

Abstract
C-H bond functionalization offers strategically novel approaches to complex organic compounds, however many C-H functionalization reactions suffer from poor compatibility with Lewis basic functional groups; especially amines, which are often essential for biological activity. This study describes a systematic examination of the substrate scope of catalytic hydroarylation in the context of complex amino coumarin synthesis. The choice of substrates was guided by the design and development of the next generation of Fluorescent False Neurotransmitters (FFNs); neuroimiaging probes we recently introduced for optical imaging of neurotransmission in the brain. Comparison of two mild protocols, using catalytic PtCl4 or Au(PPh3)Cl/AgSbF6, revealed that each method has a broad and mutually complementary substrate scope. The relatively less active platinum system out-performed the gold catalyst with indole substrates lacking substitution at the C-3 position and provided higher regioselectivity in the case of carbazole-based substrates. On the other hand, the more active gold catalyst demonstrated excellent functional group tolerance, and the ability to catalyze the formation of highly strained, helical products. The development of these two protocols offers enhanced substrate scope and provides versatile synthetic tools required for the structure-activity examination of FFN neuroimaging probes as well as for the synthesis of complex coumarins in general.
Introduction
C-H bond functionalization is an active area of research. Demonstration of novel strategic opportunities in complex organic synthesis afforded by C-H bond functionalization led to a shift of attention from simple hydrocarbons to complex organic substrates containing a diversity of functional groups.1 Indeed, the field has progressed to such an extent that C-H bond functionalization is widely considered during synthetic planning of diverse organic materials including protein ligands, drug candidates, biological probes and other functional materials. Thus, in addition to the exploration of new reactivity modes (to directly convert C-H bonds to new functionalities or C-C bonds), it is important to expand the scope and practical applicability of the C-H functionalization processes for the field to realize its full impact. Here we describe a remarkably broad substrate scope of the catalytic intramolecular hydroarylation of alkynes and its use in the development of fluorescent neuroimaging probes.
Hydroarylation of alkynes defines an overall process of the addition of an arene C-H bond across an alkyne. In an intramolecular manner, this process provides a direct route to valuable organic compounds such as benzo-fused heterocycles and carbocycles (Scheme 1). In contrast to standard cross-coupling methodologies, hydroarylation eliminates the requirement for a functional group on the participating arene ring (halide, psedohalide, boronate) and the power of hydroarylation is realized in its ability to form C-C bonds directly from C-H bonds with absolute atom economy (Scheme 1).2
Scheme 1.

Selective 6-endo Annulation via Hydroarylation
Our laboratories have been pursuing the development hydroarylation reactions, which led to the PtCl4 catalyzed hydroarylation of alkynes affording a wide variety of complex products (Scheme 1, eq. 1).3 In addition, we developed the ruthenium-catalyzed hydroarylation of alkenes providing rapid access to dihydrobenzofurans and dihydrobenzopyrans.4 We were interested in the application of the PtCl4 protocol for the cyclization of aryl alkynoate esters as a mild route to complex coumarins (Scheme 1, eq. 2) complementing the relatively harsh conditions of the classical von Pechmann condensation.5 This project was fueled by our interest in the development of fluorescent reporters for imaging metabolism and neurotransmission in the brain. The coumarins are particularly attractive in this context due to their relatively small molecular size, high brightness and good photostability.
Coumarin-based fluorescent probes for dopamine metabolism and neurotransmission
We have developed a reporter substrate for monoamine oxidases, enzymes involved in metabolism of dopamine and other monoamine neurotransmitters, based on the coumarin nucleus.6 The dopamine system plays a key role in many fundamental processes of the brain including generation of motivational forces and subjective reward, habit learning, working memory and cognition.7 Aberrations of dopaminergic neurotransmission have been implicated in numerous neuropsychiatric disorders such as Parkinson’s disease, schizophrenia, drug addiction and attention deficit hyperactive disorder (ADHD).8 It is now widely accepted that learning is dependent on plasticity of synaptic connections between neurons (i.e., changes in number and strength of synaptic connections); however, there were no methods for examining neurotransmitter release at individual synapses.
To address this problem, we have recently introduced a novel class of probes, termed Fluorescent False Neurotransmitters (FFNs) that act as fluorescent tracers of neurotransmitters (Figure 1).9 Specifically, FFNs function as fluorescent substrates of transporter proteins that accumulate physiological neurotransmitters in synaptic vesicles (vesicular monoamine transporter 2 or VMAT2 for monoamine neurotransmitters such as dopamine), and thus enable visualization of individual synapses (accumulation of FFN) and their activity (release of FFN) in the brain tissue via two-photon microscopy.
Figure 1.

Fluorescent False Neurotransmitters (FFNs) were recently introduced to enable optical imaging of neurotransmission at individual synapses in the brain. A) The structure of the first example of FFNs, compound FFN511. B) FFNs function as fluorescent substrates of vesicular monoamine transporter 2 (VMAT2), the protein that accumulates dopamine in synaptic vesicles. In this manner, FFNs act as optical tracers of the neurotransmitter, allowing for imaging of both location (accumulation of fluorescence signal) and activity of presynaptic terminals (loss of fluorescence signal).
Hydroarylation Guides the Design of Fluorescent False Neurotransmitters (FFNs)
The design of FFNs was influenced by the hydroarylation methodology developed in our laboratories. As FFNs are not fluorescent tags of biological macromolecules, but reporter substrates of monoamine transporter proteins, it is important to keep the size of the FFN condidates as small as possible, while maintaining high brightness and photostability. In this respect, coumarins are especially attractive and two design approaches were considered: 1) An endogenous neurotransmitter would be structurally modified to impart fluorescence to the product (e.g., conversion of serotonin to pyrrolocoumarin 5); and 2) A known coumarin fluorophore would be modified to incorporate the ethylamino group, the key recognition element for the relevant transporter proteins (e.g., modification of the laser dye LD49010 to the agent FFN511, Figure 2).
Figure 2.

Two design approaches to Fluorescent False Neurotransmitters (FFNs). (A) A monoamine neurotransmiter such as serotonin is modified by addition of the lactone ring to render the system fluorescent (fluorescent pyrrolocoumarin 5). (B) A known fluorophore (in this case LD490) is elaborated to include the ethylamino group, which is the key recognition element for the relevant monoamine transporter VMAT2, providing the probe FFN511.
In this context, we examined the scope of the intramolecular hydroarylation of aryl alkynoate esters catalyzed by PtCl4 and Au(PPh3)SbF6, which revealed remarkably wide and complementary applicability of these methods for the synthesis of complex coumarins. These mild catalytic methods directly affect the design and development of FFN probes by providing the flexibility of placing diverse functional groups at the coumarin core, enabling the tuning of both the fluorescence properties and the ability to function as substrates for relevant protein targets (neurotransmitter transporters). This paper focuses on the substrate scope of the hydroarylations methods, while the pharmacological and biological studies will be described in the future in a separate publication.
Results and Discussion
Search for Competent Hydroarylation Catalysts. Screening of Platinum and Gold Complexes and Salts
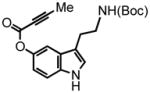
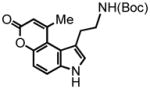
According to the first design approach to potential FFN agents (Figure 1A), aryl alkynoate ester 8 was prepared and examined with the hydroarylation condition, using PtCl4 as the catalyst developed in our laboratories (Table 1).3,4 We were pleased to find that PtCl4 (10 mol %) in a mixture of 1,4-dioxane:dichloroethane (DCE) [1:1] at 80 °C, effected the desired transformation in 8 hr, affording a 54% yield of the pyrrolocoumarin 9 (entry 1, Table 1). Formation of this strained compound as the only detectable product is noteworthy and the regioselectivity is discussed in detail below. Attempts to decrease the catalyst loading to 5 mol % led to a decrease in yield, however an increase in overall mass balance; after heating at 80 °C for 24 hr the product was isolated in 31% yield with 51% of the starting alkyne recovered (entry 2, Table 1).
Table 1.
Comparing PtCl4 Against Known Platinum and Palladium Catalytic Systems
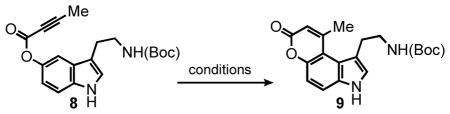
| |||||
|---|---|---|---|---|---|
| entry | catalyst | mol % | solventa | yield 9 | recov. 8 |
| 1b | PtCl4 | 10 | DCE:dioxane | 54 | 0 |
| 2b | PtCl4 | 5 | DCE:dioxane | 31 | 51 |
| 3c | Pd(OAc)2 | 5 | TFA:DCM | 0 | 0 |
| 4b | PtCl2 | 5 | Toluene | 50 | 13 |
| 5d | PtCl2 | 5 | Dioxane | 32 | 14 |
| 6d | PtCl2 | 5 | Acetone | 4 | 60 |
all reactions were run at a concentration of 0.05M.
entries 1, 2 and 4 were run at 80 °C.
entry 3 was run at room temperature.
entries 5 and 6 were heated to reflux.
For direct comparison, we examined the conditions developed by Fujiwara11 (entry 3, Table 1), Echavarren12 (entries 4 and 5, Table 1) and Fürstner13 (entry 6, Table 1) for the activation of alkynes. Fujiwara’s conditions, using Pd(OAc)2 as the catalyst and TFA as a co-solvent, afforded neither the desired product 9 nor the substrate 8, demonstrating the incompatibility of the acidic conditions with complex substrates. The loss of the Boc-protecting group in the presence of TFA exposes the primary aliphatic amine, which presumably deactivates to the palladium catalyst. The reactions employing PtCl2 (5 mol %) were dependent on the solvent; toluene gave the highest yield (50%, entry 4). We viewed the higher mass balance of the PtCl4 (5 mol %) reaction as well as our previous experience with this system as the key decision factors to explore the scope of the PtCl4 system.
In addition to platinum catalysts, reports from Reetz,14 Fürstner,15 Echavarren,16 Toste,17 Shi18 and others demonstrated high activity of gold complexes for hydroarylation and other procesess relying on alkyne activation.
Consequently, a variety of commonly employed gold salts were screened with and without silver salt additives. The most promising of the examined systems was the combination of Au(PPh3)Cl and AgSbF6 (10 mol %) which furnished the product in 92% yield in only 5 min at room temperature (entry 1, Table 2). As a control we confirmed that the catalytic amount of AgSbF6 (in the absence of gold) was inactive. Optimization of the catalyst loading for the Au(PPh3)Cl/AgSbF6 system revealed that the reaction proceeded equally well with 5 mol % (entry 2, Table 2). The product was also formed in high yield even in the presence of 1 mol % of the catalyst, however the reaction required heating to 80 °C for 6 hours. For the purpose of mapping the scope of the gold-silver system, we chose the mild room temperature conditions employing 5 mol % of the catalyst.
Table 2.
Optimization Au(PPh3)Cl/AgSbF6 Catalyst Loadinga
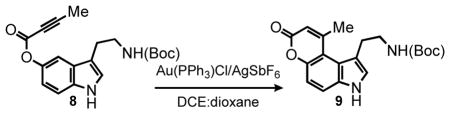
| ||||
|---|---|---|---|---|
| entry | mol % | temp. °C | time | yield 9 |
| 1 | 10 | 23 | <5 min | 92 |
| 2 | 5 | 23 | <10 min | 94 |
| 3 | 2 | 80 | 1 hr | 94 |
| 4 | 1 | 80 | 6 hr | 93 |
All reactions were run at a concentration of 0.05M.
Finally, in order to rule out the possibility of Brønsted acid catalysis by either HCl or HSbF6, formed in situ in DCE by the action of PtCl4 or AgSbF6 respectively,19 we conducted a screen of HCl, HSbF6 and TfOH against substrates of various sensitivities to acid. None of the conditions explored succeeded in furnishing the hydroarylation products (see the supporting information). These results support the role of the transition metal Lewis acids as the active catalysts in the reaction.
The Regioselectivity of Catalytic Hydroarylation of Indole-Derived Substrates
As described above, the cyclization of substrate 8 proceeded with high regioselectivity, affording a single regioisomer, namely the sterically congested product 9 under both the Pt(IV) and Au(I) conditions (Scheme 2). The other regioisomer was not detected in the crude reaction mixture. It is worth noting that these products are forced into a helical conformation due to the major steric overlap of the alkyl substituents. In addition to the electronic rationale (see discussion below and Scheme 3), we also considered the possibility that the carbamate of the ethylamino group might serve as a directing group for the catalyst, favoring cyclization to the C-4 position.
Scheme 2.

The cyclization is not directed by the substituent in 3-position
Scheme 3.

Mechanistic rationale for the observed regioselectivity in the cyclization of indole substrates
To test this hypothesis substrate 10 was prepared and submitted to the cyclization conditions (Scheme 2). The PtCl4 catalyzed reaction afforded product 11 in 54% yield, again as a single regioisomer annulated at C-4. This result demonstrates that the carbamate group is not responsible for the regioselectivity observed in the cyclization of substrates 8 and 15. Interestingly, substrate 10 under the Au(PPh3)Cl/AgSbF6 conditions decomposed rapidly even at room temperature and analysis of the crude 1H NMR failed to reveal any diagnostic signals for either regioisomer of the cyclized product. It is possible that the highly carbophilic gold cation underwent metallation of the indole nucleus at the C-3 position, followed by deleterious side reactions leading ultimately to the destruction of the substrate. In addition to putting the directing group hypothesis to rest, the substrates 8 and 10 revealed complementarity of the platinum and gold catalytic systems. The less active PtCl4 catalyst can be advantageous in the case of more reactive substrates such as compound 10.
Electronic Rationale for the Observed Regioselectivity
On this ground, we propose an electronic rationale for the regioselectivity observed with the indole substrates. Following coordination of the Lewis acid to the alkyne, the electrophilic attack can occur at either the C-4 or C-6 position of the benzene ring, affording intermediates I or IV respectively (Scheme 3). The intermediate I (and the corresponding transition state) can be stabilized via delocalization of the positive charge in the benzene ring (three resonance forms I-III) without disrupting the aromaticity of the pyrrole ring. In contrast, stabilization of intermediate IV requires delocalization of the positive charge in the pyrrole ring, thereby breaking its aromaticity (similar rationale explains the kinetic preference for aromatic electrophilic substitution of the more hindered 1-position of naphthalene). Thus, the first pathway has a lower energetic barrier, apparently by such an extent that the other regioisomer 13 is not observed. Although there is significant steric crowding between the substituents at C-3 and C-4, a close examination of the intermediate (I-III) reveals a “gauche-like” interaction due to the tetrahedral geometry at C-4. Upon deprotonation at C-4 and rehybridization of the carbon center as well as rearomatization, the product is formed and is locked in the strained conformation (following the protonative deplatination3b).
Cyclization of Highly Hindered Substrate 15
To test the limits of the hydroarylative cyclization and potentially force the formation of the other regioisomer, we prepared the sterically more demanding substrate 15 containing a phenyl ring attached to the alkyne (entry 4, Table 3). Remarkably, a single product was formed in 98% yield via the cyclization at the C-4 position under the gold catalyzed conditions, while the platinum catalyst failed completely to provide any cyclized product. These results demonstrate the remarkable robusteness of the hydroarylative cyclization under the gold catalysis, lower reactivity of the platinum catalyst and the energetic inaccessibility of the C-6 cyclization pathway. The X-ray crystallographic structure of the product 14 was obtained and confirmed a significant degree of helicity to the aromatic core, with a dihedral angle of 19.4° (bond Ca-Cb, Figure 3). As can be seen in the ORTEP plot, the phenyl ring is held above the methylene group attached to the indole ring. The upfield shift of this methylene group seen in the 1H NMR suggests significant shielding by the phenyl ring; 1.26 ppm versus 2.64 ppm in the substrate.
Table 3.
Scope Summary of Indole Derived Aryl Alkynoate Ester Substrates
| entry | substrate | product | conditions/yield | |
|---|---|---|---|---|
| 1 |
 8 |
 9 |
Pt, 80 °C | 54% |
| Au, r.t. | 93% | |||
| 2 |
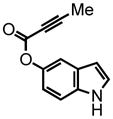 10 |
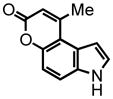 11 |
Pt, 80 °C | 54% |
| Au | decomp. | |||
| 3 |
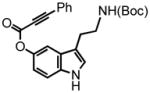 15 |
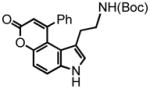 14 |
Pt | N.R. |
| Au, r.t. | 98% | |||
| 4 |
 16 |
 17 |
Pt, 80 °C | 50% |
| Au, r.t. | 80% | |||
Figure 3.

ORTEP plot of the X-ray structure of compound 14. The dihedral angle of 19.4° is labeled as θ. ORTEP graphic is oriented to see down the Ca-Cb bond.
Substrate Scope of Indole-derived Substrates
The catalytic hydroarylation of indole substrates showed an excellent tolerance for both the functional groups and steric hindrance (Table 3). Both catalytic systems are compatible with the free indole amino group, Boc-protected aliphatic amine and Boc-protected α-amino ester groups. Furthermore, complementarity of the platinum and the gold-silver system was revealed; the former one being less active which becomes advantageous with more reactive substrates such as substrate 10. Most notably, the cyclization showed exclusive regioselectivity for the 4-position, which we rationalize in terms of electronic factors, and is tolerant of considerable steric hindrance as demonstrated by the efficient formation of helical compound 14.
Substrate Scope of Aryl Alkynoate Esters Containing Aniline Amino Groups
We next investigated the hydroarylative cyclization of aniline substrates. This was driven by the need to examine the importance of the aniline nitrogen substitution in the probe FFN511 (Figure 1) on its interaction with monoamine transporters (e.g. VMAT2, see Introduction) and other CNS receptors.
The Boc-protected aniline substrate 18 underwent efficient cyclization with the Au(PPh3)Cl/AgSbF6 catalyst, providing a mixture of regioisomers 19 and 20 in 86% yield (entry 1, Table 4); whereas the same substrate failed to react in the presence of PtCl4. Presumably, the relatively low electron donating ability of the carbamate substituent renders the arene ring unreactive towards the PtCl4 activated alkyne. The free aniline 21 also failed to cyclize in the presence of PtCl4, likely due to coordination of the catalyst to the free amine (entry 2, Table 4). In contrast, the gold system was well tolerant of free aniline giving a high yield of a mixture of coumarins 22 and 23 (95%). The success of the gold catalyst in this example is attributed to its high carbophilicity that leads to preferential coordination to the alkyne.
Table 4.
Exploring the Scope of Aniline Substrates
| entry | substrate | product | conditions/yield | |
|---|---|---|---|---|
| 1 |
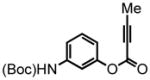 18 |
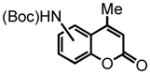 7-NH(Boc)-19 5-NH(Boc)-20 |
Pt | N.R. |
| Au, r.t. | 86% | |||
| 19:20 = 3.5:1 | ||||
| 2 |
 21 |
 7-NH2-22 5-NH2-20 |
Pt | N.R. |
| Au, r.t. | 95% | |||
| 22:23 = 1:1 | ||||
| 3 |
 24 |
 7-NH(Boc)-25 5-NH(Boc)-26 |
Pt, 80 °C | 66% |
| 25:26 = 4.8:1 | ||||
| Au, 80 °C | 80% | |||
| 25:26 = 5.5:1 | ||||
| 4 |
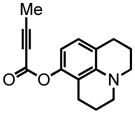 27 |
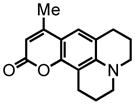 28 |
Pt, 80 °C | 88% |
| Au, r.t. | 91% | |||
| 5 |
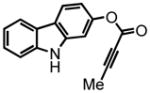 29 |
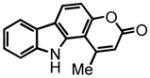 30 |
Pt, 80 °C | 69% |
| 30:31 = 5:1 | ||||
 31 |
Au, r.t | 92% | ||
| 30:31 = 1.7:1 | ||||
The tertiary anilines, on the other hand, are well tolerated under both catalytic conditions (compounds 24 and 27, entries 3 and 4, Table 4). This is due to the stronger electron-donating ability of tertiary amines (as well as poorer metal-chelating ability). In addition, the carbazole derivative 29 performed well under both conditions (entry 5, Table 4). It is noteworthy that PtCl4 catalyst afforded higher regioselectivity (1:5) in comparison to the Au(PPh3)Cl/AgSbF6 system (1:1.7). Thus, the less active catalyst is more selective for the more sterically congested product 30, illustrating once again the importance of electronic factors in this transformation.
Synthesis of FFN511 and Candidates for a New Generation of FFN Probes
As discussed in the introduction the probe FFN511 was the first example of fluorescent false neurotransmitters (FFNs) enabling for the first time visualization of neurotransmitter release from hundreds of individual presynaptic terminals in the brain.9 FFN511 is synthesized in short order from commercially available 8-hydroxyjulolidine (32) and the N-Boc-amino alkynoic acid 33 (Scheme 4). The ester coupling provides substrate 34, which is then cyclized in high yield under both sets of hydroarylation conditions; 91% and 95% employing Pt and Au systems, respectively (entry 1, Table 5). Simple deprotection with TFA furnished FFN511 as a TFA salt (Scheme 4). This procedure has successfully been scaled up to 960 mg (2.5 mmol) providing suffient quantities of material for the biological studies.
Scheme 4.

Synthesis of FFN511 via the Catalytic Hydroarylation
Table 5.
Complex Aryl Alkynoate Esters as Precursors to New FFN Probes
| entry | substrate | product | conditions/yield | |
|---|---|---|---|---|
| 1 |
 34 |
 35 |
Pt, 80 °C | 91% |
| Au/Ag, r.t. | 95% | |||
| 2 |
 36 |
 37 |
Pt, 80 °C | 88% |
| Au/Ag, r.t. | 90% | |||
| 3 |
 38 |
 39 |
Pt, 80 °C | 88% |
| Au/Ag, r.t. | 90% | |||
| 4 |
 40 |
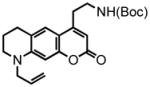 41 |
Pt, 80 °C | 76% |
| Au/Ag, r.t. | 93% | |||
| 5 |
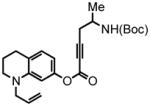 42 |
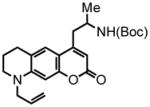 43 |
Pt, 80 °C | 75% |
| Au/Ag, r.t. | 92% | |||
| 6 |
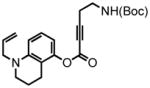 44 |
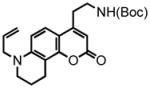 45 |
Pt, 80 °C | 74% |
| Au/Ag, r.t. | 90% | |||
Enouraging results with FFN5119 prompted us to undertake a structure-activity relationship study of FFN511 to identify the key recognition elements for the monoamine transporters while maintaining the coumarin core and the fluorescence properties. There are two major sites for modification of FFN511: 1) The aniline nitrogen in the 7-position and substitution in the adjacent positions (C-6 and C-8); and 2) the ethylamine side chain in the 4-position. The compatibility of the gold system with a range of anilines in the 7-position is described above (Table 4).
Modification of the N-Boc-protected ethylamino side chain was well tolerated, including α-methylamino substrate 36 and truncated methylamino substrate 38, offering high yields of the desired products. The results of the hydroarylation of the N-allyl protected substrates (entries 4–6, Table 5) were particularly interesting as no hydroarylation of the alkene was observed under either reaction condition. For all of these substrates the alkyne was selectively activated, leading to the formation of the targets in high yield (90–93%) for the Au(PPh3)Cl/AgSbF6 conditions and moderate yields (74–76%) for the PtCl4 system. The N-allyl group could be selectively deprotected with Pd(PPh3)4 (10 mol %) in THF in the presecence of N,N′-dimethylbarbituric acid, enabling futher modification of the secondary aniline amine.
The remarkably broad substrate scope of the hydroarylation methodology sets the stage for the preparation of a series of coumarin compounds, enabling the systematic examination of the substrate scope of monoamine transporters involved in accumulation of neurotransmitters in the presynaptic terminals. As a representative example, we show here the entire synthesis of a new FFN candidate, compound PV139, where the “upper ring” of the aniline substitution in FFN511 was removed. The properly protected aminophenol 48 was synthesized from commercially available 5-hydroxyquinoline (46, Scheme 5) by hydrogenation under acidic conditions, phenol protection, N-allylation and phenol deprotection. This phenol was then condensed with the N-Boc-amino alkynoic acid 33, to yield the aryl alkynoate substrate 44 and the hydroarylative cyclization furnished coumarin 45 (90% with the Au system). Deprotection of both amines delivered the compound PV139 as a TFA salt in excellent yield. This approach allows for selective elaboration of either the aromatic amine or the primary aliphatic amine in either order, providing a versatile platform for the structure-activity study of these compounds in the context of FFN probe development.
Scheme 5.

Synthesis of a new FFN candidate, compound PV139
a) H2 (3 atm), PtO2 EtOH, HCl (aq.), 69%. b) TBSCl, imidazole, DMF 50 °C. c) allylbromide, K2CO3, MeCN, 100 °C. d) TBAF, THF; 65% for 3 steps. e) 33, DIC, DMAP, DCM, 0 °C, 70%. f) see Table 5. g) Pd(PPh3)4 (5 mol %), N,N′-dimethylbarbituric acid, THF, 80 °C. h) TFA, DCM, 0 °C; 90% for 2 steps.
Conclusions
In summary, we have examined two catalytic systems, which afford complex coumarins in good to excellent yields under mild conditions. While the Au(PPh3)Cl/AgSbF6 system demonstrated excellent compatibility with a variety of functional groups, PtCl4 was found to be more sensitive to the electronic factors of the arene substrates. Both catalysts, however, performed well in the presence of a variety of functional groups including Boc-protected amines, tertiary anilines, allylic anilines, protected α-aminoesters, and free (NH)-indoles and carbazoles. Although the platinum system is less active, it revealed higher regioselectivity in some instances and out-performed the gold catalyst with indole substrates lacking substitution at the C-3 position.
The synthetic methods examined herein enable an SAR study of the FFN probes, where FFN511 serves as the lead compound, with the aim to develop second generation FFN probes with improved functional parameters for imaging synaptic activity in the brain. Indeed, the chemistry is in place for examining the importance of the aniline amine substitution in the 7-positions (and the adjacent positions on the arene ring) as well as the substitution and the linker of the primary amine. The results of these biological and pharmacological studies will be reported in the future. In general, the mild catalytic methods complement the von Pechman condensation and other classical methods for coumarin synthesis.
Experimental Section
General Considerations
Argon was purified by passage through Drierite. Nuclear Magnetic Resonance spectra were recorded at 300 K. 1H NMR spectra recorded in CDCl3 solutions were referenced to TMS (0.00 ppm). 13C NMR spectra recorded in CDCl3 were referenced to the residual solvent peak (77.16 ppm). 19F NMR spectra were recorded in CD3OD with α,α,α-trifluorotoluene (10 μL) as an internal standard. The spectra were referenced to the α,α,α-trifluorotoluene fluorine peak, −63.72 ppm. Several compounds containing the carbamate group were found to exist in rotameric forms. As such, NMR spectra were recorded at elevated temperatures in order to produce clearer spectra. Flash chromatography was performed on silica gel (230–400 mesh). High-resolution mass spectra (HRMS) were acquired on a high resolution sector type double focusing mass spectrometer (ionization mode: FAB+). Reactions were monitored by TLC analysis using hexanes/ethyl acetate mixtures as the eluent and visualized using permanganate stain and/or cerric ammonium molybdate stain and/or UV light. Toluene, acetonitrile, tetrahydrofuran, ethyl ether, and dichloromethane were dried via passage through a column of activated alumina. Chloroform-d1 was stored over 4Å molecular sieves. PtCl4 (99.9%-Pt), Au(PPh3)Cl (99.9+%-Au), and AgSbF6 (98%) were purchased from Strem Chemicals Inc. and used as received. 1,2-Dichloroethane (DCE) and 1,4-dioxane were used as received without any further purification.
General Procedure for the Synthesis of Aryl Alkynoates via DIC coupling of Phenols and Propiolic Acids
To a flame dried round bottom flask equipped with a magnetic stir bar was added the appropriate alkynoic acid (1.2 eq.) followed by dry DCM (0.3 M). The solution was sealed under argon with a rubber septum and cooled to 0 °C in an ice/brine bath. Neat N,N′-diisopropylcarbodiimide (DIC) (1.5 eq.) was added via syringe and stirred for 1 minute during which a white precipitate formed. The appropriate phenol (1 eq.) was dissolved in dry DCM (1 M) and added via syringe. Lastly, 4-dimethylaminopyridine (DMAP) (25 mol %) was dissolved in dry DCM (0.5 M) and added dropwise to the stirring solution. The mixture was stirred under argon at 0 °C until the phenol was consumed as determined by TLC. The reaction was then filtered through celite and concentrated in vacuo. The residue was then purified via flash column chromatography.
Representative DIC Coupling Procedure for the Formation of Substrate 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8)
To a flame dried round bottom flask equipped with a stir bar was added 2-butynoic acid (202 mg, 2.4 mmol) followed by dry DCM (8 mL). The solution was sealed under argon with a rubber septum and cooled to 0 °C in an ice/brine bath. Neat DIC (470 μL, 3.0 mmol) was added via syringe and stirred for 1 minute during which a white precipitate formed. A solution of N-Boc-serotonin20 (552 mg, 2.0 mmol) in dry DCM (1 mL) was added to the reaction mixture via syringe. Lastly, DMAP (61 mg, 0.5 mmol) was dissolved in dry DCM (1 mL) and added dropwise to the stirring solution. The reaction was stirred under argon at 0 °C until complete consumption of the phenol was observed by TLC (2 hr). The reaction was then filtered through celite and concentrated in vacuo. The residue was then purified via column chromatography, 20% EtOAc/Hexanes, to afford 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8) as an amorphous white solid (670 mg, 98%). 1H NMR (CDCl3, 300 MHz) δ 1.44 (s, 9H); 2.04 (s, 3H); 2.83 (t, J = 6.6 Hz, 2H); 3.36 (br q, J = 6.0 Hz, 2H); 4.71 (br s, 1H); 6.90-6.86 (m, 2H); 7.28-7.21 (m, 2H); 8.75 (br s, 1H); 13C NMR (CDCl3, 75 MHz) δ 4.0, 25.6, 28.5, 41.0, 72.5, 79.3, 87.9, 110.7, 111.9, 113.3, 115.8, 123.9, 127.7, 134.6, 143.5, 153.3, 156.2. HRMS (FAB+): calculated for C19H22N2O4 342.1580, measured 342.1580 [M].
1H-indol-5-yl but-2-ynoate (10) was eluted with 10% EtOAc:Hex as an amorphous white solid (480 mg, 70%). 1H NMR (CDCl3, 400 MHz) δ 2.04 (s, 3H); 6.50 (dd, J = 2.8 Hz, 0.8 Hz, 1H); 6.93 (dd, J = 8.4 Hz, J = 2.0 Hz, 1H); 7.16 (t, 2.8 Hz, 1H); 7.26 (d, J = 8.4 Hz, 1H); 7.36 (d, J = 2.4 Hz, 1H); 8.25 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.1, 72.5, 88.0, 103.0, 111.7, 112.7, 115.7, 125.9, 128.1, 134.0, 143.8, 153.3. HRMS (FAB+): calculated for C12H9NO2 199.0633, measured 199.0641 [M].
3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl 3-phenylprop-2-ynoate (15) was eluted with 20% EtOAc:Hex as an amorphous white solid (736 mg, 91%). 1H NMR (CDCl3, 300 MHz) δ 1.45 (s, 9H); 2.90 (t, J = 6.9 Hz, 2H); 3.42 (b quart, J = 6.3 Hz, 2H); 4.63 (bs, 1H); 7.00 (dd, J = 2.1 Hz, J = 8.7 Hz, 2H); 7.48-7.32 (m, 5H); 7.63 (d, J = 8.4 Hz, 2H); 8.37 (bs, 1H); 13C NMR (CDCl3, 100 MHz) δ 25.8, 28.5, 41.0, 79.4, 80.7, 88.5, 110.9, 112.0, 113.4, 115.8, 119.5, 123.9, 127.8, 128.8, 131.0, 133.2, 134.6, 143.5, 153.7, 156.2. HRMS (FAB+): calculated for C24H24N2O4 404.1736, measured 404.1730 [M].
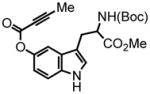
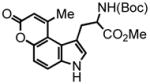
3-(2-(tert-butoxycarbonylamino)-3-methoxy-3-oxopropyl)-1H-indol-5-yl but-2-ynoate (16) was prepared from N-Boc-5-hydroxytryptophan methyl ester21 eluted with 6% EtOAc:DCM as an amorphous white solid (430 mg, 95%). 1H NMR (CDCl3, 400 MHz) δ 1.35 + 1.44 (s,s, 9H, rotomers); 2.06 (s, 3H); 3.20 (bd, J = 4.0 Hz, 2H); 3.65 (s, 3H); 4.59 (br q, J = 7.2, 1H); 5.14 (d, J = 7.6 Hz, 1H); 6.92-6.86 (m, 2H); 7.23-7.19 (m, 2H); 8.72 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.0, 27.9, 28.3, 52.4, 54.2, 72.3, 80.1, 88.0, 110.1, 110.6, 112.0, 115.8, 124.7, 127.9, 134.3, 143.6, 153.3, 155.3, 172.7. HRMS (FAB+): calculated for C21H24N2O6 400.1634, measured 400.1637 [M].
3-(tert-butoxycarbonylamino)phenyl but-2-ynoate (18) was eluted with 10% EtOAc:Hex as an amorphous white solid (1.03 g, 74%). 1H NMR (CDCl3, 400 MHz) δ 1.50 (s, 9H); 2.04 (s, 3H); 6.78 (d, J = 8.8 Hz, 2H); 7.10 (d, J = 8.4 Hz, 1H); 7.25 (t, J = 8.0 Hz, 1H); 7.25 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.0, 28.3, 72.0, 80.8, 88.3, 111.7, 115.7, 116.1, 129.7, 139.8, 150.5, 152.0, 152.5. HRMS (FAB+): calculated for C15H17NO4 275.1158, measured 275.1168 [M].
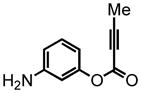
3-aminophenyl but-2-ynoate (21)
To a solution of 3-(tert-butoxycarbonylamino)phenyl but-2-ynoate (18) (812 mg, 2.9 mmol) in DCM (15 mL) at 0 °C was added trifluoroacetic acid (TFA) (5 mL) dropwise. The mixture was stirred at 0 °C for 1 hr. The solvent was then removed in vacuo and the resulting residue was dissolved in DCM (10 mL) and slowly quenched with NaHCO3 (aq. satd.). The organic layer was separated and dried over MgSO4. The suspension was filtered and the filtrate was concentrated. The residue was chromatographed on silica gel, eluting with 20% EtOAc:Hex, to yield 3-aminophenyl but-2-ynoate (21) (505 mg, 98%) as an amorphous white solid. 1H NMR (CDCl3, 400 MHz) δ 2.01 (s, 3H); 3.77 (br s, 2H); 6.38 (dd, J = 5.2 Hz, 2.0 Hz, 1H); 6.51-6.45 (m, 2H); 7.10 (dt, J = 8.0 Hz, J = 1.2 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 13.8, 72.1, 88.1, 107.8, 110.8, 113.0, 130.0, 148.0, 150.9, 152.0. HRMS (FAB+): calculated for C10H10NO2 176.0712, measured 176.0704 [M+1].
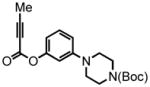
tert-butyl-4-(3-(but-2-ynoyloxy)phenyl)piperazine-1-carboxylate (24) was eluted with 10% EtOAc:Hex as an amorphous white solid (951 mg, 92%). 1H NMR (CDCl3, 400 MHz) δ 1.48 (s, 9H); 2.05 (s, 3H); 3.13 (br t, J = 6.0 Hz, 4H); 3.56 (t, J = 5.6 Hz, 4H); 6.62 (m, 1H, eclipsed by proton at 6.64 ppm); 6.64 (s, 1H); 6.79 (d, J = 9.2 Hz, 1H); 7.25 (t, J = 8.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.0, 28.5, 48.9, 72.2, 80.0, 88.0, 109.2, 112.6, 114.1, 129.9, 151.0, 152.0, 152.4, 154.7. HRMS (FAB+): calculated for C19H24N2O4 344.1736, measured 344.1746 [M].
8-(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizine)-but-2-ynoate (27) was eluted with 5% EtOAc:Hex as an amorphous beige solid (809, 79%). 1H NMR (CDCl3, 300 MHz) δ 1.99-1.89 (m, 4H); 2.04 (s, 3H); 2.58 (t, J = 6.3 Hz, 2H); 2.72 (t, J = 6.3 Hz, 2H); 3.14-3.09 (m, 4H); 6.25 (d, J = 8.1 Hz, 1H); 6.76 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.0, 21.2, 21.6, 21.9, 27.6, 49.4, 49.9, 72.3, 87.4, 108.4, 113.4, 119.6, 127.0, 144.0, 146.7, 152.3. HRMS (FAB+): calculated for C16H17NO2 255.1259, measured 255.1257 [M].
9H-carbazol-2-yl but-2-ynoate (29) was eluted with 10% EtOAc:Hex as an amorphous white solid (302 mg, 40%). 1H NMR (CDCl3, 400 MHz) δ 2.07 (s, 3H); 6.96 (dd, J = 8.4 Hz, J = 2.0 Hz, 1H); 7.15 (s, 1H); 7.25-7.21 (m, 1H); 7.40-7.33 (m, 2H); 7.96 (t, J = 6.8 Hz, 2H); 8.11 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 4.2, 72.3, 88.5, 103.8, 110.8, 113.0, 119.8, 120.3, 121.0, 121.7, 122.8, 125.9, 139.7, 140.2, 148.4, 152.8. HRMS (FAB+): calculated for C16H11NO2 249.0790, measured 249.0801 [M].
5-(tert-butoxycarbonylamino)pent-2-ynoic acid (33)
To a solution of tert-butyl but-3-ynyl(tosyl)carbamate22 (3.23 g, 10 mmol) in methanol (100 mL) was added Mg turnings (2.43 g, 100 mmol). The resulting mixture was sonicated for 2 hr at which time all of the Mg had dissolved. The reaction volume was reduced to ~20 mL in vacuo. The solution was then diluted with DCM (50 mL) and poured into aq. HCl (0.5 M, 50 mL). The resulting magnesium salts were filtered. The organic phase was separated and the aqueous solution was extracted with EtOAc. The organic layers were combined and washed with satd. NaHCO3 (aq). The organic layer was dried over MgSO4 and concentrated in vacuo. The resulting residue was purified via flash column (50% DCM/Hex) to afford tert-butyl but-3-ynylcarbamate (1.54 g, 9.1 mmol) as a colorless oil, 91%. 1H NMR (CDCl3, 400 MHz) δ 1.35 (s, 9H); 1.92 (t, J = 2.0 Hz, 1H); 2.29-2.26 (m, 2H); 3.18-3.17 (m, 2H); 5.01 (br s, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.9, 28.3, 39.3, 69.8, 79.3, 81.6, 155.8. This material (4.4 g, 26.0 mmol) in THF (150 mL) was cooled to −30 °C under argon. To the resulting solution was added MeLi (1.4 M in pentane, 40.9 mL, 57 mmol) dropwise. The cloudy solution was stirred for 30 mins. The flask was then equipped with a vent needle and a CO2 bubbler, such that the gas bubbled through the reaction solution. After 2 hrs the reaction was quenched with H2O (~100 mL) and neutralized with HCl (1 M). The solution was then extracted with EtOAc and the organic layer was dried over MgSO4. The solvent was removed in vacuo to afford a thick pale yellow oil which was placed under vacuum. While under vacuum the oil solidified. The solid was then triturated with hexanes to yield 5-(tert-butoxycarbonylamino)pent-2-ynoic acid (33) (5.05 g, 23.6 mmol) as an amorphous white solid, 91%. 1H NMR (CDCl3, 400 MHz, 350 K) δ 1.46 (s, 9H); 2.54 (t, J = 6.4 Hz, 2H); 3.32 (s, 2H); 5.23 (br s, 1H); 9.64 (br s, 1H); 13C NMR (CDCl3, 75 MHz, 350 K) δ 20.6, 28.6, 39.3, 74.6, 81.0, 88.0, 155.8, 156.7. HRMS (FAB+): calculated for C10H16NO4 214.1079, measured 214.1070 [M+1].
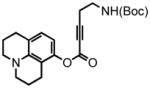
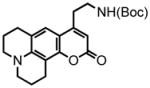
8-(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizine)-(5-(tert-butoxycarbonylamino)pent-2-ynoate) (34) was eluted with 5% EtOAc/DCM as an amorphous white solid (580 mg, 98%). 1H NMR (CDCl3, 300 MHz) δ 1.46 (s, 9H); 1.97-1.92 (m, 4H); 2.58 (t, J = 6.6 Hz, 4H); 2.72 (t, J = 6.3 Hz, 2H); 3.14-3.09 (m, 4H); 3.33 (apparent quartet, J = 6.3 Hz, 2H); 4.91 (br s, 1H); 6.25 (d, J = 7.8 Hz, 1H); 6.76 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.6, 21.2, 21.6, 21.9, 27.5, 28.4, 38.6, 49.4, 49.9, 73.8, 79.8, 88.7, 108.4, 113.4, 119.7, 127.0, 144.0, 146.7, 152.1, 155.7. HRMS (FAB+): calculated for C22H28N2O4 384.2049, measured 384.2046 [M].
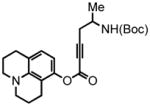
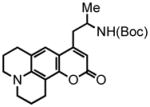
8-(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizine)-(5-(tert-butoxycarbonylamino)hex-2-ynoate) (36) was eluted with 10% EtOAc/Hex as an amorphous white solid (290 mg, 55%). 1H NMR (CDCl3, 300 MHz) δ 1.22 (d, J = 6.6 Hz, 3H); 1.45 (s, 9H); 1.96-1.91 (br m, 4H); 2.60-2.56 (m, 4H); 2.72 (t, J = 6.6 Hz, 2H); 3.14-3.09 (m, 4H); 3.91 (br s, 1H); 4.67 (br s, 1H); 6.26 (d, J = 8.1 Hz, 1H); 6.77 (d, J = 8.4 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 19.8, 21.1, 21.5, 21.8, 26.6, 27.5, 28.4, 44.4, 49.4, 49.8, 74.7, 79.6, 87.9, 108.4, 113.3, 119.6, 126.9, 144.0, 146.8, 152.0, 154.9. HRMS (FAB+): calculated for C23H30N2O4 398.2206, measured 398.2198 [M].
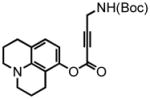
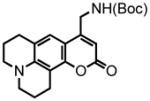
8-(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizine)-(4-(tert-butoxycarbonylamino)but-2-ynoate) (38) was eluted with 10% EtOAc/Hex as an amorphous white solid (330 mg, 40%). 1H NMR (CDCl3, 400 MHz) δ 1.47 (s, 9H); 1.94 (sext, J = 6.4 Hz, 4H); 2.56 (t, J = 6.8 Hz, 2H); 2.72 (t, J = 6.8 Hz, 2H); 3.12 (quart, J = 5.6 Hz, 4H); 4.14 (br d, J = 4.8 Hz, 2H); 4.86 (br s, 1H); 6.25 (d, J = 8.0 Hz, 1H); 6.77 (d, J = 8.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 21.2, 21.6, 21.9, 27.6, 28.4, 30.6, 49.4, 49.9, 74.8, 80.7, 85.8, 108.3, 113.3, 119.8, 127.1, 144.1, 146.5, 151.8, 155.2. HRMS (FAB+): calculated for C21H26N2O4 370.1893, measured 370.1882 [M].
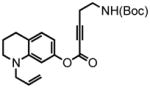
1-allyl-1,2,3,4-tetrahydroquinolin-7-yl 5-(tert-butoxycarbonylamino)pent-2-ynoate (40) was eluted with 15% EtOAc/Hex as a yellow oil (970 mg, 91%). 1H NMR (CDCl3, 300 MHz) δ 1.46 (s, 9H); 1.94 (quint, J = 6.3 Hz, 2H); 2.59 (t, J = 6.3 Hz, 2H); 2.73 (t, J = 6.3 Hz, 2H); 3.27 (t, J = 5.7 Hz, 2H); 3.34 (quart, J = 6.3 Hz, 2H); 3.82 (dd, J = 3.3 Hz, J = 1.5 Hz, 2H); 4.87 (br s, 1H) 5.20-5.13 (m, 2H); 5.87-5.74 (m, 1H); 6.24 (d, J = 2.1 Hz, 1H); 6.30 (dd, J = 8.0 Hz, J = 2.1 Hz, 1H); 6.91 (d, J = 8.1 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.4, 22.0, 27.6, 28.3, 38.5, 48.8, 53.6, 73.9, 79.6, 88.7, 103.5, 107.8, 116.1, 120.3, 129.3, 132.6, 146.1, 149.4, 152.1, 155.7. HRMS (FAB+): calculated for C22H28N2O4 384.2049, measured 384.2041 [M].
1-allyl-1,2,3,4-tetrahydroquinolin-7-yl 5-(tert-butoxycarbonylamino)hex-2-ynoate (42) was eluted with 10% EtOAc/Hex as an amorphous pale yellow solid (759 mg, 95%). 1H NMR (CDCl3, 400 MHz) δ 1.22 (d, J = 6.8 Hz, 3H); 1.44 (s, 9H); 1.92 (qunit, J = 5.6 Hz, 2H); 2.62-2.51 (m, 2H); 2.71 (t, J = 6.4 Hz, 2H); 3.25 (t, J = 5.6 Hz, 2H); 3.80 (d, J = 4.8 Hz, 2H); 3.90 (br s, 1H) 4.74 (br d, J = 7.6 Hz, 1H); 5.18-5.12 (m, 2H); 5.83-5.74 (m, 1H); 6.22 (d, J = 2.0 Hz, 1H); 6.28 (dd, J = 8.0 Hz, J = 2.0 Hz, 1H); 6.89 (d, J = 8.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.7, 22.0, 26.5, 27.6, 28.3, 44.3, 48.8, 53.6, 74.8, 79.5, 87.9, 103.5, 107.8, 116.1, 120.3, 129.3, 132.6, 146.1, 149.5, 152.1, 154.9. HRMS (FAB+): calculated for C23H30N2O4 398.2206, measured 398.2218 [M].
1-allyl-1,2,3,4-tetrahydroquinolin-5-yl 5-(tert-butoxycarbonylamino)pent-2-ynoate (44) was eluted with 15% EtOAc/Hex as an amorphous pale yellow solid (95 mg, 64%). 1H NMR (CDCl3, 400 MHz) δ 1.46 (s, 9H); 1.93 (t, J = 6.0 Hz, 2H); 2.62-2.59 (m, 4H); 3.26 (t, J = 6.0 Hz, 2H); 3.34 (quart, J = 6.0 Hz, 2H); 3.88-3.86 (m, 2H); 4.91 (s, 1H); 5.21-5.13 (m, 2H); 5.87-5.78 (m, 1H); 6.33 (dd, J = 8.0 Hz, J = 0.8 Hz, 1H); 6.45 (d, J = 8.0 Hz, 1H); 7.02 (t, J = 8.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 20.6, 21.4, 21.7, 28.5, 38.6, 48.7, 54.3, 73.8, 79.9, 88.8, 108.7, 109.3, 114.4, 116.2, 127.2, 133.1, 146.7, 148.4, 151.9, 155.7. HRMS (FAB+): calculated for C22H24N2O4 384.2049, measured 384.2044 [M].
General Procedure for the Hydroarylation of Aryl-Alkynoates
All reactions were conducted at 0.05 M (concentration of substrate) in 1:1 mixture of DCE to 1,4-dioxane with 5 mol % catalyst. The solid catalyst was weighed into an 8 mL glass vial equipped with a magnetic stir bar. A stock solution of 1:1 DCE and 1,4-dioxane was then added to the vial, followed by a solution of the substrate in 1:1 DCE and 1,4-dioxane. The vial was then sealed under air with a solid Teflon lined screw-cap and heated to the appropriate temperature. The reactions were monitored by TLC. Upon consumption of the starting material, the solvent was removed in vacuo and the residue was chromatographed to afford pure product.
Representative Procedure for the PtCl4 Catalyzed Cyclization of Substrate 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8)
To an 8 mL glass vial was added PtCl4 (7.0 mg, 0.02 mmol) followed by a magnetic stir bar. A stock solution of 1:1 DCE and 1,4-dioxane (5 mL) was then added to the vial, followed by a solution of 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8) (137 mg, 0.4 mmol) in 3 mL 1:1 DCE and 1,4-dioxane. The vial was then sealed under air with a Teflon screw cap and heated to 80 °C. The reaction was monitored by TLC. Upon complete consumption of the starting material the solvent was removed in vacuo and the residue was chromatographed, eluting with 10% EtOAc/DCM, to afford tert-butyl-9-methyl-pyrano[3,2-e]indol-7(3H)-one-1-ethylcarbamate (9) (74 mg, 54%) as an amorphous yellow solid. 1H NMR (MeOD-d4, 400 MHz) δ 1.40 (s, 9H); 2.73 (s, 3H); 3.15 (t, J = 7.6 Hz, 2H); 3.27 (br q, J = 6.0 Hz, 1H); 6.25 (s, 1H); 6.66 (s, 1H); 7.08 (d, J = 8.8 Hz, 1H); 7.35 (s, 1H); 7.61 (d, J = 8.8 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 24.4, 28.2, 30.4, 41.0, 77.5, 110.8, 112.9, 113.0, 113.2, 116.9, 120.8, 127.4, 134.4, 150.3, 154.1, 155.5, 160.0. HRMS (FAB+): calculated for C19H22N2O4 342.1580, measured 342.1582 [M]. The presence of the 2 doublets (7.08 and 7.61 ppm), but no triplet in the aromatic region of the 1H NMR spectrum indicates cyclization to the C-4 position.
Representative Procedure for the Au(PPh3)Cl/AgSbF6 Catalyzed Cyclization of Substrate 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8)
To an 8 mL glass vial was added Au(PPh3)Cl (10 mg, 0.02 mmol) and AgSbF6 (7 mg, 0.02 mmol) followed by a magnetic stir bar. A stock solution of 1:1 DCE and 1,4-dioxane (5 mL) was then added to the vial, followed by a solution of 3-(2-(tert-butoxycarbonylamino)ethyl)-1H-indol-5-yl but-2-ynoate (8) (137 mg, 0.4 mmol) in 3 mL 1:1 DCE and 1,4-dioxane. The vial was then sealed under air with a Teflon screw cap and stirred at room temperature. The reaction was monitored by TLC. Upon complete consumption of the starting material the solvent was removed in vacuo and the residue was chromatographed, eluting with 10% EtOAc/DCM, to afford tert-butyl-9-methyl-pyrano[3,2-e]indol-7(3H)-one-1-ethylcarbamate (9) (129 mg, 93%) as an amorphous yellow solid.
9-methyl-pyrano[3,2-e]indol-7(3H)-one (11) was eluted with 30% EtOAc:Hex as an amorphous white solid (22 mg, 54% with [Pt]) 1H NMR (DMSO-d6, 400 MHz) δ 2.70 (s, 3H); 6.32 (s, 1H); 6.88 (d, J = 2.4 Hz, 1H); 7.15 (d, J = 8.8 Hz, 1H); 7.60 (d, J = 2.8 Hz, 1H); 7.69 (d, J = 8.8 Hz, 1H); 11.70 (s, 1H). HRMS (FAB+): calculated for C12H9NO2 199.0633, measured 199.0643 [M].
tert-butyl-9-phenyl-pyrano[3,2-e]indol-7(3H)-one-1-ethylcarbamate (14) was eluted with 40% EtOAc:Hex as an amorphous yellow solid (79 mg, 98% with [Au]). The resulting solid was dissolved in hot EtOAc, followed by the dropwise addition of hexanes until the solution became slightly turbid. The solution was then allowed to cool undisturbed. This procedure afforded yellow crystals sufficient for X-ray crystallographic analysis. 1H NMR (DMSO-d6, 400 MHz) δ 1.26 (t, J = 7.2 Hz, 2H); 1.32 (s, 9H); 2.64 (quart, J = 6.4 Hz, 2H); 6.28-6.25 (m, 1H, eclipsed by a proton at 6.28 ppm); 6.28 (s, 1H); 7.18 (d, J = 2.0 Hz, 1H); 7.20 (d, J = 8.8 Hz, 1H); 7.48-7.37 (m, 5H); 7.70 (d, J = 8.8 Hz, 1H); 11.45 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 26.9, 28.2, 77.2, 110.3, 110.8, 113.4, 114.4, 117.3, 121.4, 127.0, 127.8, 128.7, 129.3, 134.6, 139.6, 150.7, 155.2, 155.9, 160.2 (one carbon eclipsed by the residual solvent peak). HRMS (FAB+): calculated for C24H24N2O4 404.1736, measured 404.1729 [M]. Melting point: 216.7-218.3 °C. X-ray structure was also obtained, see supporting information.
1-(2-(tert-butoxycarbonylamino)-methyl-propanoate)-9-methyl-pyrano[3,2-e]indol-7(3H)-one (17) was eluted with 15% EtOAc:DCM as an amorphous pale yellow solid (27 mg, 50% with [Pt]; 128 mg, 80% with [Au]). 1H NMR (CDCl3, 400 MHz, 350 K) δ 1.34 (s, 9H); 2.73 (s, 3H); 3.35-3.33 (m, 1H); 3.55-3.52 (m, 1H); 3.59 (s, 3H); 4.52 (s, 1H); 6.27 (s, 1H); 7.17 (d, J = 8.4, 1H); 7.25 (s, 1H); 7.51 (d, J = 8.4 Hz, 1H); 13C NMR (CDCl3, 75 MHz, 350 K) δ 25.1, 28.4, 33.6, 52.2, 55.0, 80.4, 112.1, 113.0, 114.5, 114.6, 116.3, 122.0, 127.2, 135.0, 151.8, 153.2, 155.2, 161.1, 172.6. HRMS (FAB+): calculated for C21H24N2O6 400.1634, measured 400.1622 [M].
tert-butyl 4-methyl-2-oxo-2H-chromen-7-ylcarbamate (19) was eluted with 15% EtOAc:Hex as an amorphous yellow solid (isolated as a 3.5:1 mixture of 19:20; 95 mg, 86% with [Au]). 1H NMR (CDCl3, 500 MHz) δ 1.51 (s, 9H); 2.37 (s, 3H); 6.14 (s, 1H); 7.14 (s, 1H); 7.37 (d, J = 10 Hz, 1H); 7.43 (s, 1H); 7.47 (d, J = 10 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 18.7, 28.4, 81.5, 105.7, 112.8, 114.5, 115.1, 125.3, 142.3, 152.4, 152.6, 154.5, 161.5. HRMS (FAB+): calculated for C15H17NO4 275.1158, measured 275.1160 [M].
tert-butyl 4-methyl-2-oxo-2H-chromen-5-ylcarbamate (20) was eluted with 15% EtOAc:Hex as an amorphous yellow solid (isolated as a 3.5:1 mixture of 19:20; 95 mg, 86% with [Au]). 1H NMR (CDCl3, 500 MHz) δ 1.51 (s, 9H); 2.61 (s, 3H); 6.24 (s, 1H); 6.43 (s, 1H); 7.23 (d, J = 10 Hz, 1H); 7.32 (d, J = 5 Hz, 1H); 7.46 (t, J = 10 Hz, 1H). HRMS (FAB+): calculated for C15H17NO4 275.1158, measured 275.1160 [M].
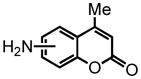
7-amino-4-methyl-2H-chromen-2-one (22) co-eluted with 23, however pure fractions of 22 were obtained via column chromatography eluting with 40% EtOAc:Hex to afford the product as an amorphous yellow solid (isolated as a 1:1 mixture of 22:23; 100 mg, 95% with [Au]). 1H NMR (MeOD-d3:CDCl3, 500 MHz, calibrated to residual MeOH peak, 3.33 ppm) δ 2.36 (s, 3H); 5.96 (s, 1H); 6.53 (s, 1H); 6.62 (d, J = 10 Hz, 1H); 7.38 (d, J = 10 Hz, 1H). 13C NMR (MeOD-d3:CDCl3, 500 MHz, calibrated to residual MeOH peak, 49.00 ppm) δ 18.1, 100.0, 108.4, 110.6, 111.9, 125.7, 151.8, 154.4, 155.3, 163.2. HRMS (FAB+): calculated for C10H10NO2 176.0712, measured 176.0710 [M+1].
5-amino-4-methyl-2H-chromen-2-one (23) eluted as an inseparable mixture with 22 eluting with 40% EtOAc:Hex, affording the product as an amorphous yellow solid (isolated as a 1:1 mixture of 22:23; 100 mg, 95% with [Au]). Given the isolation of pure 22, analysis of the spectrum containing both 22 and 23 allowed for the identification of the peaks corresponding solely to product 23. The shifts of the peaks identified in this manner, those related to 23, are reported here. 1H NMR (MeOD-d3:CDCl3, 500 MHz, calibrated to residual MeOH peak, 3.33 ppm) δ 2.68 (s, 3H); 6.02 (s, 1H); 6.65-6.60 (m, 2H, overlaps with 22 doublet 6.62); 7.23 (t, J = 10 Hz, 1H). 13C NMR (MeOD-d3:CDCl3, 500 MHz, calibrated to residual MeOH peak, 49.00 ppm) δ 24.1, 106.9, 108.7, 113.1, 114.0, 133.1, 148.9, 156.6, 156.8, 163.3. HRMS (FAB+): calculated for C10H10NO2 176.0712, measured 176.0710 [M+1].
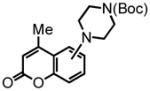
tert-butyl 4-(4-methyl-2-oxo-2H-chromen-7-yl)piperazine-1-carboxylate (25) was eluted with 30% EtOAc:Hex as a white solid (isolated as a 4.8:1 mixture of 25:26 with [Pt] 45 mg, 66%. Isolated as a 5.5:1 mixture of 25:26 with [Au] 55 mg, 80%). 1H NMR (CDCl3, 400 MHz) δ 1.49 (s, 9H); 2.37 (s, 3H); 3.31 (t, J = 4.8 Hz, 4H); 3.60 (t, J = 4.8 Hz, 4H); 6.05 (s, 1H); 6.70 (d, J = 2.4 Hz, 1H); 6.82 (dd, J = 8.8 Hz, J = 2.4 Hz, 1H); 7.45 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 28.5, 47.6, 80.2, 101.6, 111.0, 111.7, 111.9, 125.5, 152.6, 153.4, 154.6, 155.4, 161.7. HRMS (FAB+): calculated for C19H24N2O4 344.1736, measured 344.1743 [M].
tert-butyl 4-(4-methyl-2-oxo-2H-chromen-5-yl)piperazine-1-carboxylate (26) was eluted with 30% EtOAc:Hex as an amorphous white solid (isolated as a 4.8:1 mixture of 25:26 with [Pt] 45 mg, 66%. Isolated as a 5.5:1 mixture of 25:26 with [Au] 55 mg, 80%). 1H NMR (CDCl3, 400 MHz) δ 1.49 (s, 9H); 2.76 (s, 3H); 2.85-2.79 (m, 2H); 3.00 (d, J = 11.2 Hz, 2H); 3.13 (bs, 2H); 4.11 (bs, 2H); 6.19 (d, J = 0.8 Hz, 1H); 7.09 (dd, J = 8.0 Hz, J = 1.2 Hz, 1H); 7.14 (dd, J = 8.4 Hz, J = 0.8 Hz, 1H); 7.46 (t, J = 8.4 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 23.8, 28.5, 54.1, 80.2, 114.3, 116.0, 116.4, 117.6, 131.6, 152.5, 153.6, 154.8, 155.4, 160.4. HRMS (FAB+): calculated for C19H24N2O4 344.1736, measured 344.1743 [M].
2,3,6,7-tetrahydro-9-methyl-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (28) was eluted with 20% EtOAc:Hex as an amorphous yellow solid (45 mg, 88% with [Pt]; 47 mg, 91% with [Au]). 1H NMR (CDCl3, 400 MHz) δ 2.00-1.93 (m, 4H); 2.30 (s, 3H); 2.77 (t, J = 6.0 Hz, 2H); 2.87 (t, J = 6.4 Hz, 2H); 3.27-3.21 (m, 4H); 5.88 (s, 1H); 6.97 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 20.5, 20.7, 21.6, 27.8, 49.5, 50.0, 106.7, 108.1, 108.9, 118.0, 121.7, 145.8, 151.0, 153.1, 162.6. HRMS (FAB+): calculated for C16H17NO2 255.1259, measured 255.1255 [M].
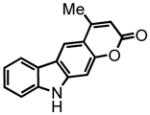
4-methyl-pyrano[2,3-a]carbazol-2(5H)-one (30) was eluted with 50% EtOAc:Hex as an amorphous pale yellow solid (isolated as a 5:1 mixture of 30:31 with [Pt] 69 mg, 69%. Isolated as a 1.7:1 mixture of 30:31 with [Au] 92 mg, 92%). 1H NMR (DMSO-d6, 400 MHz) δ 2.86 (s, 3H); 6.35 (s, 1H); 7.19 (d, J = 8.4 Hz, 1H); 7.24 (t, J = 7.6 Hz, 1H); 7.43 (dt, J = 8.0 Hz, J = 0.8 Hz, 1H); 7.71 (d, J = 8.4 Hz, 1H); 8.16 (d, J = 7.6 Hz, 1H); 8.36 (d, J = 8.4 Hz, 1H); 11.14 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.6, 106.0, 108.4, 112.2, 112.6, 119.6, 119.8, 119.9, 121.6, 124.1, 125.4, 135.0, 140.3, 152.9, 153.0, 159.9. HRMS (FAB+): calculated for C16H12NO2 250.0868, measured 250.0864 [M+1].
4-methyl-pyrano[2,3-b]carbazol-2(10H)-one (31) was eluted with 30% EtOAc:Hex as an amorphous white solid (isolated as a 5:1 mixture of 30:31 with [Pt] 69 mg, 69%. Isolated as a 1.7:1 mixture of 30:31 with [Au] 92 mg, 92%). 1H NMR (DMSO-d6, 400 MHz) δ 2.56 (s, 3H); 6.23 (d, J = 1.2 Hz, 1H); 7.23 (dt, J = 7.6 Hz, J = 0.8 Hz, 1H); 7.38 (s, 1H); 7.43 (dt, J = 8.0 Hz, J = 1.2 Hz, 1H); 7.51 (d, J = 8.0 Hz, 1H); 8.23 (d, J = 8.0 Hz, 1H); 8.55 (s, 1H); 11.63 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) 18.7, 97.2, 110.5, 111.2, 112.5, 117.2, 119.5, 120.3, 120.6, 122.2, 126.3, 140.9, 142.0, 152.1, 154.3, 160.5. HRMS (FAB+): calculated for C16H12NO2 250.0868, measured 250.0864 [M+1].
2,3,6,7-tetrahydro-9-(ethyl-2-tert-butoxycarbonylamino)-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (35) was eluted with 30% EtOAc/Hex as an amorphous pale yellow solid (229 mg, 91% with [Pt]; 73 mg, 95% with [Au]). 1H NMR (CDCl3, 300 MHz) δ 1.44 (s, 9H); 1.97 (quint, J = 6.3 Hz, 4H); 2.78 (t, J = 6.6 Hz, 2H); 2.91-2.83 (m, 4H); 3.25 (quart, J = 6.0 Hz, 4H); 3.43 (q, J = 6.3 Hz, 2H); 4.64 (br s, 1H); 5.89 (s, 1H); 7.04 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.6, 20.8, 21.7, 28.0, 28.5, 32.4, 39.8, 49.6, 50.0, 79.7, 107.0, 107.9, 118.3, 121.6, 146.0, 151.5, 154.0, 155.9, 162.4. HRMS (FAB+): calculated for C22H28N2O4 384.2049, measured 384.2048 [M].
2,3,6,7-tetrahydro-9-(propyl-2-tert-butoxycarbonylamino)-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (37) was eluted with 20% EtOAc/Hex as an amorphous pale yellow solid (70 mg, 88% with [Pt]; 72 mg, 90% with [Au]). 1H NMR (CDCl3, 400 MHz) δ 1.15 (d, J = 6.4 Hz, 3H); 1.44 (s, 9H); 1.98-1.96 (br m, 4H); 2.58-2.53 (m, 1H); 2.80 (t, J = 6.0 Hz, 2H); 2.88 (t, J = 6.4 Hz, 2H); 3.03 (br s, 1H); 3.28-3.23 (br m, 4H); 4.01-3.97 (m, 1H); 4.47 (br s, 1H); 5.87 (s, 1H); 7.27 (s, 1H, overlaps with residual solvent peak); 13C NMR (CDCl3, 75 MHz) δ. 20.6, 20.8, 21.7, 27.9, 28.5, 40.0, 46.2, 49.6, 50.1, 79.5, 106.9, 108.3, 108.8, 118.3, 122.2, 146.0, 151.5, 153.5, 155.2, 162.5. HRMS (FAB+): calculated for C23H30N2O4 398.2206, measured 398.2193 [M].
2,3,6,7-tetrahydro-9-(methyl-tert-butoxycarbonylamino)-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (39) was eluted with 30% EtOAc/Hex as an amorphous pale yellow solid (65 mg, 88% with [Pt]; 67 mg, 90% with [Au]). 1H NMR (CDCl3, 300 MHz) δ 1.47 (s, 9H); 1.96 (quint, J = 6.3 Hz, 4H); 2.75 (t, J = 6.6 Hz, 2H); 2.87 (t, J = 6.6 Hz, 2H); 3.25 (quart, J = 5.7 Hz, 4H); 4.40 (d, J = 6.0 Hz, 2H); 4.84 (br s, 1H); 6.01 (s, 1H); 6.98 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.6, 20.8, 21.7, 27.9, 28.5, 41.0, 49.7, 50.1, 80.4, 105.7, 106.8, 107.1, 118.3, 120.9, 146.1, 151.4, 152.9, 155.7, 162.6. HRMS (FAB+): calculated for C21H26N2O4 370.1893, measured 370.1879 [M]
6,7,8,9-tetrahydro-2-oxo-4-(ethyl-tert-butoxycarbonylamino)-9-(2-propen-1-yl)-2H-pyrano[3,2-g]quinoline (41) was eluted with 30% EtOAc/Hex as an amorphous pale yellow solid (290 mg, 76% with [Pt]; 72 mg, 93% with [Au]). 1H NMR (CDCl3, 400 MHz) δ 1.45 (s, 9H); 1.98 (quint, J = 6.0 Hz, 2H); 2.80 (t, J = 6.0 Hz, 2H); 2.87 (t, J = 6.8 Hz, 2H); 3.38 (t, J = 6.8 Hz, 2H); 3.46-3.42 (m, 2H); 3.91 (dd, J = 2.8 Hz, J = 2.0 Hz, 2H); 4.66 (br s, 1H) 5.21-5.14 (m, 2H); 5.84-5.76 (m, 1H); 5.90 (s, 1H); 6.40 (s, 1H); 7.16 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 22.0, 28.0, 28.5, 32.4, 39.7, 49.2, 53.8, 79.7, 97.4, 108.2, 108.5, 116.7, 119.7, 123.8, 131.5, 148.6, 153.7, 155.2, 155.9, 162.2. HRMS (FAB+): calculated for C22H28N2O4 384.2049, measured 384.2046 [M].
6,7,8,9-tetrahydro-2-oxo-4-(propyl-2-tert-butoxycarbonylamino)-9-(2-propen-1-yl)-2H-pyrano[3,2-g]quinoline (43) was eluted with 20% EtOAc/Hex as an amorphous pale yellow solid (60 mg, 75% with [Pt]; 73 mg, 92% with [Au]). 1H NMR (CDCl3, 400 MHz) δ 1.13 (d, J = 6.8 Hz, 3H); 1.40 (s, 9H); 1.94 (quint, J = 5.6 Hz, 2H); 2.55-2.50 (m, 1H); 2.79 (t, J = 6.0 Hz, 2H); 3.03-3.01 (m, 1H); 3.34 (t, J = 6.0 Hz, 2H); 3.86 (d, J = 4.4 Hz, 2H) 3.96 (quint, J = 6.8 Hz, 1H); 4.61 (br s, 1H); 5.17-5.10 (m, 2H); 5.81-5.72 (m, 1H); 5.85 (s, 1H); 6.34 (s, 1H); 7.36 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 20.4, 21.9, 27.9, 28.4, 39.9, 46.0, 49.2, 53.7, 79.4, 97.1, 108.4, 109.2, 116.5, 119.6, 124.4, 131.5, 148.4, 153.3, 155.0, 155.2, 162.2. HRMS (FAB+): calculated for C23H30N2O4 398.2206, measured 398.2215 [M].
7,8,9,10-tetrahydro-2-oxo-4-(ethyl-tert-butoxycarbonylamino)-7-(2-propen-1-yl)-2H-pyrano[3,2-f]quinoline (45) was eluted with 20% EtOAc/Hex as an amorphous pale yellow solid (71 mg, 74% with [Pt]; 86 mg, 90% with [Au]). 1H NMR (CDCl3, 400 MHz) δ 1.44 (s, 9H); 2.05-1.95 (m, 2H); 2.92-2.87 (m, 4H); 3.42-3.34 (m, 4H); 3.97-3.95 (m, 2H); 4.85 (s, 1H); 5.19-5.14 (m, 2H); 5.87-5.78 (m, 1H); 5.92 (s, 1H); 6.52 (d, J = 9.2 Hz, 1H); 7.36 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 20.6, 21.0, 28.4, 32.5, 39.9, 49.0, 53.9, 79.6, 108.0, 108.3, 108.5, 116.4, 123.0, 132.2, 148.4, 152.5, 154.2, 155.9, 162.3. HRMS (FAB+): calculated for C22H24N2O4 384.2049, measured 384.2041 [M].
7,8,9,10-tetrahydro-2-oxo-4-(ethyl-tert-butoxycarbonylamino)-2H-pyrano[3,2-f]quinoline (PV139)
To an oven dried vial was added 7,8,9,10-tetrahydro-2-oxo-4-(ethyl-tert-butoxycarbonylamino)-7-(2-propen-1-yl)-2H-pyrano[3,2-f]quinoline (45) (306 mg, 0.79 mmol) followed by THF (18 mL). To the resulting solution was added N,N′-dimethylbarbituric acid (370 mg, 2.37 mmol) and Pd(PPh3)4 (92 mg, 0.079 mmol, 10 mol%). The mixture was then sealed with a Teflon screw-cap under an atmosphere of argon and heated to 80 °C for 12 hr. Upon consumption of the starting material, the reaction mixture was concentrated and the resulting residue was purified by flash column chromatography (eluting with 30% EtOAc/Hex) to afford 7,8,9,10-tetrahydro-2-oxo-4-(ethyl-tert-butoxycarbonylamino)-2H-pyrano[3,2-f]quinoline (250 mg, 0.73 mmol) as an amorphous yellow solid, 92%. 1H NMR (CDCl3, 400 MHz) δ 1.44 (s, 9H); 1.95-1.92 (m, 2H); 2.87-2.85 (m, 4H); 3.41-3.33 (m, 4H); 4.64 (br s, 1H); 4.87 (br s, 1H); 5.92 (s, 1H); 6.39 (d, J = 8.4 Hz, 1H); 7.28 (d, J = 9.2 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 20.0, 20.7, 28.4, 32.6, 39.9, 41.2, 79.6, 107.3, 108.2, 109.1, 111.0, 122.8, 148.5, 153.2, 154.4, 155.9, 162.2. This material (222 mg, 0.65 mmol) in DCM (3.75 mL) was added TFA (1.20 mL) slowly at 0 °C. The reaction stirred for 15 min at 0 °C at which time the starting material had been fully consumed. The solvent was removed in vacuo and the residue was purified via HPLC eluting with MeCN:H2O (the water contained 1% TFA). The fractions containing the product were pooled and evaporated. The residue was then lyophilized to afford 7,8,9,10-tetrahydro-2-oxo-4-(2-aminoethyl)-2H-pyrano[3,2-f]quinoline (PV139 TFA salt) as an amorphous yellow solid (130 mg, 0.36 mmol, 55%). 1H NMR (CD3OD, 400 MHz) δ 1.90 (quint, J = 8.4 Hz, 2H); 2.79 (t, J = 8.4 Hz, 2H); 3.06 (t, J = 9.6 Hz, 2H); 3.32-3.22 (m, 4H); 5.93 (s, 1H); 6.52 (d, J = 12.0 Hz, 1H); 7.31 (d, J = 11.6 Hz, 1H); 13C NMR (CD3OD, 75 MHz) δ 20.0, 20.5, 29.5, 38.6, 40.7, 106.4, 106.8, 107.9, 111.6, 122.7, 150.2, 153.4, 153.5, 163.3. 19F-NMR (CDCl3, 300 MHz) δ −76.51. HRMS (FAB+): calculated for C14H17N2O2 245.1290, measured 245.1283 [M – trifluoroacetate].
9-(2-aminoethyl)-2,3,6,7-tetrahydro-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (FFN511 TFA salt)
To a solution of 2,3,6,7-tetrahydro-9-(ethyl-2-tert-butoxycarbonylamino)-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (35) (177 mg, 0.46 mmol) in DCM (6 mL) was added TFA (2 mL) slowly at 0 °C. The reaction stirred for 10 min at 0 °C at which time the starting material had been fully consumed. The solvent was removed in vacuo and the residue was quenched with satd. NaHCO3 (aq.) (~20 mL). The resulting solution was extracted with 10% MeOH/CHCl3 (6 × 10 mL). The organic layers were combined and dried over MgSO4. The solvent was removed in vacuo and the resulting residue was purified via HPLC eluting with MeCN:H2O (the water contained 1% TFA). The fractions containing the product were pooled and evaporated. The residue was then lyophilized to afford 9-(2-aminoethyl)-2,3,6,7-tetrahydro-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (FFN511 TFA salt) as an amorphous yellow solid (151 mg, 0.38 mmol, 82%). 1H NMR (CD3OD, 300 MHz) δ 1.99-1.95 (m, 4H); 2.82 (apparent quart, J = 6.6 Hz, 4H); 3.06 (t, J = 6.5 Hz, 2H); 3.25 (t, J = 7.8 Hz, 2H); 3.33 (eclipsed by solvent, 4H); 5.93 (s, 1H); 7.16 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.6, 20.7, 21.7, 27.8, 36.0, 41.4, 49.6, 50.0, 107.1, 107.8, 108.0, 118.1, 121.5, 145.9, 151.5, 154.4, 162.5. 19F NMR (CDCl3, 300 MHz) δ −76.38. HRMS (FAB+): calculated for C17H21N2O2 285.1603, measured 285.1601 [M – trifluoroacetate].
9-(2-aminoethyl)-2,3,6,7-tetrahydro-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (FFN511)
A solution of FFN511 TFA salt (110 mg, 0.27 mmol) in H2O (8 mL) was prepared; slight warming was required to fully dissolve all solids. To the resulting solution was added satd. NaHCO3 (aq.) (~10 mL). The solution was extracted with 10% MeOH/CHCl3 (10 × 10 mL). The organic layers were combined and dried over MgSO4. The solvent volume was reduced on a rotary evaporator at which point the product began to crystallize. The remaining solution was allowed to evaporate slowly to afford product 9-(2-aminoethyl)-2,3,6,7-tetrahydro-1H,5H,11H-[1]benzopyrano[6,7,8-ij]quinolizin-11-one (FFN511) (74 mg, 0.26 mmol) yellow crystals, 96%. 1H NMR (CDCl3, 300 MHz) δ 1.38 (br s, 2H); 1.97 (quint, J = 6.3 Hz, 4H); 2.79 (quart, J = 6.6 Hz, 4H); 2.89 (t, J = 6.6 Hz, 2H); 3.04 (t, J = 6.6 Hz, 2H); 3.25 (quart, J = 6.3 Hz, 4H); 5.92 (s, 1H); 7.02 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 20.6, 20.7, 21.5, 27.8, 35.9, 41.4, 49.5, 49.9, 106.9, 107.6, 107.9, 118.1, 121.5, 145.8, 151.4, 154.4, 162.4. HRMS (FAB+): calculated for C17H21N2O2 285.1603, measured 285.1601 [M+1]. Melting point: 160.9–162.1 °C.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Mental Health (MH086545) and the G. Harold & Leila Y. Mathers Charitable Foundation. We would also like to thank the Parkin research group for X-ray crystal analysis. The National Science Foundation (CHE-0619638) is thanked for acquisition of an X-ray diffractometer.
Footnotes
Supporting Information Available: NMR spectra for all substrates and products, and X-ray data for compound 14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; (b) Kakiuchi F, Chatani N. Adv Synth Catal. 2003;345:1077–1101. [Google Scholar]
- 2.Kitamura T. Eur J Org Chem. 2009:1111–1125. [Google Scholar]
- 3.(a) Pastine SJ, Youn SW, Sames D. Org Lett. 2003;5:1055–1058. doi: 10.1021/ol034177k. [DOI] [PubMed] [Google Scholar]; (b) Pastine SJ, Youn SW, Sames D. Tetrahedron. 2003;59:8859–8868. [Google Scholar]; (c) Pastine SJ, Sames D. Org Lett. 2003;5:4053–4055. doi: 10.1021/ol035419j. [DOI] [PubMed] [Google Scholar]
- 4.Youn SW, Pastine SJ, Sames D. Org Lett. 2004;6:581–584. doi: 10.1021/ol036385i. [DOI] [PubMed] [Google Scholar]
- 5.Milder conditions for the von Pechmann condensation using Lewis acid promoters has been demonstrated with hydroxyjulolidine. Wirtz L, Kazmaier U. Eur J Org Chem. 2011:7062.
- 6.Chen G, Yee DJ, Gubernator NG, Sames D. J Am Chem Soc. 2005;127:4544–4545. doi: 10.1021/ja0428457. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wise RA. Nature Rev Neurosci. 2004;5:1–12. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]; (b) Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- 8.Verhoeff NPLG. Neuropsychopharm. 1999;147:217–249. [Google Scholar]
- 9.Gubernator NG, Zhang H, Staal RGW, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, Sulzer D, Sames D. Science. 2009;324:1441–1444. doi: 10.1126/science.1172278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schimitschek EJ, Trias JA, Hammond RA, Atkins H, Atkins RL. Optics Commun. 1976;16:313. [Google Scholar]
- 11.(a) Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992–1995. doi: 10.1126/science.287.5460.1992. [DOI] [PubMed] [Google Scholar]; (b) Jia C, Lu W, Oyamada J, Kitamura T, Matsuda K, Irie M, Fujiwara Y. J Am Chem Soc. 2000;122:7252–7263. [Google Scholar]; (c) Jia C, Piao D, Kitamura T, Fujiwara Y. J Org Chem. 2000;65:7516–7522. doi: 10.1021/jo000861q. [DOI] [PubMed] [Google Scholar]; (d) Kitamura T, Otsubo K. J Org Chem. 2012;77:2978–2982. doi: 10.1021/jo300021a. [DOI] [PubMed] [Google Scholar]
- 12.Martín-Matute B, Nevado C, Cárdenas DJ, Echavarren AM. J Am Chem Soc. 2003;125:5757–5766. doi: 10.1021/ja029125p. [DOI] [PubMed] [Google Scholar]
- 13.(a) Fürstner A, Mamane V. J Org Chem. 2002;67:6261–6267. doi: 10.1021/jo025962y. [DOI] [PubMed] [Google Scholar]; (b) Mamane V, Hannen P, Fürstner A. Chem Eur J. 2004;10:4556–4575. doi: 10.1002/chem.200400220. [DOI] [PubMed] [Google Scholar]
- 14.Reetz MT, Sommer K. Eur J Org Chem. 2003:3485–3496. [Google Scholar]
- 15.(a) Fürstner A. Chem Soc Rev. 2009;38:3208–3221. doi: 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Davies PW. Angew Chem Int Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ferrer C, Echavarren AM. Angew Chem, Int Ed. 2006;45:1105–1109. doi: 10.1002/anie.200503484. [DOI] [PubMed] [Google Scholar]; (b) Nevado C, Echavarren AM. Chem Eur J. 2005;11:3155–3164. doi: 10.1002/chem.200401069. [DOI] [PubMed] [Google Scholar]
- 17.Gorin DJ, Toste FD. Nature. 2007;446:395–403. doi: 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- 18.Shi Z, He C. J Org Chem. 2004;69:3669–3671. doi: 10.1021/jo0497353. [DOI] [PubMed] [Google Scholar]
- 19.Dang TT, Boeck F, Hintermann L. J Org Chem. 2011;76:9353–9361. doi: 10.1021/jo201631x. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, McGowan EB, Detwiler TC. J Med Chem. 1992;35:2048–2054. doi: 10.1021/jm00089a015. [DOI] [PubMed] [Google Scholar]
- 21.Beaulieu PL, Bös M, Bousquet Y, DeRoy P, Fazal G, Gauthier J, Gillard J, Goulet S, McKercr G, Poupart MA, Valois S, Kukolj G. Bioorg Med Chem Lett. 2004;14:967–971. doi: 10.1016/j.bmcl.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 22.Becker MH, Chua P, Downham R, Douglas CJ, Garg NK, Hiebert S, Jaroch S, Matsuoka RT, Middleton JA, Ng FW, Overman LE. J Am Chem Soc. 2007;129:11987–12002. doi: 10.1021/ja074300t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.