

Abstract
Cyclohexyl ketone substrate analogue inhibitors (Ac–pSer-Ψ[C = OCH]-Pip–tryptamine) of Pin1, the cell cycle regulatory peptidyl-prolyl isomerase (PPIase), were designed and synthesized as potential electrophilic acceptors for the Pin1 active site Cys113 nucleophile to test a proposed nucleophilic addition-isomerization mechanism. Because they were weak inhibitors, models of all three stereoisomers were docked into the active site of Pin1. Each isomer consistently minimized to a trans-diaxial cyclohexane conformation. From this, we hypothesize that Pin1 stretches substrates into a trans-pyrrolidine conformation to lower the barrier to isomerization. Our reduced amide inhibitor of Pin1 adopted a similar trans-pyrrolidine conformation in the crystal structure. The molecular model of 1, which mimics the l-Ser-l-Pro stereochemistry, in the Pin1 active site showed a distance of 4.4 Å, and an angle of 31° between Cys113-S and the ketone carbon. The computational models suggest that the mechanism of Pin1 PPIase is not likely to proceed through nucleophilic addition.
Introduction
Pin1 (peptidyl-prolyl isomerase (PPIase) interacting with never-in-mitosis A kinase-1) was discovered in 1996 as a PPIase enzyme that regulates mitosis [1]. The two domains of Pin1, a WW and a PPIase domain, are connected by a flexible linker that serves as a communication conduit between the domains [2]. Both of these domains recognize the phospho-Ser/Thr-Pro bonds present in mitotic phosphoproteins [3]. Pin1 is distinct from two other PPIase families, cyclophilin and FK506 binding protein (FKBP) [4], since Pin1 only has PPIase activity for phosphorylated substrates [3]. Pin1 catalyzes prolyl cis-trans isomerization to function as a molecular timer regulating the cell cycle, cell signaling, gene expression, immune response, and neuronal function [5]. Pin1 is overexpressed in many cancer lines, and plays an important role in oncogenesis [6]. Because of its significant role in cell cycle regulation by a unique mechanism, Pin1 represents an intriguing diagnostic and therapeutic target for cancer [7], [8]. Several promising classes of Pin1 inhibitors have been synthesized as potential lead compounds [7], including designed inhibitors [9], [10], [11], [12], [13], [14], and natural products [15], [16].
The mechanisms of the PPIases, cyclophilins and FKBPs, were shown to go through a twisted amide transition state. Evidence included secondary deuterium isotope effects, molecular modeling, mutagenesis, and bound inhibitor structure [17], [18], [19], [20], [21], [22], [23], [24]. There are two proposed mechanisms for Pin1 catalysis: (1) the twisted-amide mechanism [25], and (2) the nucleophilic-addition mechanism (Figure 1) [26]. In this work, we describe the synthesis, bioassay, and docking of ketones 1, Ac–l-pSer-Ψ[C = OCH]-l-pipecolyl (Pip)–tryptamine, and rac- 2, enantiomeric Ac–d-pSer-Ψ[C = OCH]-l-Pip–tryptamine and Ac–l-pSer-Ψ[C = OCH]-d-Pip–tryptamine. These inhibitors were designed as electrophilic acceptors of the Pin1 active site Cys113 thiol nucleophile to mimic the enzyme-bound tetrahedral intermediate (Figure 1C).
Figure 1. Ketone inhibitors were designed to mimic the tetrahedral intermediate of proposed mechanism B.

(A) Proposed Pin1 hydrogen-bond assisted twisted amide mechanism [25], (B) Pin1 Cys113 nucleophilic-addition mechanism tetrahedral intermediate proposed by Ranganathan et al [26]. (C) Electrophilic ketone inhibitor designed to mimic the proposed tetrahedral intermediate upon Cys113-S nucleophilic addition.
On the other side of the coin, we have described reduced amides designed as twisted-amide transition-state analogues 3 and 4 (Figure 2) [27]. The evidence for a nucleophilic addition mechanism included the proximity of Cys113 to the substrate in the X-ray crystal structure, and the attenuation of activity for Pin1 mutants: 20-fold for C113S and 120-fold for C113A [26]. We anticipated that the ketones would be poor inhibitors, while the reduced amides, as twisted-amide analogues, would fare better. Indeed, the reduced amide 3 is a better Pin1 inhibitor than a similarly substituted substrate analogue (Z)-alkene isostere 5 (Figure 2) [13], [27]. Our crystal structure of reduced amide 4 bound to the Pin1 catalytic site adopted a trans-pyrrolidine conformation, supporting the twisted-amide mechanism [27].
Figure 2. Pin1 inhibitors discussed are cyclohexyl ketones 1 and rac-2 (this work); reduced amides 3 and 4 [27]; (Z)-alkene 5 [13]; and α-ketoamides 6a and 6b [14].

Ketones have been widely used as analogues of aldehydes or carboxylic acids to inhibit serine, cysteine [28], [29], and aspartyl proteases [30], [31]. Substrate-analogue ketones have not yet been developed as inhibitors of Pin1. Juglone is a ketone natural product that was shown to be a non-specific inhibitor of Pin1 through Michael addition to a surface Cys thiol of Pin1, resulting in unfolding [15]. Daum et al developed a series of aryl indanyl ketone inhibitors of Pin1; the best inhibitor had an IC50 value of 0.2 µM [11]. These inhibitors were reversible and cell penetrating, and they showed biological activities against p53 and β-catenin [11]. Daum et al proposed that the aryl indanyl ketones mimic the transition state of the twisted amide, based on the conformation in a crystal structure [11]. α-Ketoamides 6a and 6b were designed as potential transition state analogue inhibitors of Pin1, but their weak inhibition could not be used support either the twisted-amide or the nucleophilic-addition mechanism (Figure 2) [14].
Results
Design of Inhibitors
Ketone 1 was designed as a tetrahedral intermediate analogue, incorporating an electrophilic ketone to act as an acceptor for the Pin1 active site Cys113 thiol (Figure 1). Ketone 1 was designed based on substrate and peptide inhibitor specificities [12], [32]. The stereoisomer obtained as a side product during synthesis, rac -2, was also tested for Pin1 inhibition because Wildeman et al. found that d-Thr containing peptide inhibitors were more potent than l-Thr [12]. The carbocyclic analogue of Pip, a cyclohexyl ring, was chosen based on the 100-fold improved inhibition of peptides with a Pip instead of a Pro residue [12], [32]. Tryptamine was coupled to the C-terminus, since Pin1 binds large aromatic residues there [3], [12], [32]. An acetyl was used at the N-terminus because X-ray crystal structures of bound inhibitors showed no electron-density for residues on the N-terminal side of pSer [32], [33]. The acetyl group also improved the water solubility of the inhibitors compared with Fmoc analogues for enzyme assays [13].
Synthesis
In the synthesis of ketones 1 and rac -2, addition of cyclohexenyl lithium to a Weinreb amide was used to form the ketone functionality (Figure 3). α,β-Unsaturated ketone 7 was obtained by deprotonation of Boc-Ser(Bn)-N(OMe)Me Weinreb amide with i-PrMgCl, followed by addition of cyclohexenyl lithium [34]. The lithium reagent was prepared in situ by treating 1-iodocyclohexene with s-BuLi [34], [35].
Figure 3. Cyclohexyl ketone inhibitor 1 was synthesized by the method shown.

The Boc group was then removed with TFA, and the amine formed was acetylated with acetic anhydride to give ketone 8 (Figure 3). Michael addition to form orthothioester 9 was accomplished with LiC(SMe)3, similar to a synthesis of (+)-methylenolactocin [36]. We first attempted the Michael addition with Boc-protected α, β-unsaturated ketone 7, however a cyclic carbamate was formed as the major product instead of the desired orthothioester. We have used similar cyclic carbamates in stereochemical proofs [34]. The carbamate ring-closure cannot occur with the acetyl amide. After Michael addition, two major diastereomers of the orthothioester were obtained as a mixture; a minor diastereomer was removed during chromatography. Hydrolysis of orthothioester 9 in a mixture of THF and H2O with BF3⋅Et2O and HgO gave a mixture of diastereomeric carboxylic acids 10 [36]. Without further purification, acids 10 were coupled to tryptamine with EDC to generate the ketone diastereomeric mixture of (1 S ,3 R ,4 R )-11 and rac -11, which were separated by silica flash chromatography (Figure 3).
The two diastereomers were carried on separately to the final compounds 1 (Figure 3), and rac -2. The major diastereomer (1 S ,3 R ,4 R )-11 was treated with BCl3 to remove the benzyl group and form alcohol (1 S ,3 R ,4 R )-12 [37], [38]. Phosphorylation with dibenzylphosphoramidite gave dibenzyl phosphate (1 S ,3 R ,4 R )-13 [10], [39]. Phosphorylations were also attempted with di-tert-butyl or dicyanoethyl phosphoramidites to produce di-tert-butyl or dicyanoethyl instead of dibenzyl phosphate. Neither of these phosphates was stable on silica gel, and β-elimination products were obtained after chromatography. TFA deprotection of crude di-tert-butyl phosphate, and NH4OH deprotection of crude dicyanoethyl phosphate both gave β-elimination products as well. Thus, the dibenzylphosphate was chosen to carry through to the final products 1 and rac -2.
Hydrogenation of the crude dibenzyl phosphate (1 S ,3 R ,4 R )-13 went very slowly, giving a complex crude mixture. Thus, (1 S ,3 R ,4 R )-13 was purified by reverse-phase semi-preparative high performance liquid chromatography (HPLC). With pure dibenzyl phosphate, hydrogenation at atmospheric pressure worked very well, and gave a very clean final product 1, similar to our experience with α-ketoamides [14].
X-ray crystallography
During the synthesis of the inhibitors, Michael addition of tris-thiomethyl methide to an α,β-unsaturated ketone 8 produced three stereoisomers of 9, which could not be readily separated (Figure 3). Two diastereomers of a subsequent synthetic intermediate, (1 S ,3 R ,4 R )-11 and rac-11, were separated by chromatography. Each diastereomer was crystallized, and the relative stereochemistry was determined. The absolute configuration of the major diastereomer was assigned to be (1 S ,3 R ,4 R )-11, with the original Ser configuration intact (Figure 4). The minor isomer, rac -11, proved to be a racemic mixture. The absolute configurations were assigned as (1R,3R,4R)-11 and (1S,3S,4S)-11, in which the stereocenter of the Ser analogue was partially epimerized to the syn-Ser-trans-cyclohexyl configuration (Figure 4).
Figure 4. X-ray crystal structures of intermediates (1S,3R,4R)-11 and rac-11 are shown above as displacement ellipsoid drawings (50%).

The positional disorder of the benzyl group in rac -11 is shown as lighter lines. Hydrogen atoms are omitted for clarity. Structural depiction of the stereochemistries of (1 S ,3 R ,4 R )-11 and rac -11 are shown below each crystal structure.
Pin1 PPIase Enzyme Assays
The α-chymotrypsin protease-coupled assay was used to evaluate inhibition of Pin1 by compounds 1 and rac -2 with the same substrate concentration as described previously [10], [14]. The IC50 values of the two diastereomers were determined to be 260±30 µM for 1, and 61±8 µM for rac -2. Preincubation with Pin1 for 15 minutes did not result in improved inhibition.
Molecular modeling
Each of the three cyclohexyl ketone inhibitors was docked flexibly, with geometry minimization, into the Pin1 active site. The resulting docked stereoisomers, (1S,3R,4R)-1, (1R,3R,4R)-2, and (1S,3S,4S)-2, are shown in Figure 5. The total energies, Cys113–S—C = O ketone distances, and angles are reported in Table 1. The distance between 1,2-diequatorial carbonyl groups was 2.93 Å, while the distance between 1,2-diaxial carbonyl groups in 1,2-cyclohexanedial was 3.79 Å after geometry optimization. The distance between the carbonyl carbons of Ac–cis-Pro–OH after geometry optimization was 3.16 Å; with the trans-pyrrolidine torsion angle fixed during geometry optimization, the distance was 3.67 Å (Figure 6).
Figure 5. Models of cyclohexyl ketone inhibitors were docked with dynamic minimization.
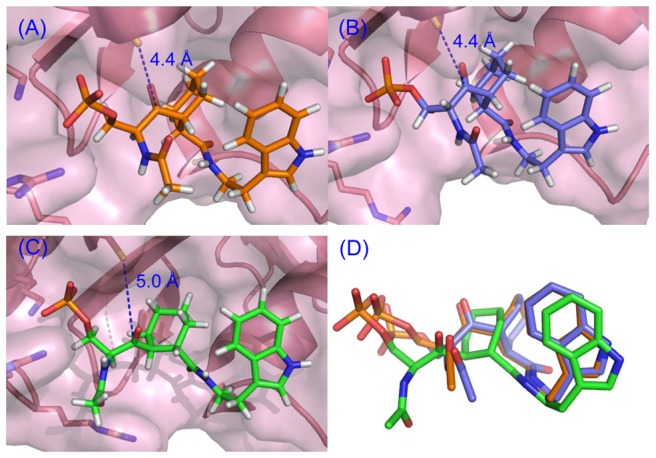
(A) (1 S, 3 R, 4 R )-1 in orange, (B) (1 R, 3 R, 4 R )-2 in blue, (C) (1 S, 3 S, 4 S )-2 in green, and (D) superposition of all atoms of 1 and rac -2. Models were based on PDB 2Q5A [32], and minimized using Sybyl 8.1.1 [42]. Images were prepared using MacPyMol [44].
Table 1. Comparison of cyclohexyl ketone inhibitor-Pin1 complex molecular models.
| Stereoisomer: | (1S,3R,4R)-1 | (1R,3R,4R)-2 | (1S,3S,4S)-2 |
| Mimics: | L-Ser-L-Pro | D-Ser-L-Pro | L-Ser-D-Pro |
| Color in Figure 5 | orange | blue | green |
| Total E (kcal/mol) | −477 | −518 | −494 |
| Cys113–S—C = O (Å) | 4.4 | 4.4 | 5.0 |
| Cys113–S—C = O ∠ | 31° | 102° | 59° |
Figure 6. Pin1 is proposed to stretch the prolyl ring by binding phosphate and C-terminal residues tightly, creating a trans-pyrrolidine conformation of the substrate and forcing pyramidalization of the prolyl nitrogen in the twisted-amide mechanism.

Distance measurements are from calculated structures of Ac–Pro–OH in the ground state and the trans-pyrrolidine transition state.
Discussion
Stereochemical results of inhibitor synthesis
Thermodynamic control in the Michael addition resulted in the anti-Ser-trans-cyclohexyl stereoisomer of 9 as the major product (Figure 4). The chiral center adjacent to the Ser carbonyl was easily epimerized due to the electron-withdrawing effects of both the α-amide and α-ketone, resulting in an enantiomeric mixture of a second diastereomer, rac-9. Because the unnatural d-Thr-containing inhibitors were more potent than the l-Thr in work by Zhang et al [32], both diastereomers 1 and rac -2 were tested for Pin1 inhibition. Inhibitor 1, corresponding to the native l-Ser-l-Pro stereochemistry of Pin1 substrates, had an IC50 value of 260 µM, while rac -2, an enantiomeric mixture of d-Ser-l-Pro and l-Ser-d-Pro analogues, had an IC50 value of 61 µM. Preincubation did not result in improved inhibition, suggesting that they are not slow-binding inhibitors. We obtained a crystal structure of the similarly substituted, reduced amide inhibitor 4 bound in the Pin1 active site, suggesting that the ketones also bind in the active site [27].
Insights into the Pin1 enzymatic mechanism
To better understand the mechanism of Pin1 PPIase activity, each of the three stereoisomers was docked into the Pin1 active site (Figure 5). Curiously, in each case the inhibitor minimized to a conformation with a trans diaxially substituted cyclohexyl ring. Attempts to force a trans diequatorial conformation on the starting structure resulted in conversion to either a twist boat or a diaxial conformation again. Clearly, the preferred conformation of these cyclohexyl substrate analogues in the Pin1 active site is diaxial. In the crystal structures of intermediates (1 S ,3 R ,4 R )-11 and rac -11, the cyclohexyl rings were in the diequatorial chair conformation (Figure 4), which are likely to be the low-energy, solution-phase conformations as well. These inhibitors would thus undergo an unfavorable diequatorial to diaxial conformational change in order to bind to the Pin1 active site.
We hypothesize that the binding interactions of the enzyme with the phosphate and the aromatic group are strong enough to stretch the cyclohexyl rings into the less stable diaxial conformation upon binding (Figure 6). The difference in the distances between diequatorial and diaxial carbonyl groups on a cyclohexane ring was 0.86 Å, an elongation of the structure. The corresponding difference between the planar Ac–cis-Pro–OH conformation, and the trans-pyrrolidine Ac–Pro–OH conformation was 0.51 Å (Figure 6). This effect of stretching the ring conformation may provide insight into the mechanism of Pin1. In either of the proposed mechanisms: (1) nucleophilic-addition [26], or (2) twisted-amide [25], the nitrogen of the prolyl ring must become pyramidalized and deconjugated from the carbonyl in the transition state [22], [24], [25]. If binding of substrate to the catalytic site forces the Pro ring into a trans-pyrrolidine conformation, the nitrogen lone pair and the carbonyl π-bond would no longer be conjugated (Figure 6). The substrate would be destabilized, lowering the barrier to rotation around the amide bond. This proposed stretching action is consistent with the twisted-amide mechanism, providing a more detailed description of how the isomerization might proceed.
Stereoisomer (R,R,R)-2, with the ketone carbonyl carbon 4.4 Å from the proposed Cys113-S nucleophile, and the S—C = O angle of 102°, had the lowest energy of the three stereoisomers (Table 1). The angle of 102° is close to the optimum angle for nucleophilic addition, i.e. close to the Bürgi-Dunitz angle of 107° [27]. Despite this, the inhibition results suggest that covalent modification, i.e. suicide inhibition, of Pin1 does not occur. Ketones 1 and rac -2 were designed as tetrahedral-intermediate analogues based on the nucleophilic-addition mechanism; they do not appear to behave as such. The IC50 values are in the range of substrate analogue inhibitors. These results argue against the proposed nucleophilic-addition mechanism for Pin1 [14].
Stereochemical effects on inhibition
The stereochemistry affected the inhibition, since the racemate rac -2 was about 4-fold more potent than diastereomer 1. Molecular modeling provides insight into the stereochemical preferences of the Pin1 active site. The relative (not absolute) energies of the three models can be compared because they are all stereoisomers bound into the same Pin1 active site (Table 1). These inhibitors are substituted with tryptamine, comparable to our ground-state alkene isostere inhibitor 5 with an IC50 value of 25 µM (Figure 2) [13], and with Ac and naphthylethylamine comparable to our α-ketoamide inhibitors 6, with IC50 values of 100 and 200 µM [14]. The Pin1-( S,R,R )-1 complex, with an intermediate energy, corresponds to the native l-Ser-l-Pro configuration, yet it had very poor inhibition (260 µM), comparable to the similarly substituted α-ketoamides 6 [14]. The Pin1-( S,S,S )-2 complex, which corresponds to the l-Ser-d-Pro configuration, had the highest energy of the three, while Pin1-( R,R,R )-2, corresponding to a d-Ser-l-Pro configuration had the lowest energy. This is consistent with the d-Thr-l-Pip in the most potent peptide inhibitors of Pin1 [12], [32]. We expect that ( R,R,R )-2 isomer would be more potent than the IC50 value of 61 µM for rac -2 indicates, and ( S,S,S )-2 is likely to be less potent than 61 µM, because the IC50 value represents a weighted average of the two. The most potent that either enantiomer could possibly be is 30 µM if the other was not an inhibitor at all. This is highly unlikely, but it serves to show that these ketone inhibitors behave as substrate analogues.
Conclusions
Three stereoisomeric ketone analogues of Pin1 substrates were synthesized, modeled, and assayed as Pin1 inhibitors. Molecular modeling shows that the inhibitors have a preference for trans-diaxial-cyclohexane conformations upon binding to Pin1. This led us to propose a stretching mechanism to attain pyramidalization of the prolyl nitrogen, consistent with the preferred twisted-amide mechanism [25]. The molecular models of the three stereoisomers in the active site of Pin1 confirmed the stereochemical preferences of Pin1 for inhibitors seen in other inhibitors [12], [14], [27], [32]. We attribute the weaker binding of these inhibitors to a combination of: (1) the conformational change required for binding, and (2) the inability of these ketones to act as electrophilic acceptors for the Pin1 Cys113 thiol. The weak inhibition of the ketones, and the correspondingly stronger inhibition by similarly substituted reduced amide inhibitors [27], provides evidence against the nucleophilic addition mechanism for Pin1.
Materials and Methods
Synthesis
Unless otherwise indicated, all reactions were carried out under dry N2 in flame-dried glassware. THF was distilled from Na-benzophenone, and CH2Cl2 was dried by passage through dry alumina. Anhydrous DMF (99.8%), MeOH, and DIEA were used directly from sealed bottles. Brine (NaCl), Na2S2O3, NaHCO3, and NH4Cl refer to saturated aqueous solutions, and HCl refers to a 1 N aqueous solution, unless otherwise noted. Flash chromatography was performed on 230–400 mesh silica gel with reagent grade solvents. Analytical HPLC were obtained on a 4.6×50 mm C18 column with 10% CH3CN/H2O for 3 min followed by a 10% to 90% CH3CN/H2O gradient over 6 min unless otherwise noted. HPLC results are reported as retention time, integrated % purity. 1H, 13C, and 31P NMR spectra were obtained at ambient temperature in CDCl3, unless otherwise noted. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS). Data are reported as follows: chemical shift, multiplicity: singlet (s), doublet (d), triplet (t), multiplet (m), broad singlet (br s), coupling constants J in Hz, and integration. HPLC chromatograms for compounds 1 and rac -2, 1H (500 MHz), 13C (125 MHz), and 31P NMR (162 MHz) NMR spectra of compounds 1, rac -2, and 7–13, are available in Dataset S1.
Boc-ketone 7
To a solution of 1-iodocyclohexene [35] (5.50 g, 26.4 mmol) in THF (60 mL) at −40°C was added sec-butyl lithium (1.4 M in cyclohexane, 37.8 mL, 52.9 mmol). The mixture was stirred at −40°C for 3 h. Boc–Ser(OBn)–N(OMe)Me [34] Weinreb amide (5.96 g, 17.6 mmol) was dissolved in THF (60 mL) in another round-bottom flask and cooled to −78°C, i-PrMgCl (2.0 M in THF, 8.64 mL, 17.3 mmol) was then added dropwise. The Weinreb amide solution was stirred at −78°C for 1 h. The cyclohexenyl lithium was added via canula at −78°C to the Weinreb amide solution. After stirring at −78°C for 1 h, the reaction was warmed to rt, stirred for 12 h, and quenched with NH4Cl (80 mL). The resulting mixture was diluted with water (40 mL) and EtOAc (100 mL). The aqueous layer was extracted with EtOAc (100 mL). The organic layers were combined, and washed with NH4Cl (2×80 mL), NaHCO3 (80 mL), and brine (80 mL). The organic layer was dried over Na2SO4, filtered and evaporated. The crude product was purified by chromatography on silica (eluant: 8% EtOAc/hexanes) to yield ketone 7 (4.3 g, 68%) as a colorless oil. Anal. HPLC, 254 nm, 7.3 min, 98.2%; 1H NMR δ 7.28 (m, 5H), 6.91 (m, 1H), 5.59 (d, J = 8.3, 1H), 5.13 (dt, J = 4.4, 8.3, 1H), 4.54 (d, J = 12.4, 1H), 4.42 (d, J = 12.4, 1H), 3.68 (dd, J = 4.4, 9.3, 1H), 3.66 (dd, J = 4.4, 9.6, 1H), 2.36 (m, 1H), 2.21 (m, 2H), 2.12 (m, 1H), 1.62 (m, 4H), 1.44 (s, 9H); 13C NMR δ 197.8, 155.5, 141.8, 137.8, 137.4, 128.4, 127.7, 127.6, 79.8, 73.1, 71.3, 54.3, 28.4, 26.2, 23.4, 21.8, 21.5; ESI+ HRMS m/z 382.1998 [M+Na]+. Calculated for C21H29NO4·Na 382.1994.
Acetyl-ketone 8
Boc-ketone 7 (1.5 g, 4.2 mmol) was dissolved in CH2Cl2 (20 mL), and iPr3SiH (0.2 mL) and TFA (20 mL) were added. The mixture was stirred at rt for 0.5 h. The reaction mixture was then concentrated under reduced pressure. The residue was triturated with hexanes (3×25 mL). After evaporation in vacuo for 2 h, the ammonium salt obtained was dissolved in CH2Cl2 (20 mL), and Ac2O (2 mL) and DIEA (2 mL) were added. The reaction mixture was stirred at rt for 1 h. After dilution with CH2Cl2 (30 mL), the mixture was washed with HCl (2×25 mL), 1 N NaOH (2×25 mL), and brine (25 mL). The organic layer was dried over Na2SO4, filtered and evaporated. The residue was purified by flash chromatography on silica (step gradient: 25% then 50% EtOAc/hexanes) to yield 8 (1.1 g, 90%) as a pale, yellow oil. Anal. HPLC, 254 nm, 5.1 min, 100%; 1H NMR δ 7.35-7.20 (m, 5H), 6.93 (m, 1H), 6.62 (br, 1H), 5.42 (m, 1H), 4.52 (d, J = 12.3, 1H), 4.40 (d, J = 12.3, 1H), 3.70 (m, 2H), 2.39-2.04 (m, 4H), 2.02 (s, 3H), 1.62 (m, 4H); 13C NMR δ 197.3, 169.8, 142.3, 137.7, 137.2, 128.5, 127.9, 127.6, 73.2, 71.1, 53.3, 26.2, 23.44, 23.38, 21.8, 21.5; ESI+ HRMS m/z 302.1760 [M+H]+. Calculated for C18H24NO3. 302.1756.
Orthothioformate 9
n-Butyl lithium (2.5 M in hexane, 6.81 mL, 17.0 mmol) was added dropwise to a solution of CH(SMe)3 (2.68 g, 17.0 mmol) dried over 4 Å molecular sieves in THF (65 mL) at −78°C. The solution was stirred at −78°C for 2 h. A solution of the acetyl ketone 8 (0.790 g, 2.62 mmol) dried over 4 Å molecular sieves in THF (50 mL) was added dropwise via canula. The reaction mixture was stirred at −78°C for 2 h, and quenched with NH4Cl (80 mL). The resulting mixture was extracted with EtOAc (3×150 mL). The organic layer was dried over Na2SO4, filtered and evaporated. The crude product was purified by chromatography on silica (step gradient: 0% then 20% EtOAc/hexanes) to yield the orthothioformate 9, a mixture of two diastereomers, (0.60 g, 50%) as a colorless oil. The mixture was used in the next reaction without separation. The major diastereomer was partially separated for characterization. Major diastereomer: 1H NMR δ 7.30 (m, 5H), 6.60 (d, J = 7.5, 1H), 5.26 (ddd, J = 3.4, 4.2, 7.6, 1H), 4.52 (d, J = 12.1, 1H), 4.49 (d, J = 11.8, 1H), 3.89 (dd, J = 3.3, 10.2, 1H), 3.79 (dd, J = 4.4, 9.9, 1H), 3.11 (ddd, J = 3.8, 10.5, 11.5, 1H), 2.39 (ddd, J = 3.6, 10.4, 11.8), 2.09 (m, 1H), 2.07 (s, 9H), 2.02 (s, 3H), 1.82 (m, 1H), 1.73 (m, 1H), 1.63 (m, 2H), 1.38 (m, 1H), 1.31 (m, 1H), 1.22 (m, 1H), 1.06 (m, 1H); 13C NMR δ 207.0, 169.4, 137.9, 128.5, 127.9, 127.8, 75.8, 73.4, 68.9, 59.5, 51.5, 48.0, 31.6, 28.6, 25.6, 25.1, 23.6, 15.2; ESI+ HRMS m/z 478.1530 [M+Na]+. Calculated for C22H33NO3S3·Na 478.1520.
Ac–Ser(OBn)–Ψ[C = OCH]–2-(indol-3-yl)-ethylamine (1S,3R,4R)-11 and rac-11
A mixture of compound 9 (0.30 g, 0.66 mmol) and HgO (0.70 g, 3.2 mmol) was suspended in 4∶1 THF∶H2O (45 mL), and BF3·Et2O (1.2 mL, 9.6 mmol) was added. The mixture was stirred at rt for 3 h. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3×50 mL). The organic layer was dried over Na2SO4, filtered and evaporated. The residue was filtered through silica to remove HgO, and the solvent was evaporated in vacuo. The crude carboxylic acid 10 was dissolved in a mixture of CH2Cl2 (100 mL) and DMF (15 mL), and tryptamine (0.27 g, 1.7 mmol), EDC (0.32 g, 1.68 mmol), HOAt (0.25 g, 1.6 mmol), DMAP (50 mg, 0.4 mmol) and DIEA (0.37 g, 2.9 mmol) were added. The reaction was stirred at rt for 16 h. The mixture was diluted with EtOAc (400 mL), washed with water (3×150 mL), HCl (3×150 mL), NaHCO3 (3×150 mL) and brine (150 mL). The crude product was purified by chromatography on silica (step gradient: 0% then 30% EtOAc/hexanes) to yield two diastereomers. The major diastereomer (120 mg, 36%) and the minor diastereomer (60 mg, 18%) were obtained as colorless oils at first, which then solidified. Both solids were recrystallized from EtOAc∶hexanes (1∶2) to determine the relative stereochemistry by X-ray crystallography.
Major isomer (1 S ,3 R ,4 R )-11: Anal. HPLC, 254 nm, 5.0 min, 98.9%; 1H NMR δ 8.10 (br, 1H), 7.57 (dd, J = 0.7, 7.8, 1H), 7.37 (dt, J = 0.8, 8.2, 1H), 7.31-7.21 (m, 5H), 7.20 (dt, J = 1.2, 7.5, 1H), 7.11 (dt, J = 0.9, 7.5, 1H), 7.02 (d, J = 2.2, 1H), 6.30 (d, J = 7.5, 1H), 5.60 (t, J = 5.8, 1H), 4.84 (dt, J = 4.2, 7.4, 1H), 4.58 (d, J = 11.8, 1H), 4.48 (d, J = 11.8, 1H), 3.86 (dd, J = 3.9, 9.9,1H), 3.76 (dd, J = 4.4, 9.9, 1H), 3.44 (m, 2H), 3.05 (ddd, J = 3.3, 10.7, 12.4, 1H), 2.84 (m, 2H), 2.37 (m, 1H), 2.13 (m, 1H), 1.99 (s, 3H), 1.80 (m, 3H), 1.49 (m, 1H), 1.26 (m, 2H), 1.08 (m, 1H); 13C NMR δ 209.5, 174.7, 170.0, 137.9, 136.5, 128.5, 128.0, 127.9, 127.4, 122.4, 122.3, 119.6, 118.9, 113.1, 111.3, 73.3, 68.4, 60.0, 49.2, 45.8, 39.4, 29.7, 29.5, 25.7, 25.4, 25.3, 23.3; ESI+ HRMS m/z 512.2530 [M+Na]+. Calculated for C29H35N3O4·Na 512.2525. Minor isomer rac -11: Anal. HPLC, 254 nm, 5.1 min, 96.3%; 1H NMR δ 8.25 (br, 1H), 7.56 (d, J = 8.0, 1H), 7.36 (d, J = 8.3, 1H), 7.35-7.25 (m, 5H), 7.19 (ddd, J = 1.1, 7.2, 8.1, 1H), 7.11 (ddd, J = 1.0, 7.4, 8.0, 1H), 7.00 (d, J = 2.2, 1H), 6.75 (d, J = 7.7, 1H), 5.56 (t, J = 5.8, 1H), 4.86 (dt, J = 4.0, 7.4, 1H), 4.49 (d, J = 12.0, 1H), 4.46 (d, J = 12.1, 1H), 3.92 (dd, J = 3.3, 9.9, 1H), 3.75 (dd, J = 4.1, 9.9, 1H), 3.48 (q, J = 6.4, 2H), 3.01 (dt, J = 3.0, 11.1, 1H), 2.86 (m, 2H), 2.33 (m, 1H), 2.01 (s, 3H), 1.78 (m, 4H), 1.43 (m, 1H), 1.19 (m, 3H); 13C NMR δ 210.9, 174.5, 170.1, 137.8, 136.5, 128.5, 127.9, 127.88, 127.4, 122.4, 122.2, 119.5, 118.8, 112.9, 111.4, 77.4, 73.4, 68.5, 58.8, 47.8, 47.7, 39.6, 30.0, 28.6, 25.3, 25.1, 23.4; ESI+ HRMS m/z 490.2710 [M+H]+. Calculated for C29H36N3O4 490.2706.
Ac–Ser–Ψ[C = OCH]–Pip–2-(indol-3-yl)-ethylamine (1S,3R,4R)-12 and rac-12
Ac–Ser(OBn)–Ψ[C = OCH]–2-(indol-3-yl)-ethylamine (1 S ,3 R ,4 R )-11 (48 mg, 0.098 mmol) was dissolved in CH2Cl2 (8 mL). The solution was cooled to −78°C, and BCl3 (1 M in CH2Cl2, 1.2 mL) was added dropwise. The reaction mixture was stirred at −78°C and warmed to 0°C over 1.5 h. The reaction mixture was cooled to −78°C, and MeOH (0.5 mL) and aq. HCl (2 N, 5 mL) were added. The solution was diluted with EtOAc (150 mL), and washed with HCl (30 mL), 5% aq. NaHCO3 (30 mL), and brine (30 mL). After filtration and evaporation, the residue was purified on silica (step gradient: %0 then 5% isopropanol/EtOAc). The product (1 S ,3 R ,4 R )-12 was obtained as a colorless oil (31 mg, 80%). 1H NMR δ 8.20 (s, 1H), 7.59 (d, J = 7.5, 1H), 7.38 (t, J = 0.8, 1H) 7.22 (app. tt, J = 7.3, 1.1, 1H), 7.14 (app. tt, J = 7.3, 1.2, 1H), 7.06 (d, J = 2.1, 1H), 6.50 (d, J = 7.3, 1H), 5.75 (t, J = 5.6, 1H), 4.87 (m, 1H), 4.39 (d, J = 11.8, 1H), 4.14 (m, 1H), 3.81 (m, 1H), 3.55 (dd, J = 13.2, 6.4, 1H), 3.49 (dd, J = 12.8, 6.4, 1H), 3.04 (dt, J = 3.4, 11.6, 1H), 2.91 (dt, J = 0.9, 6.4, 2H), 2.52 (dt, J = 3.3, 11.6, 1H), 2.06 (m, 1H), 2.02 (s, 3H), 1.81 (m, 3H), 1.42 (m, 1H), 1.26 (m, 2H), 1.06 (m, 1H); 13C NMR (100 MHz) δ 210.4, 176.0, 170.0, 136.5, 127.3, 122.5, 122.4, 119.7, 118.8, 112.7, 111.4, 63.4, 59.5, 48.7, 45.9, 39.9, 30.4, 29.2, 25.68, 25.67, 25.2, 23.4; ESI+ HRMS m/z 400.2260 [M+H]+. Calculated for C22H30N3O4 400.2236, By the same procedure, the minor isomer rac -12 was obtained as an oil (20 mg, 80%). 1H NMR δ 8.39 (s, 1H), 7.56 (d, J = 7.9, 1H), 7.37 (d, J = 8.0, 1H), 7.25 (br s, 1H), 7.20 (dt, J = 1.1, 7.6, 1H), 7.12 (dt, J = 0.8, 7.5, 1H), 7.03 (s, 1H), 5.86 (t, J = 5.1, 1H), 4.53 (ddd, J = 2.7, 4.6, 7.4, 1H), 4.08 (br s, 1H), 3.93 (dd, J = 2.4, 11.8, 1H), 3.73 (dd, J = 4.4, 11.8, 1H), 3.53 (dd, J = 6.6, 13.2, 1H), 3.48 (dd, J = 6.6, 13.2, 1H), 3.00 (dt, J = 3.2, 11.3, 1H), 2.90 (t, J = 6.6, 2H), 2.48 (dt, J = 3.3, 11.7, 1H), 2.08 (s, 3H), 1.90 (d, J = 3.2, 1H), 1.76 (m, 3H), 1.40 (m, 1H), 1.21 (m, 2H), 1.09 (m, 1H); 13C NMR δ 213.9, 175.8, 170.9, 136.6, 127.3, 122.5, 122.4, 119.6, 118.7, 112.6, 111.5, 63.2, 60.3, 48.2, 47.3, 40.0, 30.3, 28.8, 25.6, 25.4, 25.2, 23.3; ESI+ HRMS m/z 422.2063 [M+Na]+. Calculated for C22H29N3O4·Na 422.2056.
Ac–Ser(PO(OBn)2)–Ψ[C = OCH]–Pip–2-(indol-3-yl)-ethylamine (1S,3R,4R)-13 and rac-13
To a solution of (1 S ,3 R ,4 R )-12 (33 mg, 0.083 mmol) in THF (10 mL) was added 5-ethylthio–1H–tetrazole (32 mg, 0.25 mmol) and O,O-dibenzyl-N,N-diethylphosphoramidite (0.087 mL, 0.25 mmol) at rt. The mixture was stirred at rt for 16 h. The mixture was cooled to −40°C, a solution of 5–6 M tert-butyl hydroperoxide in decane (61 µL, 0.33 mmol) was added dropwise, and the mixture was stirred at −40°C for 10 min, then at rt for 30 min. The reaction was cooled to −40°C and quenched with Na2S2O3. The mixture was diluted with EtOAc (80 mL), washed with HCl (20 mL), 5% NaHCO3 (aq, 20 mL), brine (20 mL), and dried over Na2SO4. The product was concentrated in vacuo, and purified by semi-preparative HPLC (10% CH3CN/H2O for 3 min, then 10% to 90% CH3CN/H2O gradient over 10 min) to give (1 S ,3 R ,4 R )-13, ret. time 12.2 min, as a colorless oil (20 mg, 36%). 1H NMR (CD3OD) δ 7.53 (dt, J = 7.9, 1.0, 1H), 7.32 (m, 11H), 7.06 (ddd, J = 1.1, 7.0, 8.1, 1H), 7.01 (s, 1H), 6.97 (ddd, J = 0.9, 7.0, 8.0, 1H), 5.04 (d, J = 6.6, 2H), 5.02 (d, J = 6.1, 2H), 4.93 (dd, J = 3.8, 6.8, 1H), 4.53 (ddd, J = 3.9, 6.9, 10.8, 1H), 4.16 (ddd, J = 7.1, 8.2, 11.1, 1H), 3.37 (m, 2H), 2.95 (ddd, J = 3.2, 10.8, 12.2, 1H), 2.85 (m, 2H), 2.51 (m, 1H), 2.10 (m, 1H), 1.94 (s, 3H), 1.86 (m, 1H), 1.78 (m, 2H), 1.32 (m, 3H), 1.10 (m, 1H); 13C NMR δ 209.5, 177.2, 173.2, 138.1, 137.1, 137.0, 129.73, 129.68, 129.23, 129.19, 128.8, 123.5, 122.3, 119.6, 119.3, 113.2, 112.2, 71.0, 67.0 (d, 3 J P-C = 5.0), 59.1 (d, 2 J P-C = 7.5), 50.6, 47.3, 41.3, 31.1, 30.2, 26.6, 26.5, 26.3, 22.4; 31P NMR (202 MHz): δ −0.40; ESI+ HRMS m/z 660.2846 [M+H]+. Calculated for C36H43N3O7P 660.2839. By the same procedure, the minor isomer rac -13, ret. time 12.0 min, was obtained as an oil (17 mg, 37%). 1H NMR δ 8.49 (s, 1H), 7.54 (d, J = 7.7, 1H), 7.36 (d, J = 8.0, 1H), 7.33 (m, 10H), 7.18 (ddd, J = 1.1, 7.1, 8.1, 1H), 7.10 (ddd, J = 1.0, 7.0, 8.0, 1H), 6.98 (d, J = 2.2, 1H), 6.91 (d, J = 8.3, 1H), 5.50 (t, J = 5.8, 1H), 5.02 (m, 4H), 4.88 (ddt, J = 1.6, 4.2, 8.0, 1H), 4.32 (ddd, J = 4.1, 6.4, 10.5, 1H), 4.27 (ddd, J = 4.7, 6.3, 10.4, 1H) 3.58 (ddt, J = 6.0, 7.7, 13.5, 1H), 3.43 (ddt, J = 5.5, 6.5, 13.8, 1H), 2.95 (dt, J = 3.2, 11.2, 1H), 2.86 (m, 2H), 2.37 (dt, J = 3.5, 11.6, 1H), 1.94 (s, 3H), 1.84 (d, J = 11.8, 1H), 1.74 (m, 3H), 1.39 (m, 1H), 1.16 (m, 3H); 13C NMR δ 209.5, 174.7, 170.5, 136.6, 135.9, 135.88, 135.8, 128.74, 128.71, 128.69, 128.13, 128.11, 127.4, 122.5, 122.2, 119.5, 118.7, 112.6, 111.5, 69.60 (d, 2 J P-C = 3.9), 69.55 (d, 2 J P-C = 3.8), 65.4 (d, 3 J P-C = 5.4), 57.1 (d, 2 J P-C = 9.1), 47.4, 47.1, 39.5, 30.0, 28.7, 25.4, 25.3, 25.2, 23.2; 31P NMR: δ −0.53; ESI+ HRMS m/z 660.2842 [M+H]+. Calculated for C36H43N3O7P 660.2839.
Ac–Ser(PO(OH)2)–Ψ[C = OCH]–Pip–2-(indol-3-yl)-ethylamine 1 and rac-2
Dibenzyl phosphate 13 (14 mg, 0.021 mmol) and 10% Pd/C (7 mg) were dissolved in MeOH (8 mL). The reaction was stirred under H2 (1 atm) at rt for 2 h. The reaction mixture was filtered through Celite, and washed with MeOH. After evaporation, the residue was purified by semi-preparative HPLC (5% CH3CN/H2O for 3 min, then 5% to 30% CH3CN/H2O gradient over 10 min) to provide 1, ret. time 8.2 min, as a white solid after lyophilization (8.0 mg, 78%). Anal. HPLC, 254 nm, (gradient: 5% B for 3 min, then 5–90% B over 6 min), 6.0 min, 99.9%; 1H NMR (CD3OD) δ 7.54 (d, J = 7.7, 1H), 7.32 (d, J = 8.2, 1H), 7.07 (m, 2H), 6.99 (ddd, J = 0.9, 7.0, 8.0, 1H), 4.92 (m, 1H), 4.35 (m, 1H), 4.18 (m, 1H), 3.40 (m, 2H), 3.04 (dt, J = 3.0, 11.4, 1H), 2.89 (m, 2H), 2.52 (ddd, J = 3.4, 10.5, 12.2, 1H), 2.22 (dd, J = 2.2, 13.2, 1H), 2.00 (s, 3H), 1.83 (m, 3H), 1.35 (m, 3H), 1.11 (m, 1H); 13C NMR (CD3OD) δ 209.8, 177.7, 173.2, 138.1, 128.8, 123.6, 122.2, 119.5, 119.3, 113.2, 112.2, 65.5, 59.2 (d, 2 J P-C = 12), 50.4, 47.1, 41.2, 31.2, 30.3, 26.8, 26.6, 26.2, 22.3; 31P NMR (CD3OD) δ 1.44; ESI+ HRMS m/z 480.1906 [M+H]+. Calculated for C22H31N3O7P 480.1900. By the same procedure, the minor isomer rac- 2, ret. time 8.0 min, was obtained as a white powder (5.5 mg, 70%). Anal. HPLC, 254 nm, (gradient: 5% B for 3 min, then 5–90% B over 6 min), 6.8 min, 99.1%; 1H NMR (dimethylsulfoxide (DMSO)-d 6): δ 10.82 (br s, 1H), 8.25 (br s, 1H), 7.89 (t, J = 5.6, 1H), 7.50 (d, J = 7.8, 1H), 7.31 (d, J = 7.7, 1H), 7.10 (d, J = 1.5, 1H), 7.04 (t, J = 7.4, 1H), 6.96 (t, J = 7.2, 1H), 4.67 (m, 1H), 4.14 (m, 1H), 3.96 (m, 1H), 3.24 (m, 2H), 2.99 (t, J = 10.4, 1H), 2.73 (m, 2H), 2.36 (dt, J = 3.6, 11.3, 1H), 1.94 (m, 1H), 1.87 (s, 3H), 1.82 (m, 1H), 1.68 (m, 2H), 1.24 (m, 3H), 1.02 (m, 1H); 13C NMR (CD3OD): δ 210.5, 177.2, 173.1, 138.1, 128.8, 123.6, 122.2, 119.5, 119.3, 113.3, 112.2, 65.4, 60.0, 49.4, 49.3, 41.2, 31.3, 29.3, 26.5, 26.4, 26.2, 22.6; 31P NMR (DMSO-d 6): δ −2.93; ESI+ MS m/z 480.18 [M+H]+. Calculated for C22H31N3O7P 480.19.
X-ray structures
Crystal structure (1 S ,3 R ,4 R )-11: Colorless needles (0.31×0.02×0.004 mm3) were recrystallized from EtOAc∶hexanes (1∶2) at rt. The chosen crystal was centered on the goniometer of an Oxford Diffraction Nova diffractometer operating with CuKα radiation. The data collection routine, unit cell refinement, and data processing were carried out with the program CrysAlis [40]. The Laue symmetry and systematic absences were consistent with the monoclinic space groups P21 and P21/m. Since the molecule was known to be enantiomerically pure, the chiral space group, P21, was chosen. The structure was solved by direct methods and refined using SHELXTL NT [41]. The asymmetric unit of the structure comprises one crystallographically independent molecule. The final refinement model involved anisotropic displacement parameters for non-hydrogen atoms and a riding model for all hydrogen atoms. Since there were no heavy atoms, the absolute configuration could not be determined from the Friedel pairs; the Friedel pairs were therefore merged for the final refinement. The absolute configuration was assigned by reference to C(19) of known S-configuration. Relative to C(19), C(17) and C(12) are both R-configuration (Figure 4). SHELXTL NT was used for molecular graphics generation [41]. Deposited Cambridge Crystallographic Data Centre (CCDC) 782064. Crystal structure rac -11: Colorless plates (0.004×0.06×0.12 mm3) were recrystallized from EtOAc∶hexanes (1∶2) at rt. Data were collected as for (1 S ,3 R ,4 R )-11 above. The Laue symmetry and systematic absences were consistent with the monoclinic space group P21/c. The structure was solved as for (1 S ,3 R ,4 R )-11. The asymmetric unit of the structure comprised one crystallographically independent molecule. The final refinement model involved anisotropic displacement parameters for non-hydrogen atoms, and a riding model for all hydrogen atoms. The benzyl group was modeled with positional disorder, with the two positions refining to relative occupancies of 52.9(3)% and 47.1(3)% (Figure 4). Deposited CCDC 782063.
Pin1 Enzyme Assays
The Pin1 inhibition assay was performed at 4°C in 35 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) pH 7.8 in a total assay volume of 1.2 mL as published [10]. Inhibitors were dissolved in DMSO∶H2O (2∶1) and 20 µL of stock was added to give final concentrations of 1: 12, 50, 100, 200, 400, 810 µM, and rac -2: 10, 20, 40, 60, 120, 240, 480 µM, pre-equilibrated with Pin1 in HEPES at 4°C for 15 min. The Pin1 final concentration in the assay was 67 nM. The final concentration of succinyl–Ala–Glu–cis-Pro–Phe-p-nitroanilide was 34 µM. For each concentration, the assay was performed in duplicate. The plot of % Inhibition vs. log [I] (µM) produced sigmoidal curves by fitting all of the experimental data to Eq. 1 using TableCurve v3 for win32 (Dataset S2). The IC50 values were derived from the fitted equation at 50% inhibition of enzyme activity (Eq. 1), where a, b, c, and d are fitted constants given on the plots for compounds 1 and rac -2 (Dataset S2).
| (1) |
Computational Methods
Models of three stereoisomeric ketones were based on the X-ray structure of peptide inhibitor, Ac–Phe–pThr–Pip–Nal–Gln–NH2, bound to Pin1, protein data bank (PDB) 2Q5A, using Sybyl 8.1.1 (Figure 5) [32]. In each case, the Pip nitrogen was changed to a CH group with the appropriate stereochemistry. The naphthyl (Nal) side chains were modified to indoles, and the Nal carbonyls were deleted. The Thr methyl groups, the Gln, and all except the alpha-carbon and carbonyl of the Phe residues (which became acetyl groups) were deleted. Further modification of the starting structures included drafting a diequatorial chair conformation for the cyclohexyl rings, inversion of the Ser stereochemistry for (1 R, 3 R, 4 R )-2, inversion of the cyclohexyl ring stereocenters for (1 S, 3 S, 4 S )-2, and manual rotation of torsions of (1 R, 3 R, 4 R )-2 to bring the phosphate and indole groups close to these groups in the original crystal structure. Explicit waters from the crystal structure were retained. Protein termini charges, all hydrogens, and Amber FF02 atom types were added manually to the inhibitor atoms, phosphate groups, and Arg guanidines. The 3 oxygens of the phosphate groups were given formal charges of −0.67 prior to computation of Gasteiger-Marsili charges. Energy minimization, with geometry optimization of the inhibitors and all Pin1 residues within 8 Å of the inhibitors, was performed using Sybyl 8.1.1 with Gasteiger-Marsili charges, Amber FF02 force field, Powell conjugate gradient, gradient termination at 0.1 kcal/mol-Å, 8 Å non-bonded cut-off, and a dielectric constant of 1.0. Typically, gradient convergence was reached within 3000 iterations. Distances and angles were measured using Sybyl 8.1.1 [42].
Cyclohexane-1,2-cis-dial, cyclohexane-1,2-trans-dial, Ac–cis-Pro–OH, and Ac-Pro-OH with fixed trans-pyrrolidine (ω = −60°) conformation were geometry optimized using WebMO with Moller-Plesset 2, 6-31G(d), polarizable continuum model, and water as solvent [43]. For the twisted-amide conformation, the trans-pyrrolidine torsion angle was fixed to −155.6°, the angle found at the B3LYP STO-3G level of theory.
Supporting Information
HPLC chromatograms for 1 and rac -2. 1H, 13C, and 31P NMR spectra for compounds 1, rac -2, and 7–13.
(PDF)
Pin1 inhibition plots for 1 and rac -2. Crystallographic data, CCDC 782064 and 782063 for (1 S ,3 R ,4 R )-11 and rac -11 respectively, can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
(PDF)
Funding Statement
We thank National Institutes of Health (NIH) (http://www.nih.gov/) grant R01 CA110940, and NIH grant S10 RR16658 for the LC-MSMS, and Oxford Instruments for the use of the diffractometer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lu KP, Hanes SD, Hunter T (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380: 544–547. [DOI] [PubMed] [Google Scholar]
- 2. Namanja AT, Peng T, Zintsmaster JS, Elson AC, Shakour MG, et al. (2007) Substrate recognition reduces side-chain flexibility for conserved hydrophobic residues in human Pin1. Structure 15: 313–327. [DOI] [PubMed] [Google Scholar]
- 3. Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, et al. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278: 1957–1960. [DOI] [PubMed] [Google Scholar]
- 4. Etzkorn FA, Stolz LA, Chang ZY, Walsh CT (1993) Role of the cyclosporin-A-cyclophilin complex, FK506-FK506-binding protein complex and calcineurin in the inhibition of T-cell signal transduction. Curr Opin Struct Biol 3: 929–933. [Google Scholar]
- 5. Lu KP, Finn G, Lee TH, Nicholson LK (2007) Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol 3: 619–629. [DOI] [PubMed] [Google Scholar]
- 6. Ryo A, Liou YC, Lu KP, Wulf G (2003) Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J Cell Sci 116: 773–783. [DOI] [PubMed] [Google Scholar]
- 7. Xu GG, Etzkorn FA (2009) Pin1 as an anticancer drug target. Drug News Perspect 22: 399–407. [DOI] [PubMed] [Google Scholar]
- 8. Yeh ES, Means AR (2007) PIN1, the cell cycle and cancer. Nat Rev Cancer 7: 381–388. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y, Fussel S, Reimer U, Schutkowski M, Fischer G (2002) Substrate-based design of reversible Pin1 inhibitors. Biochemistry 41: 11868–11877. [DOI] [PubMed] [Google Scholar]
- 10. Wang XJ, Xu B, Mullins AB, Neiler FK, Etzkorn FA (2004) Conformationally locked isostere of phosphoSer-cis-Pro inhibits Pin1 23-fold better than phosphoSer-trans-Pro isostere. J Am Chem Soc 126: 15533–15542. [DOI] [PubMed] [Google Scholar]
- 11. Daum S, Erdmann F, Fischer G, Feaux de Lacroix B, Hessamian-Alinejad A, et al. (2006) Aryl indanyl ketones: efficient inhibitors of the human peptidyl prolyl cis/trans isomerase Pin1. Angew Chem Int Ed Engl 45: 7454–7458. [DOI] [PubMed] [Google Scholar]
- 12. Wildemann D, Erdmann F, Alvarez BH, Stoller G, Zhou XZ, et al. (2006) Nanomolar inhibitors of the peptidyl prolyl cis/trans isomerase Pin1 from combinatorial peptide libraries. J Med Chem 49: 2147–2150. [DOI] [PubMed] [Google Scholar]
- 13. Zhao S, Etzkorn FA (2007) A phosphorylated prodrug for the inhibition of Pin1. Bioorg Med Chem Lett 17: 6615–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu GG, Etzkorn FA (2010) Convergent synthesis of alpha-ketoamide inhibitors of Pin1. Org Lett 12: 696–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, et al. (1998) Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37: 5953–5960. [DOI] [PubMed] [Google Scholar]
- 16. Uchida T, Takamiya M, Takahashi M, Miyashita H, Ikeda H, et al. (2003) Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem Biol 10: 15–24. [DOI] [PubMed] [Google Scholar]
- 17. Harrison RK, Caldwell CG, Rosegay A, Melillo D, Stein RL (1990) Confirmation of the secondary deuterium isotope effect for the peptidyl prolyl cis-trans isomerase activity of cyclophilin by a competitive, double-label technique. J Am Chem Soc 112: 7063–7064. [Google Scholar]
- 18. Harrison RK, Stein RL (1990) Mechanistic Studies of Peptidyl Prolyl Cis-Trans Isomerase: Evidence for Catalysis by Distortion. Biochemistry 29: 1684–1689. [DOI] [PubMed] [Google Scholar]
- 19. Rosen MK, Standaert RF, Galat A, Nakatsuka M, Schreiber SL (1990) Inhibition of FKBP Rotamase Activity by Immunosuppressant FK506: Twisted Amide Surrogate. Science 248: 863–866. [DOI] [PubMed] [Google Scholar]
- 20. Park ST, Aldape RA, Futer O, DeCenzo MT, Livingston DJ (1992) PPIase catalysis by human FK506-binding protein proceeds through a conformational twist mechanism. J Biol Chem 267: 3316–3324. [PubMed] [Google Scholar]
- 21. Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, et al. (1992) Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci 1: 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fischer S, Michnick S, Karplus M (1993) A Mechanism for Rotamase Catalysis by the FK506 Binding Protein (FKBP). Biochemistry 32: 13830–13837. [DOI] [PubMed] [Google Scholar]
- 23. Wiederrecht G, Etzkorn FA (1994) The immunophilins. Perspect Drug Discov Des 2: 57–84. [Google Scholar]
- 24. Hur S, Bruice TC (2002) The mechanism of cis-trans isomerization of prolyl peptides by cyclophilin. J Am Chem Soc 124: 7303–7313. [DOI] [PubMed] [Google Scholar]
- 25. Schroeder OE, Carper E, Wind JJ, Poutsma JL, Etzkorn FA, et al. (2006) Theoretical and experimental investigation of the energetics of cis-trans proline isomerization in peptide models. J Phys Chem A 110: 6522–6530. [DOI] [PubMed] [Google Scholar]
- 26. Ranganathan R, Lu KP, Hunter T, Noel JP (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89: 875–886. [DOI] [PubMed] [Google Scholar]
- 27. Xu GG, Zhang Y, Mercedes-Camacho AY, Etzkorn FA (2011) A Reduced-Amide Inhibitor of Pin1 Binds in a Conformation Resembling a Twisted-Amide Transition State. Biochemistry 50: 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chatterjee S, Ator MA, Bozyczko-Coyne D, Josef K, Wells G, et al. (1997) Synthesis and biological activity of a series of potent fluoromethyl ketone inhibitors of recombinant human calpain I. J Med Chem 40: 3820–3828. [DOI] [PubMed] [Google Scholar]
- 29. Dai Y, Hedstrom L, Abeles RH (2000) Inactivation of cysteine proteases by (acyloxy)methyl ketones using S′-P′ interactions. Biochemistry 39: 6498–6502. [DOI] [PubMed] [Google Scholar]
- 30. Moore CL, Leatherwood DD, Diehl TS, Selkoe DJ, Wolfe MS (2000) Difluoro ketone peptidomimetics suggest a large S1 pocket for Alzheimer's gamma-secretase: implications for inhibitor design. J Med Chem 43: 3434–3442. [DOI] [PubMed] [Google Scholar]
- 31. Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, et al. (1999) Peptidomimetic probes and molecular modeling suggest that Alzheimer's gamma-secretase is an intramembrane-cleaving aspartyl protease. Biochemistry 38: 4720–4727. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, et al. (2007) Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem Biol 2: 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang M, Wang XJ, Chen X, Bowman ME, Luo Y, et al. (2012) Structural and kinetic analysis of prolyl-isomerization/phosphorylation cross-talk in the CTD code. ACS Chem Biol doi: 10.1021/cb3000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang XJ, Hart SA, Xu B, Mason MD, Goodell JR, et al. (2003) Serine-cis-proline and serine-trans-proline isosteres: stereoselective synthesis of (Z)- and (E)-alkene mimics by Still-Wittig and Ireland-Claisen rearrangements. J Org Chem 68: 2343–2349. [DOI] [PubMed] [Google Scholar]
- 35. Barton DHR, Chen M, Jaszberenyi JC, Taylor DK (1997) Preparation and reactions of 2-tert-butyl-1,1,3,3-tetramethylguanidine: 2,2,6-trimethylcyclohexen-1-yl iodide. Org Synth 74: 101–107. [Google Scholar]
- 36. Braukmueller S, Brueckner R (2006) Enantioselective butenolide preparation for straightforward asymmetric syntheses of gamma -lactones - paraconic acids, avenaciolide, and hydroxylated eleutherol. Eur J Org Chem 2110–2118. [Google Scholar]
- 37. Williams DR, Brown DL, Benbow JW (1989) Studies of Stemona alkaloids. Total synthesis of (+)-croomine. J Am Chem Soc 111: 1923–1925. [Google Scholar]
- 38. Xie J, Menand M, Valery J-M (2005) Regioselective debenzylation of C-glycosyl compounds by boron trichloride. Carbohydr Res 340: 481–487. [DOI] [PubMed] [Google Scholar]
- 39. de Bont HB, Liskamp RM, O'Brian CA, Erkelens C, Veeneman GH, et al. (1989) Synthesis of a substrate of protein kinase C and its corresponding phosphopeptide. Int J Pept Protein Res 33: 115–123. [DOI] [PubMed] [Google Scholar]
- 40.CrysAlis v1.171 (2004). Wroclaw, Poland: Oxford Diffraction.
- 41. Sheldrick GM (2008) A short history of SHELX. Acta Cryst A64: 112–122. [DOI] [PubMed] [Google Scholar]
- 42.(2008) SYBYL. 1699 South Hanley Rd., St. Louis, MO, 63144USA: Tripos International.
- 43.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al.. (2009) Gaussian. Wallingford CT: Gaussian, Inc.
- 44.DeLano WL (2006) MacPyMol. San Carlos, CAUSA: DeLano Scientific LLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPLC chromatograms for 1 and rac -2. 1H, 13C, and 31P NMR spectra for compounds 1, rac -2, and 7–13.
(PDF)
Pin1 inhibition plots for 1 and rac -2. Crystallographic data, CCDC 782064 and 782063 for (1 S ,3 R ,4 R )-11 and rac -11 respectively, can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
(PDF)