

Abstract
Recent clinical trials using antibodies with low toxicity and high efficiency have raised expectations for the development of next-generation protein therapeutics. However, the process of obtaining therapeutic antibodies remains time consuming and empirical. This review summarizes recent progresses in the field of computer-aided antibody development mainly focusing on antibody modeling, which is divided essentially into two parts: (i) modeling the antigen-binding site, also called the complementarity determining regions (CDRs), and (ii) predicting the relative orientations of the variable heavy (VH) and light (VL) chains. Among the six CDR loops, the greatest challenge is predicting the conformation of CDR-H3, which is the most important in antigen recognition. Further computational methods could be used in drug development based on crystal structures or homology models, including antibody–antigen dockings and energy calculations with approximate potential functions. These methods should guide experimental studies to improve the affinities and physicochemical properties of antibodies. Finally, several successful examples of in silico structure-based antibody designs are reviewed. We also briefly review structure-based antigen or immunogen design, with application to rational vaccine development.
Keywords: antibody design, antibody engineering, protein therapeutics, vaccine design
Introduction
Computational methods are almost universally accepted as important tools for the invention of small molecule drugs, helping with tasks such as optimizing affinity for a target, minimizing off-target effects and optimizing pharmacokinetic properties. In these contexts, the computational methods are generally not considered substitutes for empirical testing, but rather a way to generate testable hypotheses, helping to interpret and guide experiments. The situation with antibody therapeutics is strikingly different in this regard. While modeling has in fact contributed to the design of therapeutic antibodies, notably in early attempts at humanizing antibodies, overall the potential impact of computational methods is not as well defined, and the tools not as well developed and less broadly employed, than in small molecule drug discovery. Here we review progress in the development of computational methods that may ultimately be routinely used in antibody drug discovery.
Because we encounter a large variety of foreign molecules in daily life, ‘diversity’ is a key concept in the adaptive immune system in which antibodies take a major role. The sequences, structures and functions of antibodies have been extensively studied due to their growing importance as therapeutics (Carter, 2006; Reichert, 2008; Nelson and Reichert, 2009) and research tools (Nogi et al., 2008; Hattori et al., 2010). At the sequence level, antibody diversity is generated by (i) recombination of V(J)D gene segments (Tonegawa, 1983) and (ii) somatic mutations during antibody maturation, which leads to higher affinity (Besmer et al., 2004; Neuberger, 2008). At a structural level, antibody diversity is manifested primarily in the antigen-binding sites, comprised of six loops, and by the relative orientations between the light chain and heavy chain variable domains (VL and VH).
Our growing understanding of sequence–structure relationships in antibodies, and advances in computational protein modeling, has enabled progress toward computational methods that can assist in re-designing antibodies for higher affinity or other desired modifications (Rosenberg and Goldblum, 2006; Lippow and Tidor, 2007; Karanicolas and Kuhlman, 2009). In Fig. 1, we summarize several ways in which computational methods can be deployed in the context of antibody design. One central goal is to accurately predict the structures of antibodies from their sequences, a special case of the comparative protein modeling problem, which is valuable due to the challenges and expenses associated with experimental structure determination. In cases where the binding interaction between antibody and antigen is unknown, protein–protein docking methods can be used to predict the complex structure, although this remains challenging, especially when using homology-modeled structures for either the antibody or the antigen (Gray, 2006). Finally, using experimentally determined or predicted structures of the antibody–antigen complex, computational methods can be used to predict mutations that may improve binding affinity, specificity or other properties such as solubility.
Fig. 1.

Flow of computational antibody designs.
In addition to antibody designs, designing better antigens or immunogens is also expected to elicit neutralizing antibodies (NAbs) for viruses, such as HIV and influenza. Antigens could work as vaccines only if they elicit appropriate antibodies without showing viral activity. In traditional vaccine approaches, antigens are empirically identified and are usually isolated, inactivated or attenuated, and then injected so that they cannot cause undesirable infections, but can induce desired antibody response (Rinaudo et al., 2009). Although the principle has not changed dramatically, the accumulation of experimental data and complex structures of NAbs–antigens provides us with a better understanding of the structural basis of antigen recognition, and a structure-based rational vaccine design is now becoming possible.
In this review, we discuss successes and challenges of applying computational methods in each of these areas, focusing on areas that we view as particularly critical to achieving routine, successful use of computational methods in antibody design, especially complementarity determining region (CDR)-H3 modeling and predicting the effects of somatic or designed mutations during antibody optimization. Recent successes of structure-based antigen design based on protein scaffolds are also described. Other topics, such as canonical structures (Al-Lazikani et al., 1997; Morea et al., 2000; Vargas-Madrazo and Paz-Garcia, 2002; Chailyan et al., 2011a), especially of CDR-L3 (Kuroda et al., 2009); the antibody numbering schemes (Honegger and Pluckthun, 2001; Abhinandan and Martin, 2008); B-cell epitope predictions (Ponomarenko and Bourne, 2007; Evans, 2008); humanization (Almagro and Fransson, 2008); and details of protein–protein docking (Gray, 2006) are reviewed elsewhere.
Antibody CDR modeling
A core challenge for computational antibody engineering is predicting the structure of the antibody from sequence. This is of course a specific case of the more general protein structure prediction problem (Baker and Sali, 2001; Zhang, 2008), but antibodies present both special advantages and special challenges relative to other proteins. Before discussing methods for predicting antibody structures, we first briefly review their structural organization.
An antibody consists of two types of chains, the light and heavy chains, each of which is composed of multiple domains, all of which assume the common immunoglobulin (Ig)-fold (Murzin et al., 1995; Chothia et al., 1998). The antigen-binding site is located in the ‘variable’ domains of each chain, VL and VH. The antigen-binding sites are called CDRs and are composed primarily of six ‘hypervariable’ loops, three on the light chain (L1, L2 and L3) and three on the heavy chain (H1, H2 and H3). The remainder of the variable domains is structurally well conserved at the backbone level, and aligning antibody sequences is straightforward. For these reasons, a primary focus of antibody modeling is predicting the conformations of the CDR loops from their sequences (Whitelegg and Rees, 2004).
In the 1980s, antibody modeling started with the unexpected discovery that most of the CDR loops (all but H3) adopt a limited number of conformations called canonical structures (Chothia and Lesk, 1987; Chothia et al., 1989; Tramontano et al., 1989, 1990; Tramontano and Lesk, 1992). The most probable canonical structures can be predicted based on the length of the loop and the identities of key residues within or outside the CDR regions. This knowledge has been used in humanization procedures (Riechmann et al., 1988) where key residues should be maintained when non-human CDRs are grafted onto human frameworks; otherwise, the conformation of the antigen-binding sites could change resulting in reduced antibody–antigen-binding affinity (Morea et al., 2000; Almagro and Fransson, 2008). Canonical structures can be defined only when the same sequence motifs discovered from the Protein Data Bank (PDB) appear in the large sequence database (Kuroda et al., 2009). Recent increases in the number of antibody crystal structures have made it possible to define more canonical structures (Kuroda et al., 2009; Teplyakov et al., 2010,b; Chailyan et al., 2011a). Definitions of canonical structures are available in the literatures (Al-Lazikani et al., 1997; Morea et al., 2000). A new classification scheme for CDRs has also been proposed based on affinity propagation clustering with a large number of crystal structures (North et al., 2011).
Adopting these empirically determined sequence–structure relationships has made it possible to model the L1, L2, L3, H1 and H2 loops with high accuracy [backbone root mean square deviation (RMSD) between native structures and templates are usually <1.0 Å] and subsequent work has focused mainly on predicting CDR-H3 structures (Shirai et al., 1996; Morea et al., 1997, 1998; Oliva et al., 1998). CDR-H3 loops are located in the center of the antigen-binding site, play an important role in antigen recognitions and gain more mutations during affinity maturation than other portions of antibodies (Clark et al., 2006b). They assume diverse conformations, limiting the ability to derive simple rules for their sequence–structure relationships (Fig. 2A). Structural changes in CDR-H3 also occur upon antigen binding (Fig. 2B), which makes the problem more complex. These challenges have led to many experimental and computational studies to elucidate sequence–structure–function relationships of CDR-H3 (Zemlin et al., 2003; Brooks et al., 2010; Julien et al., 2010; Ofek et al., 2010a,b).
Fig. 2.
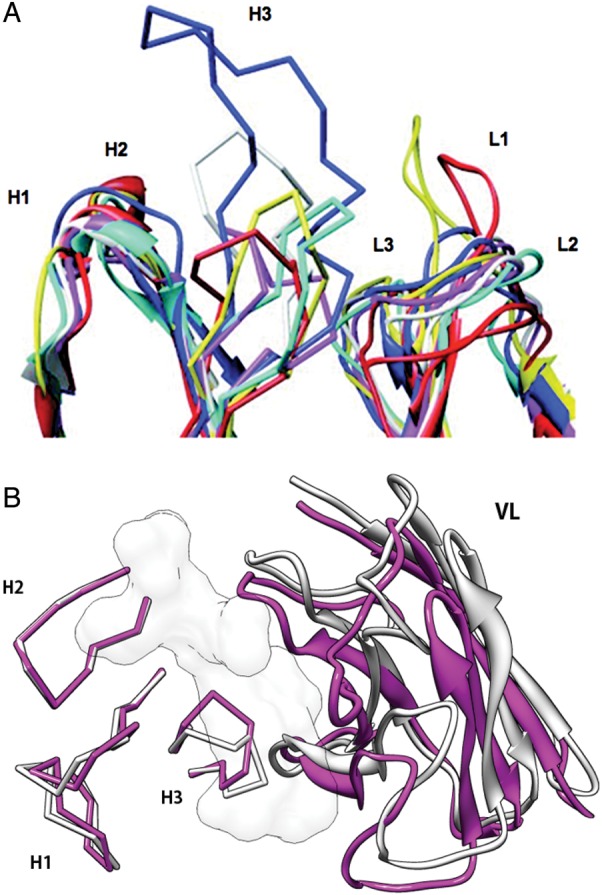
Structural diversity of antibodies. (A) Variability of six CDRs. (B) Structural changes of CDR-H3 [root mean square deviation (RMSD) of backbone N, Cα, C and O atoms is 2.3 Å] and rearrangement of VL/VH domain orientation. Anti-HIV-1 peptide antibody, Fab50.1 (magenta (1GGI) and white (1GGC) for antigen bound and free structures, respectively). The white surface model is the antigen, HIV-1 V3 loop peptide. All graphics of protein structures in this article are generated using UCSF Chimera (http://www.cgl.ucsf.edu/chimera).
Although their overall conformations are diverse, the conformations of the C-terminal base regions of CDR-H3 are limited and assume ‘kinked’ or ‘extended’ forms. Applying sequence–structure rules for this portion of the loop simplifies the problem by reducing the computational search space for the loop modeling. Shirai et al. (1996, 1999) first proposed H3-rules, which identified the base form and β-hairpin characteristics from the sequences. Recent advances in structural genomics have increased our knowledge of sequence–structure relationships in CDR-H3, leading to refinements of the H3-rules (Koliasnikov et al., 2006; Kuroda et al., 2008).
In the absence of simple sequence–structure relationships for the remainder of CDR-H3, the conformations can be predicted by exploiting the great many methods that have been developed for the more general problem of loop prediction. These methods generally work by first enumerating large numbers of plausible loop conformations, and then predicting which of these is likely to be correct by using conformational energy or a scoring function derived from the database of crystal structures (Soto et al., 2008).
One approach to enumerating plausible loop conformations for CDR-H3, and other loops for which no simple sequence–structure relationships exist, is to mine the known loop structures in the PDB (Jones and Thirup, 1986). For example, Levy and coworkers surveyed the coverage of short (7-, 9- and 15-residue) fragments in the PDB against fragments derived from a novel protein, and showed that most of them are covered with reasonable accuracy (RMSD ≤ 2.0 Å) (Du et al., 2003). Some other studies have also suggested that existing loop conformations in the PDB are sufficient to model most loops (Fernandez-Fuentes and Fiser, 2006; Choi and Deane, 2010).
With respect to CDR-H3 specifically, Kuroda et al. developed a knowledge-based sampling method using fragments derived from both CDR-H3 loops in antibodies and loops in non-homologous proteins (Kuroda et al., submitted for publication). They have also demonstrated that the usefulness of the database approach is enhanced by energy-based refinement. Similarly, Gray and coworkers proposed antibody-modeling software, RosettaAntibody, based on their Rosetta design suite (Rohl et al., 2004; Sircar et al., 2009). They modeled CDR-H3 loops using fragment assembly and the cyclic coordinate descent approach and minimized them using the Rosetta protocol (Sivasubramanian et al., 2009). They also showed that antibody models constructed by their protocols could be used in subsequent computational docking procedures using RosettaDock (Gray et al., 2003; Wang et al., 2007), although they noted a number of challenges mainly associated with CDR-H3 modeling.
A distinct approach to enumerating possible loop conformations is the so-called ‘ab initio’ approach, which uses generic ‘Ramachandran’ preferences to generate possible backbone conformations, and not specific loop conformations from the PDB. In one of the earliest antibody modeling methods, Martin et al. combined database searching and ab initio loop prediction using the CONGEN program (Bruccoleri et al., 1988; Martin et al., 1989). A neural network machine learning method was also used to tackle the H3 modeling problem (Reczko et al., 1995).
More recently, Jacobson and coworkers have developed an ab initio loop modeling protocol, which searches conformational space by backbone torsion-angle sampling with subsequent energy-based refinement and scoring based on the all-atom optimized potentials for liquid simulations force field and an implicit solvent model (Jacobson et al., 2004; Sellers et al., 2008). This method has been shown to perform well for loop modeling in generic proteins (Rossi et al., 2007, 2009) and the protocol can be applied to CDR-H3 modeling with accuracies high enough that the models could be used as surrogates for experimental structures in many applications (Sellers et al., 2010).
In general, loop prediction becomes more difficult as the loops become longer, especially for those longer than ∼12 amino acids (Zhu et al., 2006), which includes a significant fraction of CDR-H3 loops, which have a median length of 12 residues (Zemlin et al., 2003). This is largely because the sampling problem rapidly becomes more difficult with increasing loop length. However, it should also be kept in mind that long surface loops in proteins are often intrinsically flexible, such that the observed conformation can vary depending on its interactions with antigen or due to ‘artifacts’ associated with crystal packing (Rapp and Pollack, 2005). These considerations should be born in mind when evaluating or applying loop prediction methods to CDR-H3.
Theoretically, the most reliable approach should be to identify the loop conformations with the lowest free energy values at room temperature. For that purpose, Shirai et al. (1998) and Kim et al. (1999) performed the multicanonical molecular dynamics simulations (Nakajima et al., 1997; Higo et al., 2012) to probe the free energy landscapes, and they obtained the stable structural ensembles of the CDR-H3 loops at 300 K, which were found to include the corresponding crystal structures. In addition, characterizing the free energy landscapes should aid in predicting structural changes of CDR-H3 upon antigen binding based on the ‘population shift’ principle that suggests that bound conformations should be relatively close in free energy to the unbound state (Watanabe et al., 2006; Okazaki and Takada, 2008). Higo et al. (2011) recently observed both the population shift and the induced fit mechanisms during the coupled folding and binding in an intrinsically disordered protein. However, although conformational sampling methods have matured, the underlying energy models—generally all-atom force fields—remain imperfect, limiting the accuracy of the results. We expect that, in the future, with the further refinement of force field parameters, methods based on generating structural ensembles will be routinely used to accurately predict CDR-H3 loop conformations with and without antigens.
Predicting VL/VH domain orientations
Another challenge in antibody modeling is predicting the VL/VH domain orientations. The orientations of these two domains vary among different antibodies, and can also change upon antigen binding as seen in early work by Stanfield et al. (1993; Fig. 2B). When modeling antibody structures from sequence, the relative orientations of the domains will affect the structure of the antigen-binding surface. Thus, understanding sequence–structure relationships as well as the energetics of VL/VH domains interface is crucial not only to elucidate the structural diversity of antibodies, but also to accurately model antibody structures.
A three-layered structure was proposed for the VL/VH interface based on analyzing antibody crystal structures and sequences (Chothia et al., 1985; Vargas-Madrazo and Paz-Garcia, 2003). The key residues in these layers are also important for determining the conformations of CDR-H3 (Kuroda et al., 2008). Therefore it is apparent that the accuracy of domain orientation prediction can affect the accuracy of CDR-H3 modeling and vice versa, which makes sense because CDR-H3 is located close to the interface with the VL domain. In one striking example, a crystal structure of a ‘chimeric’ antibody, which had a different VL domain from the original antibody, was solved, and a structural change of CDR-H3 was observed upon the VL domain substitution (RMSD 2.88 Å) whereas the canonical structures of the parental antibodies are maintained (Fig. 3A; Pei et al., 1997). In another example, however, the VH domain of an antibody accommodated a different VL domain from another antibody without any significant structural changes of CDR-H3 (RMSD 0.54 Å) or the other portions of the VH domain (Fig. 3B; Teplyakov et al., 2010a). These studies, in which artificial VH/VH domain pairs form stable complexes, shed light on the possibility of multi-specific antibodies (Bostrom et al., 2009), which recognize two distinct antigens with non-overlapping functional paratopes. That is, variable domains of light and heavy chains, which recognize an antigen, respectively, independently of the other chain, can be combined, leading to a multi-specific antibody. Considering the large number of VL and VH sequences in nature, computer-guided approaches to explore the potential pairing of these two domains and the domain-wise contributions to antigen binding, which would be difficult to examine by experiments, are expected.
Fig. 3.
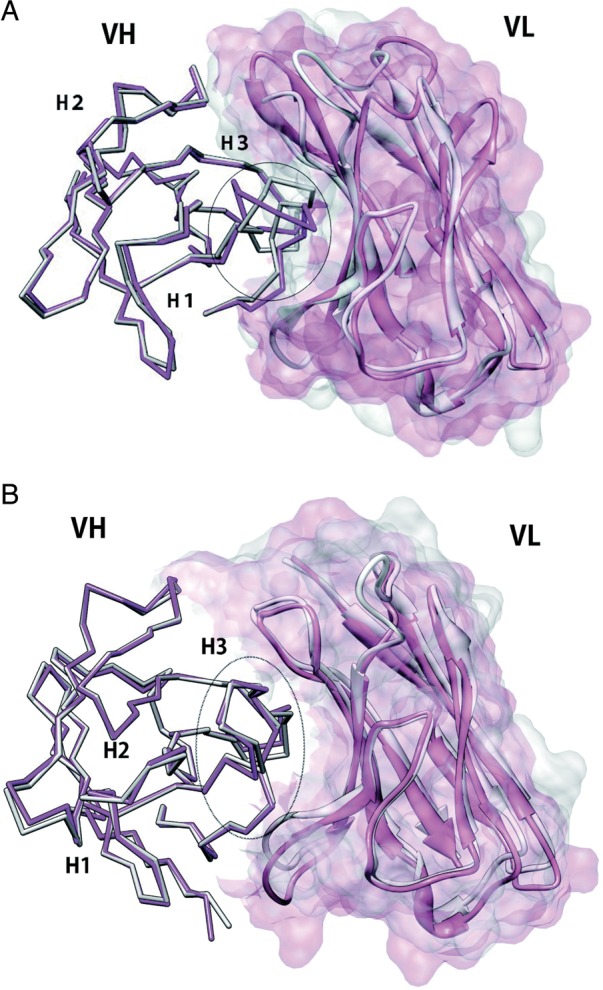
Effect of VL domain substitution on CDR-H3 conformation. (A) B1–8 (1A6V; magenta (VH) and pink (VL)) and B1–8/NQ11 (1NQB; gray (VH) and white (VL)). The VL domain of B1–8 (pink) was replaced by that of another antibody, NQ11, which results in a ‘chimeric’ antibody termed B1–8/NQ11. Structural change of CDR-H3 is observed (RMSD 2.9 Å). (B) C836 (3L7E; magenta (VH) and pink (VL)) and X836 (3MBX; gray (VH) and white (VL)). The VL domain of C836 (pink) was replaced by that of another antibody, which results in a ‘chimeric’ antibody termed X836. Structural change of CDR-H3 is not observed (RMSD 0.5 Å).
Exploring the possibility of the prediction of correct domain orientations based on energetics, Jacobson and coworkers examined the variations of VL/VH domain orientations in crystal structures and how they relate to sequence (Narayanan et al., 2009). In this study, the difference in domain orientation was defined as the non-CDR Cα RMSD of the VL domain when structural superposition is performed using the VH domain. They also calculated molecular mechanics energies between the two domains, showing that the native VL/VH orientations correspond to those of their energy minima in conformational space.
In another study, Abhinandan and Marin defined the VL/VH packing angles as a torsion angle at the interface using four pseudo-coordinates derived from a set of structurally conserved residues, and determined the distribution of the packing angle, which varies from −31.0 to −60.8° following approximately a normal distribution with a mean of −45.6° (Abhinandan and Martin, 2010). The authors proposed a method to predict the VL/VH packing angles using neural networks trained against several interface residues in antibody crystal structures. This work also identified some potential key residues (L38, L40, L41, L44, L46, L87, H33, H42, H45, H60, H62, H91, H105), which are located in the framework regions and are extracted by the genetic algorithm, for the prediction of the packing angle and for antibody engineering (Abhinandan and Martin, 2010). Here it is noteworthy that the evaluation of VL/VH domain orientations only by the packing angles could neglect another aspect of the variations: translation of each domain.
Tramontano and coworkers also analyzed the variability of VL/VH domain angles using cluster analysis based on a distance by LGA software (Chailyan et al., 2011b). The number of antibody crystal structures with κ light chain in the PDB is much larger than that of antibodies with λ light chain, and the authors used a ‘balanced light chain set’, which included similar numbers of Vκ and Vλ antibody sequences, respectively. They found that VL/VH pairings can be divided into two major clusters where one cluster is only observed in mouse antibodies and the other is observed both in mouse and in human, indicating the importance of template selection in humanization procedure. They pointed out that these clusters could be assigned from the sequences by looking at some specific positions (L8, L28, L36, L41, L42, L43, L44, L66) in the sequences. Interestingly, only two of these residues, L41 and L44, are shared with the residues proposed by Abhinandan and Marin as the packing determinants. The AbNum scheme (Abhinandan and Martin, 2008) was used in both studies for antibody numbering. Structural positions of these residues are described in Fig. 4. In another finding of Tramontano and coworkers, the antibodies in one of the clusters tend to recognize smaller antigens, such as haptens, whereas ones in the other cluster tend to show more promiscuous binding, indicating the correlation between the relative orientation and the antigen-binding capability.
Fig. 4.

Structural positions of residues that were identified as important for VL/VH packing. AbNum numbering scheme was used both in Abhinandan and Martin (2010) and in Chailyan et al. (2011b). Consensus residues proposed in both studies are shown as yellow spheres while residues proposed only by Abhinandan and Martin (2010) and Chailyan et al. (2011b) are shown as green and red spheres, respectively. (A) VL domain. (B) VH domain.
The studies discussed above imply that VL/VH domain orientations are encoded by the sequence of each domain, but the dependence seems to be subtle. The domain orientations can also change somewhat upon antigen binding and may fluctuate in solution. Another approach might be to use the predicted domain orientation to select the best templates for antibody modeling. Also currently unexplored is the correlation between the packing angle and other properties such as solubility and stability.
Wang et al. (2009) performed covariation analyses using a large multiple sequence alignment of Ig-fold derived from NR-database at National Center for Biotechnology Information as well as from the PDB. Their calculations suggested the existence of conserved amino acid networks in VH (H37, H38, H44, H45, H47, H103) and VL (L36, L37, L43, L44, L46, L98) domains, respectively, and the networks clustered in VL/VH domain interface. They also identified the conserved network in VH/CH domain interface, but not in VL/CL domain interface. Protein designs based on these conserved amino acid networks showed some successes in the context of generating thermostabile antibodies.
Some software for antibody modeling is available on the web. Tramontano and coworkers developed the PIGS web server, which provides various options for choosing templates for the light and heavy chains (Marcatili et al., 2008). It constructs CDRs simply by grafting canonical structures and CDR-H3 loops of other antibodies onto the modeled framework. RosettaAntibody uses a VL/VH docking procedure based on the Rosetta energy function and rigid-body minimization to refine the orientations following CDR-H3 modeling (Sircar et al., 2009; Sivasubramanian et al., 2009). Web Antibody Modeling (WAM), which was a pioneering software package for antibody modeling developed by Rees and coworkers, is also available on the web (Whitelegg and Rees, 2000). Recently, Almagro and coworkers performed blind tests of antibody modeling methods including PIGS, RosettaAntibody and the other two modeling strategies independently developed in Chemical Computer Group and Accerlys Inc., using nine unpublished antibody crystal structures at that time (Almagro et al., 2011). They demonstrated that, overall, the results of each method showed similar trends. In each case, the conformations of the framework regions were predicted quite well (average RMSDs are ∼1.0 Å) whereas they tended to fail to predict the conformations of CDR-H3 (average RMSDs > 3.0 Å). These results reconfirmed the difficulty of modeling CDR-H3, and there seems to be much room to improve the methods. More importantly, this work may lead to additional blind-prediction exercises, analogous to CASP (Critical Assessment of protein Structure Prediction) and CAPRI (Critical Assessment of PRediction of Interactions).
There are a few public databases that contain antibody data from the PDB (Allcorn and Martin, 2002; Kaas et al., 2004) or from the large sequence database (Retter et al., 2005; Giudicelli et al., 2006). Considering the rapid increase in antibody structural and sequence data, these public databases will play important roles in organizing the information.
Antibody–antigen recognition
Needless to say, the best way to gain atomic-level insights into antibody–antigen recognition is to determine crystal structures of the complexes by X-ray crystallography. However solving the structures of antibody–antigen complexes is frequently challenging. Computational protein–protein docking methods provide an alternative way to study antibody–antigen interactions when structures are not available. Although computational protein–protein docking methods are rapidly improving, there are still limitations, especially when modeled structures are used as starting points (Gray, 2006). On the other hand, there are a few advantages in applying protein–protein docking to antibody–antigen complexes. First, we know that antigens bind to the CDRs of antibody structures. In addition, the residues in antigens that the antibody binds to, called epitopes, can be predicted by computational methods to some extent (Evans, 2008). This information can help to more accurately predict antigen–antibody complex structures, along with any other experimental data such as from mutagenesis that can be used to restrain the protein–protein docking.
In general, antigenic epitopes in proteins are discontinuous in the protein sequences (Barlow et al., 1986; Van Regenmortel, 1996). One problem in epitope prediction is that many existing methods focus on protein sequences to predict continuous epitopes. The increase in the number of antibody–antigen complex structures has enabled us to better understand the amino acid compositions of antibody–antigen interfaces and how they differ from those of other heterodimer proteins. Structure-based epitope prediction algorithms have begun to be developed based on such information (Liang et al., 2009). Another problem is that current methods, including structure-based algorithms (Haste Andersen et al., 2006), are not designed to predict epitopes for specific antibodies. Recently Soga et al. (2010) have developed a method to predict epitope residues for individual antibodies from the sequence composition of the antibody–antigen interfaces.
In general, protein–protein interactions can be classified into permanent and transient interactions (Ozbabacan et al., 2011). Antibody–antigen interactions are often considered to be transient (Cho et al., 2006). Although we might expect high complementarity of antibody–antigen interfaces since antibodies often show high specificities and affinities, a previous analysis based on a small dataset of available structures showed that the shape complementarity of antibody–antigen interfaces was lower on average than that of protein–protein interfaces in general (Lawrence and Colman, 1993). From an evolutionary perspective, antibodies could evolve independently of antigens in our body, while other proteins involved in protein–protein interactions can evolve with their counterparts, which is often called coevolution, so that the shape complementarity of general proteins could be optimized against their partners. However, a later analysis with high-resolution crystal structures suggested the possibility that the previous observation that antibody–antigen complexes had lower shape complementarity was due to the low quality of the crystal structures (Cohen et al., 2005). It has been also found that overall topography of antigen-binding sites is roughly classified into a limited number of groups associated with different types of antigens (MacCallum et al., 1996; Lee et al., 2006). For example, anti-protein antibodies tend to have flat antigen-binding sites while those of anti-peptide antibodies have a groove-like shape, in which peptides are buried. Electrostatic complementarity of antibody–antigen interfaces seemed to show a similar trend to that of protein–protein interface in general (McCoy et al., 1997). Since the discussions above were based on a limited number of antibody–antigen structures, a more comprehensive analysis of the complementarities of the interfaces is required to understand the essential nature of antibody–antigen recognitions, helping to guide the rational design of antibody therapeutics.
Lysozyme has frequently been used as a model antigen to gain insights into antibody–antigen interactions (Davies and Cohen, 1996). In computational studies, Smith-Gill and coworkers investigated antibody–antigen interactions using homology-modeled anti-lysozyme antibody structures, HyHEL8 and HyHEL26, and a crystal structure, HyHEL10 (Sinha et al., 2002; Mohan et al., 2003). They calculated the binding energies and performed kinetics experiments, showing that a greater electrostatic contribution to antigen binding leads to higher specificity while hydrophobic interactions favor conformational flexibility and cross-reactivity. Their subsequent work based on molecular dynamics (MD) simulations with a crystal structure, HyHEL63, also supported the importance of electrostatic forces in the antibody–antigen association (Sinha et al., 2007).
In a recent application of protein–protein docking to antibody–antigen complexes, Gray and coworkers used antibody models generated by WAM and antigen structures to propose the potential binding modes of antibody–antigen complexes using RosettaDock (Sivasubramanian et al., 2006, 2008). The protein–protein docking used a rigid-body Monte Carlo (MC) search and a low-resolution interaction potential including an alignment score specific for antibodies in an initial stage, and, in the subsequence refinement stage, an all-atom scoring function was used to select a solution (Gray et al., 2003; Gray, 2006). In one of the examples, an EGFR-binding antibody, they also incorporated computational and experimental mutagenesis information into their docking procedures (Sivasubramanian et al., 2006).
The flexibility of protein–protein interactions should be incorporated when considering the possibility of structural change upon binding (Zacharias, 2010). Structural errors in comparative models could also lead to serious errors in the subsequent steps in protein–protein docking. Methods are available for treating side chain flexibility, for instance by taking advantage of rotamer libraries, while dealing with backbone flexibility is much more difficult. Although implicit, one approach to incorporate backbone flexibility in protein–protein docking is the so-called ensemble docking, which uses several structures generated by sampling methods as starting points. MD simulations can also be used to generate such conformational ensembles. An advantage of ensemble docking is that it uses rapid rigid protein–protein docking methods, which have shown good performance in CAPRI contests so far, to identify the correct near-native models (Bonvin, 2006).
Gray and coworkers examined how ensemble docking is effective by varying the treatment of backbone flexibilities in each ensemble in protein–protein docking simulations in general (Chaudhury and Gray, 2008). They took advantage of two ensembles for docking: computational ensembles generated by RosettaRelax (Misura and Baker, 2005) and nuclear magnetic resonance (NMR) ensembles provided by NMR solution state studies. Although the backbone flexibilities were restricted only to the ligand proteins, they showed better performance using ensemble dockings than using a rigid-body protein–protein docking. Recently, for the application of modeling antibody–antigen complexes, they also developed a novel docking procedure called SnugDock, which simultaneously optimizes paratopes, i.e. CDR loops, and VL/VH domain orientations during antibody–antigen docking (Sircar and Gray, 2010). In a subsequent study, this method was applied to protein–protein docking in general, showing the versatility of this protocol (Sircar et al., 2010). In another innovative study, Simonelli et al. (2010) combined experimental data with a computational protein–protein docking method. They used several antibody models created by the PIGS and RosettaAntibody programs as an ensemble, and docked them to the antigen, a surface protein of dengue virus, using RosettaDock. They took advantage of NMR chemical shift data to validate the docking results, showing that NMR epitope mapping improved the accuracy of computational docking.
Looking forward, incorporating backbone flexibility in the docking procedure is a promising and an essential approach to take into account the structural change upon binding and to overcome the small structural errors expected when using homology-modeled structures.
Affinity maturation by somatic mutations and computational design
Antibodies can evolve in a short time in response to antigens, so that they are more specific to their antigens and have higher affinity, mainly by improving the complementarity of the antibody–antigen interfaces (Li et al., 2003; Cauerhff et al., 2004; Acierno et al., 2007). Somatic mutations can occur not only in the antigen-binding site, but also in the framework region, which antigens usually do not contact physically (Lavoie et al., 1992). Computational methods have played an important role in understanding how these mutations improve binding affinity, and more recently, have also been used to predict novel mutations to improve affinity or specificity.
Based on the analysis of a large number of sequences, Clark et al. (2006) examined trends in amino acid substitutions during the somatic maturation process. Specifically, using a gene-fitting procedure with codon probability tables, they examined mutation probabilities in 23 116 heavy chains and 11 095 light chains. By comparing the probabilities with the conventional BLOSUM matrix, they concluded that amino acid substitutions in somatic mutation process have similar trends to those observed in protein evolution in general. They also showed that mutations tend to occur in antibody–antigen interfaces and in exposed surface regions, rather than the framework region. Finally, they examined changes of amino acid composition in the antibody–antigen interface, showing that the numbers of tyrosine, serine and tryptophan residues decrease while those of histidine, proline and phenylalanine increase during the maturation process.
Several computational studies have been conducted to investigate the relationship between somatic mutations and conformational flexibilities of antibodies. A plausible hypothesis is that antibodies acquire rigidity during maturation in order to increase binding affinity (by minimizing entropic losses upon binding) and to improve specificity. In fact, crystal structures of antibody drugs available in the PDB are predicted to assume rigid conformations based on their sequence features (Kuroda et al., 2008). In these studies, the rigidification was hypothesized to occur within the entire variable domains rather than within only CDRs, by forming regular secondary structures, such as β-turns, and hydrogen bonds (Kuroda et al., 2008), or by making residue contact networks, during the maturation process (Zimmermann et al., 2010). Thermodynamic experiments and MD simulations demonstrated that all residues in variable domains may participate in the rigidification rather than the limited number of CDR residues (Jimenez et al., 2003; Thielges et al., 2008; Zimmermann et al., 2010).
Kollman and coworkers (Chong et al., 1999) used MD simulations to study the germline and matured anti-hapten antibodies, 48G7 (Patten et al., 1996; Wedemayer et al., 1997). They showed that maturation of 48G7 was driven by electrostatic optimization of the antigen-binding site, which resulted in geometric reorganization and rigidification, as manifested by smaller root mean square fluctuations of matured 48G7 compared with those of the germline antibody. Demirel and Lesk (2005) investigated the same antibodies using an elastic network model and concluded that the fluctuations of the germline antibody were similar to those of matured one. Rather, the structural difference observed in crystal structures was thought to result from the different binding mechanisms of germline and matured antibodies; induced fit could occur upon hapten binding in the germline antibody while hapten binding to the mature one was explained by a lock-and-key model. A second well-studied pair of germline and mature antibodies are those of the anti-hapten antibody, 4-4-20. Experimental results and MD simulations again suggested that the somatic mutations, several of which are not in direct contact with the antigen, restrict the conformational fluctuations in the mature antibody (Zimmermann et al., 2006; Thorpe and Brooks, 2007). More recently, Babor and Kortemme analyzed several germline and matured antibody structures in the PDB using their multi-constraint computational design protocol (Humphris and Kortemme, 2007) with the Rosetta scoring function, and confirmed that native sequences of germline CDR-H3 are designed for conformational flexibility while those of matured ones are highly optimized for single conformations (Babor and Kortemme, 2009). On the other hand, rigidification upon maturation is most likely not universal, because a matured anti-hapten antibody, SPE-7, is known to assume several different conformations in response to binding different antigens (James et al., 2003; James and Tawfik, 2003). In addition, a series of studies of anti-hapten antibodies has demonstrated that rapid maturation based on CDR rigidification tend to arrive at a moderate affinity ceiling whereas flexibilities in CDR-H3 may play an important role in higher affinity maturation (Furukawa et al., 1999, 2001).
To more directly determine how somatic mutations modulate CDRs flexibility in nature, Jacobson and coworker also analyzed a set of antibodies, whose crystal structures were solved both in germline and matured forms, without their antigens in the crystal structures, using MD simulations with an implicit solvent model (Wong et al., 2011). Significant rigidification was observed for CDR residues that contact the antigen, mainly in CDR-H3, while the other five CDRs frequently displayed some increase in flexibility. The observed rigidification was explained by new hydrogen bonds, salt bridges and side chain packings that were improved by somatic mutations. Interestingly, conformations similar to the germline antigen free- and bound forms of 7G12 (1NGZ and 1N7M, respectively) were reproduced when using the germline free form (1NGZ) as the initial structure in the simulation, whereas the germline bound form was not sampled when the matured free form (1NGY) was used as the initial structure. This observation suggested a population shift mechanism of antigen binding in the 7G12 antibody. Consistent with his hypothesis, a structural change of CDR-H3 upon antigen binding was observed in the germline crystal structures (RMSD of CDR-H3 between the antigen bound- and free forms is 2.22 Å) while that of matured form is not observed (RMSD 0.39 Å), and CDR-H3 of the germline bound form is almost identical to those of matured antigen free- and bound forms (RMSD of CDR-H3 in the germline form against those in matured antigen free- and bound forms is 0.39 and 0.42 Å, respectively) (Fig. 5). In this antibody, the S97M mutation in the VH domain contributed to improve hydrophobic contacts and modulated the flexibility of CDR-H3 by anchoring the loop to its antigen free from, thus shifting the equilibrium toward the conformation most suitable for binding, so that the matured form could bind the antigen without entropic penalty. Another mutation that modulates flexibility in this antibody was the mutation of Ser to Asn at the position 76 in the VH domain. This S76N mutation anchored CDR-H1 loop via hydrogen bonds during the simulations. In the simulations of the germline form, the Ser residue at the position 76 rarely formed hydrogen bonds with the CDR-H1 whereas, in the simulations of the matured form, the Asn at the corresponding position often formed multiple hydrogen bonds via the side chain with the CDR-H1, which results in the rigidification of the CDR. It is noteworthy that position 76 in the VH domain is far from the antigen-binding site (Fig. 5). Therefore, this example shows how mutations distant from the antigen-binding site can modulate binding.
Fig. 5.
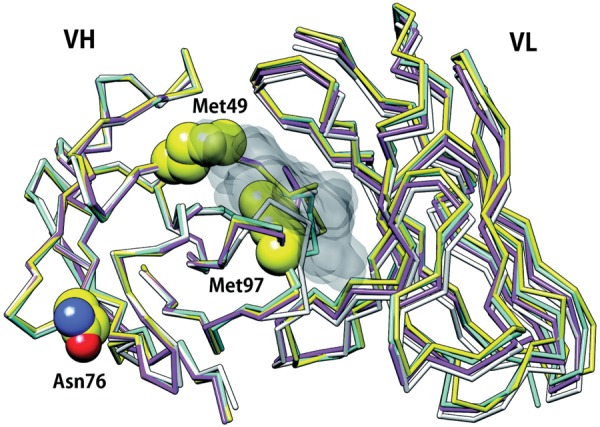
Positions of somatic mutations in a metal chelatase catalytic antibody, 7G12. The mutations, R49M, S76N and S97M in VH domain, are represented as spheres. Germline and matured antigen bound forms are represented by magenta (1N7M) and yellow (3FCT) model, respectively. Germline and matured antigen-free forms are represented by white (1NGZ) and cyan (1NGY) model, respectively. The antigen is shown as a black surface model. The residues at position 49 and 97 contact the antigen whereas the residue at position 76 is far from the antigen-binding site. Structural change of CDR-H3 is observed only in the germline form (magenta and white; RMSD 2.2 Å. See text.).
Although antibodies can evolve in vivo through affinity maturation, arguments have emerged that binding affinities and specificity could be further improved by using artificial systems, such as synthetic antibody libraries (Soderlind et al., 2000; Borrebaeck and Ohlin, 2002; Bond et al., 2005; Sidhu and Fellouse, 2006; Michnick and Sidhu, 2008; Babor et al., 2011) and computational protein designs (Babor and Kortemme, 2009). Here we review some recent successes of the latter approach, in which the affinities of natural antibodies were improved to varying extents (Table I). Details of protein design methodologies are reviewed elsewhere (Rosenberg and Goldblum, 2006; Lippow and Tidor, 2007; Karanicolas and Kuhlman, 2009).
Table I.
Results of computational designs
| Initial structuresa | Resolution (Å) | Antigen | KDWT,b | KDMut,b | Affinity improvementc | Designed mutationsd | Ref. |
|---|---|---|---|---|---|---|---|
| AQC2 (1MHP) | 2.80 | Integrin | 7 nM | 850 pM | 10 | L: S28Q, N52E, H: T50V, K64E | Clark et al. (2006b) |
| D44.1 (1MLC) | 2.50 | Lysozyme | 4.4 nM | 30 pM | 140 | L: N92A, H: T28D, E35S, S57V, T58D, G99D | Lippow et al. (2007) |
| TA4 | N/Ae | Gastrin | 6 mM | 13.2 nM | 454 | L: H91F, Q92F, R94P, V96A, H: I100L, S102V | Barderas et al. (2008) |
| E2 (3BN9) | 2.17 | Protease | 4.8 nM | 340 pM | 14 | H: T98R | Farady et al. (2009) |
aAntibody structures to be designed. PDB codes are shown in parentheses.
bReported values in the papers. For E2, KI value is shown.
cReported values in the papers.
dThere are a number of designed mutations in each works. Only representatives are shown. L and H represent light and heavy chains, respectively.
eHomology modeled structure was used.
Most of the current examples of computational antibody design start from antigen–antibody complex structures in the PDB, that is, the studies can be described as ‘re-design’ of antigen–antibody interfaces. Protein interface re-design relies on estimating free energy changes due to amino acid substitutions, which can be accomplished using physics-based force fields (Huang et al., 2006) or knowledge-based potentials derived from the structural database (Ota et al., 2001; Russ and Ranganathan, 2002; Clark and van Vlijmen, 2008). Similar to comparative modeling methods, re-design also requires efficient conformational search algorithms, such as dead-end elimination (DEE) (Desmet et al., 1992; Dahiyat and Mayo, 1996, 1997; De Maeyer et al., 2000) and MC searches (Koehl and Levitt, 1999; Kuhlman and Baker, 2000), to sample conformations of modified structures. In most studies, protein backbones have been fixed during the procedures due to computational limitations. However, it has become possible to incorporate explicit backbone flexibility in computational protein design (Hu et al., 2007; Murphy et al., 2009).
Clark et al. optimized an antibody AQC2 for binding to the I domain of human integrin VLA1 using the CHARMM force field (Brooks et al., 1983), the ICE software (Kangas and Tidor, 1998), which calculates electrostatic interactions and desolvation energies, and a DEE-based side chain repacking software, DEZYMER, developed by Hellinga and coworkers (Looger and Hellinga, 2001). This approach succeeded in improving the binding affinity even though the original antibody has the relatively high affinity (KD ∼ 7 nM) (Clark et al., 2006a). They tried >80 designed variants including multiple mutations, in which the combination of four mutations (L: S28Q, N52E, H: T50V, K64E; Fig. 6A) displayed the highest affinity of 850 pM, which was a 10-fold improvement over the wild type. The designed structure was solved and the rearrangement of a hydrogen bond network in the interface was elucidated.
Fig. 6.
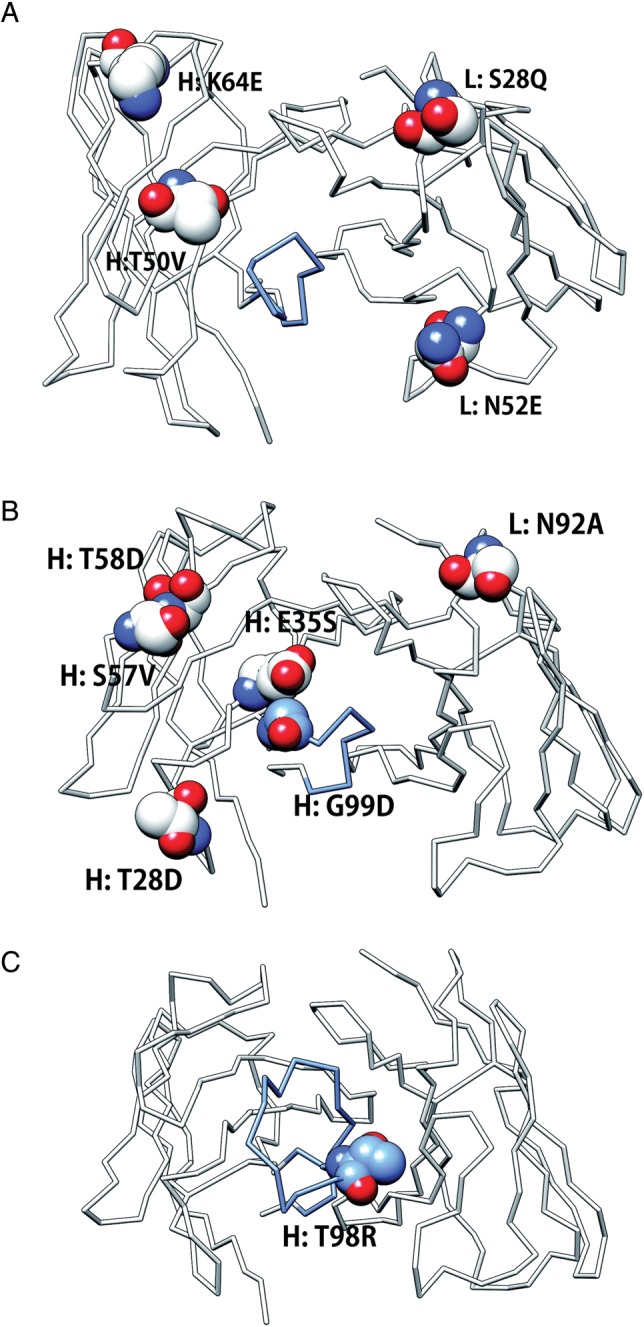
Positions of designed mutations in antibodies. CDR-H3 is colored blue. Positions of designed mutations are represented as sphere models. Original residues are shown. (A) Anti-integrin antibody, AQC2 (1MHP). (B) Anti-lysozyme antibody, D44.1 (1MLC). (C) Anti-protease antibody, E2 (3BN9).
In another successful antibody–antigen re-design, Lippow et al. (2007) focused on electrostatic interactions. One of the obvious advantages to optimize electrostatic forces is that long-range interactions could be incorporated into design process. They designed single mutations in all CDR positions using the DEE and A* search algorithms with the CHARMM force field, ranked them by Poisson–Boltzmann electrostatics and demonstrated that four mutations (L: N92A, H: T28D, S57V, T58D; Fig. 6B) together generate 100-fold improvement of the affinity over wild type (43 pM vs. 4.4 nM for wild type).
A somewhat different use of computational modeling is to identify favorable positions for experimental random mutagenesis, rather than relying on computational prediction to identify specific favorable mutations. In such a study, Barderas et al. (2008) used computational modeling, including constrained antigen docking and MD simulations, to select favorable residues for random mutagenesis in CDR-H3 and -L3 by phage display. This approach resulted in mature anti-gastrin antibodies with binding affinities as potent as 13.2 nM, a 454-fold improvement over wild type based on six mutations.
In some cases, specificity, and not affinity, is the major challenge addressed by computational design, as in the work by Farady et al. (2009), which aimed to improve the species cross-reactivity of an anti-protease antibody, E2. The problem is that antibodies derived from mice immunized by human antigens often display low efficacy in pre-clinical animal models because the structures and sequences of the antigen targets are not completely conserved between humans and mice. In the case of the protease antigen, called MT-SP1 in human and epithin in mouse, they share 87% sequence identity, and three surface residues that contact the antibody differ. Based on the complex structure of MT-SP1/E2, an epithin/E2 structure was modeled. Residues in the antibody that contacted the antigen were selected for in silico mutations and several mutations were predicted to improve the affinity for epithin on the basis of a molecular mechanics energy function. The T98R mutation (Fig. 6C) in CDR-H3 showed the largest predicted free energy change, and subsequent experimental tests confirmed that the mutation results in a 14-fold affinity improvement over wild type (KI reduced from 4.8 nM to 340 pM). The mutation is believed to improve epithin binding by introducing new hydrogen bonds in the antigen–antibody interface.
What can we learn from these successes? First, high-resolution crystal structures are not necessarily required for computational design. As shown in Table I, initial structures, which have moderate resolution from 2.17 to 2.80 Å, could be exploited for computational design, as well as homology models of antibodies and antigens. These successes imply that current antibody modeling methods might be sufficiently accurate to be used in computational antibody design. Second, computational protein–protein docking could be used as a first step in design procedure, particularly in cases where experimental information can be used to help to guide the docking. Third, significant improvement in binding affinity frequently requires multiple amino acid substitutions. Finally, as seen in Figs 5 and 6, it is clear that not only CDR-H3 but also the other five CDR loops can be candidates for affinity-enhancing mutations. Conformational changes that occur upon antigen binding, especially in CDR-H3, present a challenge for antibody design. In addition, current protocols mainly focus on optimizing antigen-contacting residues in CDRs. Mutations during somatic maturations, however, often occur in the framework regions, which antigens cannot directly contact (Lavoie et al., 1992). Exploiting mutations of framework regions might be possible when incorporating backbone flexibility during computational designs.
Stability design and aggregation in antibodies
Another challenging application of computational designs in antibody engineering is to predict aggregate-prone regions (APRs) from amino acid sequences and to design aggregate-resistant antibodies by introducing mutations in those regions. One mechanism of aggregation is the formation of amyloid fibrils, which are rich in β-sheet structure, and have been associated with diseases such as Alzheimer's or Parkinson's diseases (Dobson, 1999). Experimental observations suggest that short segments of a protein could facilitate aggregation (Ventura et al., 2004), and these are termed as APRs. Currently, several computational methods are available to predict APRs and rates of aggregations, mainly based on the sequence composition and on propensities such as hydrophobicity, charge and secondary structure propensity (Caflisch, 2006; Conchillo-Sole et al., 2007; Trovato et al., 2007; Garbuzynskiy et al., 2010). Empirical or phenomenological models have been proposed for the effect of mutations on the aggregation rate on the basis of the experimental data (Chiti et al., 2003; DuBay et al., 2004). MD simulations have also been used to interpret and complement experimental studies of protein aggregation and amyloidosis (Ma and Nussinov, 2006; Sharma et al., 2008). A review of aggregation in protein therapeutics is available elsewhere (Agrawal et al., 2011), so here we limit ourselves to recent studies on design of aggregation-resistant antibodies.
Aggregation in therapeutic proteins can lead to problems with immunogenicity (Rosenberg, 2006; Kumar et al., 2011) and in addition, antibodies have been known to aggregate in high concentration of formulations for therapy and in storage (Shire et al., 2004). Many experimental studies have been performed to investigate antibody stability and resistance to aggregation, primarily using single-chain Fv fragments (scFv) (Bird et al., 1988; Glockshuber et al., 1990; Worn and Pluckthun, 1998, 1999, 2001).
Designed mutations for aggregation-resistant antibodies may induce the loss of desired affinities. Wang et al. examined how APRs in antibody structures contribute to antigen recognition using TANGO (Fernandez-Escamilla et al., 2004) and PAGE (Tartaglia et al., 2005), which are computational tools to predict APRs in protein sequences, based on 29 antigen–antibody complexes in the PDB (Wang et al., 2010). They found that the APRs most frequently appeared in CDR-H2, and less frequently in CDR-H3, although residues in the N-terminal framework region of CDR-H3 are predicted to be APRs. Based on changes of buried surface area upon antigen binding, they also showed that aromatic residues, Tyr and Trp, are favored both in antigen-binding sites and APRs, indicating that aggregation may be associated with antigen binding via these residues.
In a different, structure-based approach to predicting APRs in antibodies, Trout and coworkers proposed a novel measure called spatial aggregation propensity (SAP), which quantifies the exposure of hydrophobic residues, averaged over snapshots from a MD simulations with explicit water (Chennamsetty et al., 2009a,b; Voynov et al., 2009). Using this measure, they identified 14 aggregation-prone motifs in constant regions of human IgG molecules and, by comparing the amino acid sequences, they noticed that those APRs are not conserved among the other antibody classes (IgA1, IgD, IgE and IgM) (Chennamsetty et al., 2009a). In these works, antibody structures were used to derive SAP values but antibody models could potentially be used when crystal structures are unavailable. Subsequent work from the same lab demonstrated that the SAP measure could be applied to antibody fragments, such as Fab or Fc, and homology-modeled structures, and that MD simulations could be performed with implicit solvent models to obtain SAP values with reasonable computational time and tolerable accuracies (Chennamsetty et al., 2010).
In an experimental study, Perchiacca et al. (2011) performed mutational analysis of a human VH domain antibody to investigate the molecular origin of the aggregation by comparing the biophysical properties of an aggregation-prone antibody and the aggregation-resistant counterpart. The difference between the two antibodies is only in the sequence composition of each CDR. Although the introduction of charged mutations distant from the CDRs failed to confer aggregation-resistance property for the aggregation-prone antibody, the mutations within the first CDR loop (H31–H33) or near the CDR (H29) successfully produced an aggregation-resistant antibody. It is noteworthy that, while the results of some computational methods that were used to predict APRs in this antibody contain several false positives, the consensus APR of those prediction tools (H28–H32) is almost identical to the aggregation hot spots in this experiment (H29, H31–H33). This indicates that computational meta-approaches that integrate prediction results from several methods may help to improve prediction of APRs in antibodies.
Wang and Duan (2011) performed MD simulations with the aim of proposing a strategy for improving the thermal stability of an anti-VEGF scFv antibody. The simulations demonstrated that, at least in the scFv antibody, when the temperature is elevated, the dissociation of VL/VH domain interface occurred at first followed by unfolding of VL domain and then partial unfolding of VH domain, suggesting that stability of the VL/VH domain interface may govern the stability of entire antibody structure. If this is correct, then engineering of the VL/VH domain interface is the first choice for improving the thermal stability of the antibody. The extensive analyses of VL/VH domain interfaces reviewed in the previous section may provide guidance for producing more stable antibodies based on VL/VH interface designs in silico.
Antigen design to elicit NAbs
Advances in computational protein design methods, and the increasing number of protein crystal structures, may make it possible to develop vaccine candidates more rationally and efficiently. Vaccine injections elicit desired NAbs for antigens or viruses targeted in therapy (Burton, 2002). HIV-1 has attracted much attention because of the lack of appropriate vaccines and because of the increasing number of patients. NAbs work either by binding to the virus surface and then blocking subsequent cellular response, or by binding after virion attachment to a target cell so that they can inhibit viral entry. HIV-1 shows significant variability of the sequences and glycosylation patterns, which results in its escaping from human immune systems (Burton et al., 2004). A few regions in the HIV-1 envelope spike regions are structurally conserved among diverse isolates, including the CD4-binding site on gp120, the coreceptor binding site on gp120 and the membrane proximal external region (MPER) of gp41, all of which can be targets for binding by antibodies. Thus, eliciting broadly NAbs by those conserved epitopes or by their mimics is challenging, and advanced computational approaches could conceivably help tackle this issue.
Ofek et al. (2010a,b) used computational-guided approach to design epitope scaffolds to elicit anti-HIV-1 antibodies. They introduced a known linear epitope of MPER, which is usually recognized by 2F5 antibody, into scaffolds selected from the PDB. The existing scaffolds were re-designed by RosettaDesign suit to introduce mutations so that the targeted epitope is accommodated in the scaffolds, leading to stable proteins that consist of the HIV-1 epitope and the scaffolds derived from unrelated proteins. These designs succeeded in eliciting some antibodies with similar binding modes of 2F5 with the peptide epitope but with shorter CDR-H3 loops than 2F5. Despite this apparent success, the resulting antibodies did not neutralize the virus in an experimental test probably due to the lack of a long CDR-H3 loop with the appropriate hydrophobic nature. However, crystal structures demonstrated that the elicited antibodies induced the desired conformational change of the HIV epitope, which is encouraging for computational immunogen design based on epitope scaffolds already existing in the PDB. They also suggested a correlation between epitope flexibility and immunogenicity based on the structures obtained.
In a related study, Lapelosa et al. (2009, 2010) analyzed the MPER epitope with human rhinovirus (HRV) scaffolds using replica exchange molecular dynamics (REMD) simulation techniques with an implicit solvent model. Their intention was to identify the conformational preferences of the epitope, which are considered to be recognized by 2F5 antibody, and to identify the virus scaffold that maximizes the epitope exposure to solvent. They proposed a structural model of the induced antibody and HRV complex. This model was then used for further design of virus constructs. Using a focused library, the anchor of each end of the 2F5 epitope in the modeled HRV sequence was optimized, and the binding affinity of the optimized models was evaluated experimentally. Subsequently, the authors used REMD simulations of the optimized models to show a correlation between the binding affinity and the conformational preferences of the epitope, showing that conformations that closely mimicked ones in crystal structures displayed high affinity, confirming the validity of their proposed computational model and the strategy used.
Another NAb that recognizes the MPER of gp41 is the 4E10 antibody. Correia et al. (2010b) designed an epitope scaffold based on the binding mode of this antibody, again by using Rosetta. They searched for appropriate backbone scaffolds from the PDB, and designed the amino acid sequences with the goal of designing scaffolds that are soluble and stable with the epitope peptide in solution. Their subsequent experimental validation showed that the designed proteins consisting of the MPER epitope and the scaffolds bind to 4E10 with higher affinities (from pM to nM levels) compared with only the MPER peptide epitope itself (nM level). Then, they determined the crystal structures, demonstrating the similarity between the inserted epitope in the scaffold and the epitope recognized by 4E10 in the antibody–peptide crystal structures. Based on these designed chimeric proteins, they also proposed two distinct computational technologies: flexible backbone remodeling and resurfacing (Correia et al., 2010a). These methods were used to modify the structure of the designed proteins with the goal of the minimizing immunogenicity while retaining the thermal stability, solubility and high affinity toward cognate antibody obtained in the earlier study (Correia et al., 2010b). By using the backbone remodeling method, a globular domain, which assumes α–β–α topology, of the designed protein scaffold was trimmed, then the sequence of the remaining scaffold was designed by the resurfacing method or side chain replacement, resulting in a chimeric protein having a better melting temperature and a similar affinity to the parent protein. They also determined the crystal structure of the re-designed proteins, showing close agreement between the structure and the model. However they also pointed out that the loop region that was replaced from a domain in the parent protein in the remodeling stage had a slightly different backbone conformation from the crystal structure, which results in packing errors in the region, demonstrating that even a small displacement of a backbone conformation could lead to an inaccurate prediction.
In a related study, Oomen et al. (2003) designed an immunogen by synthesizing cyclic peptides, which reduce the peptide flexibility and mimic the β-turn conformation in the peptide–antibody complex structure, as an alternative to using an epitope scaffold to elicit an antibody. They assessed conformational preferences and the stability of four designed peptides using MD simulations. The most stable and the least flexible peptide evaluated by the simulations actually induced a cross-reactive potent antibody in an experiment.
In the studies discussed here, the common strategy for antigen designs is to produce epitope mimics of the NAbs identified from crystal structures, and a challenge is maximizing the exposure of the mimics, which is supposed to be recognized by an antibody, to solvent in a certain protein environment. Advanced computational techniques have been utilized in this context. Currently, a huge number of protein structures are available in public, and the number is still growing due to the structural genomics efforts. Hence searching for a protein scaffold from the structural database seems to be a promising approach for rational vaccine designs. De novo scaffolds for protein design were also proposed (MacDonald et al., 2010). In addition, MD simulations are required to analyze conformational preferences of epitopes in solution, ideally in conjunction with experiments such as NMR.
Looking forward, we envision that computational methods may be particularly useful to study antibody–peptide complexes, that is, the recognition of sequential epitopes. The peptides often assume β-turn conformations in an antigen-binding site, and the recognition mechanism seems to depend on the length of CDR-H3 (Bates et al., 1998). Considering their smaller size compared with protein antigens, the evaluation of free energy of binding for designed peptides by computer simulations is a promising approach for rational vaccine design. Furthermore, information concerning the behavior of mutant viral antigens may be quite useful for rational vaccine design since pandemic could occur because of antigenic drifts. Structures and simulations of antibody–antigen complexes provide essential information to analyze such behavior.
Perspectives and concluding remarks
Recent advances in computational technologies combined with dramatic advances in high-throughput technology (Reddy and Georgiou, 2011) have the potential to make important contributions to the development of biological therapeutics, including those based on antibodies and antigens. A central challenge is accurately predicting antibody structures from their sequences, and significant progress has been made toward this goal. The prediction of antibody–antigen-binding modes, by computational protein–protein docking, has also made great progress, especially by exploiting knowledge of the antigen-binding site and experimental data. Accurate prediction of epitopes and paratopes should further improve accuracy. Finally, we have reviewed several examples of computational affinity maturation (Clark et al., 2006a; Lippow et al., 2007; Farady et al., 2009), all of which have taken advantage of crystal structures of antibody–antigen complexes in the PDB except for Barderas et al. (2008).
However, there are still no striking examples where all of these computational capabilities have been combined, i.e. both an antibody and an antigen are modeled from their sequences, their binding mode is predicted by protein-protein docking, and then the resultant model is used to predict mutations that improve binding affinity and/or specificity. In such an ambitious undertaking, relatively small errors in each step could be amplified in subsequent steps. However, we anticipate continuing improvements in all of these computational approaches; for example, the recent emergence of methods for incorporating backbone flexibility in protein docking and protein engineering will be potentially useful in computational antibody design. Treating loop flexibility is particularly important in antibody design because large conformational changes can occur upon antigen binding, and affinity maturation can modulate both the conformations and dynamics of the CDR loops.
Beyond improvements in conformational sampling methods, continuing improvements in ‘scoring functions’ used to estimate free energies are needed. Approximations in the scoring function can lead to deleterious effects, especially when using implicit solvent models, and quantitative estimations of free energy changes upon antigen binding are still very difficult, even in the case of small ligands. Most current force fields for protein design include many heuristic terms. Although qualitative estimations might be sufficient for some applications, rigorous free energy evaluations explicitly including the contributions of conformational entropy and solvent polarization are challenging issues in computational chemistry and biology. Knowledge-based potentials will likely also continue to improve as the number of protein structures and sequences continues to increase exponentially.
In terms of improvements of the properties in antibodies, such as immunogenicity and solubility, sequence-based methods have shown promise in antibody engineering (Abhinandan and Martin, 2007; David et al., 2010; Thullier et al., 2010), in addition to structure-based methods. The sequence-based methods can take advantage of the rapid advances in sequencing technology.
Structure-based vaccine design approaches will also be enhanced with increasing knowledge of antibody–antigen structures as well as improved computational methods. Scaffold-based vaccine design is a promising approach. Although high specificity is desired for an antibody drug, the importance of poly-reactivity of antibodies as a vaccine candidate has been pointed out (Dimitrov et al., 2011) because such antibodies might accommodate high variability of single antigens. Thus, understanding the origins of high specificity and cross-reactivity of antibodies is important in terms of developing protein therapeutics, and further structural analyses of antibody–antigen complex structures should provide hints to elucidate such mechanisms.
Overall, although there has been much progress in understanding sequence–structure relationships in antibodies, there is still much to learn, especially regarding the mechanisms of antibody affinity maturation, such as the relationship between somatic mutations and conformational heterogeneity. Other types of protein scaffolds have been explored as an alternative to antibodies, and computational methods are likely to play an important role in these undertakings. Another area that requires greater attention is the role of water molecules in antibody–antigen interfaces (Yokota et al., 2003). Water molecules can improve the complimentary of the interface as seen in the case of affinity maturations by somatic mutations. Explicit treatment of water molecules in the design process will require novel theories or technologies, and some such efforts are underway (Jiang et al., 2005; Schymkowitz et al., 2005). Finally, it will be challenging to simultaneously optimize binding affinity, specificity, stability, solubility and other properties needed for a successful therapeutic protein.
Acknowledgements
D.K. was supported by a Research Fellowship for Young Scientists and an Excellent Young Researcher Overseas Visit Program of the Japan Society for the Promotion of Science. M.P.J. thanks the Sandler Program in Basic Sciences for supporting work in his lab on antibody modeling, and NIH GM81710 for supporting work on homology modeling. M.P.J. is a consultant to Schrodinger LLC.
Footnotes
Edited by Dan Tawfik
Conflict of interest
We have read the journal's policy and have the following conflicts: M.P.J. is a member of the scientific advisory board of Schrodinger LLC.
Funding
Funding to pay the Open Access publication charges for this article was provided by Japan Biological Informatics Consortium.
References
- Abhinandan K.R., Martin A.C. J. Mol. Biol. 2007;369:852–862. doi: 10.1016/j.jmb.2007.02.100. [DOI] [PubMed] [Google Scholar]
- Abhinandan K.R., Martin A.C. Mol. Immunol. 2008;45:3832–3839. doi: 10.1016/j.molimm.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Abhinandan K.R., Martin A.C. Protein Eng. Des. Sel. 2010;23:689–697. doi: 10.1093/protein/gzq043. [DOI] [PubMed] [Google Scholar]
- Acierno J.P., Braden B.C., Klinke S., Goldbaum F.A., Cauerhff A. J. Mol. Biol. 2007;374:130–146. doi: 10.1016/j.jmb.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Agrawal N.J., Kumar S., Wang X., Helk B., Singh S.K., Trout B.L. J. Pharm. Sci. 2011;100:5081–5095. doi: 10.1002/jps.22705. [DOI] [PubMed] [Google Scholar]
- Al-Lazikani B., Lesk A.M., Chothia C. J. Mol. Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- Allcorn L.C., Martin A.C. Bioinformatics. 2002;18:175–181. doi: 10.1093/bioinformatics/18.1.175. [DOI] [PubMed] [Google Scholar]
- Almagro J.C., Beavers M.P., Hernandez-Guzman F., et al. Proteins. 2011;79:3050–3066. doi: 10.1002/prot.23130. [DOI] [PubMed] [Google Scholar]
- Almagro J.C., Fransson J. Front. Biosci. 2008;13:1619–1633. doi: 10.2741/2786. [DOI] [PubMed] [Google Scholar]
- Babor M., Kortemme T. Proteins. 2009;75:846–858. doi: 10.1002/prot.22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babor M., Mandell D.J., Kortemme T. Protein Sci. 2011;20:1082–1089. doi: 10.1002/pro.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D., Sali A. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Barderas R., Desmet J., Timmerman P., Meloen R., Casal J.I. Proc. Natl Acad. Sci. USA. 2008;105:9029–9034. doi: 10.1073/pnas.0801221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D.J., Edwards M.S., Thornton J.M. Nature. 1986;322:747–748. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- Bates P.A., Dokurno P., Freemont P.S., Sternberg M.J. J. Mol. Biol. 1998;284:549–555. doi: 10.1006/jmbi.1998.2210. [DOI] [PubMed] [Google Scholar]
- Besmer E., Gourzi P., Papavasiliou F.N. Curr. Opin. Immunol. 2004;16:241–245. doi: 10.1016/j.coi.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Bird R.E., Hardman K.D., Jacobson J.W., et al. Science. 1988;242:423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- Bond C.J., Wiesmann C., Marsters J.C., Jr, Sidhu S.S. J. Mol. Biol. 2005;348:699–709. doi: 10.1016/j.jmb.2005.02.063. [DOI] [PubMed] [Google Scholar]
- Bonvin A.M. Curr. Opin. Struct. Biol. 2006;16:194–200. doi: 10.1016/j.sbi.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Borrebaeck C.A., Ohlin M. Nat. Biotechnol. 2002;20:1189–1190. doi: 10.1038/nbt1202-1189. [DOI] [PubMed] [Google Scholar]
- Bostrom J., Yu S.F., Kan D., et al. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- Brooks C.L., Blackler R.J., Sixta G., et al. Glycobiology. 2010;20:138–147. doi: 10.1093/glycob/cwp150. [DOI] [PubMed] [Google Scholar]
- Bruccoleri R.E., Haber E., Novotny J. Nature. 1988;335:564–568. doi: 10.1038/335564a0. [DOI] [PubMed] [Google Scholar]
- Burton D.R. Nat. Rev. Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- Burton D.R., Desrosiers R.C., Doms R.W., et al. Nat. Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- Caflisch A. Curr. Opin. Chem. Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Carter P.J. Nat. Rev. Immunol. 2006;6:343–357. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- Cauerhff A., Goldbaum F.A., Braden B.C. Proc. Natl Acad. Sci. USA. 2004;101:3539–3544. doi: 10.1073/pnas.0400060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailyan A., Marcatili P., Cirillo D., Tramontano A. Proteins. 2011a;79:1513–1524. doi: 10.1002/prot.22979. [DOI] [PubMed] [Google Scholar]
- Chailyan A., Marcatili P., Tramontano A. FEBS J. 2011b;278:2858–2866. doi: 10.1111/j.1742-4658.2011.08207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S., Gray J.J. J. Mol. Biol. 2008;381:1068–1087. doi: 10.1016/j.jmb.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennamsetty N., Helk B., Voynov V., Kayser V., Trout B.L. J. Mol. Biol. 2009a;391:404–413. doi: 10.1016/j.jmb.2009.06.028. [DOI] [PubMed] [Google Scholar]
- Chennamsetty N., Voynov V., Kayser V., Helk B., Trout B.L. Proc. Natl Acad. Sci. USA. 2009b;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennamsetty N., Voynov V., Kayser V., Helk B., Trout B.L. J. Phys. Chem. B. 2010;114:6614–6624. doi: 10.1021/jp911706q. [DOI] [PubMed] [Google Scholar]
- Chiti F., Stefani M., Taddei N., Ramponi G., Dobson C.M. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- Cho K.I., Lee K., Lee K.H., Kim D., Lee D. Proteins. 2006;65:593–606. doi: 10.1002/prot.21056. [DOI] [PubMed] [Google Scholar]
- Choi Y., Deane C.M. Proteins. 2010;78:1431–1440. doi: 10.1002/prot.22658. [DOI] [PubMed] [Google Scholar]
- Chong L.T., Duan Y., Wang L., Massova I., Kollman P.A. Proc. Natl Acad. Sci. USA. 1999;96:14330–14335. doi: 10.1073/pnas.96.25.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C., Gelfand I., Kister A. J. Mol. Biol. 1998;278:457–479. doi: 10.1006/jmbi.1998.1653. [DOI] [PubMed] [Google Scholar]
- Chothia C., Lesk A.M. J. Mol. Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- Chothia C., Lesk A.M., Tramontano A., et al. Nature. 1989;342:877–883. doi: 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- Chothia C., Novotny J., Bruccoleri R., Karplus M. J. Mol. Biol. 1985;186:651–663. doi: 10.1016/0022-2836(85)90137-8. [DOI] [PubMed] [Google Scholar]
- Clark L.A., Boriack-Sjodin P.A., Eldredge J., et al. Protein Sci. 2006a;15:949–960. doi: 10.1110/ps.052030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L.A., Ganesan S., Papp S., van Vlijmen H.W. J. Immunol. 2006b;177:333–340. doi: 10.4049/jimmunol.177.1.333. [DOI] [PubMed] [Google Scholar]
- Clark L.A., van Vlijmen H.W. Proteins. 2008;70:1540–1550. doi: 10.1002/prot.21694. [DOI] [PubMed] [Google Scholar]
- Cohen G.H., Silverton E.W., Padlan E.A., Dyda F., Wibbenmeyer J.A., Willson R.C., Davies D.R. Acta Crystallogr. D Biol. Crystallogr. 2005;61:628–633. doi: 10.1107/S0907444905007870. [DOI] [PubMed] [Google Scholar]
- Conchillo-Sole O., de Groot N.S., Aviles F.X., Vendrell J., Daura X., Ventura S. BMC Bioinformatics. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia B.E., Ban Y.E., Friend D.J., et al. J. Mol. Biol. 2010a;405:284–297. doi: 10.1016/j.jmb.2010.09.061. [DOI] [PubMed] [Google Scholar]
- Correia B.E., Ban Y.E., Holmes M.A., et al. Structure. 2010b;18:1116–1126. doi: 10.1016/j.str.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Dahiyat B.I., Mayo S.L. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiyat B.I., Mayo S.L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- David M.P., Concepcion G.P., Padlan E.A. BMC Bioinformatics. 2010;11:79. doi: 10.1186/1471-2105-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies D.R., Cohen G.H. Proc. Natl Acad. Sci. USA. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maeyer M., Desmet J., Lasters I. Methods Mol. Biol. 2000;143:265–304. doi: 10.1385/1-59259-368-2:265. [DOI] [PubMed] [Google Scholar]
- Demirel M.C., Lesk A.M. Phys. Rev. Lett. 2005;95:208106. doi: 10.1103/PhysRevLett.95.208106. [DOI] [PubMed] [Google Scholar]
- Desmet J., Demaeyer M., Hazes B., Lasters I. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- Dimitrov J.D., Kazatchkine M.D., Kaveri S.V., Lacroix-Desmazes S. PLoS Pathog. 2011;7:e1002095. doi: 10.1371/journal.ppat.1002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C.M. Trends Biochem. Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- Du P., Andrec M., Levy R.M. Protein Eng. 2003;16:407–414. doi: 10.1093/protein/gzg052. [DOI] [PubMed] [Google Scholar]
- DuBay K.F., Pawar A.P., Chiti F., Zurdo J., Dobson C.M., Vendruscolo M. J. Mol. Biol. 2004;341:1317–1326. doi: 10.1016/j.jmb.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Evans M.C. Curr. Opin. Drug Discov. Devel. 2008;11:233–241. [PubMed] [Google Scholar]
- Farady C.J., Sellers B.D., Jacobson M.P., Craik C.S. Bioorg. Med. Chem. Lett. 2009;19:3744–3747. doi: 10.1016/j.bmcl.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla A.M., Rousseau F., Schymkowitz J., Serrano L. Nat. Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fuentes N., Fiser A. BMC Struct. Biol. 2006;6:15. doi: 10.1186/1472-6807-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K., Akasako-Furukawa A., Shirai H., Nakamura H., Azuma T. Immunity. 1999;11:329–338. doi: 10.1016/s1074-7613(00)80108-9. [DOI] [PubMed] [Google Scholar]
- Furukawa K., Shirai H., Azuma T., Nakamura H. J. Biol. Chem. 2001;276:27622–27628. doi: 10.1074/jbc.M102714200. [DOI] [PubMed] [Google Scholar]
- Garbuzynskiy S.O., Lobanov M.Y., Galzitskaya O.V. Bioinformatics. 2010;26:326–332. doi: 10.1093/bioinformatics/btp691. [DOI] [PubMed] [Google Scholar]
- Giudicelli V., Duroux P., Ginestoux C., Folch G., Jabado-Michaloud J., Chaume D., Lefranc M.P. Nucleic Acids Res. 2006;34:D781–784. doi: 10.1093/nar/gkj088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockshuber R., Malia M., Pfitzinger I., Pluckthun A. Biochemistry. 1990;29:1362–1367. doi: 10.1021/bi00458a002. [DOI] [PubMed] [Google Scholar]
- Gray J.J. Curr. Opin. Struct. Biol. 2006;16:183–193. doi: 10.1016/j.sbi.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Gray J.J., Moughon S.E., Kortemme T., Schueler-Furman O., Misura K.M., Morozov A.V., Baker D. Proteins. 2003;52:118–122. doi: 10.1002/prot.10384. [DOI] [PubMed] [Google Scholar]
- Haste Andersen P., Nielsen M., Lund O. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T., Umetsu M., Nakanishi T., Togashi T., Yokoo N., Abe H., Ohara S., Adschiri T., Kumagai I. J. Biol. Chem. 2010;285:7784–7793. doi: 10.1074/jbc.M109.020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo J., Ikebe J., Kamiya N., Nakamura H. Biophys. Rev. 2012 doi: 10.1007/s12551-011-0063-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo J., Nishimura Y., Nakamura H. J. Am. Chem. Soc. 2011;133:10448–10458. doi: 10.1021/ja110338e. [DOI] [PubMed] [Google Scholar]
- Honegger A., Pluckthun A. J. Mol. Biol. 2001;309:657–670. doi: 10.1006/jmbi.2001.4662. [DOI] [PubMed] [Google Scholar]
- Hu X., Wang H., Ke H., Kuhlman B. Proc. Natl Acad. Sci. USA. 2007;104:17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N., Kalyanaraman C., Bernacki K., Jacobson M.P. Phys. Chem. Chem. Phys. 2006;8:5166–5177. doi: 10.1039/b608269f. [DOI] [PubMed] [Google Scholar]
- Humphris E.L., Kortemme T. PLoS Comput. Biol. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.P., Pincus D.L., Rapp C.S., Day T.J., Honig B., Shaw D.E., Friesner R.A. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- James L.C., Roversi P., Tawfik D.S. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- James L.C., Tawfik D.S. Protein Sci. 2003;12:2183–2193. doi: 10.1110/ps.03172703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Kuhlman B., Kortemme T., Baker D. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- Jimenez R., Salazar G., Baldridge K.K., Romesberg F.E. Proc. Natl Acad. Sci. USA. 2003;100:92–97. doi: 10.1073/pnas.262411399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Thirup S. EMBO J. 1986;5:819–822. doi: 10.1002/j.1460-2075.1986.tb04287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien J.P., Huarte N., Maeso R., Taneva S.G., Cunningham A., Nieva J.L., Pai E.F. J. Virol. 2010;84:4136–4147. doi: 10.1128/JVI.02357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas Q., Ruiz M., Lefranc M.P. Nucleic Acids Res. 2004;32:D208–210. doi: 10.1093/nar/gkh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangas E., Tidor B. J. Chem. Phys. 1998;109:7522–7545. [Google Scholar]
- Karanicolas J., Kuhlman B. Curr. Opin. Struct. Biol. 2009;19:458–463. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.T., Shirai H., Nakajima N., Higo J., Nakamura H. Proteins. 1999;37:683–696. [PubMed] [Google Scholar]
- Koehl P., Levitt M. J. Mol. Biol. 1999;293:1161–1181. doi: 10.1006/jmbi.1999.3211. [DOI] [PubMed] [Google Scholar]
- Koliasnikov O.V., Kiral M.O., Grigorenko V.G., Egorov A.M. J. Bioinform. Comput. Biol. 2006;4:415–424. doi: 10.1142/s0219720006001874. [DOI] [PubMed] [Google Scholar]
- Kuhlman B., Baker D. Proc. Natl Acad. Sci. USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Singh S.K., Wang X., Rup B., Gill D. Pharm. Res. 2011;28:949–961. doi: 10.1007/s11095-011-0414-9. [DOI] [PubMed] [Google Scholar]
- Kuroda D., Shirai H., Kobori M., Nakamura H. Proteins. 2008;73:608–620. doi: 10.1002/prot.22087. [DOI] [PubMed] [Google Scholar]
- Kuroda D., Shirai H., Kobori M., Nakamura H. Proteins. 2009;75:139–146. doi: 10.1002/prot.22230. [DOI] [PubMed] [Google Scholar]
- Lapelosa M., Arnold G.F., Gallicchio E., Arnold E., Levy R.M. J. Mol. Biol. 2010;397:752–766. doi: 10.1016/j.jmb.2010.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapelosa M., Gallicchio E., Arnold G.F., Arnold E., Levy R.M. J. Mol. Biol. 2009;385:675–691. doi: 10.1016/j.jmb.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie T.B., Drohan W.N., Smith-Gill S.J. J. Immunol. 1992;148:503–513. [PubMed] [Google Scholar]
- Lawrence M.C., Colman P.M. J. Mol. Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Lee M., Lloyd P., Zhang X., Schallhorn J.M., Sugimoto K., Leach A.G., Sapiro G., Houk K.N. J. Org. Chem. 2006;71:5082–5092. doi: 10.1021/jo052659z. [DOI] [PubMed] [Google Scholar]
- Li Y., Li H., Yang F., Smith-Gill S.J., Mariuzza R.A. Nat. Struct. Biol. 2003;10:482–488. doi: 10.1038/nsb930. [DOI] [PubMed] [Google Scholar]
- Liang S., Zheng D., Zhang C., Zacharias M. BMC Bioinformatics. 2009;10:302. doi: 10.1186/1471-2105-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippow S.M., Tidor B. Curr. Opin. Biotechnol. 2007;18:305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippow S.M., Wittrup K.D., Tidor B. Nat. Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looger L.L., Hellinga H.W. J. Mol. Biol. 2001;307:429–445. doi: 10.1006/jmbi.2000.4424. [DOI] [PubMed] [Google Scholar]
- Ma B.Y., Nussinov R. Curr. Opin. Chem. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- MacCallum R.M., Martin A.C., Thornton J.M. J. Mol. Biol. 1996;262:732–745. doi: 10.1006/jmbi.1996.0548. [DOI] [PubMed] [Google Scholar]
- MacDonald J.T., Maksimiak K., Sadowski M.I., Taylor W.R. Proteins. 2010;78:1311–1325. doi: 10.1002/prot.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcatili P., Rosi A., Tramontano A. Bioinformatics. 2008;24:1953–1954. doi: 10.1093/bioinformatics/btn341. [DOI] [PubMed] [Google Scholar]
- Martin A.C., Cheetham J.C., Rees A.R. Proc. Natl Acad. Sci. USA. 1989;86:9268–9272. doi: 10.1073/pnas.86.23.9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Chandana Epa V., Colman P.M. J. Mol. Biol. 1997;268:570–584. doi: 10.1006/jmbi.1997.0987. [DOI] [PubMed] [Google Scholar]
- Michnick S.W., Sidhu S.S. Nat. Chem. Biol. 2008;4:326–329. doi: 10.1038/nchembio0608-326. [DOI] [PubMed] [Google Scholar]
- Misura K.M., Baker D. Proteins. 2005;59:15–29. doi: 10.1002/prot.20376. [DOI] [PubMed] [Google Scholar]
- Mohan S., Sinha N., Smith-Gill S.J. Biophys. J. 2003;85:3221–3236. doi: 10.1016/S0006-3495(03)74740-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morea V., Lesk A.M., Tramontano A. Methods. 2000;20:267–279. doi: 10.1006/meth.1999.0921. [DOI] [PubMed] [Google Scholar]
- Morea V., Tramontano A., Rustici M., Chothia C., Lesk A.M. Biophys. Chem. 1997;68:9–16. doi: 10.1016/s0301-4622(96)02266-1. [DOI] [PubMed] [Google Scholar]
- Morea V., Tramontano A., Rustici M., Chothia C., Lesk A.M. J. Mol. Biol. 1998;275:269–294. doi: 10.1006/jmbi.1997.1442. [DOI] [PubMed] [Google Scholar]
- Murphy P.M., Bolduc J.M., Gallaher J.L., Stoddard B.L., Baker D. Proc. Natl Acad. Sci. USA. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nakajima N., Nakamura H., Kidera A. J. Phys. Chem. 1997;101:817–824. [Google Scholar]
- Narayanan A., Sellers B.D., Jacobson M.P. J. Mol. Biol. 2009;388:941–953. doi: 10.1016/j.jmb.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Nelson A.L., Reichert J.M. Nat. Biotechnol. 2009;27:331–337. doi: 10.1038/nbt0409-331. [DOI] [PubMed] [Google Scholar]
- Neuberger M.S. Immunol. Cell Biol. 2008;86:124–132. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- Nogi T., Sangawa T., Tabata S., Nagae M., Tamura-Kawakami K., Beppu A., Hattori M., Yasui N., Takagi J. Protein Sci. 2008;17:2120–2126. doi: 10.1110/ps.038299.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North B., Lehmann A., Dunbrack R.L., Jr J. Mol. Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G., Guenaga F.J., Schief W.R., Skinner J., Baker D., Wyatt R., Kwong P.D. Proc. Natl Acad. Sci. USA. 2010a;107:17880–17887. doi: 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G., McKee K., Yang Y., et al. J. Virol. 2010b;84:2955–2962. doi: 10.1128/JVI.02257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K., Takada S. Proc. Natl Acad. Sci. USA. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva B., Bates P.A., Querol E., Aviles F.X., Sternberg M.J. J. Mol. Biol. 1998;279:1193–1210. doi: 10.1006/jmbi.1998.1847. [DOI] [PubMed] [Google Scholar]
- Oomen C.J., Hoogerhout P., Bonvin A.M., Kuipers B., Brugghe H., Timmermans H., Haseley S.R., van Alphen L., Gros P. J. Mol. Biol. 2003;328:1083–1089. doi: 10.1016/s0022-2836(03)00377-2. [DOI] [PubMed] [Google Scholar]
- Ota M., Isogai Y., Nishikawa K. Protein Eng. 2001;14:557–564. doi: 10.1093/protein/14.8.557. [DOI] [PubMed] [Google Scholar]
- Ozbabacan S.E., Engin H.B., Gursoy A., Keskin O. Protein Eng. Des. Sel. 2011;24:635–648. doi: 10.1093/protein/gzr025. [DOI] [PubMed] [Google Scholar]
- Patten P.A., Gray N.S., Yang P.L., Marks C.B., Wedemayer G.J., Boniface J.J., Stevens R.C., Schultz P.G. Science. 1996;271:1086–1091. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- Pei X.Y., Holliger P., Murzin A.G., Williams R.L. Proc. Natl Acad. Sci. USA. 1997;94:9637–9642. doi: 10.1073/pnas.94.18.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perchiacca J.M., Bhattacharya M., Tessier P.M. Proteins. 2011;79:2637–2647. doi: 10.1002/prot.23085. [DOI] [PubMed] [Google Scholar]
- Ponomarenko J.V., Bourne P.E. BMC Struct. Biol. 2007;7:64. doi: 10.1186/1472-6807-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp C.S., Pollack R.M. Proteins. 2005;60:103–109. doi: 10.1002/prot.20492. [DOI] [PubMed] [Google Scholar]
- Reczko M., Martin A.C., Bohr H., Suhai S. Protein Eng. 1995;8:389–395. doi: 10.1093/protein/8.4.389. [DOI] [PubMed] [Google Scholar]
- Reddy S.T., Georgiou G. Curr. Opin. Biotechnol. 2011;22:584–589. doi: 10.1016/j.copbio.2011.04.015. [DOI] [PubMed] [Google Scholar]
- Reichert J.M. Curr. Pharm. Biotechnol. 2008;9:423–430. doi: 10.2174/138920108786786358. [DOI] [PubMed] [Google Scholar]
- Retter I., Althaus H.H., Munch R., Muller W. Nucleic Acids Res. 2005;33:D671–674. doi: 10.1093/nar/gki088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann L., Clark M., Waldmann H., Winter G. Nature. 1988;332:323–327. doi: 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- Rinaudo C.D., Telford J.L., Rappuoli R., Seib K.L. J. Clin. Invest. 2009;119:2515–2525. doi: 10.1172/JCI38330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C.A., Strauss C.E., Misura K.M., Baker D. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg A.S. AAPS J. 2006;8:E501–507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M., Goldblum A. Curr. Pharm. Des. 2006;12:3973–3997. doi: 10.2174/138161206778743655. [DOI] [PubMed] [Google Scholar]
- Rossi K.A., Nayeem A., Weigelt C.A., Krystek S.R., Jr J. Comput. Aided Mol. Des. 2009;23:411–418. doi: 10.1007/s10822-009-9274-3. [DOI] [PubMed] [Google Scholar]
- Rossi K.A., Weigelt C.A., Nayeem A., Krystek S.R., Jr Protein Sci. 2007;16:1999–2012. doi: 10.1110/ps.072887807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W.P., Ranganathan R. Curr. Opin. Struct. Biol. 2002;12:447–452. doi: 10.1016/s0959-440x(02)00346-9. [DOI] [PubMed] [Google Scholar]
- Schymkowitz J.W., Rousseau F., Martins I.C., Ferkinghoff-Borg J., Stricher F., Serrano L. Proc. Natl Acad. Sci. USA. 2005;102:10147–10152. doi: 10.1073/pnas.0501980102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers B.D., Nilmeier J.P., Jacobson M.P. Proteins. 2010;78:2490–2505. doi: 10.1002/prot.22757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers B.D., Zhu K., Zhao S., Friesner R.A., Jacobson M.P. Proteins. 2008;72:959–971. doi: 10.1002/prot.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Ding F., Dokholyan N.V. Front. Biosci. 2008;13:4795–4807. doi: 10.2741/3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai H., Kidera A., Nakamura H. FEBS Lett. 1999;455:188–197. doi: 10.1016/s0014-5793(99)00821-2. [DOI] [PubMed] [Google Scholar]
- Shirai H., Kidera A., Nakamura H. FEBS Lett. 1996;399:1–8. doi: 10.1016/s0014-5793(96)01252-5. [DOI] [PubMed] [Google Scholar]
- Shirai H., Nakajima N., Higo J., Kidera A., Nakamura H. J. Mol. Biol. 1998;278:481–496. doi: 10.1006/jmbi.1998.1698. [DOI] [PubMed] [Google Scholar]
- Shire S.J., Shahrokh Z., Liu J. J. Pharm. Sci. 2004;93:1390–1402. doi: 10.1002/jps.20079. [DOI] [PubMed] [Google Scholar]
- Sidhu S.S., Fellouse F.A. Nat. Chem. Biol. 2006;2:682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- Simonelli L., Beltramello M., Yudina Z., Macagno A., Calzolai L., Varani L. J. Mol. Biol. 2010;396:1491–1507. doi: 10.1016/j.jmb.2009.12.053. [DOI] [PubMed] [Google Scholar]
- Sinha N., Li Y., Lipschultz C.A., Smith-Gill S.J. Cell Biochem. Biophys. 2007;47:361–375. doi: 10.1007/s12013-007-0031-8. [DOI] [PubMed] [Google Scholar]
- Sinha N., Mohan S., Lipschultz C.A., Smith-Gill S.J. Biophys. J. 2002;83:2946–2968. doi: 10.1016/S0006-3495(02)75302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar A., Chaudhury S., Kilambi K.P., Berrondo M., Gray J.J. Proteins. 2010;78:3115–3123. doi: 10.1002/prot.22765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar A., Gray J.J. PLoS Comput. Biol. 2010;6:e1000644. doi: 10.1371/journal.pcbi.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar A., Kim E.T., Gray J.J. Nucleic Acids Res. 2009;37:W474–479. doi: 10.1093/nar/gkp387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasubramanian A., Chao G., Pressler H.M., Wittrup K.D., Gray J.J. Structure. 2006;14:401–414. doi: 10.1016/j.str.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Sivasubramanian A., Maynard J.A., Gray J.J. Proteins. 2008;70:218–230. doi: 10.1002/prot.21595. [DOI] [PubMed] [Google Scholar]
- Sivasubramanian A., Sircar A., Chaudhury S., Gray J.J. Proteins. 2009;74:497–514. doi: 10.1002/prot.22309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlind E., Strandberg L., Jirholt P., et al. Nat. Biotechnol. 2000;18:852–856. doi: 10.1038/78458. [DOI] [PubMed] [Google Scholar]
- Soga S., Kuroda D., Shirai H., Kobori M., Hirayama N. Protein Eng. Des. Sel. 2010;23:441–448. doi: 10.1093/protein/gzq014. [DOI] [PubMed] [Google Scholar]
- Soto C.S., Fasnacht M., Zhu J., Forrest L., Honig B. Proteins. 2008;70:834–843. doi: 10.1002/prot.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield R.L., Takimoto-Kamimura M., Rini J.M., Profy A.T., Wilson I.A. Structure. 1993;1:83–93. doi: 10.1016/0969-2126(93)90024-b. [DOI] [PubMed] [Google Scholar]
- Tartaglia G.G., Cavalli A., Pellarin R., Caflisch A. Protein Sci. 2005;14:2723–2734. doi: 10.1110/ps.051471205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyakov A., Obmolova G., Carton J.M., Gao W., Zhao Y., Gilliland G.L. Mol. Immunol. 2010a;47:2422–2426. doi: 10.1016/j.molimm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Teplyakov A., Obmolova G., Rogers A., Gilliland G.L. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010b;66:229–232. doi: 10.1107/S1744309109054141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielges M.C., Zimmermann J., Yu W., Oda M., Romesberg F.E. Biochemistry. 2008;47:7237–7247. doi: 10.1021/bi800374q. [DOI] [PubMed] [Google Scholar]
- Thorpe I.F., Brooks C.L., III Proc. Natl Acad. Sci. USA. 2007;104:8821–8826. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thullier P., Huish O., Pelat T., Martin A.C. J. Mol. Biol. 2010;396:1439–1450. doi: 10.1016/j.jmb.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Tonegawa S. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Tramontano A., Chothia C., Lesk A.M. Proteins. 1989;6:382–394. doi: 10.1002/prot.340060405. [DOI] [PubMed] [Google Scholar]
- Tramontano A., Chothia C., Lesk A.M. J. Mol. Biol. 1990;215:175–182. doi: 10.1016/S0022-2836(05)80102-0. [DOI] [PubMed] [Google Scholar]
- Tramontano A., Lesk A.M. Proteins. 1992;13:231–245. doi: 10.1002/prot.340130306. [DOI] [PubMed] [Google Scholar]
- Trovato A., Seno F., Tosatto S.C. Protein Eng. Des. Sel. 2007;20:521–523. doi: 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel M.H.V. Methods. 1996;9:465–472. [Google Scholar]
- Vargas-Madrazo E., Paz-Garcia E. Proteins. 2002;47:250–254. doi: 10.1002/prot.10187. [DOI] [PubMed] [Google Scholar]
- Vargas-Madrazo E., Paz-Garcia E. J. Mol. Recognit. 2003;16:113–120. doi: 10.1002/jmr.613. [DOI] [PubMed] [Google Scholar]
- Ventura S., Zurdo J., Narayanan S., et al. Proc. Natl Acad. Sci. USA. 2004;101:7258–7263. doi: 10.1073/pnas.0308249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voynov V., Chennamsetty N., Kayser V., Helk B., Trout B.L. MAbs. 2009;1:580–582. doi: 10.4161/mabs.1.6.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Schueler-Furman O., Andre I., London N., Fleishman S.J., Bradley P., Qian B., Baker D. Proteins. 2007;69:758–763. doi: 10.1002/prot.21684. [DOI] [PubMed] [Google Scholar]
- Wang N., Smith W.F., Miller B.R., Aivazian D., Lugovskoy A.A., Reff M.E., Glaser S.M., Croner L.J., Demarest S.J. Proteins. 2009;76:99–114. doi: 10.1002/prot.22319. [DOI] [PubMed] [Google Scholar]
- Wang T., Duan Y. Protein Eng. Des. Sel. 2011;24:649–657. doi: 10.1093/protein/gzr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Singh S.K., Kumar S. Pharm. Res. 2010;27:1512–1529. doi: 10.1007/s11095-010-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.S., Fukunishi H., Nakamura H. BIOPHYSICS. 2006;2:1–12. doi: 10.2142/biophysics.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemayer G.J., Wang L.H., Patten P.A., Schultz P.G., Stevens R.C. J. Mol. Biol. 1997;268:390–400. doi: 10.1006/jmbi.1997.0974. [DOI] [PubMed] [Google Scholar]
- Whitelegg N., Rees A.R. Methods Mol. Biol. 2004;248:51–91. doi: 10.1385/1-59259-666-5:51. [DOI] [PubMed] [Google Scholar]
- Whitelegg N.R., Rees A.R. Protein Eng. 2000;13:819–824. doi: 10.1093/protein/13.12.819. [DOI] [PubMed] [Google Scholar]
- Wong S.E., Sellers B.D., Jacobson M.P. Proteins. 2011;79:821–829. doi: 10.1002/prot.22920. [DOI] [PubMed] [Google Scholar]
- Worn A., Pluckthun A. Biochemistry. 1998;37:13120–13127. doi: 10.1021/bi980712q. [DOI] [PubMed] [Google Scholar]
- Worn A., Pluckthun A. Biochemistry. 1999;38:8739–8750. doi: 10.1021/bi9902079. [DOI] [PubMed] [Google Scholar]
- Worn A., Pluckthun A. J. Mol. Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- Yokota A., Tsumoto K., Shiroishi M., Kondo H., Kumagai I. J. Biol. Chem. 2003;278:5410–5418. doi: 10.1074/jbc.M210182200. [DOI] [PubMed] [Google Scholar]
- Zacharias M. Curr. Opin. Struct. Biol. 2010;20:180–186. doi: 10.1016/j.sbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Zemlin M., Klinger M., Link J., Zemlin C., Bauer K., Engler J.A., Schroeder H.W., Jr, Kirkham P.M. J. Mol. Biol. 2003;334:733–749. doi: 10.1016/j.jmb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K., Pincus D.L., Zhao S., Friesner R.A. Proteins. 2006;65:438–452. doi: 10.1002/prot.21040. [DOI] [PubMed] [Google Scholar]
- Zimmermann J., Oakman E.L., Thorpe I.F., Shi X., Abbyad P., Brooks C.L., III, Boxer S.G., Romesberg F.E. Proc. Natl Acad. Sci. USA. 2006;103:13722–13727. doi: 10.1073/pnas.0603282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J., Romesberg F.E., Brooks C.L., III, Thorpe I.F. J. Phys. Chem. B. 2010;114:7359–7370. doi: 10.1021/jp906421v. [DOI] [PMC free article] [PubMed] [Google Scholar]