

Abstract
Escherichia coli is a common Gram-negative organism that causes bacteremia. Prc, a bacterial periplasmic protease, and its homologues are known to be involved in the pathogenesis of Gram-negative bacterial infections. The present study examined the role of Prc in E. coli bacteremia and characterized the ability of the prc mutant of the pathogenic E. coli strain RS218 to cause bacteremia and survive in human serum. The prc mutant of RS218 exhibited a decreased ability to cause a high level of bacteremia and was more sensitive to serum killing than strain RS218. This sensitivity was due to the mutant's decreased ability to avoid the activation of the antibody-dependent and -independent classical complement cascades as well as its decreased resistance to killing mediated by the membrane attack complex, the end product of complement system activation. The demonstration of Prc in the evasion of classical complement-mediated serum killing of pathogenic E. coli makes this factor a potential target for the development of therapeutic and preventive measures against E. coli bacteremia.
INTRODUCTION
Escherichia coli is one of the most common Gram-negative organisms that cause bacteremia (35). The mortality and morbidity associated with E. coli bacteremia and sepsis remain substantial (23, 24, 35). This is due mainly to our incomplete understanding of the microbial factors contributing to bacteremia and the underlying mechanisms by which the pathogen causes bacteremia. Thus, the identification and characterization of bacterial factors that contribute to the survival of E. coli in the bloodstream are critical for an understanding of the pathogenesis of E. coli bacteremia as well as the development of preventive and therapeutic interventions against this disease.
Prc (also named Tsp) and its homologues are bacterial factors shown to be involved in the pathogenesis of several Gram-negative bacterial infections. The Prc of Salmonella enterica serovar Typhimurium as well as the Prc homologue CtpA of Brucella suis and Burkholderia mallei have been shown to be required for the survival of these pathogens within macrophages and for full virulence in mice (3–5, 11). However, the role of Prc in the pathogenesis of E. coli infection remains to be elucidated. Prc, originally identified in E. coli as a periplasmic protease, has been shown to be responsible for the C-terminal processing of a periplasmic protein, penicillin-binding protein 3 (PBP-3), in E. coli and to selectively degrade proteins with nonpolar C termini in vitro (16, 21, 22, 37). In addition, E. coli with an inactivated prc gene exhibits periplasmic protein leakage suggestive of increased outer membrane (OM) permeability (16), which may be responsible for the mutant's growth defect under conditions of osmotic stress (low osmolarity) at 42°C and its increased susceptibility to multiple antibiotics (16, 36). In this study, we demonstrate a new function of Prc in pathogenic E. coli, namely, its involvement in the bacterium's evasion of complement attack.
To cause bacteremia, pathogenic bacteria must escape from serum killing, which is mediated mainly by the complement system. The activation of the complement cascade in serum can be triggered by the classical, alternative, and mannose-binding lectin (MBL) pathways (45). These pathways converge at the action of depositing the C3 derivative C3b onto the target pathogens, leading to the activation of the common terminal complement pathway and the formation of the membrane attack complex (MAC), which results in the killing of the pathogen. The activation of the classical complement pathway is initiated by the deposition of a complement component, C1q, onto the surface of target bacteria. C1q deposition could be antibody dependent or independent; i.e., C1q deposition can occur through C1 binding to the Fc portion of the antibody, which binds to the antigen on the bacterial surface, or C1q can bind directly to the bacterial surface.
In the present study, we report that Prc is involved in E. coli evasion of serum killing that is mediated by the classical complement pathway, resulting in a high level of bacteremia. This finding suggests that Prc is a potential target for the prevention and therapy of invasive diseases caused by E. coli.
MATERIALS AND METHODS
Bacterial strains.
E. coli K1 strain RS218 (O18:K1:H7) is a cerebrospinal fluid isolate from a neonate with meningitis (2, 38). The spontaneous streptomycin-resistant mutant of RS218 and its derivatives were used in this study (Table 1). The prc and lacZ deletion mutants of RS218 were constructed by a PCR-based method described previously (9). Primers NK-prc-F and NK-prc-R were used for the prc deletion, while NK-lacZ-F and NK-lacZ-R were used for the lacZ deletion (Table 1).
Table 1.
E. coli strains, primers, and plasmids used in this study
| Strain, plasmid, or primer | Relevant information or sequence | Reference(s) |
|---|---|---|
| E. coli strains | ||
| RS218 | E. coli K1 RS218 isolated from the cerebrospinal fluid of a neonate with meningitis | 43, 49 |
| L346 | RS218 with a lacZ deletion | This study |
| L365 | RS218 with a prc lacZ double deletion | This study |
| Δprc-RS218 | RS218 with a prc deletion | This study |
| Plasmids | ||
| pCL1920 | Low-copy-no. plasmid | 26, 42 |
| pCL1920-prc | pCL1920 harboring the prc gene, which is under the control of the promoter on the plasmid | This study |
| pTR147 | pTrc99A harboring a DNA fragment encoding C-terminally His6-tagged Prc | 39 |
| pTR163 | pTrc99A harboring a DNA fragment encoding the C-terminally His6-tagged Prc K455A variant | 39 |
| Primers | ||
| NK-prc-F | 5′-AGGCTTACCGCGTTAGCTGGCCTGCTTGCAATAGCAGGCCAGACCTTCGCCATATGAATATCCTCCTTAG-3′ | This study |
| NK-prc-R | 5′-CAAGCTTCGCCAGATCGAGTGCGATATTCACCGTCTCATCCAGATAAGGAGTGTAGGCTGGAGCTGCTTC-3′ | This study |
| NK-lacZ-F | 5′-GTATTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCATATCAATATCCTCCTTAG-3′ | This study |
| NK-lacZ-R | 5′-GACACCAGACCAACTGGTAATGGTAGCGACCGGCGCTCAGCTGGAATTCCGTGTAGGCTGGAGCTGCTTC-3′ | This study |
Human sera, rabbit OmpA antiserum, C1q, IgG, IgM, and C3b.
The normal human serum (NHS) used in this study was pooled from the sera of 10 healthy adults and stored in aliquots at −80°C. Heat-inactivated NHS (HI-NHS) was prepared by heating NHS at 56°C for 30 min. The C1q-depleted and factor B-depleted sera and human IgG and IgM were purchased from Calbiochem (Darmstadt, Germany), and purified C1q and C3b were purchased from Complement Technology, Inc. (Tyler, TX). To prepare the MBL pathway-inhibited serum, NHS was treated with 100 mM mannose (31). The rabbit OmpA antiserum was described previously (43).
The procedures for the collection of human serum were approved by the Institutional Reviewer Board (IRB) of National Cheng Kung University Hospital, Tainan City, Taiwan (approval no. ER-98-143). Informed consents were obtained from healthy volunteers according to the relevant guidelines of the IRB.
Preparation of the IgG/IgM double-depleted serum.
One milliliter of 60% NHS diluted with phosphate-buffered saline (PBS) was incubated with 600 μl of anti-human IgM agarose (Sigma-Aldrich, St. Louis, MO) at 4°C for 1 h. The resulting serum was then incubated with 600 μl of recombinant protein G-Sepharose (Invitrogen, Grand Island, NY) at 4°C for 1 h. More than 90% of the IgG and IgM was removed in the double-depleted serum based on Western blot analyses (data not shown).
Mouse serum and mouse model of E. coli bacteremia.
Normal mouse serum (NMS) was collected from 8-week-old BALB/c mice. Heat-inactivated NMS (HI-NMS) was prepared by heating NMS at 56°C for 30 min.
E. coli bacteremia was induced in 8-week-old BALB/c mice by the intraperitoneal injection of 2 × 107 CFU/mouse. For coinfections, each mouse was inoculated with a mixture of equal numbers (1 × 107 CFU) of two bacterial strains. The two bacteria in the blood were differentiated by colors (blue and white) after cultivation on LB agar containing 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and 20 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). The lower detection limit of these experiments was 33 CFU/ml of blood. Thus, for statistical analysis, this value (33 CFU/ml) was assigned to blood samples with undetectable bacterial counts.
All of the experimental procedures involving animals adhered to the rules of the Animal Protection Act of Taiwan and were approved by the Institutional Animal Care and Use Committee (IACUC) of National Cheng Kung University, Tainan City, Taiwan (approval no. 99228). All surgery was performed under chloral hydrate anesthesia, and all efforts were made to minimize any suffering.
Serum survival assay.
E. coli cells used in these assays were obtained from cultures grown overnight for 16 h in LB medium. The bacteria (1 × 106 CFU) were incubated at 37°C in 100 μl of 60% serum diluted with PBS. After different lengths of time, the survival of the bacteria was determined by culturing on LB agar. For coincubation experiments, a mixture of two bacterial strains, each at 5 × 105 CFU, was incubated in 100 μl of 60% NHS. The two E. coli strains were differentiated as described above for the mouse coinfection experiments. Because it was reported previously that the process used to delete complement components in serum may chelate calcium and magnesium irons, which are the ions required for the activation of the complement system (1), the C1q-depleted and factor B-depleted sera (Calbiochem) were supplemented with 5 mM CaCl2 and 2 mM MgCl2 and then diluted with PBS.
For the serum survival assays with inhibitor peptide treatment, the inhibitor peptide (GRGYALAA), which was synthesized on an automatic peptide synthesizer, was purchased from Kelowna International Scientific, Inc. (Taipei City, Taiwan). The peptide was dissolved in PBS before incubation with bacteria. The bacteria were grown in 2 ml LB medium containing 300 μM peptide for 16 h at 37°C. In the serum survival assays, the bacteria were incubated in serum containing 300 μM peptide.
Flow cytometry analysis.
E. coli cells (3 × 107 CFU) were incubated at 37°C in 100 μl of 40% NHS diluted with Veronal buffer (Lonza, Walkersville, MD) or in 100 μl of Veronal buffer containing 25 μg/ml of C1q protein for different time periods. After 3 washes with PBS, the bacteria were incubated with antibodies against the MAC (rabbit IgG from Abcam, Cambridge, MA), C1q (goat IgG from Calbiochem), C4bp (sheep IgG from Abcam), IgG (goat IgG from Calbiochem), or IgM (rabbit IgG from Abcam) at room temperature for 30 min, followed by 3 washes with PBS. The primary-antibody-labeled bacteria were then incubated with their corresponding fluorescein isothiocyanate (FITC)-conjugated secondary antibodies for 30 min at room temperature, followed by 3 washes with PBS. To determine the deposition of C3b, an FITC-conjugated anti-C3 antibody (Abcam) was used according to the above-mentioned protocol, without secondary antibody treatment. The surface deposition of the molecules was analyzed with a FACSCalibur flow cytometer (Becton Dickinson).
Purification and quantification of outer membrane proteins.
The outer membrane proteins of the E. coli strains were prepared as described previously (43). The bacterial cells used for this experiment were obtained from cultures grown overnight for 16 h in LB broth. The concentration of the purified outer membrane proteins was determined by using a Pierce BCA protein assay kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer's instructions.
Purification and quantification of the K1 capsule and LPS.
The lipopolysaccharides (LPSs) on the E. coli strains were purified according to methods described previously by Kariyawasam et al. (19). Purified LPS was analyzed by silver staining in polyacrylamide gels, as described previously (13).
The purification of the K1 capsule from E. coli was performed as described previously by Vermeulen et al. (44), while the quantification of the purified K1 capsule by colorimetric assays was performed as described previously by Warren (46).
Overexpression and purification of Prc, and Prc proteolysis assays.
To overexpress and purify the Prc protease, E. coli strain DH5α transformed with pTR147, which encodes C-terminally His6-tagged Prc (39), was induced with 0.1 mM IPTG for 5 h at 37°C. The overexpressed recombinant protein was purified by immobilized metal affinity chromatography, as described previously (39).
For the Prc proteolysis assays, 3 μg of substrate (β-casein, IgG, C1q, or C3b) and 2 μg of Prc were incubated in 30 μl of reaction buffer (20 mM Tris-HCl [pH 8.0]) at 37°C for 2.5 h (39). β-Casein was purchased from Sigma-Aldrich. For the serum survival assays with the addition of the purified Prc protease, 0.67 μg/ml of Prc was added to the mixture of the bacteria and 60% NHS.
RNase leakage analysis.
Bacterial strains were initially grown on LB plates. After culturing overnight, isolated bacterial colonies were stab inoculated onto an RNase test agar plate, as described previously (34). The RNase test agar plate was incubated at 30°C for 48 h. The leaked periplasmic RNase from bacteria would cause pick halos around the colonies of the bacteria (16).
Statistical analysis.
For the mouse model of E. coli bacteremia, independent infections were analyzed by using an unpaired, nonparametric Mann-Whitney test, and coinfection experiments were analyzed by using a nonparametric Wilcoxon matched-pair test (27). For the remaining experiments, Student's t test was used. A P value of <0.05 was set as the threshold for statistical significance.
RESULTS
The prc mutant of the pathogenic E. coli K1 strain RS218 is defective in inducing a high degree of bacteremia.
To determine whether prc plays a role in E. coli bacteremia, a prc deletion mutant was generated in E. coli K1 strain RS218. This prc mutant (Δprc-RS218) and wild-type strain RS218 (WT-RS218) exhibited similar growths in LB broth at 37°C and 41°C (data not shown). However, as shown in Fig. 1A and B, Δprc-RS218 induced a significantly lower level of bacteremia in mice than did WT-RS218, while trans-complementation with prc restored the mutant's ability to induce bacteremia to the level of WT-RS218. When coinoculated with the wild-type strain in mice, Δprc-RS218 still exhibited a decreased ability to survive in the bloodstream, and this defect was also restored by the trans-complementation of prc (Fig. 1C and D). These results indicated that prc contributes to the induction of a high degree of E. coli bacteremia and that the coexistence of WT-RS218 cannot restore the ability of Δprc-RS218 to induce a high level of bacteremia.
Fig 1.

Effect of the prc deletion on the ability of E. coli to induce bacteremia. (A) Independent infections of mice with WT-RS218 and Δprc-RS218. Blood counts were enumerated at 8 h postinfection. (B) trans-Complementation of Δprc-RS218 with the prc gene in independent infection experiments. pCL1920 is the empty vector, while pCL1920-prc is the vector harboring the prc gene. (C) Coinfection of mice with equal numbers of the otherwise wild-type strain L346 (the lacZ deletion mutant of RS218) and Δprc-RS218. Blood counts were differentiated and enumerated at 8 h postinfection on LB agar containing IPTG and X-Gal. (D) trans-Complementation of Δprc-RS218 with the prc gene in coinfection experiments. Equal numbers of L365 (the lacZ prc double-deletion mutant of RS218) harboring pCL1920 and Δprc-RS218 harboring pCL1920-prc were coinoculated into mice. P values for independent infections were determined by using the Mann-Whitney test, while those for coinfections were determined by using the Wilcoxon matched-pair test. *, P values of <0.05; **, P values of <0.01. Bars indicate the median level of the blood bacterial count. The dotted lines indicate the lower limit of detection.
Prc is involved in E. coli resistance to serum killing.
To assess whether serum-mediated killing was responsible for the decreased ability of Δprc-RS218 to induce a high degree of bacteremia, WT-RS218 and Δprc-RS218 bacteria were incubated in normal mouse serum (NMS) or heat-inactivated normal mouse serum (HI-NMS), and survival was determined. Δprc-RS218 exhibited a decreased ability to survive in NMS compared to WT-RS218 after 3 h or 4 h of incubation, while both strains showed similar abilities to survive in HI-NMS, in which the complement system was inactivated (Fig. 2A). We further investigated the abilities of WT-RS218 and Δprc-RS218 to survive in normal human serum (NHS). Δprc-RS218 exhibited a more dramatically decreased ability to survive in NHS than did WT-RS218 after only 15 min of incubation, while both strains showed similar abilities to survive in heat-inactivated normal human serum (HI-NHS) (Fig. 2B). Moreover, trans-complementation with prc restored the ability of Δprc-RS218 to resist serum killing (Fig. 2C). When coincubated with the wild-type strain, Δprc-RS218 exhibited lower rates of survival in NHS (Fig. 2D). These results suggested that prc is involved in the resistance of E. coli to complement-mediated serum killing and that the coexistence of WT-RS218 does not restore the ability of Δprc-RS218 to survive in NHS.
Fig 2.

Effect of the prc deletion on the abilities of E. coli to resist serum killing. (A) Survival of WT-RS218 and Δprc-RS218 in 60% NMS or HI-NMS. (B) Survival of WT-RS218 and Δprc-RS218 in 60% NHS or HI-NHS. (C) Survival of Δprc-RS218 trans-complemented with pCL1920-prc or pCL1920 and of WT-RS218 containing pCL1920 in 60% NHS. (D) Survival of otherwise wild-type strain L346 and Δprc-RS218 coincubated in 60% NHS. For all results, the data are presented as the survival rates in the original inoculums. The data are representative of three independent experiments performed in triplicate and are shown as the means ± standard deviations.
The classical complement pathway is responsible for the serum killing of Δprc-RS218.
Because the serum killing of Δprc-RS218 occurred within 15 min of exposure to NHS (Fig. 2B), we further investigated the role of Prc in complement-mediated serum killing.
The levels of C3b and MAC deposition on bacteria reflect the intensity of the complement activation that occurred on the bacteria in NHS. As shown in Fig. 3A and B (see also Fig. S1A and S1B in the supplemental material), the levels of C3b and MAC deposition on Δprc-RS218 were significantly higher than those on WT-RS218 after incubation in NHS, demonstrating that complement activation was stronger on Δprc-RS218 than on WT-RS218.
Fig 3.

Interaction of WT-RS218 and Δprc-RS218 with complement components in human serum. (A, B, and F) Deposition of C3b (A), MAC (B), and C1q (F) on WT-RS218 and Δprc-RS218 in NHS. The levels of deposition were measured by flow cytometry after incubation of the bacteria in 40% NHS. The data are presented as the mean fluorescent intensity (MFI). The bacteria in the control group of the C3b deposition assay (A) were incubated in PBS instead of 40% NHS for 15 min and stained with the FITC-conjugated anti-C3 antibody. The bacteria in the control groups of MAC (B) and C1q (F) deposition assays were stained with the nonspecific isotypic antibodies of the corresponding primary antibodies after incubation in 40% NHS for 15 min. (C) Survival of E. coli strains after 15 min of incubation in 60% factor B-depleted human serum [FB(−)] or heat-inactivated factor B-depleted human serum [HI-FB(−)]. (D) Survival of the E. coli strains after 15 min of incubation in 60% mannose-treated NHS [MLB(−)] or heat-inactivated mannose-treated NHS [HI-MLB(−)]. (E) Survival of E. coli strains after 15 min of incubation in 60% C1q-depleted human serum with or without a supplement of 60% of the physiological concentration of purified C1q (the physiological concentration of C1q is about 80 μg/ml in NHS [18]). For panels C to E, the results are presented as relative survival rates compared to the survival rates of WT-RS218 in factor B-depleted serum, mannose-treated NHS, or C1q-depleted serum. For all of the results, the data shown are representative of three independent experiments performed in triplicate. The results are shown as the means ± standard deviations. *, P values of <0.05; **, P values of <0.01; ***, P values of <0.001.
To investigate which complement pathways are responsible for the serum killing of Δprc-RS218, the survival rates of WT-RS218 and Δprc-RS218 were examined and compared in factor B-depleted serum and mannose-treated NHS, in which the alternative and MBL pathways, respectively, are blocked (29, 31). The rate of survival of Δprc-RS218 was significantly lower than that of WT-RS218 in both sera, while the survival rates of the wild-type and mutant strains were similar in the two sera after heat inactivation (Fig. 3C and D). These results suggest that the alternative and MBL pathways may not play a major role in the serum killing of Δprc-RS218. Conversely, WT-RS218 and Δprc-RS218 exhibited similar survival rates in C1q-depleted human serum, in which the classical pathway is blocked (Fig. 3E). The reconstitution of this serum with a physiological concentration of C1q significantly decreased the survival rate of Δprc-RS218 but not that of WT-RS218 (Fig. 3E). Taken together, these results indicated that the classical complement pathway is mainly responsible for the serum killing of Δprc-RS218. This finding can be further corroborated by examining the level of C1q deposition on bacteria, which reflects the intensity of the classical complement pathway triggered by the bacteria in NHS. As shown in Fig. 3F (see also Fig. S1C in the supplemental material), Δprc-RS218 exhibited significantly higher levels of C1q deposition than did WT-RS218 in NHS, suggesting that Δprc-RS218 triggers a stronger activation of the classical complement pathway than the wild-type strain.
In addition, it is known that E. coli can recruit C4bp, a serum regulator of the classical complement pathway, on bacterial surfaces to suppress the activation of the classical complement pathway (33). However, we found that WT-RS218 and Δprc-RS218 exhibited similar levels of C4bp deposition in NHS (data not shown), suggesting that the decreased ability of Δprc-RS218 to survive in NHS is not related to the ability to recruit C4bp.
Δprc-RS218 triggers classical complement activation through both antibody-dependent and antibody-independent mechanisms.
The classical complement pathway can be initiated through antibody-dependent and/or -independent mechanisms. Antibody-dependent activation of the classical complement pathway is initiated by the binding of IgG or IgM, while antibody-independent activation is initiated by direct C1q deposition on the bacteria. We found that Δprc-RS218 exhibited significantly higher levels of IgG and IgM binding than did WT-RS218 in NHS (Fig. 4A and B; see also Fig. S1D in the supplemental material), suggesting that Δprc-RS218 may trigger a higher level of antibody-dependent activation in the classical complement pathway. In addition, the level of serum C1q deposition on the bacteria after incubation in NHS reflects the sum level of antibody-dependent and -independent C1q deposition. Therefore, to measure antibody-independent C1q deposition, the levels of C1q deposition on the bacteria were determined after incubation with purified C1q. As shown in Fig. 4C (see also Fig. S1E in the supplemental material), Δprc-RS218 recruited significantly higher levels of purified C1q than did WT-RS218, indicating that the mutant strain was able to cause a higher level of C1q binding on the surface, without the involvement of IgG and IgM. These results suggest that Δprc-RS218 triggers a higher level of classical pathway activation through both antibody-independent and -dependent mechanisms.
Fig 4.

Roles of IgG, IgM, and C1q in complement-mediated serum killing. (A) Deposition of serum IgG. The levels of IgG deposition were measured by flow cytometry after incubation of the bacteria in 40% NHS. The bacteria in the control group were stained with the nonspecific isotypic antibody of the corresponding primary antibody after incubation in 40% NHS for 15 min. (B) Deposition of serum IgM. The levels of IgM deposition were determined by Western blot assays of similar amounts of bacteria after incubation in 40% NHS. Because the deletion of prc did not affect the expression of the OmpA protein in E. coli (data not shown), OmpA served as a loading control for bacteria. The levels of OmpA were determined by Western blot analysis with rabbit OmpA antiserum. (C) Deposition of purified C1q. The levels of C1q deposition were measured by flow cytometry after incubation of the bacteria with purified C1q protein (25 μg/ml). The bacteria in the control group were stained with the nonspecific isotypic antibody of the corresponding primary antibody after incubation in 40% NHS for 15 min. For panels A and C, the data are presented as mean fluorescent intensities. (D) Survival of WT-RS218 and Δprc-RS218 after 15 min of incubation in 60% IgG/IgM double-depleted NHS with supplementation with different concentrations of purified IgG. A concentration of 4 mg/ml of IgG is about 60% of that in NHS (40). (E) Survival of WT-RS218 and Δprc-RS218 after 15 min of incubation in 60% IgG/IgM double-depleted NHS with supplementation with anti-C1q antibodies (10% heat-inactivated rabbit antiserum) to block direct C1q binding. For panels D and E, the data are presented as relative survival rates compared to the survival rates of WT-RS218. All of the data shown are representative of three independent experiments performed in triplicate. The results are shown as the means ± standard deviations. *, P values of <0.01; **, P values of <0.01; ***, P values of <0.001.
To further investigate the roles of IgG and IgM in the complement-mediated killing of Δprc-RS218, the survival of WT-RS218 and Δprc-RS218 in IgG/IgM double-depleted NHS was assessed (Fig. 4D). After 15 min of incubation, the rate of survival of Δprc-RS218 was significantly higher in depleted NHS (65% ± 4% of the survival of WT-RS218) (Fig. 4D) than in NHS (22% ± 7% of the survival of WT-RS218) (Fig. 2B), although the rate of survival of Δprc-RS218 was still significantly lower than that of WT-RS218 in depleted NHS. The resupplementation of purified IgG to the depleted NHS decreased the survival rate of Δprc-RS218 (Fig. 4D), while it did not significantly affect that of WT-RS218 (data not shown). The resupplementation of purified IgM in the double-depleted NHS did not affect the survival of WT-RS218 and Δprc-RS218 (data not shown). These results indicated that IgG is likely to be the major immunoglobulin responsible for the antibody-dependent complement-mediated killing of Δprc-RS218 in NHS.
In addition, it is known that anti-C1q antibodies are able to block the function of C1q (17). We found that the addition of anti-C1q antibodies to the depleted serum restored the survival of Δprc-RS218 to the level of WT-RS218 (Fig. 4E). This finding suggests that the serum killing mediated by antibody-independent C1q binding may be responsible for the lower serum survival rate of Δprc-RS218 (than that of WT-RS218) in the depleted serum (Fig. 4D). This result, along with the data shown in Fig. 4C, indicated that the activation of the classical complement pathway by an antibody-independent mechanism also contributed to the serum killing of Δprc-RS218.
Prc proteolytic activity dose not damage IgG, C1q, and C3b.
Because Prc is a protease, it is possible that the Prc proteolytic activity may directly damage antibodies and complement proteins in NHS to facilitate the serum survival of E. coli. To assess this possibility, we investigated whether the Prc protease cleaves IgG, C1q, and C3b by incubating these serum components with purified recombinant Prc. However, these components were not sensitive to Prc cleavage, while β-casein, which is a substrate of Prc (39), was sensitive to degradation by the Prc protease (positive control) (Fig. 5). We also found that the addition of the purified Prc protease did not affect the survival of WT-RS218 and Δprc-RS218 in NHS (data not shown). These results suggest that the proteolytic activity of Prc does not directly affect IgG or complement components to facilitate the E. coli evasion of complement-mediated killing.
Fig 5.
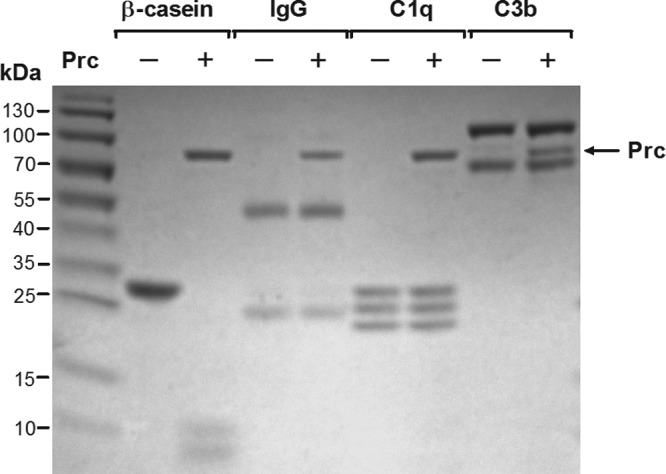
SDS-PAGE analysis of Prc proteolytic activity on β-casein, IgG, C1q, or C3b. Proteolysis assays were performed by using the C-terminally His6-tagged Prc protease with β-casein, IgG, C1q, or C3b as the substrate. Reaction mixtures were incubated at 37°C for 2.5 h and subjected to SDS-PAGE analysis. The molecular masses of β-casein and the recombinant Prc protease are 25.3 kDa and 75.2 kDa, respectively. Note that IgG contains light (25 kDa) and heavy (55 kDa) chains. C1q contains A (26 kDa), B (25 kDa), and C (24 kDa) chains. C3b contains α (101 kDa) and β (75 kDa) chains.
Deletion of prc changes the outer membrane protein profile of E. coli and increases bacterial sensitivity to MAC.
The components of the bacterial envelope, including the capsule, lipopolysaccharides (LPSs), and outer membrane (OM) protein (OMP), have been shown to be involved in the resistance of E. coli to complement-mediated serum killing (32, 33). We found that WT-RS218 and Δprc-RS218 expressed similar levels of K1 capsule (data not shown) and O18 LPS (Fig. 6A), suggesting that these two components are not likely to be involved in the decreased ability of Δprc-RS218 to survive in serum. However, as shown in Fig. 6B, WT-RS218 and Δprc-RS218 exhibited distinct OMP profiles. In addition, the peptide GRGYALAA was shown previously to be a competitive inhibitor of the Prc protease in vitro (6). The incubation of WT-RS218 with the inhibitor caused this strain's OMP profile to change, which partially resembled the change in the OMP profile of Δprc-RS218. For example, a protein close to 70 kDa, which apparently appeared in the OM of WT-RS218 but was undetectable in that of Δprc-RS218, almost disappeared in the OM of inhibitor-treated WT-RS218 bacteria. These results suggest that the deletion of prc is likely to affect the properties of the OM.
Fig 6.

Effects of the prc deletion on LPS, OMP, and MAC sensitivity of E. coli. (A) LPS of WT-RS218 and Δprc-RS218. The LPS samples derived from equal amounts of bacteria were analyzed by silver staining after separation by SDS-PAGE. (B) Silver-stained SDS-PAGE gels of OMPs of WT-RS218, Δprc-RS218, and pCL1920-prc-transformed Δprc-RS218 as well as of WT-RS218 bacteria grown with 300 μM the inhibitor peptide GRGYALAA. The arrow indicates a protein band close to 70 kDa that was differentially expressed in the OMs of Δprc-RS218 and WT-RS218 with or without inhibitor treatment. (C) WT-RS218 and Δprc-RS218 exhibited similar levels of MAC deposition after incubation in 80% and 10% NHS for 15 min (experimental group). The levels of MAC deposition were measured by flow cytometry and are presented as mean fluorescent intensities. The bacteria in the control group were stained with the nonspecific isotypic antibody of the corresponding primary antibody after incubation in NHS for 15 min. (D) Survival of WT-RS218 and Δprc-RS218 incubated in 80% and 10% NHS for 15 min. The results of serum survival are presented as relative survival rates compared to the survival rates of WT-RS218. All of the data shown are representative of three independent experiments performed in triplicate. The results are shown as the means ± standard deviations. *, P values of <0.05.
Because the OM is where the MAC, the final product of the complement cascade, exerts its bactericidal effect, MAC deposition and serum survival were examined and compared between WT-RS218 and Δprc-RS218. As shown in Fig. 6C (see also Fig. S1F in the supplemental material), the level of MAC deposition on the mutant after 15 min of incubation with 10% NHS was similar to that on the wild type after incubation with 80% NHS. However, despite the similar levels of MAC deposition, the rate of survival of Δprc-RS218 in 10% NHS was decreased to 62% of the WT-RS218 survival rate in 80% NHS (Fig. 6D). This result suggests that Δprc-RS218 may be more sensitive to MAC-mediated killing than WT-RS218.
We also found that, like the prc mutant of E. coli K-12 (16), Δprc-RS218 showed a leakage of periplasmic RNase (see Fig. S2 in the supplemental material), suggesting that the deletion of prc increases the OM permeability of pathogenic E. coli RS218.
The protease function of Prc is critical for the serum survival of E. coli.
We next investigated whether the protease function of Prc contributes to the serum survival of E. coli. As shown in Fig. 7A, the preincubation of WT-RS218 with the inhibitor peptide GRGYALAA decreased the serum survival of the bacteria in NHS but not in HI-NHS (Fig. 7B). Preincubation with the inhibitor peptide did not significantly affect the serum survival of Δprc-RS218 in NHS and HI-NHS (data not shown). Furthermore, the Prc residue Lys-455 is essential for the catalytic activity of this protease. Prc with an alanine substitution at this position has no protease activity but still has a structure and a substrate-binding ability similar to those of the wild-type protein (20, 39). As shown in Fig. 7C, the serum survival of Δprc-RS218 trans-complemented with plasmid pTR147, which encodes wild-type Prc, was restored to the level of WT-RS218, while the serum survival rate of Δprc-RS218 trans-complemented with plasmid pTR163, which encodes the K455A variant of Prc, was significantly lower than that of WT-RS218. Collectively, these results indicated that the protease function of Prc is critical for E. coli resistance to serum killing mediated by the classical complement pathway.
Fig 7.

Effect of the protease activity of Prc on serum survival of E. coli. (A) Survival of WT-RS218 preincubated with or without the inhibitor peptide GRGYALAA in NHS. (B) Survival of WT-RS218 preincubated with or without the inhibitor peptide in HI-NHS. For panels A and B, the results of serum survival are presented as the survival rates of the original inoculums. (C) Survival of Δprc-RS218 strains that were complemented with plasmid pTR147 or pTR163 after incubation in 60% NHS for 15 min. The results of serum survival are presented as relative survival rates compared to the survival rates of WT-RS218. All of the data shown are representative of three independent experiments performed in triplicate. The results are shown as the means ± standard deviations. *, P values of <0.05; ***, P values of <0.001.
DISCUSSION
This study is the first to demonstrate that Prc is critical for pathogenic E. coli to induce a high degree of bacteremia. Our results also support the involvement of Prc in the resistance of E. coli to serum killing mediated by the complement system. Without Prc, E. coli triggered a higher level of activation of both the antibody-dependent and -independent classical complement pathway and became more sensitive to MAC deposition in NHS. Serum killing mediated by the classical complement pathway is likely to play a major role in clearing the E. coli prc mutant in the bloodstream, because the classical pathway-mediated bactericidal effect on Δprc-RS218 appeared within 15 min of exposure to NHS. The loss of the protease function of Prc is likely to contribute to this phenotype of the prc mutant.
It seems unlikely that the prc mutant is deficient in the production or secretion of a bacterial factor in the bloodstream to facilitate the evasion of complement-mediated killing by E. coli. This is supported by our finding that coinfection with WT-RS218 did not restore the ability of Δprc-RS218 to survive in NHS and the bloodstream. Also, it is unlikely that the Prc protease directly cleaves antibodies or complement components to facilitate bacterial evasion of serum killing, because the addition of this protease in NHS did not affect the serum survival of WT-RS218 and Δprc-RS218, and the protease did not proteolytically damage IgG, C1q, and C3b. However, as a periplasmic protease, Prc may act on target periplasmic proteins and/or OMPs that are directly or indirectly involved in regulating the properties of the OM after processing by this periplasmic protease. This concept is supported by our finding that the prc deletion causes the alteration of the OMP profiles of E. coli as well as our findings and those described previously by Hara et al. showing that the deletion of prc increases E. coli OM permeability (16). The altered OM properties are likely to be responsible for the decreased ability of Δprc-RS218 to avoid activating the classical complement cascades as well as this mutant's increased susceptibility to MAC-mediated killing.
The OM is where the complement system initiates its activation and exerts its bactericidal activity. The altered OM properties of Δprc-RS218 may increase the accessibility of the bacterial binding targets of IgG and C1q, allowing higher levels of IgG and C1q binding to trigger the antibody-dependent and antibody-independent activation of the classical pathway. In addition, the MAC-mediated bactericidal effect occurs through disruption of the OM, thereby increasing its permeability and then inducing lethal changes in the inner membrane (28, 48). The prc mutant's increased OM permeability may facilitate MAC-mediated bactericidal activity, thus increasing the mutant's susceptibility to the MAC.
IgM was less likely to be important for the complement-mediated killing of Δprc-RS218, as the resupplementation of IgM did not affect the survival of Δprc-RS218 in IgM/IgG-depleted serum, although this mutant recruited a higher level of IgM binding than did WT-RS218 in NHS. It is likely that the level of IgM binding to the mutant was not sufficient to trigger significant classical complement-mediated killing in NHS. This notion may be consistent with our finding that the difference in IgM binding between WT-RS218 and Δprc-RS218 could be detected only by Western blot analysis and not by flow cytometry analysis (data not shown). Flow cytometry analysis measures IgM binding at the level of each individual bacterium, while Western blot analysis measures the sum total of large numbers of bacteria. Thus, after the incubation of the bacteria with NHS, the level of serum IgM binding on each bacterial cell may be below the detection limit of the flow cytometer.
Two lines of our results support that the protease function of Prc contributes to the serum survival of E. coli: (i) the serum survival of Δprc-RS218 in NHS was not restored by trans-complementation with a plasmid encoding the Prc K455A variant, which has no protease activity (Fig. 7C), and (ii) the preincubation of WT-RS218 with the peptide inhibitor of the Prc protease decreased the serum survival of WT-RS218 in NHS (Fig. 7A). However, the inhibitor peptide's effect is relatively small compared to the marked reduction of serum survival with the prc deletion. Consistently, the peptide partially changed the OMP profile of WT-RS218, compared to the change caused by the deletion of prc. The peptide inhibitor acts by competing for the protease with its substrates (6). It is likely that this inhibitor's affinity for Prc may not be strong enough to compete with the protease substrates in E. coli so that the inhibitory effect of the peptide is modest in E. coli. Alternatively, the peptide may not effectively enter the bacteria to access Prc in the periplasmic space. Nevertheless, this finding suggests the concept that the Prc protease function can be inhibited by the addition of an inhibitor in E. coli.
The increased emergence of antibiotic resistance among E. coli strains has led to a desperate need for new measures to treat or prevent invasive diseases caused by E. coli. The deletion and inhibition of Prc rendered E. coli more susceptible to serum killing, and Prc may be a novel target for developing such measures against E. coli bacteremia. prc was also shown previously to be common in clinical isolates of E. coli, and the inactivation of prc increases the susceptibilities of E. coli to multiple antibiotics (36). Thus, a drug able to inhibit the protease function of Prc may facilitate the host's complement system and increase the efficiency of antibiotics in eliminating invading E. coli bacteria.
Mutations in the tolA and tolB genes of bacteria have also been shown to change bacterial OM permeability and induce increased bacterial susceptibility to complement-mediated killing. The Tol-Pal system, which is well conserved among Gram-negative bacteria (41), consists of at least five interacting envelope proteins, TolQ, TolR, TolA, TolB, and Pal (lipoprotein) (15). Because the Tol-Pal system is involved in maintaining OM integrity, mutations in the component genes of this system are commonly associated with increased OM permeability (12, 25, 47). In addition, a tolA deletion in E. coli and S. Typhimurium and a tolB deletion in S. Typhimurium were shown previously to cause increased bacterial susceptibility to complement-mediated killing (8, 14, 30). Therefore, it was proposed that the Tol-Pal system is a potential target in the development of novel attenuated live vaccines against Gram-negative pathogens (30). The Prc mutant of E. coli strains may also be a candidate for an attenuated vaccine. In addition to the attenuated phenotypes identified in the present study, the greater susceptibility of the Prc mutant to antibiotics and its growth defect at 42°C under conditions of low osmolarity (16) may be attractive properties for controlling and monitoring the attenuated strains as potential vaccines.
The substrate-binding and catalytic activities of the Prc protease are separated in distinct domains in the protein (7, 20). A block of the function of the two domains may lead to the inactivation of the protease activity (or the two sites are the candidate target for blocking the protease activity). It was found that three of the Prc residues (Ser-430, Asp-411, and Lys-455) are essential for the catalytic activity (20). Based on this finding, it was proposed that Prc may belong to the family of serine proteases that use a serine-lysine dyad mechanism for catalysis (20). However, Prc is not sensitive to serine protease inhibitors, such as diisopropylfluorophosphate (DFP) and phenylmethylsulfonyl fluoride (PMSF) (20). The substrate-binding site of Prc is located in a PDZ domain (amino acids 206 to 334) (7). PDZ domains span about 100 amino acids and were initially found in eukaryotic proteins, in which they are involved in protein-protein interactions (10). The PDZ domain of Prc binds to the nonpolar C-terminal sequence of the substrates (21, 37). Because the inhibitor peptide GRGYALAA contains a nonpolar C-terminal sequence and is able to block the protease function of Prc, we speculate that the peptide is able to compete for the PDZ domain of Prc with the substrates (6).
In conclusion, we identified the involvement of the Prc protease in E. coli bacteremia. Prc contributed to the E. coli evasion of classical complement-mediated serum killing. These findings suggest that Prc is a potential target for the development of therapeutic and/or preventive strategies against invasive diseases caused by E. coli. Studies are in progress to investigate whether altered OMPs are involved in serum killing and also to identify the substrates of the Prc protease, which will facilitate the identification of novel inhibitors of this protease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alastair R. Hawkins for kindly providing plasmids pTR147 and pTR163.
This work was supported by the National Science Council, Taiwan (grant no. NSC99-2320-B-006-005-MY3), and the Multidisciplinary Center of Excellence for Clinical Trial and Research, Department of Health, Executive Yuan, Taiwan (grant no. DOH100-TD-B-111-002).
Footnotes
Published ahead of print 23 July 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abdullah M, et al. 2005. Killing of dsrA mutants of Haemophilus ducreyi by normal human serum occurs via the classical complement pathway and is initiated by immunoglobulin M binding. Infect. Immun. 73:3431–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Achtman M, et al. 1983. Six widespread bacterial clones among Escherichia coli K1 isolates. Infect. Immun. 39:315–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bandara AB, et al. 2008. A disruption of ctpA encoding carboxy-terminal protease attenuates Burkholderia mallei and induces partial protection in CD1 mice. Microb. Pathog. 45:207–216 [DOI] [PubMed] [Google Scholar]
- 4. Bandara AB, Sriranganathan N, Schurig GG, Boyle SM. 2005. Carboxyl-terminal protease regulates Brucella suis morphology in culture and persistence in macrophages and mice. J. Bacteriol. 187:5767–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baumler AJ, Kusters JG, Stojiljkovic I, Heffron F. 1994. Salmonella typhimurium loci involved in survival within macrophages. Infect. Immun. 62:1623–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beebe KD, Pei D. 1998. A continuous fluorimetric assay for tail-specific protease. Anal. Biochem. 263:51–56 [DOI] [PubMed] [Google Scholar]
- 7. Beebe KD, et al. 2000. Substrate recognition through a PDZ domain in tail-specific protease. Biochemistry 39:3149–3155 [DOI] [PubMed] [Google Scholar]
- 8. Bowe F, et al. 1998. At least four percent of the Salmonella typhimurium genome is required for fatal infection of mice. Infect. Immun. 66:3372–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fanning AS, Anderson JM. 1996. Protein-protein interactions: PDZ domain networks. Curr. Biol. 6:1385–1388 [DOI] [PubMed] [Google Scholar]
- 11. Fields PI, Swanson RV, Haidaris CG, Heffron F. 1986. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc. Natl. Acad. Sci. U. S. A. 83:5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fognini-Lefebvre N, Lazzaroni JC, Portalier R. 1987. tolA, tolB and excC, three cistrons involved in the control of pleiotropic release of periplasmic proteins by Escherichia coli K12. Mol. Gen. Genet. 209:391–395 [DOI] [PubMed] [Google Scholar]
- 13. Fomsgaard A, Freudenberg MA, Galanos C. 1990. Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J. Clin. Microbiol. 28:2627–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaspar JA, Thomas JA, Marolda CL, Valvano MA. 2000. Surface expression of O-specific lipopolysaccharide in Escherichia coli requires the function of the TolA protein. Mol. Microbiol. 38:262–275 [DOI] [PubMed] [Google Scholar]
- 15. Godlewska R, Wisniewska K, Pietras Z, Jagusztyn-Krynicka EK. 2009. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol. Lett. 298:1–11 [DOI] [PubMed] [Google Scholar]
- 16. Hara H, Yamamoto Y, Higashitani A, Suzuki H, Nishimura Y. 1991. Cloning, mapping, and characterization of the Escherichia coli prc gene, which is involved in C-terminal processing of penicillin-binding protein 3. J. Bacteriol. 173:4799–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herpers BL, et al. 2009. Hemolytic assay for the measurement of functional human mannose-binding lectin: a modification to avoid interference from classical pathway activation. J. Immunol. Methods 343:61–63 [DOI] [PubMed] [Google Scholar]
- 18. Ikeda F, et al. 1998. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J. Immunol. 161:5712–5719 [PubMed] [Google Scholar]
- 19. Kariyawasam S, Wilkie BN, Hunter DB, Gyles CL. 2002. Systemic and mucosal antibody responses to selected cell surface antigens of avian pathogenic Escherichia coli in experimentally infected chickens. Avian Dis. 46:668–678 [DOI] [PubMed] [Google Scholar]
- 20. Keiler KC, Sauer RT. 1995. Identification of active site residues of the Tsp protease. J. Biol. Chem. 270:28864–28868 [DOI] [PubMed] [Google Scholar]
- 21. Keiler KC, Sauer RT. 1996. Sequence determinants of C-terminal substrate recognition by the Tsp protease. J. Biol. Chem. 271:2589–2593 [DOI] [PubMed] [Google Scholar]
- 22. Keiler KC, et al. 1995. C-terminal specific protein degradation: activity and substrate specificity of the Tsp protease. Protein Sci. 4:1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim KS. 2001. Escherichia coli translocation at the blood-brain barrier. Infect. Immun. 69:5217–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim KS. 2003. Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat. Rev. Neurosci. 4:376–385 [DOI] [PubMed] [Google Scholar]
- 25. Lazzaroni JC, Portalier R. 1992. The excC gene of Escherichia coli K-12 required for cell envelope integrity encodes the peptidoglycan-associated lipoprotein (PAL). Mol. Microbiol. 6:735–742 [DOI] [PubMed] [Google Scholar]
- 26. Lerner CG, Inouye M. 1990. Low copy number plasmids for regulated low-level expression of cloned genes in Escherichia coli with blue/white insert screening capability. Nucleic Acids Res. 18:4631 doi:10.1093/nar/18.15.4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lloyd AL, Smith SN, Eaton KA, Mobley HL. 2009. Uropathogenic Escherichia coli suppresses the host inflammatory response via pathogenicity island genes sisA and sisB. Infect. Immun. 77:5322–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. MacKay SL, Dankert JR. 1990. Bacterial killing and inhibition of inner membrane activity by C5b-9 complexes as a function of the sequential addition of C9 to C5b-8 sites. J. Immunol. 145:3367–3371 [PubMed] [Google Scholar]
- 29. Matsumoto M, et al. 1997. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc. Natl. Acad. Sci. U. S. A. 94:8720–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paterson GK, et al. 2009. Deletion of tolA in Salmonella Typhimurium generates an attenuated strain with vaccine potential. Microbiology 155:220–228 [DOI] [PubMed] [Google Scholar]
- 31. Petersen SV, et al. 2000. Control of the classical and the MBL pathway of complement activation. Mol. Immunol. 37:803–811 [DOI] [PubMed] [Google Scholar]
- 32. Pluschke G, Mayden J, Achtman M, Levine RP. 1983. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. Infect. Immun. 42:907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC. 2002. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J. Immunol. 169:6352–6360 [DOI] [PubMed] [Google Scholar]
- 34. Quaas R, Landt O, Grunert HP, Beineke M, Hahn U. 1989. Indicator plates for rapid detection of ribonuclease T1 secreting Escherichia coli clones. Nucleic Acids Res. 17:3318 doi:10.1093/nar/17.8.3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Russo TA, Johnson JR. 2003. Medical and economic impact of extraintestinal infections due to Escherichia coli: focus on an increasingly important endemic problem. Microbes Infect. 5:449–456 [DOI] [PubMed] [Google Scholar]
- 36. Seoane A, Sabbaj A, McMurry LM, Levy SB. 1992. Multiple antibiotic susceptibility associated with inactivation of the prc gene. J. Bacteriol. 174:7844–7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Silber KR, Keiler KC, Sauer RT. 1992. Tsp: a tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc. Natl. Acad. Sci. U. S. A. 89:295–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silver RP, Aaronson W, Sutton A, Schneerson R. 1980. Comparative analysis of plasmids and some metabolic characteristics of Escherichia coli K1 from diseased and healthy individuals. Infect. Immun. 29:200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spiers A, et al. 2002. PDZ domains facilitate binding of high temperature requirement protease A (HtrA) and tail-specific protease (Tsp) to heterologous substrates through recognition of the small stable RNA A (ssrA)-encoded peptide. J. Biol. Chem. 277:39443–39449 [DOI] [PubMed] [Google Scholar]
- 40. Stoop JW, Zegers BJ, Sander PC, Ballieux RE. 1969. Serum immunoglobulin levels in healthy children and adults. Clin. Exp. Immunol. 4:101–112 [PMC free article] [PubMed] [Google Scholar]
- 41. Sturgis JN. 2001. Organisation and evolution of the tol-pal gene cluster. J. Mol. Microbiol. Biotechnol. 3:113–122 [PubMed] [Google Scholar]
- 42. Tadokoro A, et al. 2004. Interaction of the Escherichia coli lipoprotein NlpI with periplasmic Prc (Tsp) protease. J. Biochem. 135:185–191 [DOI] [PubMed] [Google Scholar]
- 43. Teng CH, et al. 2010. NlpI contributes to Escherichia coli K1 strain RS218 interaction with human brain microvascular endothelial cells. Infect. Immun. 78:3090–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vermeulen C, Cross A, Byrne WR, Zollinger W. 1988. Quantitative relationship between capsular content and killing of K1-encapsulated Escherichia coli. Infect. Immun. 56:2723–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walport MJ. 2001. Complement. First of two parts. N. Engl. J. Med. 344:1058–1066 [DOI] [PubMed] [Google Scholar]
- 46. Warren L. 1959. The thiobarbituric acid assay of sialic acids. J. Biol. Chem. 234:1971–1975 [PubMed] [Google Scholar]
- 47. Webster RE. 1991. The tol gene products and the import of macromolecules into Escherichia coli. Mol. Microbiol. 5:1005–1011 [DOI] [PubMed] [Google Scholar]
- 48. Wright SD, Levine RP. 1981. How complement kills E. coli. I. Location of the lethal lesion. J. Immunol. 127:1146–1151 [PubMed] [Google Scholar]
- 49. Xie Y, Yao Y, Kolisnychenko V, Teng CH, Kim KS. 2006. HbiF regulates type 1 fimbriation independently of FimB and FimE. Infect. Immun. 74:4039–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.