

Abstract
The rational design of artificial enzymes either by applying physio-chemical intuition of protein structure and function or with the aid of computation methods is a promising area of research with the potential to tremendously impact medicine, industrial chemistry and energy production. Designed proteins also provide a powerful platform for dissecting enzyme mechanisms of natural systems. Artificial enzymes have come a long way, from simple α-helical peptide catalysts to proteins that facilitate multi-step chemical reactions designed by state-of-the-art computational methods. Looking forward, we examine strategies employed by natural enzymes which could be used to improve the speed and selectivity of artificial catalysts.
In the fifties and sixties, the advent of the semiconductor transistor and the integrated circuit transformed digital computers from powerful curiosities into pragmatic, cost-effective tools. Along with advances in numerical methods, computers revolutionized the design and construction of aircraft, allowing engineers to simulate complex, non-linear systems that integrated aerodynamics, propulsion, control, etc., thereby pushing aircraft technology well beyond what was possible with previous analytical models. Today, a Boeing 747 is an incredibly complex machine with over 6,000,000 parts. As such, computers have become indispensable in the aerospace industry. Although much smaller in size, the mechanistic complexity of enzymes and challenges associated with their design (Box 1) argue that they are as sophisticated as passenger airliners, and it is expected that computational methods in chemistry and biology will promote a similar revolution in the design of artificial catalysts.
BOX 1.
Hen Egg White Lysozyme (HEWL) was the first enzyme atomic structure to be solved by X-ray crystallography in 1965 110. The three dimensional structure highlights many of the physical characteristics of enzymes which make them unusually challenging proteins to design. HEWL functions in antibacterial defense and cleaves glycosidic linkages found in bacterial cell walls. The active site consists of two amino acids, a glutamic acid which functions as a general acid/base and an aspartic acid nucleophile. These are placed at the bottom of a deep substrate cleft which confers specificity and poises the substrate over the active site. Many small molecule catalysts function better in organic solvents, where the low bulk dielectric enhances electrostatic interactions. The cleft mimics this by isolating catalytic groups from bulk water, strengthening local electrostatic interactions. Accurate modeling of catalytic residue conformations and local electrostatics are key in designing effective artificial enzymes. Quantum mechanics methods have been useful in moving this area of design forward.
The protein fold must be sufficiently stable to form this cleft and preorganize active site residues, which is why enzymes are much larger than natural catalysts. The computational design of proteins with partially buried polar active sites is especially challenging. The protein fold must be able to absorb the energetic cost of desolvating polar active site groups and stabilizing electrostatic interactions that favor catalysis.
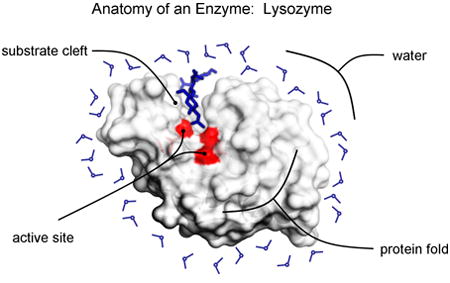
The promise of constructing enzymes that are capable of efficiently catalyzing virtually any chemical reaction is a tremendous motivator for researchers in the protein design field. Enzymes catalyze difficult chemical reactions in mild, aqueous environments, often with a speed and specificity unrivaled by synthetic catalysts. Designing an enzyme from scratch is also the most rigorous way to test our understanding of how natural enzymes function. Several recent designs have been stripped-down or rebuilt versions of natural enzymes, providing powerful tools for dissecting molecular contributions to enzyme structure and reactivity.
Enzyme design is inextricably linked with the design of protein structure. Advances in protein design are often rapidly followed by attempts to apply new technologies to artificial enzymes. Therefore, this is as much a review of protein fold design as of catalyst design. However, it should be noted that complex protein topologies are not a prerequisite for catalysis. Proline alone can catalyze a remarkable array of reactions including aldolase-like formations of carbon-carbon bonds through enamine intermediates with high yields and substantial product enantiomeric excess. Other processes including asymmetric epoxidations and acylations are achievable using short peptides. The impressive catalytic properties of proline and small peptides have been extensively reviewed previously1, 2 and are not covered here. Few designed enzymes have achieved the catalytic utility of such small peptides, and much remains to be done before designer enzymes find practical applications. However, the remarkable selectivity, rate-enhancements and product specificity of natural enzymes under aqueous conditions warrants more work in developing powerful molecular design technologies.
The complexities of enzyme design can be quite daunting. Examining high-resolution structures of natural enzyme-substrate complexes, reveals that the conformation of active site amino acids is exquisitely poised to facilitate catalysis. Second-shell interactions tune the reactivity of the active site through networks of direct interactions with primary ligands and long range electrostatic forces. Designing such molecules from scratch presents a host of computational challenges (see Box 1). To accurately model important forces in the active site requires quantum mechanics (QM) calculations. Unfortunately, it is not feasible to perform QM calculations on molecules the size of even the smallest enzymes. Design also requires the rapid evaluation of a large number of candidate structure/sequence combinations and QM calculations are very demanding on computational resources. Effective treatment of water molecules and their interactions with active site residues and reactants also can significantly increase the complexity of calculations. Along with high resolution design of the active site residues, candidate enzymes must maintain overall structural integrity, and in some cases, incorporate large scale protein motions which may support catalysis. Integrating all of these factors into a design is a formidable challenge.
It is reasonable to ask what one gains through such sophisticated computation. After all, a number of novel folds and catalytically active proteins have been built without such tools. In this review, we survey several designed, artificial enzymes that have been developed with varying degrees of computational involvement. These include de novo enzymes, where both the protein topology and the active site are built from scratch, and active site design, where surfaces and cavities on existing proteins are repurposed for catalysis. We build on previous reviews of this field3 by including a deeper discussion of computational challenges associated with enzyme design. Additionally, we look to the future of design, such as introducing multiple substrates, protein motion, allostery into artificial enzymes, and expanding protein design principles to a broader class of structured, catalytically active polymers.
The Helix and the Enzyme
A fundamental paradigm in biochemistry is the link between a protein’s function and its three dimensional fold, which in turn depends on its amino acid sequence. Getting from sequence to structure to function is the common goal unifying all work in the field of protein design. Some of the earliest model proteins were built with the α-helix as the fundamental unit of structure. A seven-residue repeating sequence, ●○○●●○○, where the first and fourth amino acids are non-polar (●) and the rest polar (○), will characteristically form multimeric, left-handed coiled-coil assemblies of amphipathic α-helices; non-polar sidechains associate between helical elements to form a hydrophobic core.4-8 Specific homo and heterooligomers of α-helices can be achieved by rational design of core packing interactions and surface electrostatics.9-11 The power of this simple idea was demonstrated in a clever design where the super-helical twist was inverted by changing the motif to an eleven residue repeat, ●○○●○○○●○○○, thus maintaining a continuous hydrophobic core in a right handed coiled-coil.12 This was an example of a true de novo design with no known natural counterpart at the time it was constructed.
The majority of early attempts at enzymes used an amphipathic α-helix as the principle structural component. ‘Helichrome’ was designed to function as a hydrolase, using four α-helices to form a hydrophobic substrate binding pocket over one face of an iron porphoryn to which the helices were covalently tethered.13 Helichrome could convert aniline to p-aminophenol in the presence of NADPH with a kcat of 0.02 min-1 and Km of 5.0 mM.
In work by Barbier and Brack, it was found that regular copolymers of leucine and lysine could hydrolyse polyribonucleotides.14, 15 Poly(Lys-Leu) and poly(Leu-Lys-Lys-Leu) had superior hydrolysis rates to random copolymers or those incorporating both d,l-Leu and d,l-Lys, demonstrating that either β-sheet structure (formed by a poly-●○ pattern16) or α-helical structure played a significant role in improving activity. The regular structure provided a cationic surface to support binding of negatively charged substrates such as nucleic acids. Repulsive electrostatic interactions between adjacent lysine sidechains served to depress the pKa of the amino group, making it a better catalytic base.
These features were exploited in the design of ‘oxaldie’, a fifteen-residue α-helical peptide comprised mostly of leucines and lysines that catalyzed decarboxylation of oxaloacetate to pyruvate through an imine intermediate (Figure 1).17 Possible amine donors in the covalent intermediate were lysine sidechains and the backbone amino terminus. The activity of the peptide was concentration dependent, suggesting that a helical multimer was the active form. Inserting a helix-breaking proline into the center of the peptide significantly reduced activity, affirming the relationship between structure and activity. Rate enhancements of 103 to 104 over free amines were observed.
Figure 1. From Sequence to Structure to Function.
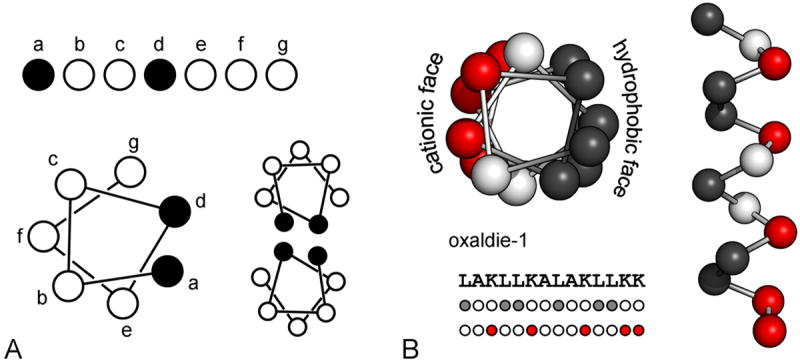
(A) A repeating seven residue pattern of nonpolar (●) and polar (○) residues will create a hydrophobic surface on one face of the helix. These surfaces can drive associations of helices, forming a hydrophobic core. (B) The oxaldie enzymatic peptides17 make dual use of this repeating pattern, creating one hydrophobic, leucine rich face and one cationic, lysine rich face. Spatial clustering of lysines lowers their pKa and provides a surface for the negatively charged oxaloacetate substrate to bind. L = leucine (grey), K = lysine (red), A = alanine(white).
Oxaldie was designed on the premise that the α-helix would raise the basicity of the active site, whether it was the amino terminus through interactions with helix macrodipole, or the amino groups of lysine through electrostatic interactions between sidechains. This made it possible to rationally improve activity by targeting structure. In one strategy, lysines were placed on the solvent exposed face of avian pancreatic peptide, a 36-amino acid protein with a helix packing against an extended polyproline chain. The resulting oxaldie-3 was monomeric unlike its predecessor, eliminating the need for high peptide concentrations to elicit catalysis.18 Modest improvements in kcat and Km were also achieved. In oxaldie-4, a bovine pancreatic peptide scaffold with intrinsic disulfides further stabilized the fold, resulting in even better kinetic parameters and activity at higher temperatures.19 The same scaffold was adapted to make a miniature esterase.20
The self-replicating peptides are an interesting case of α-helical enzymes where the catalyst does not form covalent intermediates with reactants.21, 22 The enzyme is a 32-residue amphipathic helix with a hydrophobic Leu/Val rich face which binds two 16-residue peptides that are N and C-terminal halves of the same sequence. Binding reduces the entropic costs of chemical ligation between a C-terminal thioester leaving group and an N-terminal cysteine. The product also serves as a template, resulting in progressive amplification of catalytic activity. Although computational methods have not been applied in the design of these systems, they could potentially be used to improve catalytic efficiency, by developing sequences that balance the competing processes of template formation and product inhibition.
The α-helix continues to be a useful tool in the rational design of enzymes without the need for sophisticated computation. The polar/nonpolar sequence periodicity of the helix serves as a simple mechanism for promoting multimer associations, and for spatially clustering active site residues. β-hairpins and small sheets have not been used extensively as a platform for developing small peptide catalysts, given the increased difficulty of designing folded β-structures, which are stabilized by interactions well-separated in the primary sequence. Significant progress in β-sheet design may soon change this.23-25 There are limitations to the chemical complexity one can achieve on the face of a single helix. As such a number of enzyme designs have made use of more complex protein topologies, combining multiple helical elements.
Enzyme Models with Tertiary Structure
Valuable progress has been made in the design of catalytic proteins where active site and second shell residues are donated by multiple structural elements. This significantly enhances the potential for chemical diversity, as well as providing space for binding sites to improve affinity and specificity. One productive scaffold is the helix-loop-helix motif, where a short turn connects two amphipathic α-helices, which then dimerize into a four-helix bundle.26 Because the two helices are part of the same peptide, they can host a greater diversity of chemical groups. The Baltzer lab has used this motif to target a number of reactions involving model RNA-like substrates (Figure 2). Functionalizing one face of a helix-loop-helix at four sites with a zinc-triazacyclononane amino acid resulted in a peptide capable of catalyzing transesterification of 2-hydroxypropyl-p-nitrophenyl phosphate 380 times faster under saturating conditions.27 The same scaffold has been used to hydrolyze phosphodiester bonds using histidine as a general acid/base instead of metals.28, 29
Figure 2. Helical designs with tertiary structure.
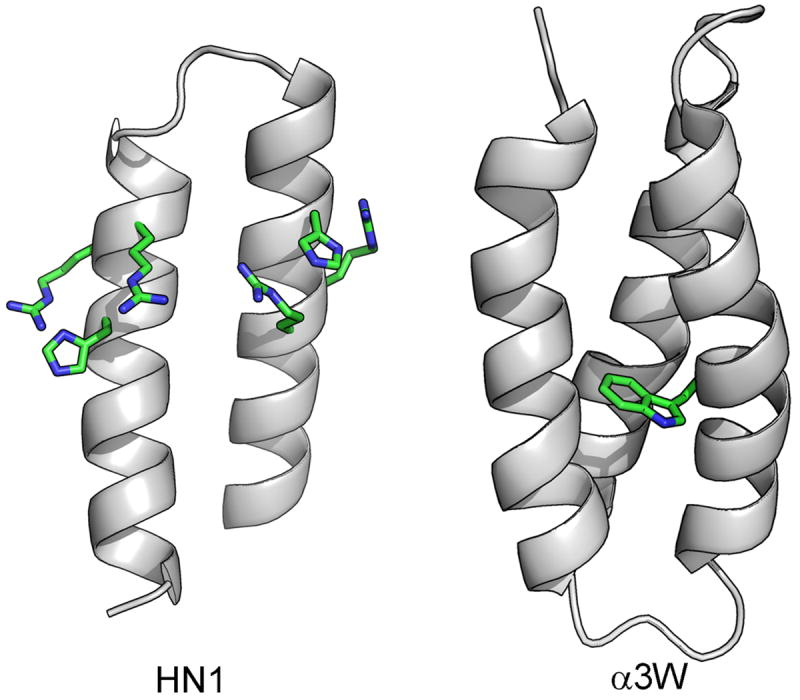
Using short connecting loops, multi-helical structures provide the potential for greater chemical diversity at the active site, a binding surface for defining substrate specificity and a core for controlling the microenvironment of key catalytic residues. Two examples are the HN1 ribonuclease29 with an active site of two histidines and four arginines (model structure), and α3W 31, a three helix bundle which tunes the reduction potential of a tryptophan radical in the bundle core.
A three helix design (helix-loop-helix-loop-helix) with a single tryptophan or tyrosine in the hydrophobic core was designed to measure how the protein environment modulates the stability of amino acid radicals, as inferred from electrochemical measurements. Amino acid radicals are important in a number of fundamental biochemical reactions including the evolution of molecular oxygen from water by photosystem II and the biosynthesis of DNA from ribonucleotides by ribonucleotide reductase. The low dielectric environment inside the protein was shown to significantly raise the reduction potential of both Tyr and Trp.30 The structure of α3W also uncovered a cation-π interaction between the Trp and an Arg, which modeling suggested would also raise its reduction potential.31 Although the α3Y/W peptides have not yet been developed into a catalyst, they showcase an important function of protein design – to provide minimal systems to help us understand the complex molecular forces involved in enzyme function.
Binary Patterning-Based Design of an Artificial Oxygen Transporter
As discussed above for helichrome, oxalide and others, simple folds such as helical bundles can be designed non-computationally with a high probability of success merely by placing polar amino acid side chains at solvent exposed positions and nonpolar amino acids at core positions, a process termed binary patterning.6 Such design experiments typically result in molten globules which exhibit stable secondary structure but lack a unique three dimensional structure, most likely because the randomly selected core residues lack ‘knobs and holes’ type intercalation, allowing these stable elements of secondary structure to move independently.32-34 Binary patterning coupled with explicitly designed polar interactions buried within the protein core can fix these elements of secondary structure, lifting these proteins into unique three dimensional conformations, either using internal hydrogen bonding or metal-ligand interactions.34, 35
This design approach been exploited in an artificial oxygen transport protein, HP-7.36 In this protein one of the two ligand histidines of a bis-histidine heme binding site in a binary patterned four helix bundle scaffold was destabilized by the addition of core glutamic acid residues on the same helix. This intentional violation of the rules of binary patterning results in the stabilization of an alternate conformation wherein the ligand histidine detaches and the core glutamate residues rotate out into solution, opening a heme iron coordination site for oxygen binding (Figure 3). This mechanism is similar to that observed for other hexacoordinate hemoglobins such as cytoglobin, neuroglobin or leghemoglobin,37 although the structure and sequence of HP-7 is unrelated to any natural oxygen carrier.
Figure 3. Step-wise oxygen binding to a binary-patterned four helix bundle.
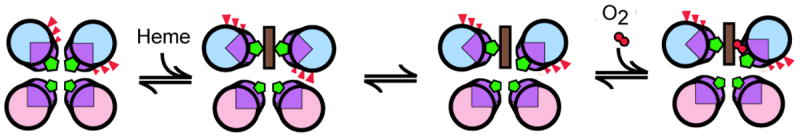
In the HP-7 maquette36, heme binding requires rotation of a-helices to present histidines (green) in the proper geometry. This forces the unfavorable burial of glutamates (red triangles) inside the nonpolar protein core. Release of one of the axial histidines allows the glutamates to interact with solvent and provides an open coordination site on the heme for oxygen binding. (Figure adapted from reference 36)
Measurement of the kinetic and thermodynamic constants for oxygen and carbon monoxide binding by HP-7 demonstrates that it performs equivalently to natural globins, with the unprecedented exception that HP-7 binds molecular oxygen tighter than carbon monoxide. This work not only demonstrates that an intuition-led approach like the modified binary patterning method described here can lead to sophisticated biological function, it presages protein design’s ability to exceed the capabilities of the proteins found in nature.
Computational Design of a de novo Enzyme
Designing effective enzymes requires the ability to design structure. This is an area where computational methods have proven most useful. Fully automated designs of sequences to achieve target folds have been demonstrated multiple times over the past decade, from small proteins such as a zinc-finger like structure that folds without metal38 to Top7,39 a novel mixed α/β fold with no natural counterpart. Although the fully automated design of an enzyme from the ground up has yet to be accomplished, recent work on computationally designed metalloenzymes have made significant advances in this area.
Computational design played an appreciable role in the DF series of proteins (due ferro, Italian for “two iron”). These were inspired by a class of natural oxygen activating enzymes with dinuclear iron clusters.40 A retrostructural analysis of metalloenzymes such as the R2 subunit of ribonucleotide reductase and methane monooxygenase revealed a simple underlying D2 symmetry of the protein backbone, an antiparallel four helix bundle, which reflected the symmetry of the active site metals and substrates (Figure 4).41 This made it possible to generate a scaffold de novo using an ideal α-helix and symmetry operations.42 Key metal ligands (called keystone interactions) and second-shell interactions were placed on the scaffold. The protein was designed using a combination of manual and automated modeling. One of the computational tools used was ROC (Repacking of Cores), a program that optimizes a design by evaluating structure/sequence candidates on van der Waals energies of sidechain packing.43 The final model was a four helix bundle composed a homodimer of helix-loop-helix chains, much like those of Baltzer. The atomic structure of the design was solved by X-ray crystallography in the presence of zinc. Comparison of the structure and the model demonstrated that many of the design elements were successfully implemented. DF1 provided a clear link between sequence, structure and function, making it a rich platform to understand how this class of proteins functions.
Figure 4. Retrostructural analysis and design of a dinuclear metalloprotein.
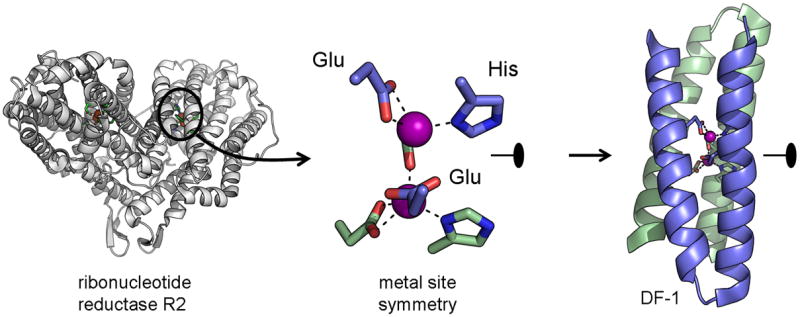
The DF series of designed metalloenzymes were built from structural analysis of dinuclear metal sites in proteins such as ribonucleotide reductase 41. The two metals, two histidines and four glutamates that formed the active site could be described by two half-sites related by a C2 symmetry axis. The same symmetry was found locally in the natural metalloenzymes and was used in the de novo design of a helix-loop-helix dimer, DF-1.
The structure of DF1 suggested that any potential catalytic activity was hampered by the presence of two leucines at the metal site. These were mutated to alanine in DF2, providing a substrate channel capable of binding small aromatic molecules.44 Upon adding 4-aminophenol, it was shown that DF2 could catalyze two-electron oxidation to benzoquinone monoimine through a diferric intermediate in the presence of oxygen; multiple turnovers and thousand-fold rate enhancements over the uncatalyzed reaction were observed.45 Using an electron-rich phenol such as 4-AP provided a facile substrate for establishing structural features necessary for binding and catalysis. Additional optimization through combinatorial libraries or directed evolution could be used to develop more powerful metalloenzymes.
The DF series has also proven to be a useful tool in understanding how natural metalloenzymes may fold and function. One important feature of natural enzymes recapitulated by DF is preorganization. Preorganization of active site residues improves catalytic activity by reducing the entropic cost of forming the enzyme-substrate complex. It also allows the enzyme to dictate the configuration of the complex, which may be important for targeting transition states and high-energy intermediates. In the absence of metal, the fold was highly similar and key metal binding residues were constrained close to the active site.46 Another feature that distinguishes enzymes from small molecule catalysts is their faculty to couple reactivity to protein motion, such as allosteric conformational changes that toggle an enzyme between active and inactive states. In high resolution structures of DF1 bound to manganese, two coordination environments were found in the asymmetric unit: one where a solvent molecule bridge the two metals, and a second where two solvent molecules bound trans to protein ligands.47, 48 This was coupled with a shift between the two helix-loop-helix motifs, suggesting that a large-scale sliding motion could tune active site reactivity.
DF has also proved useful in furthering general computational methods in enzyme design and simulation. In work to create a single chain protein, DFsc, it was found that correctly modeling the turn residues between helices could significantly improve design stability. This lead to an in-depth characterization of helix-turn-helix structures in the Protein Data Bank (PDB), identifying key turn motifs that correlated to specific geometries of helix-helix pairs. A de novo computational simulation of charge-pair interactions on the surface of DF was used to design sequences that formed a 2A:2B heterotetramer between four separate helices.49 It was even possible to build an A:B:2C heterotetramer from three peptides using surface electrostatics.50 This will facilitate combinatorial approaches to develop better enzymes by mixing libraries of peptides and screening for activity. High resolution active site design has been initiated with DF1, subjecting a chemically simplified metal site to Car-Parrinello molecular dynamics simulations.51 Snapshots from these simulations correspond well to high-resolution structures of DF1. Simulation methods could be used to generate structures of transition states and high-energy intermediates in catalysis. These could then be included as constraints in the redesign of DF1 for novel catalytic activity.
The retrostructural analysis strategy has also been applied to the computational design of a β-sheet metalloprotein and a four-helix bundle that binds arrays of non-natural porphyrin cofactors.52, 53 Key features such as focusing on keystone interactions, binding site preorganization by second-shell ligands, and mirroring of protein fold and active site symmetry have been implemented in these systems. Integration of computation and chemical intuition in the de novo design of proteins will further our understanding of the basic relationship between sequence, structure and reactivity.
Computational de novo Active Site Design
Although the concurrent design of structure and catalysis promises to broadly expand the scope of artificial enzymes, this area is still in its infancy. Even state of the art designs such as DF still depend heavily on biological motifs and helical topologies. Thus, a number of labs have pursued the more tractable target of designing novel enzymatic functionality into existing protein scaffolds. Two major challenges in de novo active site design are (1) identifying optimal locations on a protein to introduce the active site residues and (2) modeling the active site with sufficient accuracy to enable appropriate reactivity. These issues represent a long standing conflict in protein design between speed and accuracy. Approximations in energy calculations and coarse-grained sampling methods may speed simulations, allow broader sampling of sequence/structure combinations. However, the tradeoff with accuracy may reduce the ability to discriminate between successful and unsuccessful designs.
Software such as METAL SEARCH54-56 and DEZYMER57 successfully introduced metal binding sites into novel locations on protein surfaces. Starting with a high resolution structure of the target protein, sites in the backbone were selected based on their capacity to present metal binding residues presenting thiol, carboxylate or imidazole ligands. These were then modeled onto the structure in various rotameric configurations to determine whether the coordination geometry of the metal was satisfied.
Despite the comparatively large energy of metal-ligand bond formation relative to other intramolecular forces in proteins, accurate design of metalloproteins presents a number of challenges, particularly with regards to specificity. For example, DEZYMER was used to convert the substrate binding region of Maltose Binding Protein (MBP) into a zinc-binding site. MBP has two large domains connected by a flexible hinge region, allowing a cavity between the two domains to open and close. Maltose binds to this cavity and stabilizes the closed state (Figure 5). Marvin and Hellinga systematically modeled groups of amino acids in the domain interface as potential zinc ligands, until a set of four amino acids were identified, two from each domain, which could potentially bind zinc when MBP was in the closed conformation.58 The best design bound zinc with micromolar affinity. Subsequent biophysical and structural characterization revealed an unanticipated mode of zinc binding which involved the open state.59 Two histidines of the four designed amino acid substitutions were shown to bind the zinc. A nearby aspartate carboxylate sidechain was found within four angstroms of the metal, too far to form direct, strong ligand-metal bonds. However, mutation of this Asp to Ala abolished affinity for zinc, indicating some role in binding. Thus a key challenge in metalloprotein design is understanding the role of second shell and potentially weak first shell interactions in affinity and specificity. The large-scale conformational transitions in MBP between open and closed states highlights another challenge – that of negative design. By optimizing sidechain metal-ligand geometries, programs seek to maximize the stability of a target state, usually using a crystal structure as a starting point. Negative design seeks to account for competing, off-target conformations and either explicitly or implicitly destabilize them. This is extremely difficult without a detailed knowledge of the structure of conformational microstates. Optimization of sequence over an ensemble of target and off-target protein conformations also exacerbates the already formidable number of states to be sampled. This is currently a very active area in protein design.
Figure 5. Design versus Structure of a Zn2+ Binding MBP.
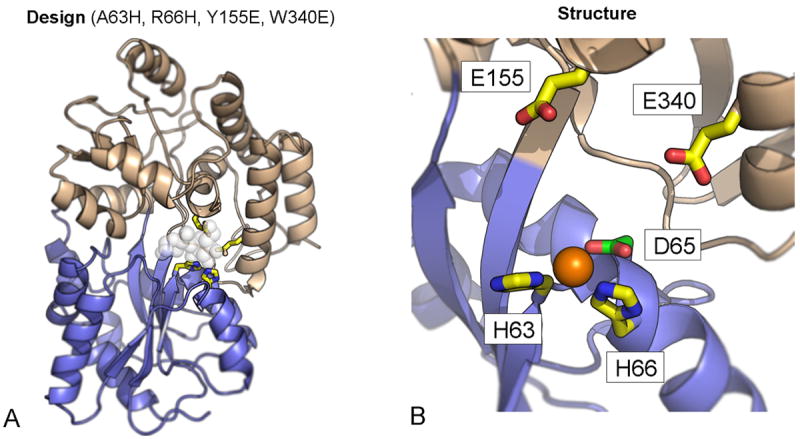
(A) Using the computer program DEZYMER57, four substitutions were designed into closed form of the Maltose Binding Protein (MBP) which conferred affinity for zinc. Mutations (in yellow) were in the maltose binding site (white spheres). (B) Crystal structures of the MBP variant demonstrated binding was in fact to a conformation closer to the open form, only involving half of the residues and an additional aspartate ligand (green). A = alanine, R = arginine, Y = tyrosine, W = tryptophan, E = glutamate, D = aspartate, H = histidine.
A series of His3Fe sites were introduced to thioredoxin in various environments classified as grooves, shallow pockets, and a deep pocket, allowing them to study the effect of the protein microenvironment on ‘nascent’ enzymatic activity.60, 61 Superoxide dismutase (SOD) activity, the conversion of superoxide radical into molecular oxygen and hydrogen peroxide, correlated with local electrostatic interactions, where a net positive charge at the binding site was hypothesized to attract the O2.˜ species. Further structural characterization of these designs could be very informative, as the previous MBP example emphasizes. Where possible, an atomic resolution structure of a de novo functional protein is an important step in understanding how design elements correlate to mechanism.
A similar approach was used to build a non-metal proto-enzyme site into a thioredoxin scaffold that catalyzed the hydrolysis of paranitrophenol acetate (PNPA) into PNP and acetate.62 This was accomplished by a histidine nucleophile that formed a high-energy transition state with PNPA. In order to computationally model this reaction, a composite sidechain comprised of the histidine covalently linked to PNPA was introduced and conformationally sampled around accessible bond rotations. Adjacent amino acids to the site of the His-PNPA were allowed to mutate to alanine to facilitate substrate binding and recognition. The conformations of His-PNPA and surrounding sidechains were optimized using Dead End Elimination, a powerful algorithm which significantly reduces the combinatorial complexity of multi-site optimizations by eliminating pairwise states that are provably incompatible with the global energy minimum.63 The top two scoring candidates, PZD1 and 2 were synthesized. PZD2 demonstrated significant rate enhancements over the uncatalyzed reaction and saturation kinetics with increasing substrate concentration.
Although the thioredoxin derived metalloenzymes and PDZ2 had kinetic parameters well below those of natural enzymes and even catalytic antibodies, they were key first steps in developing computational methods for enzyme design. Important advances in the efficiency of computational methods such as Dead End Elimination are proving crucial in making these design problems tractable.64 Although initial extension of these computational methods to the design of a triose phophate isomerase turned out to be unsuccessful, many important ideas put forth in these studies were incorporated into the recent, successful design of chemically ambitious artificial enzymes.
In parallel with computational advances, active site designs continue to progress using rational, intuition-based strategies. Esterase activity was introduced to human carbonic anhydrase (HCAIII) through both protein and substrate engineering.65 HCAIII affinity for benzenesulfonamide containing molecules was used to model a substrate such that the scissile bond was positioned within a cleft in the protein. Grafting a His dyad from previous de novo helix-loop-helix designs resulted in an HCAIII variant with enhanced esterase activity over wild-type.
The ROSETTA Enzymes
ROSETTA is a suite of computational tools developed in the laboratory of David Baker for protein structure prediction, protein-protein complex prediction and protein design. One of the key innovations in ROSETTA is its utilization of high-resolution structures in the PDB as a ‘parts list’. Small fragments around ten residues in length are assembled into larger molecules, drastically reducing the conformational degrees of freedom to be sampled. This approach rests on the assumption that structure and stability information is implicitly encoded locally within each fragment.34 The global stability of a design is evaluated based on a scoring potential that combines physics based energy terms such as van der Waals packing and knowledge based energy terms derived from statistical analysis of amino acid interactions the PDB.66 Recently, ROSETTA was used to develop artificial enzymes that catalyzed a retro-aldol reaction (Figure 6) and a Kemp elimination.67, 68 These designs were impressive in the extent to which the relationship between structure and reactivity was modeled and characterized.
Figure 6. Assembly line for the ROSETTA Enzymes.
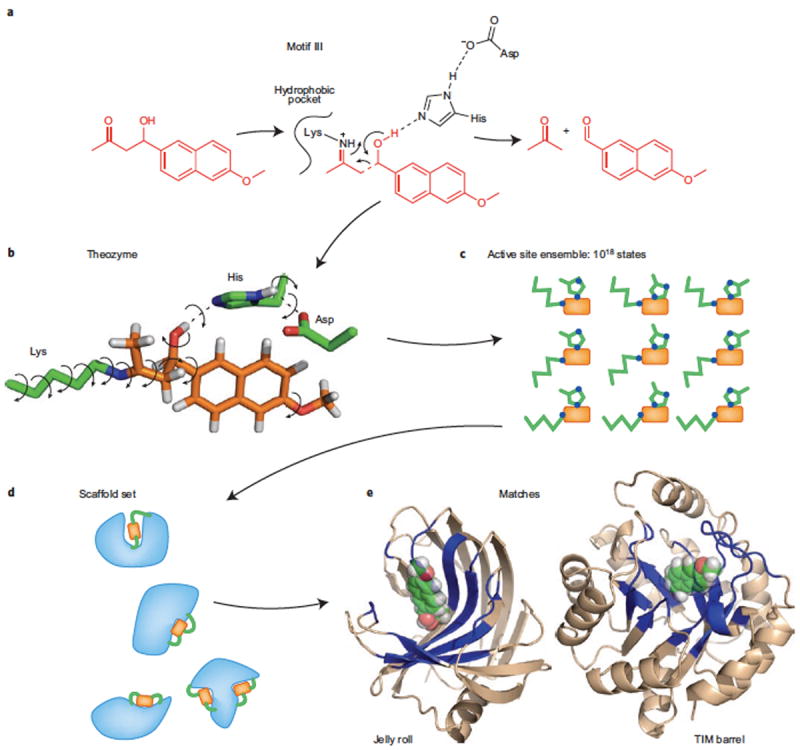
(A) A reaction motif is sketched, outlining key intermediates, general acid base ligands, and strategies for modulating catalytic residue pKas, (B) QM calculations are used to optimize the geometry of a transition state model including truncated active site residues. (C) Elaboration of catalytic residue rotamers creates an ensemble of active sites. (D) These are matched to complementary surfaces on a family of target protein scaffolds. (E) Promising designs are synthesized and characterized for activity. (Figure adapted from reference 111).
In the retro-aldolase, the goal was to break a carbon-carbon bond in a non-natural substrate, 4-hydroxy-4-(6-methoxy-2-napthyl)-2-butanone.69 The intended reaction was significantly more complex than previous designs, involving multiple transition states and intermediates to cleave the substrate and regenerate the active site. The reaction proceeded through an imine intermediate involving a lysine as a Schiff base, similar to the oxaldie decarboxylation of oxaloacetate. In oxaldie, the lysine nucleophile was stabilized through electrostatic interactions with other charged sidechains or the helix macrodipole. The same mechanism was used in the first of four reaction motifs attempted by placing a second lysine in the vicinity of the first. The other three used a hydrophobic pocket to lower the pKa of the lysine. A general acid/base was included to trigger cleavage of the carbon-carbon bond. Each motif utilized a different base: I – a Lys/Asp dyad, II – tyrosine, III – a His/Asp and IV – a water molecule. Attempting designs based on several reaction motifs not only increases the chance of a successful outcome, but also demonstrates how design can be used to test various hypotheses for catalysis.
As previously discussed, two challenges in computational enzyme design are accurate modeling of active site interactions and sufficient sampling of candidate backbone templates on protein scaffolds. In order to meet the first challenge, quantum mechanics calculations were performed on a minimal chemical representation of the protein-substrate complex.70 To simplify active site construction, a composite transition state was constructed that combined optimal geometries of the carbinolamine alcohol and the bond breaking state. The remainder of the active site sidechains were then built and sampled for rotameric degrees of freedom. After all active site and second shell residues were modeled in, a total of anywhere from 1013 to 1018 potential sequence/structure combinations were generated, depending on the reaction motif considered.
At this point in the design, the researchers had built a huge ensemble of disembodied active sites. The next challenge was to compare these active sites to a set of potential scaffolds in seventy protein structures. These structures represented a diverse set of protein folds including triose phosphate isomerase (TIM) barrels, jelly rolls, β-propellors, lipocalins and periplasmic binding proteins. To model 1018 active sites onto all possible combinations of backbone positions would be computationally intractable. An algorithm called RosettaMatch, made use of geometric hashing to reduce the problem to one that scaled linearly with the number of states.71, 72 For a very readable review of geometric hashing, we recommend reference 73. Still, this was an immense calculation, and other resources such as distributed computing over thousands of personal computers volunteered through the Rosetta@Home project made this possible. After the first stage of matching, the number of designs was around 180,000. This was further reduced by computational redesign of adjacent amino acids surrounding the active site, in order to maintain structural integrity of the overall protein fold.
After an extensive computational vetting process, searching through four reaction motifs, a billion billion active site configurations and a diverse set of scaffolds, seventy proteins were synthesized. Each design contained anywhere from eight to twenty mutations relative to the wild-type protein. Impressively, nearly half showed some aldolase activity. Interestingly, the successful enzymes were not equally distributed over mechanism or fold. Only motifs III and IV which used histidine or water as the general acid/base were successful, suggesting the other mechanisms were either chemically unfeasible or difficult to design. Similarly, only the jelly-roll and TIM barrel were productive, indicating these folds may have intrinsic geometric properties favoring the design of catalytic sites (the capacity for a fold to accommodate multiple sequences is referred to as designability, and quantifying this parameter has been attempted for simple systems).74, 75 Rate enhancements of up to 104 were achieved. Structures of designs solved by X-ray crystallography showed the atomic accuracy of the computational models, an essential feature for furthering rational design of these enzymes. Designs were verified by mutagenesis of active site residues to ablate catalysis. Careful analysis verified saturation kinetics. Although these designs fall short of retro-aldolase activity seen in catalytic antibodies,69, 76 they remove a number of constraints on the design of artificial enzymes, opening the possibility for reactivity on a broad spectrum of protein scaffolds.
This approach can also be combined with in vitro evolution to improve catalytic efficiency, as was shown with a set of Kemp elimination enzymes also designed with the ROSETTA platform.68 The computational approach to designing Kemp elimination enzymes was very similar to the retro-aldolase. Kemp elimination is a model reaction for proton abstraction from a carbon. Successful designs were again found in TIM Barrel folds, with rate enhancements of up to 105, saturation kinetics in certain cases and greater than seven turnovers. In vitro evolution of one the design KE07 by cycles of mutagenesis and activity screening resulted in a 200-fold improvement in kcat/Km. Residues involved in catalysis were unchanged; rather, most mutations were found in amino acids adjacent to the active site. High resolution crystal structures of KE07 were superimposable on the computational model, allowing the researchers to develop specific hypotheses regarding the mechanism of improved enzymatic function by evolution. This highlights the potential advantage of combining rational design with directed evolution.
As with retro-aldolase designs, KE07 only demonstrates kinetic parameters equivalent to or below that of catalytic antibodies77, or even nonspecific reaction rates with ‘off the shelf’ proteins such as bovine and human serum albumins78. The real success of the ROSETTA enzymes is not in the computational design of catalysts of practical value, a goal which has still to be achieved by any group. Rather, they are among the earliest demonstrations of the ability to harness the three-dimensional protein fold to stabilize energetically unfavorable active sites,79 for example by locating a general base in a hydrophobic microenvironment. This is an important component of enzyme design.
A two pronged approach combining laboratory evolution with structure-based computation may increase the likelihood of designing synthetic enzymes with activities approaching those of natural counterparts. One strategy is to reduce the number of sequences to be experimentally characterized. This has been approached in multiple ways: identifying positions in a protein that can tolerate mutations80, identifying protein domain boundaries in order to generate libraries of permuted chimeric species81 and predicting amino acid substitutions that preserve function while maximizing chemical diversity of sampled sequences.82 The Tidor laboratory took an inverse approach, using computational sequence optimization to enhance antibody binding after affinity maturation against two targets (lysozyme and human epidermal growth factor receptor).83
Moving enzyme design out of the active site
KE07 models a potentially powerful strategy of combining computational design and laboratory evolution toward novel enzymes. Below, we highlight other areas where rational methods may introduce new functionality that would be difficult using laboratory evolution alone.
The designed enzymes to date have each focused on the creation of a transition state-stabilizing active site as the basis for their catalytic function. Thus, they necessarily have the same limitations in scope as has been noted for catalytic antibodies elicited with transition state analogues 76, in particular their modest catalytic rate accelerations which pale in comparison to the values of up to 1023 observed in natural enzymes 84. This is at least in part due to the fact that catalysts which bind to transition states too tightly suffer from product inhibition, setting a limit on the maximal rate acceleration possible using this method alone 85. For rationally designed enzymes to surpass these limitations, additional strategies, many derived from natural enzyme mechanism, must be employed 86. While these strategies incur an additional level of complexity in rational design, they are also responsible for much of the additional catalytic power observed in protein catalysts. Some of these additional mechanistic enhancements may offer an entryway into catalytic complexity that is not only impossible in catalytic antibodies but also not accessible to directed evolution approaches. Furthermore, it seems likely that many of these strategies are themselves open to implementation using chemical intuition.
Multiple substrates
Perhaps the simplest mechanism by which enzymes accelerate chemical reactions is by utilizing binding energy to compensate for the entropy loss incurred in bimolecular reactions 87. Merely holding two substrates in proximity to each other at the correct orientation for a productive reaction can engender a 108–fold rate acceleration in the absence of any transition state-stabilizing protein-substrate interactions. Such a strategy proved successful in the self-replicating peptide system 21. This, coupled with transition state-stabilization, could elevate rate enhancements to levels similar to those observed in natural enzymes. Designing a single active site which will bind, orient and activate two substrates simultaneously is, however, a more difficult problem. It seems likely that simpler bi-substrate mechanisms, either an ordered mechanism with a covalent intermediate, or a single protein with two active sites coupled by the channeling of an intermediate, would be more accessible to current design technology 3.
Timing in multiple substrate reactions
An important facet of the mechanism of many of nature’s more complicated catalysts is the temporal control of substrate binding and transfer events. One merely has to look at the exquisite engineering apparent in the multiple electron and proton transfers in photosynthesis, respiration and nitrogen fixation to see the advantages conferred by the ability to control the timing and energetics of the intermediates in multiple substrate reactions 88. As these reaction mechanisms would require the simultaneous optimization of several different catalytic events at several different sites, it is unlikely that such would arise from laboratory time scale evolution. A more likely scenario is one where an initial enzymatic scaffold is designed explicitly and then the kinetics and thermodynamics of the intermediates are further optimized using directed evolution.
Small scale enzyme motions
The analysis of hydrogen tunneling in enzymes has established unequivocally that protein dynamics can play a large role in rates of enzymatic catalysis 89. Computational analysis of enzyme-substrate complexes using molecular mechanics has estimated the role of the dynamics of ‘near attack complexes’ in catalytic function in the absence of atomic tunneling to be as high as 103 90. However, these nanosecond time-scale motions are an intrinsic property of all biopolymers. It is not clear to what extent choreographed fast dynamics promotes catalysis in different natural enzymes. A recent report of the dynamic analysis of each intermediate state in the catalytic mechanism of dihydrofolate reductase 91 demonstrated that in each kinetic intermediate, the protein accessed only the conformations present in the current state and the states immediately before and after. These findings indicate that for at least some enzymes such dynamics are considerably choreographed.
Because the origins and even the consequences of these motions are not well understood, this contribution to catalysis will be difficult to reproduce. As the optimization brought about by directed evolution techniques is often a result of mutations distal to the active site which may impact protein motions, it seems that this method is currently the best approach for increasing catalytic efficiency with small scale dynamics. Designed enzymes offer a uniquely adaptable scaffold upon which to test our ideas about such dynamic motions, and artificial proteins will no doubt prove to be central in the development of our understanding of enzyme dynamics.
Large scale enzyme motions
Multi-Angstrom, millisecond time scale protein motions are a critical component of many natural enzyme mechanisms 92. One well-characterized example is the Ω loop governing the function of adenylate kinase 93, 94, where this loop has an open conformation, competent for substrate binding and product release, and a closed conformation, competent for catalysis (see Figure 7). This hinge opening motion on a ‘lid’ over the active site is a common motif in protein function, particularly the TIM-barrel family of enzymes 95. There are even larger motions, involving entire protein domains, utilized by much complex multidomain enzymes – the ‘escapement’ mechanisms which have been well characterized in the cytochromes bc1 and the flavocytochrome P450 BM3 96 are simple large scale motions. Intuitive approaches can be envisioned for incorporating large scale motions into a protein design, such as the introduction of flexible loops. Such features are unlikely to result directly from laboratory time scale evolution approaches without some degree of initial design.
Figure 7. Millisecond time-scale motions in adenylate-cyclase.
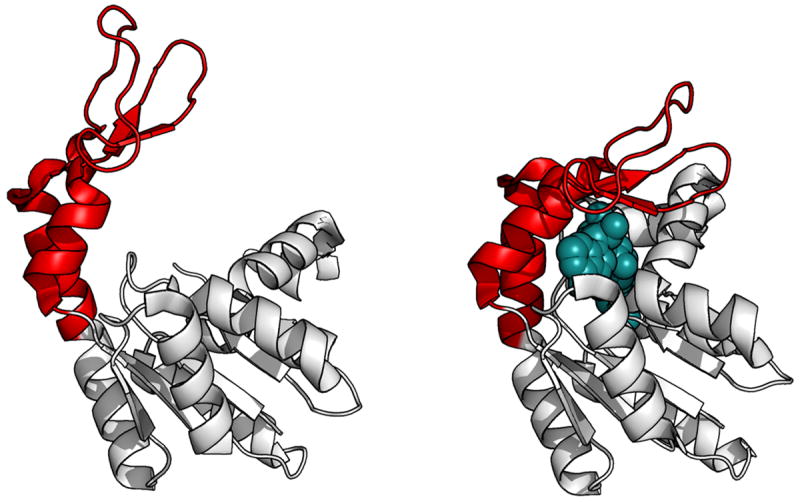
Adenylate cyclase maintains intracellular concentrations of adenylate nucleotides by converting an ATP and an AMP into two ADP molecules. Binding and catalysis requires a significant conformational rearrangement.93, 94
The helical rotation discussed above in the mechanism of the artificial neuroglobin HP-7, is exemplary of the types of problems de novo enzyme design can target relative to catalytic antibody formation or directed evolution. Antibodies are relatively inflexible and evolutionary methods are unlikely to generate multiple conformations simultaneously. As such conformational switching is an essential component of the function of many protein catalysts; these motions are best incorporated at the design level.
Allostery
Cooperative phenomena are fundamental in many biological functions such as oxygen transport, metabolic and transcriptional regulation97. The ability to incorporate allosteric regulation into artificial enzymes will enable the creation of medicinally useful in vivo catalysts which can be regulated either by metabolites or using exogenous small molecule effectors. Such behavior is in most cases a more complicated version of the large scale conformational switching described above, only in this case the large scale protein motion is actuated by the binding of an effector molecule.
Keeping energetic intermediates from the cellular environment
Anyone who has observed a bioinorganic chemist at work in a dry glovebox can appreciate the fragility of metalloenzyme active sites. Such enzymes employ reactive intermediates that must be screened from water, oxygen and reactive species in the cellular environment such as glutathione and superoxide. In fact, the failure of many initial attempts at metalloenzyme design can be attributed to the desire to make these model proteins as small as possible 98. Larger proteins have sufficiently sized hydrophobic cores that they can completely encapsulate these intermediates in a nonreactive environment, screening them from solution and lengthening their lifetimes.
Catalytic Foldamers
Despite the intensive focus on protein enzymes, the first catalytic biomolecules may have been based on RNA rather than amino acids 99. Nucleotides can carry out metal-assisted reactions, act as general acid-bases and function to orient substrates and isolate them from solvent, much like the most sophisticated catalytic proteins 100. Catalytic activity has also been found in certain bacterial carbohydrates 101. Evidently, proteins do not have a monopoly on biological catalysis. Although the successful design of a new protein is a rewarding experience in itself, often real advances are in our understanding of the basic molecular forces that guide structure and function. Given that catalysis is not limited to proteins, it is important for us to ask whether insights gained from current de novo proteins are idiosyncratic to proteins, or whether we are learning broader lessons about molecular design. This is the goal of ‘foldamer’ research, the development of novel polymeric systems that fold into unique, three dimensional structures 102, 103.
Toward designing a functional foldamer, it is important to remember the lessons of oxaldie and other catalytic peptides – a firm grasp of molecular structure is an important prerequisite to introducing function. Computational studies of foldamers are still in their infancy. Several groups have used simulation approaches to study the conformational space available to peptidomimetics incorporating D-amino acids104, 105 or β-amino acids106, 107, as well as completely non-biological foldamers such as m-phenylene ethylenes (mPE) 108. These studies establish non-natural scaffolds which can then be functionalized to facilitate reactivity. mPEs are promising due to their capacity to form helical cavities which can be functionalized with catalytic residues and serve to isolate substrate from solvent 109. This is an exciting new direction for molecular design.
Although the Wright brothers did not use a computer to build the first airplane, the aerospace industry has since greatly benefitted from sophisticated simulation and design software. Clearly, both the chemist and the computer will play important roles in the future of artificial enzymes. Design by chemical intuition tests our understanding of basic rules, i.e. binary patterning, molecular forces, metal coordination, catalytic mechanisms. These rules are then codified to allow the computational design of increasingly complex systems. Designer enzymes are a useful test of our understanding of how proteins fold and function. They also provide a rational path toward inexpensive, non-toxic catalysts that perform novel chemistry.
Contributor Information
Vikas Nanda, Robert Wood Johnson Medical School - UMDNJ Biochemistry, Center for Advanced Biotechnology and Medicine, 679 Hoes Lane West, Piscataway, New Jersey 08854, USA.
Ronald L. Koder, City College of New York Physics, 160 Convent Avenue, New York, New York 10031, USA
References
- 1.Davie EAC, Mennen SM, Xu Y, Miller SJ. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- 2.List B. Proline-catalyzed asymmetric reactions. Tetrahedron. 2002;58:5573–5590. [Google Scholar]
- 3.Koder RL, Dutton PL. Intelligent design: the de novo engineering of proteins with specified functions. Dalton Transactions. 2006;25:3045–3051. doi: 10.1039/b514972j. [DOI] [PubMed] [Google Scholar]
- 4.Lim V. Protein Folding. In: Jaenicke R, editor. 28th Conference of the German Biochemical Society; West Germany: Elservier, Regensburg; 1979. pp. 149–166. [Google Scholar]
- 5.Crick FHC. The Packing of alpha-Helices: Simple Coiled Coils. Acta Crystallographica. 1953;6:689–697. [Google Scholar]
- 6.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Protein Design by Binary Patterning of Polar and Nonpolar Amino Acids. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 7.Lau SYM, Taneja AK, Hodges RS. Synthesis of a Model Protein of Defined Secondary and Quaternary Structure. Journal of Biological Chemistry. 1984;259:13253–13261. [PubMed] [Google Scholar]
- 8.DeGrado WF, Lear JD. Induction of Peptide Conformation at Apolar/Water Interfaces. 1. A Study with Model Peptides of Defined Hydrophobic Periodicity. Journal of the American Chemical Society. 1985;107:7684–7689. [Google Scholar]
- 9.Bryson JW, et al. Protein Design: A Hierarchic Approach. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 10.Handel TM, Williams SA, DeGrado WF. Metal Ion-Dependent Modulation of the Dynamics of a Designed Protein. Science. 1993;261:879–885. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 11.Lovejoy B, et al. Crystal Structure of a Synthetic Triple-Stranded alpha-Helical Bundle. Science. 1993;259:1288–1293. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 12.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–7. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki T, Kaiser ET. Helichrome: Synthesis and Enzymatic Activity of a Designed Hemeprotein. Journal of the American Chemical Society. 1989;111:380–381. [Google Scholar]
- 14.Barbier B, Brack A. Basic Polypeptides Accelerate the Hydrolysis of Ribonucleic Acids. Journal of the American Chemical Society. 1988;110:6880–6882. [Google Scholar]
- 15.Barbier B, Brack A. Conformation-Controlled Hydrolysis of Polyribonucleotides by Sequential Basic Polypeptides. Journal of the American Chemical Society. 1992;114:3511–3515. [Google Scholar]
- 16.Brack A, Spach G. Multiconformational Synthetic Polypeptides. Journal of the American Chemical Society. 1981;103:6319–6323. [Google Scholar]
- 17.Johnsson K, Allemann RK, Widmer H, Benner SA. Synthesis, structure and activity of artificial, rationally designed catalytic polypeptides. Nature. 1993;365:530–532. doi: 10.1038/365530a0. [DOI] [PubMed] [Google Scholar]
- 18.Taylor SE, Rutherford TJ, Allemann RK. Design, synthesis and characterisation of a peptide with oxaloacetate decarboxylase activity. Bioorg Med Chem Lett. 2001;11:2631–5. doi: 10.1016/s0960-894x(01)00519-4. [DOI] [PubMed] [Google Scholar]
- 19.Taylor SE, Rutherford TJ, Allemann RK. Design of a folded, conformationally stable oxaloacetate decarboxylase. J Chem Soc, Perkins Trans. 2002;2:751–755. [Google Scholar]
- 20.Nicoll AJ, Allemann RK. Nucleophilic and general acid catalysis at physiological pH by a designed miniature esterase. Organic Biomolecular Chemistry. 2004;2:2175–2180. doi: 10.1039/B404730C. [DOI] [PubMed] [Google Scholar]
- 21.Lee DH, Severin K, Yokobayashi Y, Ghadiri MR. Emergence of symbiosis in peptide self-replication through a hypercyclic network. Nature. 1997;390:591–4. doi: 10.1038/37569. [DOI] [PubMed] [Google Scholar]
- 22.Saghatelian A, Yokobayashi Y, Soltani K, Ghadiri MR. A chiroselective peptide replicator. Nature. 2001;409:797–801. doi: 10.1038/35057238. [DOI] [PubMed] [Google Scholar]
- 23.Butterfield SM, Cooper WJ, Waters ML. Minimalist protein design: a beta-hairpin peptide that binds ssDNA. J Am Chem Soc. 2005;127:24–5. doi: 10.1021/ja045002o. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield SM, Goodman CM, Rotello VM, Waters ML. A peptide flavoprotein mimic: flavin recognition and redox potential modulation in water by a designed beta hairpin. Angew Chem Int Ed Engl. 2004;43:724–7. doi: 10.1002/anie.200352527. [DOI] [PubMed] [Google Scholar]
- 25.Hughes RM, Waters ML. Model systems for beta-hairpins and beta-sheets. Curr Opin Struct Biol. 2006;16:514–24. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Olofsson S, Baltzer L. Structure and dynamics of a designed helix-loop-helix dimer in dilute aqueous trifluoroethanol solution. A strategy for NMR spectroscopic structure determination of molten globules in the rational design of native-like proteins. Fold Des. 1996;1:347–356. doi: 10.1016/S1359-0278(96)00050-8. [DOI] [PubMed] [Google Scholar]
- 27.Rossi P, Tecilla P, Baltzer L, Scrimin P. De novo metallonucleases based on helix-loop-helix motifs. Chemistry. 2004;10:4163–70. doi: 10.1002/chem.200400160. [DOI] [PubMed] [Google Scholar]
- 28.Razkin J, Lindgren J, Nilsson H, Baltzer L. Enhanced complexity and catalytic efficiency in the hydrolysis of phosphate diesters by rationally designed helix-loop-helix motifs. Chembiochem. 2008;9:1975–84. doi: 10.1002/cbic.200800057. [DOI] [PubMed] [Google Scholar]
- 29.Razkin J, Nilsson H, Baltzer L. Catalysis of the cleavage of uridine 3’-2,2,2-trichloroethylphosphate by a designed helix-loop-helix motif peptide. J Am Chem Soc. 2007;129:14752–8. doi: 10.1021/ja075478i. [DOI] [PubMed] [Google Scholar]
- 30.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. De novo proteins as models of radical enzymes. Biochemistry. 1999;38:9495–507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 31.Dai QH, et al. Structure of a de novo designed protein model of radical enzymes. J Am Chem Soc. 2002;124:10952–3. doi: 10.1021/ja0264201. [DOI] [PubMed] [Google Scholar]
- 32.Gibney BR, Rabanal F, Skalicky JJ, Wand AJ, Dutton PL. Design of a unique protein scaffold for maquettes. Journal of the American Chemical Society. 1997;119:2323–2324. [Google Scholar]
- 33.Gibney BR, Rabanal F, Skalicky JJ, Wand AJ, Dutton PL. Iterative protein redesign. Journal of the American Chemical Society. 1999;121:4952–4960. [Google Scholar]
- 34.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of Protein Tertiary Structures from Fragments with Similar Local Sequences using Simulated Annealing and Bayesian Scoring Functions. Journal of Molecular Biology. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 35.Koder RL, et al. Native-like structure in designed four helix bundles driven by buried polar interactions. Journal of the American Chemical Society. 2006;128:14450–14451. doi: 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- 36.Koder RL, et al. Design and engineering of an O2 transport protein. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kundu S, Trent JT, Hargrove MS. Plants, humans and hemoglobins. 2003;8:387–393. doi: 10.1016/S1360-1385(03)00163-8. [DOI] [PubMed] [Google Scholar]
- 38.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–7. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 39.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 40.Wallar BJ, Lipscomb JD. Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 41.Lombardi A, et al. Inaugural article: retrostructural analysis of metalloproteins: application to the design of a minimal model for diiron proteins. Proc Natl Acad Sci U S A. 2000;97:6298–305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Summa CM, Lombardi A, Lewis M, DeGrado WF. Tertiary templates for the design of diiron proteins. Curr Opin Struct Biol. 1999;9:500–8. doi: 10.1016/S0959-440X(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 43.Lazar GA, Desjarlais JR, Handel TM. De novo design of the hydrophobic core of ubiquitin. Protein Sci. 1997;6:1167–78. doi: 10.1002/pro.5560060605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Costanzo L, et al. Toward the de novo design of a catalytically active helix bundle: a substrate-accessible carboxylate-bridged dinuclear metal center. J Am Chem Soc. 2001;123:12749–57. doi: 10.1021/ja010506x. [DOI] [PubMed] [Google Scholar]
- 45.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc Natl Acad Sci U S A. 2004;101:11566–70. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Preorganization of molecular binding sites in designed diiron proteins. Proc Natl Acad Sci U S A. 2003;100:3772–7. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geremia S, et al. Response of a designed metalloprotein to changes in metal ion coordination, exogenous ligands, and active site volume determined by X-ray crystallography. J Am Chem Soc. 2005;127:17266–76. doi: 10.1021/ja054199x. [DOI] [PubMed] [Google Scholar]
- 48.DeGrado WF, et al. Sliding helix and change of coordination geometry in a model di-MnII protein. Angew Chem Int Ed Engl. 2003;42:417–20. doi: 10.1002/anie.200390127. [DOI] [PubMed] [Google Scholar]
- 49.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. Computational de novo design, and characterization of an A(2)B(2) diiron protein. J Mol Biol. 2002;321:923–38. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 50.Marsh EN, DeGrado WF. Noncovalent self-assembly of a heterotetrameric diiron protein. Proc Natl Acad Sci U S A. 2002;99:5150–4. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papoian GA, DeGrado WF, Klein ML. Probing the configurational space of a metalloprotein core: an ab initio molecular dynamics study of Duo Ferro 1 binuclear Zn cofactor. J Am Chem Soc. 2003;125:560–9. doi: 10.1021/ja028161l. [DOI] [PubMed] [Google Scholar]
- 52.Cochran FV, et al. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J Am Chem Soc. 2005;127:1346–7. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 53.Nanda V, et al. De novo design of a redox-active minimal rubredoxin mimic. J Am Chem Soc. 2005;127:5804–5. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 54.Clarke ND, Yuan SM. Metal search: a computer program that helps design tetrahedral metal-binding sites. Proteins. 1995;23:256–63. doi: 10.1002/prot.340230214. [DOI] [PubMed] [Google Scholar]
- 55.Klemba M, Gardner KH, Marino S, Clarke ND, Regan L. Novel metal-binding proteins by design. Nat Struct Biol. 1995;2:368–73. doi: 10.1038/nsb0595-368. [DOI] [PubMed] [Google Scholar]
- 56.Regan L, Clarke ND. A tetrahedral zinc(II)-binding site introduced into a designed protein. Biochemistry. 1990;29:10878–83. doi: 10.1021/bi00501a003. [DOI] [PubMed] [Google Scholar]
- 57.Hellinga HW, Richards FM. Construction of new ligand binding sites in proteins of known structure. I. Computer-aided modeling of sites with pre-defined geometry. J Mol Biol. 1991;222:763–85. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 58.Marvin JS, Hellinga HW. Conversion of a maltose receptor into a zinc biosensor by computational design. Proc Natl Acad Sci U S A. 2001;98:4955–60. doi: 10.1073/pnas.091083898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Telmer PG, Shilton BH. Structural studies of an engineered zinc biosensor reveal an unanticipated mode of zinc binding. J Mol Biol. 2005;354:829–40. doi: 10.1016/j.jmb.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 60.Benson DE, Wisz MS, Hellinga HW. Rational design of nascent metalloenzymes. Proc Natl Acad Sci U S A. 2000;97:6292–7. doi: 10.1073/pnas.97.12.6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto AL, Hellinga HW, Caradonna JP. Construction of a catalytically active iron superoxide dismutase by rational protein design. Proc Natl Acad Sci U S A. 1997;94:5562–7. doi: 10.1073/pnas.94.11.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci U S A. 2001;98:14274–9. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Desmet J, Demaeyer M, Hazes B, Lasters I. The Dead-End Elimination Theorem and Its Use in Protein Side-Chain Positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 64.Looger LL, Hellinga HW. Generalized dead-end elimination algorithms make large-scale protein side-chain structure prediction tractable: implications for protein design and structural genomics. J Mol Biol. 2001;307:429–45. doi: 10.1006/jmbi.2000.4424. [DOI] [PubMed] [Google Scholar]
- 65.Host GE, Razkin J, Baltzer L, Jonsson BH. Combined enzyme and substrate design: grafting of a cooperative two-histidine catalytic motif into a protein targeted at the scissile bond in a designed ester substrate. Chembiochem. 2007;8:1570–6. doi: 10.1002/cbic.200600540. [DOI] [PubMed] [Google Scholar]
- 66.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–8. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–91. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–5. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 69.Tanaka F, Fuller R, Shim H, Lerner RA, Barbas CF., 3rd Evolution of aldolase antibodies in vitro: correlation of catalytic activity and reaction-based selection. J Mol Biol. 2004;335:1007–18. doi: 10.1016/j.jmb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 70.Tantillo DJ, Chen J, Houk KN. Theozymes and compuzymes: theoretical models for biological catalysis. Current Opinion in Chemical Biology. 1998;2:743–750. doi: 10.1016/s1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]
- 71.Zanghellini A, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–94. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ladman Y, Schwartz JT, Wolfson HJ. Affine Invariant Model-Based Object Recognition. IEEE Transactions on Robotics and Automation. 1990;6:578–589. [Google Scholar]
- 73.Wolfson HJ, Rigoutsos I. Geometric Hashing: An Overview. IEEE Computational Science & Engineering. 1997:10–21. [Google Scholar]
- 74.Li H, Helling R, Tang C, Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science. 1996;273:666–9. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 75.Li H, Tang C, Wingreen NS. Are protein folds atypical? Proc Natl Acad Sci U S A. 1998;95:4987–90. doi: 10.1073/pnas.95.9.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hilvert D. Critical analysis of antibody catalysis. Annu Rev Biochem. 2000;69:751–792. doi: 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- 77.Thorn SN, Daniels RG, Auditor MT, Hilvert D. Large rate accelerations in antibody catalysis by strategic use of haptenic charge. Nature. 1995;373:228–30. doi: 10.1038/373228a0. [DOI] [PubMed] [Google Scholar]
- 78.Hollfelder F, Kirby AJ, Tawfik DS. Off-the-shelf proteins that rival tailor-made antibodies as catalysts. Nature. 1996;383:60–2. doi: 10.1038/383060a0. [DOI] [PubMed] [Google Scholar]
- 79.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. Journal of Biological Chemistry. 1998;273:27035–8. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 80.Voigt CA, Mayo SL, Arnold FH, Wang ZG. Computational method to reduce the search space for directed protein evolution. Proc Natl Acad Sci U S A. 2001;98:3778–83. doi: 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voigt CA, Martinez C, Wang ZG, Mayo SL, Arnold FH. Protein building blocks preserved by recombination. Nat Struct Biol. 2002;9:553–8. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 82.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc Natl Acad Sci U S A. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–6. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 85.Lienhard GE. enzymatic catalysis and transition-state theory. Science. 1973;180:149–154. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 86.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annual Review Of Biochemistry. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 87.Jencks WP. Catalysis in Chemistry and Enzymology. McGraw-Hill; New York: 1969. [Google Scholar]
- 88.Noy D, Moser CC, Dutton PL. Design and engineering of photosynthetic light-harvesting and electron transfer using length, time, and energy scales. Biochimica Et Biophysica Acta-Bioenergetics. 2006;1757:90–105. doi: 10.1016/j.bbabio.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 89.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chemical Reviews. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 90.Bruice TC. Computational approaches: Reaction trajectories, structures, and atomic motions. Enzyme reactions and proficiency Chemical Reviews. 2006;106:3119–3139. doi: 10.1021/cr050283j. [DOI] [PubMed] [Google Scholar]
- 91.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 92.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 93.Henzler-Wildman KA, et al. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–U27. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 94.Henzler-Wildman KA, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–U13. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 95.Xiang JY, Jung JY, Sampson NS. Entropy effects on protein hinges: The reaction catalyzed by triosephosphate isomerase. Biochemistry. 2004;43:11436–11445. doi: 10.1021/bi049208d. [DOI] [PubMed] [Google Scholar]
- 96.Munro AW, et al. P450BM3: the very model of a modern flavocytochrome. Trends In Biochemical Sciences. 2002;27:250–257. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 97.Wyman J, Gill SJ. Binding and Linkage. University Science Books; Mill Valley, CA: 1990. [Google Scholar]
- 98.Anderson JLR, Koder RL, Moser CC, Dutton PL. Controlling complexity and water penetration in functional de novo protein design. Biochemical Society Transactions. 2008;36:1106–1111. doi: 10.1042/BST0361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joyce GF. The antiquity of RNA-based evolution. Nature. 2002;418:214–21. doi: 10.1038/418214a. [DOI] [PubMed] [Google Scholar]
- 100.Doudna JA, Lorsch JR. Ribozyme catalysis: not different, just worse. Nat Struct Mol Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- 101.Lee S, Jung S. Cyclosophoraose as a catalytic carbohydrate for methanolysis. Carbohydr Res. 2004;339:461–8. doi: 10.1016/j.carres.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. A Field Guide to Foldamers. Chem Rev. 2001;101:3893–4011. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 103.Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as versatile frameworks for the design and evolution of function. Nature Chemical Biology. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nanda V, DeGrado WF. Computational design of heterochiral peptides against a helical target. J Am Chem Soc. 2006;128:809–16. doi: 10.1021/ja054452t. [DOI] [PubMed] [Google Scholar]
- 105.Nanda V, Degrado WF. Simulated evolution of emergent chiral structures in polyalanine. J Am Chem Soc. 2004;126:14459–67. doi: 10.1021/ja0461825. [DOI] [PubMed] [Google Scholar]
- 106.Baldauf C, Gunther R, Hoffmann HJ. Helix Formation in alpha, gamma- and beta,gamma-hybrid peptides: Theoretical Insights into Mimicry of alpha- and beta-peptides. Journal of Organic Chemistry. 2006;71:1200–1208. doi: 10.1021/jo052340e. [DOI] [PubMed] [Google Scholar]
- 107.Sandvoss LM, Carlson HA. Conformational Behavior of beta-Proline Oligomers. Journal of the American Chemical Society. 2003;125:15855–15862. doi: 10.1021/ja036471d. [DOI] [PubMed] [Google Scholar]
- 108.Lee O-S, Saven JG. Simulation Studies of a Helical m-Phenylene Ethylene Foldamer. Journal of Physical Chemistry B. 2004;108:11988–11994. [Google Scholar]
- 109.Smaldone RA, Moore JS. Reactive Sieving with Foldamers: Inspiration from Nature and Directions for the Future. Chemistry - A European Journal. 2008;14:2650–2657. doi: 10.1002/chem.200701503. [DOI] [PubMed] [Google Scholar]
- 110.Blake CC, et al. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature. 1965;206:757–61. doi: 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- 111.Nanda V. Do-it-yourself Enzymes. Nature Chemical Biology. 2008;4:273–275. doi: 10.1038/nchembio0508-273. [DOI] [PubMed] [Google Scholar]