

Abstract
Lipid nanoparticles (LNP) are the leading systems for in vivo delivery of small interfering RNA (siRNA) for therapeutic applications. Formulation of LNP siRNA systems requires rapid mixing of solutions containing cationic lipid with solutions containing siRNA. Current formulation procedures employ macroscopic mixing processes to produce systems 70-nm diameter or larger that have variable siRNA encapsulation efficiency, homogeneity, and reproducibility. Here, we show that microfluidic mixing techniques, which permit millisecond mixing at the nanoliter scale, can reproducibly generate limit size LNP siRNA systems 20 nm and larger with essentially complete encapsulation of siRNA over a wide range of conditions with polydispersity indexes as low as 0.02. Optimized LNP siRNA systems produced by microfluidic mixing achieved 50% target gene silencing in hepatocytes at a dose level of 10 µg/kg siRNA in mice. We anticipate that microfluidic mixing, a precisely controlled and readily scalable technique, will become the preferred method for formulation of LNP siRNA delivery systems.
Keywords: lipid nanoparticle, microfluidics, nanomedicine, siRNA, synthesis and formulation
Introduction
Lipid nanoparticles (LNP) are the leading systems for in vivo delivery of small interfering RNA (siRNA) for therapeutic applications.1,2,3 LNP siRNA systems can silence therapeutically relevant genes in a variety of animal models4,5,6,7 following intravenous injection and are in clinical trials for treatment of cardiovascular disease, liver cancer and other disorders.8 Methods that have been developed to manufacture LNP siRNA include mixing preformed vesicles (PFVs) with siRNA in ethanol–water solutions9,10 (PFV process) or mixing lipid dissolved in ethanol with aqueous solutions of siRNA using a T-tube mixer.4,11 Both of these methods generate LNP with reported diameters of 70 nm or larger6 that have siRNA encapsulation efficiencies of 65–95%. The physical properties of individual LNP formed by mixing processes are sensitive to local mixing rates, particularly slower mixing rates where mass transport effects may result in larger LNP with variable composition. Macroscopic mixing methods give rise to a range of local mixing rates, often leading to LNP with high polydispersity and poor batch-to-batch reproducibility. Microfluidic mixing techniques permit millisecond mixing at the nanoliter scale.12,13,14 Here, we demonstrate the use of microfluidic mixing to enable the reliable synthesis of LNP siRNA systems not accessible by previous methods employing a simple microfluidic mixing device [staggered herringbone micromixer (SHM), see Figure 1]. This SHM design enables dramatically higher throughput relative to other micromixer geometries such as hydrodynamic flow focusing,15,16 making it suitable for the synthesis of substantial amounts of LNP-siRNA.
Figure 1.

Schematic of lipid nanoparticle (LNP) small interfering RNA (siRNA) formulation strategy employing the staggered herringbone micromixer (SHM). (a) Lipid in ethanol and siRNA in aqueous solution is pumped into the two inlets of the microfluidic mixing device using a syringe pump. Herringbone structures induce chaotic advection of the laminar streams causing rapid mixing of the ethanol and aqueous phases and correspondingly rapid increases in the polarity experienced by the lipid solution. At a critical polarity precipitates form as LNP. (b) Parallelization of microfluidic mixers to enable formulation scale-up while maintaining identical production conditions. This is achieved through vertical (i = 1, 2,..) and horizontal (j = 1, 2,…) replication of the mixers with fluid handling through on-chip plumbing. Dimensions of the mixing channel were 200 µm × 79 µm, and the herringbone structures were 31-µm high and 50-µm thick.
Results
Microfluidic mixing results in production of monodisperse LNP
The SHM provides a means of reproducible and very rapid mixing of two input streams using microstructure-induced chaotic advection17 at intermediate Reynolds number (2 < Re < 500). Following the combination of two fluid streams they pass through a series of herringbone structures that induce rotational flow which wraps the fluids onto each other, with an orientation that changes between half cycles. This results in a chaotic flow profile, characterized by an exponentially shrinking characteristic diffusion length between the streams, and rapid advective mixing. At total flow rates of 2 ml/min the SHM employed here can provide complete mixing in 3 msec (Supplementary Figure S1 and Supplementary Materials and Methods).
In order to develop the SHM for the manufacturing of LNP siRNA, we employed a test formulation based on previous work6 in which LNP-siRNA systems were made using both the PFV and the T-tube mixing processes. The lipid composition consisted of an ionizable cationic lipid (DLinKC2-DMA), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol and a PEG-lipid where the cationic lipid contents ranged from 40 to 60 mol% and PEG-lipid contents from 1 to 5 mol%. The siRNA/total lipid ratio was maintained at 0.06 (wt/wt). DLinKC2-DMA, which resulted in particularly potent LNP siRNA systems for silencing target genes in vivo, has an apparent pKa of 6.76 allowing formulation with siRNA at low pH (e.g., pH 4.0) but also enabling LNP to exhibit near-neutral surface charge at physiological pH, reducing toxic side effects.3,10 The test formulation consisted of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA at 40:11.5:47.5:1 mol ratios and an siRNA/lipid ratio of 0.06 (wt/wt). The PEG-lipid content, the cationic lipid content and/or the siRNA/lipid ratio was subsequently varied to optimize LNP size or potency.
We first investigated the ability of the SHM to produce monodisperse LNP siRNA systems of defined size over a range of solute concentrations. We postulated that at sufficiently fast mixing rates LNP, formed by precipitation in response to the rapidly rising polarity of the medium, will adopt a minimum or “limit size” compatible with the physical properties of component molecules. Thus, at faster flow rates, providing progressively shorter mixing times, limit size LNP siRNA systems that are increasingly monodisperse would be expected. LNP siRNA systems were generated from the test formulation at a total flow rate of 0.02 to 4 ml/min. LNP diameters, as measured by dynamic light scattering remained constant at ~55 nm (number-weighted mode; reflecting the mean diameter derived from the apparent LNP molecular weight) for flow rates above 0.2 ml/min. However, at the highest total flow rates of 4 ml/min, highly monodisperse “limit size” LNP siRNA systems were achieved with polydispersity indices (PDI) of 0.03 or smaller (Figure 2) for final siRNA concentrations ranging from 0.25 mg/ml to 0.59 mg/ml.
Figure 2.

Higher total flow rates through the staggered herringbone micromixer (SHM) reduce lipid nanoparticle (LNP) small interfering RNA (siRNA) polydispersity. Dependence of the polydispersity index (PDI) on the total flow rate and the concentration of lipid and siRNA in the ethanol and aqueous phases, respectively. The PDI was determined from the second order coefficient in the cumulants analysis provided by dynamic light scattering (DLS) (PDI = (σ/µ)2). The total flow rate was varied from 0.02 to 4 ml/min keeping the aqueous buffer to ethanol volumetric flow rate ratio constant at 3:1. The aqueous siRNA concentration was varied from 0.25 to 0.59 mg/ml while the lipid concentration was varied from ~4 to 10 mg/ml to keep the siRNA/total lipid ratio constant at 0.06 wt/wt. PDI values represent averages from 4 measurements. The lipid composition employed was DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA at mol ratios of 40:11.5:47.5:1. The aqueous buffer was 25 mmol/l acetate, pH 4. DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine.
Microfluidic mixing results in production of “limit size” systems where the size is dictated by the PEG-lipid content
We next tested whether rapid mixing enabled by the SHM mixer could result in the production of LNP siRNA systems having sizes controlled by the lipid composition employed, rather than by mass transport effects occurring during synthesis. In particular, we sought to generate very small LNP with diameters 20–50 nm, since these have the potential to greatly improve tissue penetration.18,19,20,21 Given that the LNP siRNA systems composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA at 40:11.5:47.5:1 mol ratios had a limit size of ~55 nm, we reasoned that smaller limit size systems should be possible using higher proportions of relatively polar lipids, such as PEG-lipids, which may deposit preferentially on the LNP surface during LNP formation.22 We therefore varied the PEG-lipid content over the range 1–5 mol% using the lipid composition DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA; 40:11.5:47.5–x:1+x, mol/mol and monitored LNP siRNA size for a 4 ml/min total flow rate. As shown in Figure 3a, progressively smaller LNP siRNA systems were achieved as the PEG-lipid content was increased, with the 5 mol% systems exhibiting mean diameters of ~25 nm as determined by light scattering (number mode). In order to determine whether the measured size of LNP siRNA systems with 5 mol% PEG-c-DMA was influenced by the presence of PEG micelles, the size of these LNP siRNA systems was measured after passage through a size exclusion column to remove any potential micellar component. The size of the LNP siRNA fraction compared favorably with the measured size before passage through the column (see Supplementary Materials and Methods, Supplementary Figure S6, and commentary). It may be noted that LNP siRNA systems with diameters of 50 nm and smaller represent the smallest reported to date. Importantly, siRNA encapsulation efficiencies in excess of 95% and polydispersity indices well below 0.1 were maintained for these smaller systems (see Figure 3b,c).
Figure 3.

Increasing PEG-c-DMA content produces progressively smaller lipid nanoparticle (LNP) small interfering RNA (siRNA) systems. (a) Influence of PEG-c-DMA content on LNP size as produced under rapid mixing conditions (4 ml/min total flow rate with an siRNA-buffer:lipid–ethanol volumetric flow rate ratio of 3:1). LNP were composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA at mol ratios of 40:11.5:47.5:1, 40:11.5:46:2.5, and 40:11.5:43.5:5 for the 1, 2.5, and 5 mol% PEG-c-DMA, respectively. LNP were produced with an siRNA-total lipid ratio of 0.06 wt/wt. (b) Encapsulation efficiency as LNP size is decreased from 42 to 26 nm by increasing the PEG-c-DMA content from 1 to 5 mol%. LNP samples were dialyzed against phosphate-buffered saline (PBS) before measurement of encapsulation. Encapsulation refers to the percentage siRNA present in the LNP following removal of free siRNA using an anionic exchange spin column. (c) Polydispersity of LNP as the size was reduced from 54 to 28 nm by increasing the PEG-c-DMA content from 1 to 5 mol%. The polydispersity index (PDI) was determined as described in legend to Figure 2. (d) Size of empty LNP as a function of PEG-lipid content, which was varied from 0.25–5 mol%. LNP were composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA, with DLinKC2-DMA and DSPC maintained at 40 and 11.5 mol%, respectively. Titration of PEG-c-DMA was compensated by adjustment of cholesterol. All LNP were produced at an initial lipid concentration of 20 mmol/l in the lipid-ethanol phase prior to mixing with 25 mmol/l acetate buffer, pH 4. Number-weighted mean diameters are shown for the LNP following dialysis against PBS to remove residual ethanol and increase the pH to 7.4. Error bars represent standard deviation from mean (n = 3). DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine.
The observation that the limit size LNP siRNA systems produced by rapid microfluidic mixing is dependent on the PEG-lipid content is consistent with the proposal that the PEG-lipid resides in the outer monolayer of the LNP siRNA systems, thus allowing higher surface-to-volume ratios and correspondingly smaller limit sizes as the PEG-lipid content is increased. In this regard recent work indicates that LNP siRNA systems exhibit a nanostructured interior consisting of inverted lipid micelles, some of which contain siRNA.23 In order to determine whether the limit sizes achievable by microfluidic mixing are consistent with this model we generated LNP systems (in the absence of siRNA) for PEG-lipid contents ranging from 0.25 to 5 mol% using the lipid composition DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA; 40:11.5:47.5–x:1+x, mol/mol. As shown in Figure 3d this resulted in LNP with sizes ranging from 108 to 18 nm diameter. We then attempted to fit this data to a model where the LNP structure consists of a PEG-lipid monolayer coating a hydrophobic core of radius r that contains tightly packed inverted micelles with an average aqueous internal radius rm. We assumed a lipid monolayer thickness (tm) of 2 nm and that the core lipids consist of cholesterol, DLinKC2-DMA, and DSPC with an average molecular weight, (MW), of 535 g/mol and a lipid density, (ρ) of 0.9 g/ml. The amount of PEG-lipid required to coat the external surface of an LNP of a given size in this limit size model is given by Equation (1). As shown in Figure 3d, by using the area per PEG-lipid (Ap) and rm as variables, an excellent fit to the experimental data could be achieved for rm = 3 nm and Ap = 26 nm.24 The mean particle diameter represents the LNP core, including the outer lipid monolayer, plus the additional thickness provided by the PEG-lipid (tPEG) coating which is determined using an adjusted Flory radius.24 It may be concluded that the limit size of the LNP systems generated by rapid microfluidic mixing can be varied in a predictive manner by adjusting the PEG-lipid content.
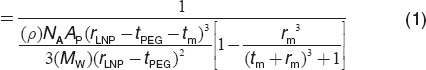 |
LNP siRNA systems generated by microfluidic mixing exhibit high encapsulation efficiencies and an electron dense core
We next investigated the ability of the SHM formulation process to provide efficient encapsulation over a range of siRNA/lipid ratios. As shown in Figure 4, at a total flow rate of 2 ml/min, siRNA encapsulation efficiencies of ~100% were achieved as the siRNA/lipid ratio was varied over the range 0.01 to 0.2 (wt/wt). Encapsulated siRNA was fully protected from external nucleases as demonstrated in Supplementary Figure S2. Above an siRNA/ total lipid ratio of 0.21 (wt/wt), which corresponds to charge balance between the cationic lipid and anionic siRNA, the encapsulation efficiency decreased (data not shown). This can be attributed to insufficient charged cationic lipid available for encapsulation of the siRNA.
Figure 4.

Formulation of lipid nanoparticle (LNP) small interfering RNA (siRNA) employing microfluidic mixing results in highly efficient encapsulation over a wide range of siRNA-to-cationic charge ratios. LNP were composed of Dlin-KC2-DMA/DSPC/cholesterol/PEG-c-DMA at mol ratios of 40:11.5:47.5:1 employing an siRNA- total lipid ratio of 0.06 wt/wt. The total flow rate was maintained at 2 ml/min employing a 10 mmol/l lipid- in-ethanol phase mixed with aqueous buffer (25 mmol/l acetate, pH 4) containing siRNA. Encapsulation refers to the percentage siRNA present in the LNP following removal of free siRNA using an anionic exchange spin column. Error bars represent standard deviation of encapsulation as measured from three LNP formulations. DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine.
In order to gain information concerning the morphology of LNP siRNA systems produced by microfluidic mixing, and to validate the sizes and size distributions obtained by light scattering techniques, the LNP siRNA systems containing 1 and 5 mol% PEG-lipid were analyzed by cryo-transmission electron microscopy as shown in Figure 5a and b. LNP sizes measured from these micrographs indicate diameters of 44.5 ± 6.8 nm and 22.4 ± 3.9 nm for the 1 and 5 mol% formulations, respectively, in agreement with the light scattering measurements (Supplementary Table S1). We note that the LNP siRNA systems exhibit an electron dense interior core similar to that obtained elsewhere for LNP siRNA systems generated employing the T-tube method.25 Such morphology is distinctly different than observed for bilayer LNP systems and has been ascribed to a nanostructured lipid interior.23
Figure 5.

Cryo-transmission electron microscopy (cryo-TEM) micrographs of lipid nanoparticle (LNP) small interfering RNA (siRNA) systems containing 1 and 5 mol% PEG-c-DMA. (a) Cryo-TEM micrograph of LNP siRNA composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA (mol ratios 40:11.5:47.5:1) and siRNA at an siRNA to total lipid ratio 0.06 (wt/wt). (b) Cryo-TEM micrograph of LNP siRNA composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA (mol ratios 40:11.5:43.5:5) and siRNA at an siRNA-to-total lipid ratio 0.06 (wt/wt). LNP were imaged at 50K magnification. LNP formulation was performed under rapid mixing conditions (4 ml/min total flow rate with an siRNA-buffer:lipid–ethanol volumetric flow rate ratio of 3:1) with the staggered herringbone micromixer (SHM), with an ethanol phase containing 30 mmol/l lipid. The LNP siRNA dispersion was concentrated before imaging. The scale bar represents 100 nm. DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine.
Parallelization of microfluidic mixers allows scaled-up LNP manufacture
A viable LNP siRNA formulation process must also be scalable.26 Scaling-up can be challenging due to differences in mass transport that arise as the total volume of a reaction is increased. An intrinsic advantage of microfluidic mixing is that it is easily scaled by parallelization of mixing devices13 (Figure 1b). To demonstrate this we used a device with six parallel SHM elements to produce limit size LNP composed of 1-palmitoyl, 2-oleoyl PC (POPC)/cholesterol at 72 ml/min or 580 mg LNP/min (see Supplementary Figures S3 and S4). This is to our knowledge the highest rate of LNP synthesis using microfluidic methods yet demonstrated and compares favorably to alternative scale-up processes.26
LNP siRNA systems generated by microfluidic mixing exhibit potent gene silencing in vivo
It is important to show that LNP siRNA systems produced by microfluidic mixing exhibit equivalent or improved potency as those produced by previous techniques. To date the most potent siRNA-based agents reported are LNP systems made by T-tube synthesis using DLinKC2-DMA cationic lipids with optimized cationic lipid proportions and siRNA/lipid ratios. These LNP siRNA systems have been shown to achieve 50% gene silencing in a mouse factor VII model6 at dose levels (ED50) as low as 0.02 mg siRNA/kg body weight. The gene silencing potencies of LNP FVII-siRNA systems produced by microfluidic mixing were therefore measured in the mouse FVII model first as a function of the amount of cationic lipid and second as a function of the siRNA/lipid ratio.
As shown in Figure 6a, increasing the cationic (DLinKC2-DMA) lipid content from 40 to 60 mol% while holding the siRNA/total lipid ratio constant at 0.06 (wt/wt) improved the ED50 from 0.2 to ~0.03 mg siRNA/kg. Higher levels (70 mol % DLinKC2-DMA) did not provide further gains (data not shown). Further improvements in potency could, however, be achieved by decreasing the siRNA-to-lipid ratio. This is illustrated in Figure 6b which shows FVII gene silencing when the siRNA-to-lipid ratio was varied from 0.01 to 0.35 (wt/wt), corresponding to a siRNA-to-cationic lipid charge ratio of ratio of ~0.025, 0.25, 0.5, and 1, respectively. The LNP FVII-siRNA formulation with an siRNA-to-lipid ratio of 0.01 (wt/wt) achieved 50% FVII silencing at a 0.01 mg siRNA/kg dose. While it is difficult to make direct comparisons due to differences in LNP size and siRNA, the potency is clearly equal or superior to current gold standards using DLinKC2-DMA.4,6 The importance of the rapid mixing rates provided by the SHM mixer to increased potency are demonstrated by the fact that LNP siRNA prepared at slower mixing rates give rise to larger particles that exhibit poor gene silencing potency (Supplementary Figure S5).
Figure 6.

Optimization of lipid nanoparticle (LNP) small interfering RNA (siRNA) gene silencing potency in a FVII mouse model as a function of cationic lipid content and siRNA/total lipid ratio. (a) Influence of LNP DLinKC2-DMA content on FVII gene silencing in the FVII mouse model. FVII expression was monitored 24 hours after intravenous injection of LNP siRNA systems containing between 40 and 60 mol% DLinKC2-DMA. The PEG-c-DMA content was held constant at 1 mol% and the addition of cationic lipid was compensated for by reduction in the DSPC and cholesterol content, holding the DSPC to cholesterol ratio constant at 0.25 (mol/mol). (b). Influence of variation of siRNA/total lipid ratio on FVII gene silencing in the FVII mouse model. The lipid composition used was DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA (mol ratios 60:7.5:31.5:1). The siRNA/total lipid ratio was varied from 0.01 to 0.35 (wt/wt), corresponding to a siRNA-to-cationic lipid charge ratio of 0.025, 0.25, 0.5, and 1, respectively. Systemic administration of LNP siRNA to mice was performed by tail vein injection (n = 3 per dose level). Blood was collected 24 hours postinjection and factor VII levels were determined by colorimetric assay. DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine.
Discussion
The results presented in this work demonstrate that a microfluidic mixing device containing a SHM can be used to generate LNP systems of predictable “limit” size, that siRNA is efficiently encapsulated in these systems, and that the LNP siRNA systems produced exhibit excellent gene silencing capabilities in vivo. Here, we distinguish the results presented here from previous work using microfluidic mixing technology to form LNP systems, then discuss the mechanisms whereby LNP and LNP siRNA systems are formed using the microfluidic mixing device and finally indicate the advantages of microfluidic mixing using the SHM geometry as compared to previous macroscopic mixing procedures used to formulate LNP siRNA systems.
Considerable efforts have been made to utilize microfluidic mixing (primarily using hydrodynamic flow focusing geometry) to generate lipid and polymer nanoparticles,15,16,28,29 or lipoplex systems containing plasmid DNA.30 Results, while promising, show limitations in terms of the sizes and amounts of material generated. For example limit size 50 nm diameter systems produced by hydrodynamic flow focusing are only achieved at flow rate ratios of 30 or higher,15,28 resulting in substantial material dilutions as compared to the flow rate ratios of three employed here. The results presented here demonstrate the ability of the SHM to produce high quantities (≥500 mg/min) of limit size LNP siRNA systems where the limit size is controlled over the 20–100 nm size range by simple variation of the PEG-lipid content. Finally, while the present work was in the last stages of preparation, a paper by Chen et al.31 appeared demonstrating the use of microfluidic mixing employing an SHM micromixer to produce small amounts of a large variety of LNP siRNA formulations in order to identify promising new compositions for in vivo delivery. This work employed low flow rates (~0.3 ml/min), lower encapsulation efficiencies (~80%) and LNP sizes of ~70-80 nm.
With regard to the mechanism(s) whereby the microfluidic process allows the formation of LNP and LNP-containing siRNA, two points of interest concern first the mechanism whereby LNP of 100 nm size or smaller are formed and second the mechanism whereby siRNA can be encapsulated with efficiencies approaching 100%. With regard to formation of LNP, the rate of mixing is clearly an important parameter as shown here and elsewhere.15,16,28,29,31,32 Rapid mixing of the ethanol–lipid solution with aqueous buffer results in a rapid increase in the polarity of the medium which causes the solution to quickly achieve a state of high supersaturation of lipid monomers throughout the entire mixing volume, resulting in the rapid and homogeneous nucleation of nanoparticles. These nucleation events are very rapid compared to the time-scale for particle formation/aggregation and result in the formation of sub-limit size particles that are not thermodynamically stable. These sublimit size particles then coalesce to form limit size particles with the size of the particles determined by the steric and energetic constraints of the components provided. Appropriate selection of lipid components and their relative amounts then allow for control of the resulting size of the LNP. This was shown here by varying the amount of PEG-lipid which preferentially resides on the LNP exterior; providing stability to the LNP and limiting subsequent growth through further lipid monomer incorporation.
The observation that LNP siRNA systems formulated by the microfluidic mixing technique exhibit siRNA encapsulation efficiencies approaching 100% suggests an initial precipitation of nucleates of siRNA with cationic lipid as the polarity of the medium increases, which are subsequently coated by PEG-lipid as the polarity increases further. As detailed elsewhere23 such a model is consistent with the “solid core” electron dense appearance of the LNP siRNA systems as visualized by cryo-transmission electron microscopy (see Figure 5) which is different from that observed for bilayer vesicular systems which exhibit a circular morphology with a less electron dense interior.33 The presence of a nanostructured lipid core is also supported by molecular modeling approaches as well as the densities and other physical characteristics exhibited by these LNP siRNA systems.23
It should be noted that in line mixing techniques such as the SHM mixer require that the lipid be soluble in the organic solvent employed. Lipids that are less soluble in ethanol may require solvent heating to achieve adequate solubility, or alternative water-miscible organic solvents could be employed. Microfluidic mixers such as the SHM can be constructed from a variety of materials that are resistant to most solvents.34,35
The microfluidic formulation procedure offers several advantages over previous LNP siRNA synthesis techniques employing macroscopic mixing techniques, which include the PFV method,9 and the T-tube mixing11 technique. With regard to the PFV process, the microfluidic process results in higher encapsulation efficiencies, the ability to produce smaller LNP systems, and the ability to produce small scale batches with little loss due to the small dead volume (~1 µl) of the apparatus. The production of PFVs is also not required. Advantages of the microfluidics approach as compared to the T-tube mixer again includes the ability to produce smaller systems below 50-nm diameter (the smallest reported using the T-tube approach are of 50-nm diameter25) as well as the fact that high flow rates (>1 ml/sec) are required to achieve the velocities required for rapid mixing to occur using the T-tube mixer. The micromixer allows LNP siRNA formulation under well defined, reproducible conditions at much lower flow rates resulting in reduced losses due to dead volumes and straightforward preparation of small scale batches for LNP optimization and in vitro testing. The ability to move from bench-scale preparations to larger scale LNP siRNA manufacture by parallelization of microfluidic mixing devices constitutes a further major advantage.
In summary, the results presented here indicate that microfluidic mixing using the SHM micromixer enables routine production of LNP siRNA systems in the size range 20–100 nm and also offers advantages of ease of design, low polydispersity, high siRNA encapsulation efficiencies, improved scalability, and equivalent or better gene silencing potency as compared to previous formulation processes. The ability to synthesize LNP with diameters of 50 nm or smaller is important as such systems can exhibit improved abilities to penetrate target tissue such as tumors,18,21 as is the ability to form LNP systems at flow rate ratios as low as three, which significantly enables LNP production at reasonable scales. It is anticipated that microfluidic mixing employing the SHM will become the technique of choice for LNP synthesis at laboratory and clinical scales.
Materials and Methods
Materials. DSPC, was purchased from Avanti Polar Lipids (Alabaster, AL), whereas cholesterol was obtained from Sigma (St Louis, MO). N-[(methoxy poly(ethylene glycol)2000)carbamyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-C-DMA) was synthesized by AlCana Technologies (Vancouver, British Columbia, Canada). Factor VII (FVII) and low GC negative control siRNA were purchased from Invitrogen (Carlsbad, CA). Factor VII siRNA: 5′-GGAucAucucAAGucuuAcT*T-3′ (FVII sense), 5′-GuAAGAcuuGAGAuGAuccT*T-3′ (FVII antisense). DLinKC2-DMA was purchased from AlCana Technologies.
Preparation of LNP systems employing the microfluidic SHM. The oligonucleotide (siRNA) solution was prepared in 25 mmol/l acetate buffer at pH 4.0. Depending on the desired formulation, an ethanol solution containing DLinKC2-DMA, DSPC, cholesterol, and a PEG-lipid at the appropriate molar ratio solutions were prepared at concentrations of 1 to 39 mg/ml total lipid.
The microfluidic apparatus used in this work was produced by soft lithography, the replica molding of microfabricated masters in elastomer.36 The device (Figure 1) features a 200-µm wide and 79 µm high-mixing channel with herringbone structures formed by 31 µm high and 50-µm thick features on the roof of the channel. Fluidic connections were made with 1/32” I.D., 3/32” O.D. tubing that was attached to 21G1 needles for connection with syringes. One ml or 3 ml syringes were used for inlet streams. Two syringe pumps (KD200; KD Scientific, Holliston, MA) were used to control the flow rate through the device. The total flow rate of was varied from 0.02 to 4 ml/min, corresponding to a Reynolds number between 2 and 476. The syringe pump introduces the two solutions into the microfluidic device (see Figure 1), where they come into contact at the Y-junction. For each formulation siRNA-in-aqueous buffer (pH 4.0) was mixed with lipids-in-ethanol at a volumetric flow rate ratio of 3:1 (aqueous to ethanol) at room temperature. The product was then dialyzed against 50 mmol/l citrate/ 50 mmol/l MES, pH 6.7, to remove residual ethanol and then against phosphate-buffered saline to raise the pH to 7.4.
Characterization of LNP. Particle size was determined by dynamic light scattering using a Nicomp model 370 Submicron Particle Sizer (Particle Sizing Systems, Santa Barbara, CA) following buffer exchange into phosphate-buffered saline. Number-weighted size and distribution data was used. Lipid concentrations were verified by measuring total cholesterol using the Cholesterol E enzymatic assay from Wako Chemicals USA (Richmond, VA). Removal of free siRNA was performed employing VivaPureD MiniH columns (Sartorius Stedim Biotech, Goettingen, Germany). The eluants were then lysed in 75% ethanol and siRNA was quantified by measuring absorbance at 260 nm. Encapsulation efficiency was determined from the ratio of oligonucleotide before and after removal of free oligonucleotide content, normalized to lipid content.
Cyro-TEM. LNP samples were first concentrated to 20-40 mg/ml with Amicon Ultra-4 centrifugal filter units (Millipore, Billerica, MA). Samples were prepared by applying 3 µl of phosphate-buffered saline containing LNP to a glow discharged Lacey Carbon 300 mesh copper TEM grid (Canemco, Canton de Gore, Quebec, Canada). Excess liquid was removed by blotting with a Vitrobot system (FEI, Hillsboro, OR) and then plunge-freezing the LNP suspension in liquid ethane to rapidly freeze the vesicles in a thin film of amorphous ice. Images were taken under cryogenic conditions at a magnification of 50K with an AMT HR CCD side mount camera. Samples were loaded using a Gatan 70 degree cryo-transfer holder in an FEI G20 Lab6 200kV TEM under low dose conditions with an underfocus of 3–6 um to enhance image contrast. Mean particle diameter was determined by measuring 120 LNP for each sample using Adobe Photoshop (Adobe Systems, San Jose, CA).
In vivo studies to characterize FVII gene silencing activity. Six to eight-week-old, female C57Bl/6 mice were obtained from Charles River Laboratories (Wilmington, MA). LNP-siRNA containing factor VII siRNA were filtered through a 0.2-µm filter and diluted to the required concentrations in sterile phosphate-buffered saline before use. The formulations were administered intravenously via the lateral tail vein at a volume of 10 ml/kg. After 24 hours, animals were anesthetized with Ketamine/Xylazine and blood was collected by cardiac puncture. Samples were processed to serum (Microtainer Serum Separator Tubes; Becton Dickinson, Franklin Lakes, NJ) and tested immediately or stored at –70 °C for later analysis of serum Factor VII levels. All procedures were approved by the University of British Columbia institutional animal care committee and performed in accordance with guidelines established by the Canadian Council of Animal Care.
Serum Factor VII levels were determined using the colorimetric Biophen VII assay kit (Anaira). Control serum was pooled and serially diluted (200–3.125%) to produce a calibration curve for calculation of FVII levels in treated animals. Appropriately diluted plasma samples from treated animals (n = 3 per dosage) and a saline control group (n = 4) were analyzed using the Biophen VII kit according to manufacturer's instructions. Analysis was performed in 96-well, flat bottom, non-binding polystyrene assay plates (Corning, Corning, NY) and absorbance was measured at 405 nm. Factor VII levels in treated animals were determined from a calibration curve produced with the serially diluted control serum.
Acknowledgments
This work was supported by the National Science and Engineering Research Council of Canada under grant F09-04486. We thank Lindsay Heller at the Center for Drug Research and Development (CDRD) for use of confocal microscopy. We also thank B. Ross (UBC BioImaging Facility) for assistance with cryo-TEM. This work was supported by the National Science and Engineering Research Council of Canada under grant F09-04486. N.M.B., J.H., A.W., T.J.L., R.J.T., C.L.H., and P.R.C. have a financial interest in Precision Nanosystems.
Supplementary Material
Influence of volumetric flow rate on extent of mixing in SHM.
Protection of siRNA from degradation during incubation in serum.
Schematic of 6× scale-up microfluidic device for formulation of lipid nanoparticles using individual microfluidic SHM mixers.
Scale-up of 1-palmitoyl, 2-oleoyl PC (POPC)/cholesterol vesicles by parallelization of individual microfluidic SHM mixers.
Impact of formulation conditions and resulting LNP size on FVII gene silencing in mice.
Elution profiles of LNP containing 5 mol% PEG-c-DMA and PEG-c-DMA micelles.
LNP Size as determined by DLS and CryoTEM imaging.
References
- Allen TM., and, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- Whitehead KA, Langer R., and, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenske DB, Chonn A., and, Cullis PR. Liposomal nanomedicines: an emerging field. Toxicol Pathol. 2008;36:21–29. doi: 10.1177/0192623307310960. [DOI] [PubMed] [Google Scholar]
- Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR.et al. (2010Lipid-like materials for low-dose, in vivo gene silencing Proc Natl Acad Sci USA 1071864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN.et al. (2006RNAi-mediated gene silencing in non-human primates Nature 441111–114. [DOI] [PubMed] [Google Scholar]
- Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK.et al. (2010Rational design of cationic lipids for siRNA delivery Nat Biotechnol 28172–176. [DOI] [PubMed] [Google Scholar]
- Basha G, Novobrantseva TI, Rosin N, Tam YY, Hafez IM, Wong MK.et al. (2011Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells Mol Ther 192186–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D. The business of RNAi therapeutics in 2012. Mol Ther - Nucleic acids. 2012;1 doi: 10.1038/mtna.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer N, Wong KF, Stark H, Louie L, McIntosh D, Wong T.et al. (2001Spontaneous entrapment of polynucleotides upon electrostatic interaction with ethanol-destabilized cationic liposomes Biophys J 802310–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple SC, Klimuk SK, Harasym TO, Dos Santos N, Ansell SM, Wong KF.et al. (2001Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures Biochim Biophys Acta 1510152–166. [DOI] [PubMed] [Google Scholar]
- Jeffs LB, Palmer LR, Ambegia EG, Giesbrecht C, Ewanick S., and, MacLachlan I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res. 2005;22:362–372. doi: 10.1007/s11095-004-1873-z. [DOI] [PubMed] [Google Scholar]
- Song Y, Hormes J., and, Kumar CS. Microfluidic synthesis of nanomaterials. Small. 2008;4:698–711. doi: 10.1002/smll.200701029. [DOI] [PubMed] [Google Scholar]
- DeMello AJ. Control and detection of chemical reactions in microfluidic systems. Nature. 2006;442:394–402. doi: 10.1038/nature05062. [DOI] [PubMed] [Google Scholar]
- Song H, Chen DL., and, Ismagilov RF. Reactions in droplets in microfluidic channels. Angew Chem Int Ed Engl. 2006;45:7336–7356. doi: 10.1002/anie.200601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn A, Vreeland WN, DeVoe DL, Locascio LE., and, Gaitan M. Microfluidic directed formation of liposomes of controlled size. Langmuir. 2007;23:6289–6293. doi: 10.1021/la070051a. [DOI] [PubMed] [Google Scholar]
- Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W.et al. (2008Microfluidic platform for controlled synthesis of polymeric nanoparticles Nano Lett 82906–2912. [DOI] [PubMed] [Google Scholar]
- Stroock AD, Dertinger SK, Ajdari A, Mezic I, Stone HA., and, Whitesides GM. Chaotic mixer for microchannels. Science. 2002;295:647–651. doi: 10.1126/science.1066238. [DOI] [PubMed] [Google Scholar]
- Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M.et al. (2011Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size Nat Nanotechnol 6815–823. [DOI] [PubMed] [Google Scholar]
- Li SD., and, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- Huang L, Sullenger B., and, Juliano R. The role of carrier size in the pharmacodynamics of antisense and siRNA oligonucleotides. J Drug Target. 2010;18:567–574. doi: 10.3109/10611861003734019. [DOI] [PubMed] [Google Scholar]
- Perrault SD, Walkey C, Jennings T, Fischer HC., and, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- Woodle MC., and, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta. 1992;1113:171–199. doi: 10.1016/0304-4157(92)90038-c. [DOI] [PubMed] [Google Scholar]
- Leung A, Hafez I, Baoukina S, Belliveau N, Zhigaltsev I, Afshinmanesh E.et al. (2012Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core J Phys Chem Cin press). [DOI] [PMC free article] [PubMed]
- Soong R., and, Macdonald PM. PEG molecular weight and lateral diffusion of PEG-ylated lipids in magnetically aligned bicelles. Biochim Biophys Acta. 2007;1768:1805–1814. doi: 10.1016/j.bbamem.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Crawford R, Dogdas B, Keough E, Haas RM, Wepukhulu W, Krotzer S.et al. (2011Analysis of lipid nanoparticles by Cryo-EM for characterizing siRNA delivery vehicles Int J Pharm 403237–244. [DOI] [PubMed] [Google Scholar]
- Wagner A., and, Vorauer-Uhl K. Liposome technology for industrial purposes. J Drug Deliv. 2011;2011:591325. doi: 10.1155/2011/591325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N.et al. (2008A combinatorial library of lipid-like materials for delivery of RNAi therapeutics Nat Biotechnol 26561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn A, Stavis SM, Hong JS, Vreeland WN, DeVoe DL., and, Gaitan M. Microfluidic mixing and the formation of nanoscale lipid vesicles. ACS Nano. 2010;4:2077–2087. doi: 10.1021/nn901676x. [DOI] [PubMed] [Google Scholar]
- Zook JM., and, Vreeland WN. Effects of temperature, acyl chain length, and flow-rate ratio on liposome formation and size in a microfludic hydrodynamic focusing device. Soft Matter. 2010;6:1352–1360. [Google Scholar]
- Hsieh AT, Hori N, Massoudi R, Pan PJ, Sasaki H, Lin YA.et al. (2009Nonviral gene vector formation in monodispersed picolitre incubator for consistent gene delivery Lab Chip 92638–2643. [DOI] [PubMed] [Google Scholar]
- Chen D, Love KT, Chen Y, Eltoukhy AA, Kastrup C, Sahay G.et al. (2012Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation J Am Chem Soc 1346948–6951. [DOI] [PubMed] [Google Scholar]
- Zhigaltsev IV, Belliveau N, Hafez I, Leung AK, Huft J, Hansen C.et al. (2012Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing Langmuir 283633–3640. [DOI] [PubMed] [Google Scholar]
- Zhigaltsev IV, Winters G, Srinivasulu M, Crawford J, Wong M, Amankwa L.et al. (2010Development of a weak-base docetaxel derivative that can be loaded into lipid nanoparticles J Control Release 144332–340. [DOI] [PubMed] [Google Scholar]
- Ren K, Dai W, Zhou J, Su J., and, Wu H. Whole-Teflon microfluidic chips. Proc Natl Acad Sci USA. 2011;108:8162–8166. doi: 10.1073/pnas.1100356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedair MF., and, Oleschuk RD. Fabrication of porous polymer monoliths in polymeric microfluidic chips as an electrospray emitter for direct coupling to mass spectrometry. Anal Chem. 2006;78:1130–1138. doi: 10.1021/ac0514570. [DOI] [PubMed] [Google Scholar]
- Xia Y., and, Whitesides GM. Soft Lithography. Angewandte Chemie International Edition. 1998;37:550–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Influence of volumetric flow rate on extent of mixing in SHM.
Protection of siRNA from degradation during incubation in serum.
Schematic of 6× scale-up microfluidic device for formulation of lipid nanoparticles using individual microfluidic SHM mixers.
Scale-up of 1-palmitoyl, 2-oleoyl PC (POPC)/cholesterol vesicles by parallelization of individual microfluidic SHM mixers.
Impact of formulation conditions and resulting LNP size on FVII gene silencing in mice.
Elution profiles of LNP containing 5 mol% PEG-c-DMA and PEG-c-DMA micelles.
LNP Size as determined by DLS and CryoTEM imaging.