stability of murine leukemia viruses containing insertions at the Env-3' untranslated region boundary. J Virol. 2001;75:6989–6998. doi: 10.1128/JVI.75.15.6989-6998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paar M, Schwab S, Rosenfellner D, Salmons B, Günzburg WH, Renner M.et al. (2007Effects of viral strain, transgene position, and target cell type on replication kinetics, genomic stability, and transgene expression of replication-competent murine leukemia virus-based vectors J Virol 816973–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AJ, Ardiani A, Sanchez-Bonilla M., and, Black ME. Comparative analysis of enzyme and pathway engineering strategies for 5FC-mediated suicide gene therapy applications. Cancer Gene Ther. 2011;18:533–542. doi: 10.1038/cgt.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaün C, Gyan E, Rodrigues P., and, Heard JM. Pit2 assemblies at the cell surface are modulated by extracellular inorganic phosphate concentration. J Virol. 2002;76:4304–4311. doi: 10.1128/JVI.76.9.4304-4311.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Zhong C, Chang Z., and, Roy-Burman P. A potential therapeutic strategy to combat leukemia virus infection. Cancer Biol Ther. 2003;2:92–99. doi: 10.4161/cbt.236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Configuration of plasmid constructs encoding RRV's constructed from Moloney MLV with an amphotropic envelope and various CD and Hybrid CD genes.
Stability profile of vectors by serial passage.