

Abstract
The crystal structure of the β2-adrenergic receptor in complex with an agonist and its cognate G protein has just recently been solved. It is now possible to explore in molecular detail the means by which this paradigmatic transmembrane receptor binds agonist, communicates the impulse or signalling event across the membrane and sets in motion a series of G protein-directed intracellular responses. The structure was determined using crystals of the ternary complex grown in a rationally designed lipidic mesophase by the so-called in meso method. The method is proving to be particularly useful in the G protein-coupled receptor field where the structures of thirteen distinct receptor types have been solved in the past five years. In addition to receptors, the method has proven useful with a wide variety of integral membrane protein classes that include bacterial and eukaryotic rhodopsins, a light harvesting complex II (LHII), photosynthetic reaction centers, cytochrome oxidases, β-barrels, an exchanger, and an integral membrane peptide. This attests to the versatility and range of the method and supports the view that the in meso method should be included in the arsenal of the serious membrane structural biologist. For this to happen however, the reluctance in adopting it attributable, in part, to the anticipated difficulties associated with handling the sticky, viscous cubic mesophase in which crystals grow must be overcome. Harvesting and collecting diffraction data with the mesophase-grown crystals is also viewed with some trepidation. It is acknowledged that there are challenges associated with the method. Over the years, we have endeavored to establish how the method works at a molecular level and to make it user-friendly. To these ends, tools for handling the mesophase in the pico- to nano-liter volume range have been developed for highly efficient crystallization screening in manual and robotic modes. Methods have been implemented for evaluating the functional activity of membrane proteins reconstituted into the bilayer of the cubic phase as a prelude to crystallogenesis. Glass crystallization plates have been built that provide unparalleled optical quality and sensitivity to nascent crystals. Lipid and precipitant screens have been designed for a more rational approach to crystallogenesis such that the method can now be applied to an even wider variety of membrane protein types. In this Current Topics article, these assorted advances are outlined along with a summary of the membrane proteins that have yielded to the method. The prospects for and the challenges that must be overcome to further develop the method are described.
Keywords: complex, crystallization, enzyme, G protein-coupled receptor, G protein, high-throughput, in meso method, lipid metabolism, lipidic cubic phase, macromolecular X-ray crystallography, membrane protein structure, mesophase, robot, structure-function, transporter
INTRODUCTION
Whenever the lead author teaches a course on Introductory Biochemistry he suggests that the students come up with their own definition of biochemistry and encourages them to reflect on that definition at the end of term in light of the material covered. At the same time, he offers his own somewhat contrived definition, an abbreviated version of which is that biochemistry is the scientific endeavour which seeks to establish how an organism produces the right chemicals in the right place at the right time and in the right amounts. It is contrived in the sense that it is used to alert the students to the range of topics that will be covered. The definition includes the word ‘how’ which is there to introduce the concepts of mechanism and function. By relating these to structure it is possible to engage the students in a discussion of how structure relates to function or activity, and to describe the myriad ways in which biomolecular structure is revealed.
In that discussion of structure and function the treatment is usually broadened to include the work of Louis Sullivan who, in reference to buildings and architecture, introduced the dictum “form ever follows function.” The concept is nicely illustrated by considering the amphitheatres and odeons of old, designed for effective communication of one or a few with many. In the biological world, the form, or structure, and function relationship can be viewed in a similar light. However, the directionality is a little different in that usually what we seek, as biochemists and structural biologists, is insight into function as revealed by knowing the structure of the corresponding biomolecule, usually a protein. By structure is meant the arrangement in 3-dimensional space and the connectivity of all (usually non-hydrogen) atoms in the protein. Insight has two primary uses. One is to establish the chemical, physical and structural or spatial bases of a particular function or activity. An activity could be a reaction catalyzed by an enzyme, the creation of an ion gradient, or the binding of a ligand leading to signal transduction by a receptor. The other is the exploitation of such understanding, for the purpose of rational drug design for example.
For the level of detail required, the structure revealing method most relevant to integral membrane proteins, the focus of this Current Topics article, is macromolecular X-ray crystallography (MX). To perform a successful MX study however, diffraction quality 3-dimensional crystals of the target protein are needed. There are several methods by which to crystallize membrane proteins and these are divided here into two major categories. The first and most successful, hereafter referred to as the in surfo method,1 was introduced some three decades ago.2 It uses surfactants to produce mixed micelles that incorporate the pure target protein, residual lipid if present, and detergent. These soluble protein-detergent complexes, with or without added small amphiphiles, are treated in essentially the same way as water soluble proteins for the production of crystals by vapor diffusion, counter-diffusion, micro-dialysis or batch methods. The target protein can originate from native membranes, from the membranes or inclusion bodies of recombinant organisms, from cell-free expression3,4 and from chemical synthesis.5
Difficulties in getting membrane proteins to crystallize by the in surfo approach have been attributed to inherent protein flexibility and to conformational inhomogeneity. At fault too can be the relatively diminutive polar surface that is simply too small to extend beyond the surfactant swath and to make lasting molecular contacts with neighboring proteins in the crystal. Shorter chain detergents and amphiphile additives have been used to advantage here.2,6 Protein fusions,7–23 antibodies,12,24–27 and designed ankyrin repeat protein binders28 have been introduced which also make good these deficits. They can be tailored, to a degree, to create stable protein-protein polar contacts within the crystal. Furthermore, by using high-affinity recombinant antibodies raised against a discontinuous epitope on the native protein surface, flexibility in the protein-antibody complex co-crystal is minimized and conformational homogeneity is favored. All contribute to producing a well-diffracting crystal. These technically challenging approaches have been used successfully in structure studies of cytochrome c oxidase,29 the cytochrome bc1 complex (with and without cytochrome c),24 ion channels,30–35 transporters,36 G-protein coupled receptors (GPCR),25,26,37 and a receptor-Gs protein complex.12
The second category of membrane protein crystallization methods includes those that make use of a lipidic bicontinuous mesophase,38–40 a discoidal lipid/detergent bicelle,41 or vesicle fusion.42 In all three cases, an extended bilayer composed of lipid, detergent and target protein is presumed to form. For this reason, these are referred to collectively here as the bilayer methods of membrane protein crystallization. Unfortunately, we know little of the molecular mechanism whereby crystals form by the assorted in surfo protocols. Accordingly, the possibility cannot be excluded that a bilayered structure also forms as an intermediate in the crystallization pathway that begins with a solution of mixed micelles in the in surfo method. The focus of the rest of this article is on the method which employs a lipidic mesophase, the cubic phase in particular. This is referred to hereafter as the in meso method. It is proving to be a useful approach for crystallizing a broad range of membrane protein types (Table 1), having had extraordinary success recently with GPCRs (Table 2). The cubic phase is a lyotropic liquid crystal.43,44 It consists of a highly curved lipid bilayer, the mid-plane of which is draped over a periodic minimal surface with cubic symmetry. The bilayer separates two interpenetrating but non-contacting aqueous channels. Both the aqueous and bilayer compartments are continuous in three dimensions. As a result, the mesophase is described as being bicontinuous (Figure 1).
Table 1.
In Meso Structures of Non-GPCR Membrane Proteins and Peptides
| Protein (Organism) [PDB Record Count]a | Protein Size (Number of Amino Acids)b | Detergentc,d | Protein Concentration (mg/mL)e | Hosting Lipidsc,f | PDB IDg (Resolution, Å) [Solvent Content, %] |
|---|---|---|---|---|---|
| β-Helix | |||||
| Gramicidin D (Brevibacillus brevis) [3] | 16 | Not Applicable | 130 | 7.7 MAG; 8.8 MAG; 9.9 MAG | 2Y5M (1.08) [52.3]; 2Y6N (1.26) [52.0]; 2XDC (1.70) [51.0] |
| β-Barrel | |||||
| ButB (Escherichia coli) [1] | 594 | LDAO | 15 | 9.9 MAG | 2GUF (1.95) [53.1] |
| OpcA (Neisseria meningitidis) [1] | 253 | DDM | 12 | 9.9 MAG | 2VDF (1.95) [43.0] |
| OmpF (Escherichia coli) [3] | 340 | DDM | 10–55 | 9.9 MAG | 3POQ (1.90) [52.0]; 3POX (2.00) [64.7]; 3POU (2.80) [61.2] |
| Intimin (Escherichia coli) [1] | 242 | LDAO | 20 | 9.9 MAG | 4E1S (1.86) [50.1] |
| Invasin (Yersinia pseudotuberculosis) [1] | 245 | LDAO | 20 | 9.9 MAG | 4E1T (2.26) [57.6] |
| α-helical | |||||
| Bacterorhodopsin (Halobacterium salinarum) [38] | 248 | OG | 10–30 | 9.9 MAG; β-XylOC16+4h | 1M0K (1.43) [42.1]; 1M0M (1.43) [41.3]; 1M0L (1.47) [41.3]; 1P8H (1.52) [30.8]; 2NTU (1.53) [44.1]; 2NTW (1.53) [44.1]; 1C3W (1.55) [53.2]; 1O0A (1.62) [42.4]; 1P8U (1.62) [30.9]; 1KGB (1.65) [46.9]; 1F50 (1.70) [46.7]; 3NS0 (1.78) [36.6]; 3NSB (1.78) [43.9]; 1C8R (1.80) [42.3]; 1F4Z (1.80) [46.7]; 1KG9 (1.81) [46.9]; 2I21 (1.84) [44.0]; 1P8I (1.86) [32.3]; 1QHJ (1.90) [48.0]; 1C8S (2.00) [54.2]; 1KG8 (2.00) [46.0]; 2I1X (2.00) [47.9]; 1JV6 (2.00) [43.7]; 1MGY (2.00) [43.7]; 3MBV (2.00) [44.7]; 2I20 (2.08) [44.1]; 1E0P (2.10) [51.0]; 1QKO (2.10) [48.0]; 1QKP (2.10) [48.0]; 2WJL (2.15) [33.1]; 1CWQ (2.25) [nulli]; 1JV7 (2.25) [52.1]; 1BRX (2.30) [43.1]; 2WJK (2.30) [35.5]; 1S8J (2.30) [48.7]; 1S8L (2.30) [47.2]; 1VJM (2.30) [46.7]; 1AP9 (2.35) [48.0] |
| Halorhodopsin (Halobacterium salinarum) [3] | 274 | OG | 8–10 | 9.9 MAG | 2JAF (1.70) [47.4];1E12 (1.80) [41.5]; 2JAG (1.93) [48.2] |
| Sensory Rhodopsin II (Natronomonas pharaonis) [6] | 217 | DDM | 11.2 | 9.9 MAG | 3QAP (1.90) [55.8]; 1H68 (2.10) [null]; 1GU8 (2.27) [53.0]; 1GUE (2.27) [53.0]; 1JGJ (2.40) [60.8]; 3QDC (2.50) [57.0] |
| Sensory Rhodopsin II/Transducer Complex (Natronomonas pharaonis) [3] | 225 + 82; 248 + 122; 248 + 163 | OG | Not Available | 11.7 MAG | 1H2S (1.93) [null]; 2F93 (2.00) [38.9]; 2F95 (2.20) [31.6] |
| Sensory Rhodopsin (Nostoc sp. pcc 7120) [1] | 261 | DDM | 9 | 9.9 MAG | 1XIO (2.00) [55.9] |
| Channelrhodopsin (Chlamydomonas reinhardtii) [1] | 333 | DDM | 10 | 9.9 MAG | 3UG9 (2.30) [50.6] |
| Acetabularia Rhodopsin II (Acetabularia acetabulum) [1] | 229 | DDM | 45.4 | 9.9 MAG +10%(w/w)Cholesterol | 3AM6 (3.20) [58.1] |
| Light Harvesting Complex II (Rhodoblastus acidophilus) [1] | 53 + 41 | LDAO | 10 | 9.9 MAG | 2FKW (2.45) [66.0] |
| Photosynthetic Reaction Center (Blastochloris viridis) [5] | 336 + 258 + 274 + 324 | LDAO | 20 – 25 | 9.9 MAG | 2WJN (1.86) [54.0]; 2WJM (1.95) [54.0]; 2X5U (3.00) [54.0]; 2X5V (3.00) [68.7]; 4AC5 (8.20) [64.0] |
| Photosynthetic Reaction Center (Rhodobacter sphaeroides) [4] | 235 + 281 +300; 250 + 281 + 307; 281 + 307 + 260; 281 + 307 + 260 | LDAO | 20 – 25 | 9.9 MAG | 2GNU (2.20) [51.6]; 1OGV (2.35) [55.6]; 2BNS (2.50) [59.1]; 2BNP (2.70) [55.6]; |
| ba3 Cytochrome c Oxidase (Thermus thermophilus) [2] | 569 + 168 +34 | DDM | 15 | 9.9 MAG | 3S8F (1.80) [60.8]; 3S8G (1.80) [61.7] |
| caa3 Cytochrome c Oxidase (Thermus thermophilus) [1] | 791 + 337 + 66 | DM | 10 | 7.7 MAG | 2YEV (2.36) |
| Na+-Ca2+ Exchanger (Methanocaldococcus jannaschii) [1] | 320 | DM | 10 | 9.9 MAG | 3V5U (1.90) [51.5] |
As of June 2012, the total count of non-GPCR structure records in the PDB attributable to the in meso method is 77.
Protein size refers to the number of amino acid residues in the protein. Where the protein consists of multiple subunits or protomers the number of residues in each is provided. Protein size information is listed in the same order as the corresponding PDB ID.
Abbreviations used for detergents and lipids: LDAO, lauryldimethylamine-N-oxide; DDM, n-dodecyl-β-D-maltopyranoside; DM, n-decyl-β-D-maltopyranoside; OG, n-octyl-β-D-glucopyranoside; b-XylOC16+4, 1-O-(3,7,11,15-tetramethylhexadecyl)-β–d-xyloside.
This refers to the detergent in which the protein was solubilized prior to its use in creating the protein-laden mesophase for in meso crystallogenesis.
Refers to the protein concentration of the solution used to prepare the hosting mesophase with one exception. In the case of gramicidin, concentration refers to the peptide concentration in the mesophase after reconstitution.
Refers to the composition of the lipid used to prepare the hosting mesophase. Where different hosting lipids are used with a single protein their identities are given in the same order as the corresponding PDB IDs.
PDB records are linked to the corresponding record in the Membrane Protein Data Bank (www.mpdb.tcd.ie) which links to the PDB and to other useful sites.
This lipid was used in the work associated with PDB ID: 3MBV.
Null is used in the PDB to indicate that the relevant information is not available.
Table 2.
In Meso Structures of G Protein-Coupled Receptors and Receptor Complexes
| Receptor (Pharmacological State) | Liganda | Covalent/Non-Covalent Partner | Detergent/Lipidb | Protein Concentrationc (mg/mL) | Hosting Lipidd | PDB IDe (Resolution, Å) [Solvent Content, %] |
|---|---|---|---|---|---|---|
| μ-Opioid (Inactive) | β-FNA | T4 Lysozyme | MNG-DDM/CHS | 30 | 9.9 MAG + 10 %(w/w) Cholesterol | 4DKL (2.80) [67.8] |
| κ-Opioid (Inactive) | JDTic | T4 Lysozyme | DDM/CHS | 40 | 9.9 MAG + 10 %(w/w) Cholesterol | 4DJH (2.90)[67.8] |
| Nociceptin (Inactive) | Compound-24 | Apo-cytochrome b562RIL | DDM/CHS | 40 | 9.9 MAG + 10 %(w/w) Cholesterol | 4EA3 (3.01) [48.4] |
| δ-Opioid (Inactive) | Naltrindole | T4 Lysozyme | MNG/CHS | 50 | 9.9 MAG + 10 %(w/w) Cholesterol | 4EJ4 (3.40) [69.3] |
| Sphingosine 1-Phosphate Subtype 1 (Inactive) | ML056 | T4 Lysozyme | DDM/CHS | 100 | 9.9 MAG + 10 %(w/w) Cholesterol | 3V2Y (2.80) [52.9] |
| ML056 | T4 Lysozyme | DDM/CHS | 100 | 9.9 MAG + 10 %(w/w) Cholesterol | 3V2W (3.35) [52.9] | |
| Muscarinic Acetylcholine M2 (Inactive) | QNB | T4 Lysozyme | MNG | 50 | 9.9 MAG + 10 %(w/w) Cholesterol | 3UON (3.00) [57.7] |
| Muscarinic Acetylcholine M3 (Inactive) | Tiotropium | T4 Lysozyme | MNG/CHS | 60 | 9.9 MAG + 10 %(w/w) Cholesterol | 4DAJ (3.40) [54.2] |
| Histamine H1 (Inactive) | Doxepin | T4 Lysozyme | DDM/CHS | 40 | 9.9 MAG + 10 %(w/w) Cholesterol | 3RZE (3.10) [60.0] |
| Dopamine D3 (Inactive) | Eticlopride | T4 Lysozyme | DDM/CHS | 30 | 9.9 MAG + 10 %(w/w) Cholesterol | 3PBL (3.15) [63.5] |
| CXCR4 Chemokine (Inactive) | IT1t | T4 Lysozyme | DDM/CHS | 60–70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3ODU (2.50) [55.8] |
| CVX15 | T4 Lysozyme | DDM/CHS | 60–70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3OE0 (2.90) [66.0] | |
| IT1t | T4 Lysozyme | DDM/CHS | 60–70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3OE8 (3.10) [55.8] | |
| IT1t | T4 Lysozyme | DDM/CHS | 60–70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3OE9 (3.10) [60.3] | |
| IT1t | T4 Lysozyme | DDM/CHS | 60–70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3OE6 (3.20) [58.0] | |
| A2A Adenosine (Inactive) | TBAf | Apo-cytochrome b562RIL | TBAf | TBAf | TBAf | 4EIY |
| ZM241385 | T4 Lysozyme | DDM/CHS | 70 | 9.9 MAG + 10 %(w/w) Cholesterol | 3EML (2.60) [56.79] | |
| A2A Adenosine (Active) | UK-432097 | T4 Lysozyme | DDM/CHS | 60 | 9.9 MAG + 10 %(w/w) Cholesterol | 3QAK (2.70) [58.11] |
| β2 Adrenergic (Inactive) | Carazolol | T4 Lysozyme | DDM/CHS | 50 | 9.9 MAG + 10 %(w/w) Cholesterol | 2RH1 (2.40) [59.98] |
| Timolol | T4 Lysozyme | DDM/CHS | 30 | 9.9 MAG + 10 %(w/w) Cholesterol | 3D4S (2.80) [47.36] | |
| Not Available | T4 Lysozyme | DDM/CHS | >60 | 9.9 MAG + 10 %(w/w) Cholesterol | 3NY9 (2.80) [48.68] | |
| ICI 118,551 | T4 Lysozyme | DDM/CHS | >60 | 9.9 MAG + 10 %(w/w) Cholesterol | 3NY8 (2.84) [49.02] | |
| Alprenolol | T4 Lysozyme | DDM/CHS | >60 | 9.9 MAG + 10 %(w/w) Cholesterol | 3NYA (3.10) [48.33] | |
| FAUC50 | T4 Lysozyme | MNG-3 | 50 | 9.9 MAG + 10 %(w/w) Cholesterol | 3PDS (3.50) [65.00] | |
| β2 Adrenergic - Gs Protein (Active) | BI-167107 | T4 Lysozyme/Nanobody 35 | MNG-3 | 50 | 9.9 MAG + 10 %(w/w) Cholesterol | 3SN6 (3.20) [60.05] |
| β2 Adrenergic (Active) | BI-167107 | T4 Lysozyme/Nanobody 80 | MNG-3 | Not Available | 7.7 MAG + 10 %(w/w) Cholesterol | 3P0G (3.50) [53.97] |
Ligands are reported as in the source article.
Abbreviations for detergents and lipids: DDM, n-dodecyl-β-D-maltopyranoside; CHS, cholesteryl hemi-succinate; MNG, maltose-neopentyl glycol.
Refers to the protein concentration in the solution used to prepare the hosting mesophase.
Refers to the composition of the lipid used to prepare the hosting mesophase.
PDB records are linked to the corresponding record in the Membrane Protein Data Bank (www.mpdb.tcd.ie) which links to the PDB and to other useful sites.
To be added upon release of coordinates in the PDB.
Figure 1.
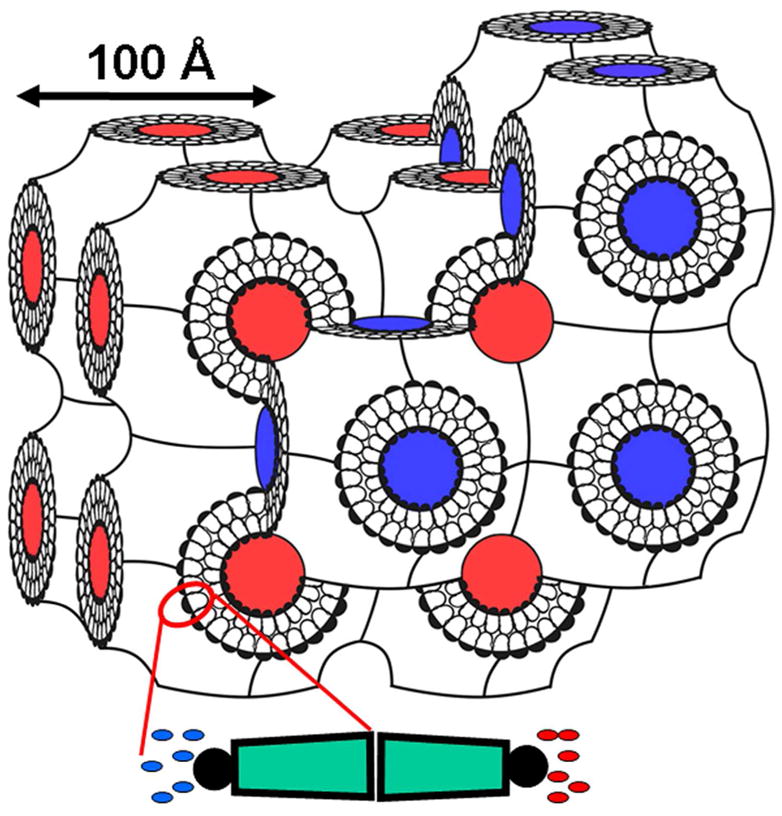
Cartoon representation of a bicontinuous lipidic cubic mesophase. At its simplest, the cubic phase is formed by homogenizing lipid, typically monoolein (9.9 MAG), and water in approximately equal parts at 20 °C. An expanded view of the lipid component that forms the continuous curved bilayer is shown at the bottom of the figure. Water channels, on either side of the bilayer, that interpenetrate but never contact one another as they permeate the mesophase, are colored blue and red for clarity. The lattice parameter of the cubic phase, in this case of space group Im3m, obtained using small-angle X-ray scattering, is indicated.48,131
The in meso method is particularly appealing because it offers the prospect that crystallization takes place from within a lipid bilayer, akin to the native environment encountered in a biomembrane. This is in contrast to the more traditional in surfo crystallogenesis methods which involve using potentially destabilizing surfactant micelles. Our research team, in the Membrane Structural and Functional Biology (MS&FB) Group, has been working on the in meso method since its introduction in the late 90’s.43 The thrust of our work in this area has three major themes. First, to decipher the molecular basis of in meso crystallogenesis. Second, to automate and miniaturize the method and to make it more user-friendly and generally accessible. And third, to use the method to solve the structures of membrane proteins that are critical to human health. In what follows, the origins of the in meso method, its development as a high-throughput technique, the membrane proteins that have yielded to it, and the prospects and the challenges ahead are described. Practical and strategic issues that the membrane protein crystallographer might consider ahead of launching into and during the course of in meso crystallization trials are summarized in Chart 1.
Chart 1.

Issues to consider when undertaking an in meso crystallogenesis study assuming little prior knowledge about the crystallization potential of the membrane protein target. Items with an asterisk should be given priority.
EXPERIMENTAL ASPECTS
Performing an in meso crystallization trial is simple and straightforward. Typically, it involves combining two parts protein solution with three parts lipid at 20 °C.45,46 The lipid most commonly used is the monoacylglycerol (MAG), monoolein (9.9 MAG)1. According to the monoolein/water phase diagram (Figure 2B),48 and assuming there is no major influence on phase behaviour of the protein solution components, this mixing process should generate, by spontaneous self-assembly, the cubic mesophase (Figure 2) at or close to full hydration. The pure cubic phase is colorless, optically isotropic (non-birefringent), transparent and viscous. The last three characteristics can be used conveniently to indicate that the proper phase has been accessed2.
Figure 2.

Temperature-composition phase diagrams for (A) 7.7 MAG and (B) 9.9 MAG, two monoacylglycerols that have proven to be particularly useful hosting lipids for the in meso crystallization of membrane proteins, complexes and peptides (Table 1). Cartoon representations of the different solid (Lc), liquid (FI) and liquid crystalline phases (Lα, HII, cubic-Pn3m, cubic-Ia3d) accessed in the temperature and composition range studied are shown along the top of the figure. The phase diagrams were constructed based on small-angle X-ray scattering measurements.48,133
The cubic phase is sticky and viscous akin to thick toothpaste. Without the proper tools, it is not particularly easy to handle. In the course of earlier lipid phase science work in the MS&FB Group we had developed tools (Figure 3) and procedures for preparing and manipulating such rheologically refractory materials. One of these, the coupled syringe mixing device49, was ideally suited to the task of combining micro-liter volumes of lipid with membrane protein solution in a way that produces protein-laden mesophase for direct use in crystallization trials with minimal waste and change in composition. The mixer consists of two Hamilton micro-syringes connected by a narrow bore coupler. Lipid is placed in one syringe, protein solution in the other. Mixing is achieved by repeatedly moving the contents of the two syringes back and forth through the coupler.46 The coupler is replaced by a needle for convenient dispensing of the homogenous mesophase into wells of custom-designed glass sandwich crystallization plates.50,51 Precipitant solutions of varying compositions are placed over the mesophase and the wells are sealed with a cover-glass. The plates are incubated at 20 °C and monitored for crystal growth. Optical quality is the best it can be given that the mesophase is sandwiched between two glass plates and the mesophase itself, ideally, is transparent. This means that crystals, just a few micrometres in size, can be seen readily with a good quality microscope, whether or not the protein is coloured. The use of cross-polarizers can help with visualizing small crystals which usually appear birefringent on a dark background; as noted, the cubic phase itself is optically isotropic and non-birefringent. If the target protein is naturally colored or has been labelled with a colored or a fluorescent tag,52,53 visibility will be enhanced. However, it is important to stress that crystals of colorless protein are readily seen in sandwich plates (Figure 4).45,46 An added feature of the glass sandwich plate is that the double-sided tape used to create the wells provides almost hermetic sealing ensuring minimal change in well contents during the course of trials that can last for months. Step-by-step instructions, complete with an on-line video demonstration of the entire in meso crystallization process just described, have been published.45,46 Additional video articles illustrating harvesting (Li, D., Boland, C., Aragao, D., Walsh, K., and Caffrey, M. Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. In review) and use of a robot (Li, D., Boland, C., Walsh, K., and Caffrey, M. Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. In press) are in preparation.
Figure 3.

The tools and supplies used to set up in meso crystallization trials in manual mode. Full details of the in meso method are available in print45 and in an online video.46 Key: A. Laboratory notebook. B. Temperature-composition phase diagram. C. Milli-Q water. D. Methanol. E. Paper towels. F. Pipeting devices covering volumes in the microliter range. G. Hamilton syringes (removable needle type, gas-tight) of varying sizes (10 and 100 μL usually). H. Narrow bore coupler. I. Repeat dispenser. J. Screwdriver. K. Glass slides and cover slips. L. Perforated double stick tape. M. Tweezers. N. Coupled syringes loaded with lipid and proteins solution, as indicated.
Figure 4.

Crystals of membrane proteins in mesophases prepared with different hosting lipids. Examples of proteins and peptides are shown that either did not produce crystals or the crystals that grew were of lesser diffraction quality in the benchmark lipid, 9.9 MAG (monoolein), compared to the identified MAG. Diffraction quality (in brackets) is identified as is crystal growth temperature if other than 20 °C. Images for the gramicidin and β2AR-Gs complex are from references 74 and 12, respectively. In all cases, the protein is colorless but the crystals are clearly visible in the hosting mesophase in wells of the glass sandwich crystallization plate.
PROPOSED MOLECULAR MECHANISM
The Model
A proposal has been advanced for how in meso crystallogenesis takes place at the molecular level (Figure 5).1,43,44,54 It begins typically with an isolated biological membrane that is treated with detergent to solubilize the target protein. The protein-detergent complex is purified by standard wet-lab biochemical methods. Homogenizing with a MAG effects a uniform reconstitution of the purified protein into the bilayer of the cubic phase. As noted, the latter is bicontinuous in the sense that both the aqueous and bilayer compartments are continuous in 3-dimensional space (Figure 1). Upon reconstitution, the protein ideally retains its native conformation and activity and has partial or complete mobility within the plane of the cubic phase bilayer. A precipitant is added to the mesophase, which triggers a local alteration in mesophase properties that include phase identity, microstructure, long-range order and phase separation. Under conditions leading to crystallization, one of the separated phases is enriched in protein, which nucleates and develops into a bulk crystal. The hypothesis envisions a local lamellar phase that acts as a medium in which nucleation and 3-dimensional crystal growth occur. Molecular dynamics simulations highlight the hydrophobic/hydrophilic mismatch between the protein and the surrounding bilayer in the lamellar phase as a driving force for oligomerization in the membrane plane.55 The local lamellar phase also serves as a conduit or portal for proteins on their way from the cubic phase reservoir to the growing face of the crystal. Initially at least, the proteins leave the lamellar conduit and ratchet into the developing crystal to generate a layered-type (Type I) 2 packing of protein molecules (Figures 5 and 6). Given that proteins reconstitute across the bilayer of the cubic phase with no preferred orientation and the 3-dimensional continuity of the mesophase, it is possible for the resulting crystals to be polar or nonpolar.44 These correspond to situations in which adjacent proteins in a layer have their long-axis director oriented in the same or in the opposite directions.
Figure 5.

Cartoon representation of the events proposed to take place during the crystallization of an integral membrane protein from the lipidic cubic mesophase. The process begins with the protein reconstituted into the curved bilayer of the “bicontinuous” cubic phase (tan). Added “precipitants” shift the equilibrium away from stability in the cubic membrane. This leads to phase separation wherein protein molecules diffuse from the bicontinuous bilayered reservoir of the cubic phase into a sheet-like or lamellar domain (A) and locally concentrate therein in a process that progresses to nucleation and crystal growth (B). Co-crystallization of the protein with native or additive lipid (cholesterol) is shown in this illustration. As much as possible, the dimensions of the lipid (tan oval with tail), detergent (pink oval with tail), cholesterol (purple), protein (blue and green; β2-adrenergic receptor-T4 lysozyme fusion; PDB code 2RH1), bilayer, and aqueous channels (dark blue) have been drawn to scale. The lipid bilayer is ~40 Å thick.44 Figure 5 is from reference 138.
Figure 6.

Layered or Type I packing is observed in all crystals of membrane proteins produced to date by the in meso method. In the case of the β2-adrenoreceptor-Gs protein complex shown here (PDB code 3SN6),12 the transmembrane receptor (tan) drives the layering process by the proposed mechanism outlined in Figure 4. The approximate location of the bilayer supporting the receptor is indicated by the paired horizontal white lines.
The proposal for how nucleation and crystal growth occur in meso relies absolutely on the 3-dimensional continuity of the mesophase. Under the assumption that the sample exists as a single liquid crystallite or mono-domain, continuity ensures that the mesophase acts essentially as an infinite reservoir from which all protein molecules in the sample can end up in a bulk crystal. Neither the lamellar liquid crystal (Lα) nor the inverted hexagonal (HII) phases, both of which are accessible mesophases in lipidic systems (Figure 2), have 3-dimensional continuity and alone are unlikely to support membrane protein crystallogenesis by the in meso method.44
Because of the proposed need for the diffusion of proteins in the bilayer and of precipitant components in the aqueous channels of the mesophase, the expectation is that crystal growth rates might be tardy in meso. However, crystals have been seen to form within an hour3, which suggests that the slowness associated with restricted diffusion can be compensated for by a reduction in dimensionality. The latter is a result of the protein being confined to a lipid bilayer with its long axis oriented perpendicular to the membrane plane. Thus, the number of orientations that must be sampled to effect nucleation and crystal growth is few in meso compared with its in surfo counterpart, in which all of 3-dimensional space is accessible.2
That crystal growth takes place in a mesophase implies it is happening in a convection-free environment. This is analogous to growth under conditions of microgravity or in a gel, which offers the advantage of a stable zone of depletion around the growing crystal and thus a slower and more orderly growth.56 Settling of crystals and subsequent growth into one another are also avoided under these conditions, as is the likelihood that impurities are wafted in from the surrounding solution to poison the face of the crystal and limit growth. For all these reasons in meso crystallogenesis is similar to crystallization in space with the prospect of producing high-quality, structure-grade crystals.
Evidence for the Model
Experimental evidence in support of aspects of the hypothesis outlined above follow.
Reconstitution
The in meso method begins with what is assumed to be a uniform reconstitution of the protein into the lipid bilayer of the cubic phase (Figure 5). The protein is combined with MAG, typically in a ratio that should produce the cubic phase provided the detergent concentration of the protein solution is not too high. When this was done with the vitamin B12 transporter BtuB,57 the invasin/adhesin OpcA,58 and gramicidin D59 for example, the expected cubic-Pn3m phase was produced as evidenced by small-angle X-ray scattering (SAXS). The lattice parameter of the cubic phase was similar to the value observed with control, protein-free samples. Upon addition of precipitant solution to trigger nucleation and crystal growth in the case of BtuB, the cubic phase swelled and formed the sponge phase.60 It is from this swollen, bicontinuous mesophase that the crystals of BtuB were harvested. These data are consistent with the view that the protein reconstitutes into the cubic phase in a way that is homogenous and that does not perturb the original mesophase.
That reconstitution is uniform throughout the cubic mesophase is obvious when working with highly colored proteins such as bacteriorhodopsin,38 the photosynthetic reaction center,61 and the light-harvesting complex II (LHII).60 After the lipid and protein solution is homogenized, an optically clear mesophase is produced that, to the naked eye, is uniformly colored.
The electronic fluorescence properties of the gramicidin molecule directly reconstituted into the lipid bilayer of the cubic phase suggest that it resides in an apolar environment.33 Thus, the yield and wavelength of maximum intensity of the fluorescence from the tryptophans in gramicidin were increased and blue-shifted, respectively, compared with tryptophan in aqueous solution.
Quenching of intrinsic tryptophan fluorescence by a lipid with a dibrominated acyl chain (bromo-MAG) has been used to demonstrate reconstitution of BtuB,57 OpcA,58 diacylglycerol kinase (DgkA)62 and gramicidin59 in the lipidic mesophase. Respectively, these have 13, 4, 5 and 4 tryptophans, of which 12, 3, 3 and 4 should be directly accessible to quenching by bromo-MAG, provided the target is reconstituted into the cubic phase bilayer. The extent (>80%) of quenching observed (Figure 7) is consistent with this expectation and supports the view that the targets are reconstituted prior to crystallization.
Figure 7.

Intrinsic tryptophan fluorescence quenching curve of gramicidin D in the cubic phase of hydrated monoolein. Bromo-MAG is the quenching lipid and its concentration is expressed as mole% in monoolein. Flourescence data were corrected for background fluorescence from buffer and lipid and for the inner filter effect, and were normalized to the quencher-free (Fc,0) value. The quenching profile is consistent with the peptide being reconstituted into the bilayer of the cubic phase. Data from reference 59.
Some additional evidence in support of a bilayer location derives from the fact that the quenching behavior of gramicidin was sensitive to acyl chain identity of the accompanying, non-quenching MAG.59 Because the chains are confined to the bilayer interior, some property(ies) of the bilayer itself changes with the different MAGs. This is sensed by gramicidin presumably only when it is associated with that same lipid bilayer. In so doing, it responds differently to the quenching effect of the brominated lipid, which has a distinct character imprinted on it by the different non-quenching MAGs. One of the properties that changes with MAG identity is bilayer thickness. This, in turn, defines the relative positions of the apolar/polar interface across the membrane that will affect the fluorescence behavior of the tryptophans that sample such an environment.
A final piece of evidence for bilayer location hinges on the logic that gramicidin is so apolar that it is unfavorable for it to reside anywhere else within the confines of the mesophase. SAXS data show that the cubic phase can accommodate gramicidin up to a point. Beyond that limit, it triggers a transformation to the HII phase.59 This presumably reflects a change in the energetics associated with mismatch between the peptide and the lipid/water interface, which is a result of a gramicidin that is bilayer bound.
Conformation
Spectroscopic measurements were made to examine the conformational state of three membrane proteins and gramicidin reconstituted in the cubic phase. The UV-visible spectra of the BtuB57 and OpcA58 preparations in micellar form and in meso were, within experimental error, the same regardless of the protein dispersion state. In the case of LHII a similar observation held, with the exception of a slight change in bacteriochlorophyll absorption in the 780- to 900-nm range.60 Circular dichroism spectra showed that the gross secondary structure of both BtuB57 and OpcA58 was insensitive to whether they were in a micellar or a bilayer environment. Together these data suggest that cubic phase reconstitution does not dramatically alter the conformation of protein targets consistent with the hypothesis.
Functional Activity
It is assumed that proteins reconstituted prior to crystallization retain functionality in meso. In the case of BtuB, this was examined by measuring substrate (cyanocobalamin, CNCbl) binding to the protein reconstituted into the cubic phase.57 Protein-free control samples exhibited no binding, whereas test in meso BtuB-containing samples showed convincing evidence of substrate uptake. Binding was shown by quenching of intrinsic fluorescence of aromatic residues by CNCbl and by direct ligand binding to be tight with an apparent Kd value of ~1 nM. Similar Kd values have been reported for the native membrane-bound and micellarized form of the protein. Sialic acid binding to OpcA, measured by fluorescence quenching as with BtuB, was identical in meso and in detergent solution.58 Taken together, the data support the view that these β-barrel proteins reconstitute into the bilayer of the cubic phase in an active form prior to in meso crystallization.
Functional activity assays in meso have been extended to include membrane protein enzymes.62 In the case of diacylglycerol kinase (DgkA), a coupled enzyme assay was used (Figure 8). With phosphatidylglycerol phosphate synthase (PgsA), activity was quantified by direct assay. In both cases, the viscous, sticky and porous nature of the cubic phase was used to advantage in enabling continuous spectrophotometric activity assays to be performed in a high throughput microplate format. With both enzymes, the cubic mesophase served as a useful and a convenient nanoporous membrane mimetic that supported native-like activity. Recent studies with the dopamine 2 long (D2L) and histamine 1 (H1) GPCRs indicate ligand binding in the nanomolar range based on radio-labeled assays.63 In this study, the receptors were reconstituted into the cubic phase by a passive method (see below) and showed significantly enhanced specific binding compared to their detergent solubilized counterpart.
Figure 8.
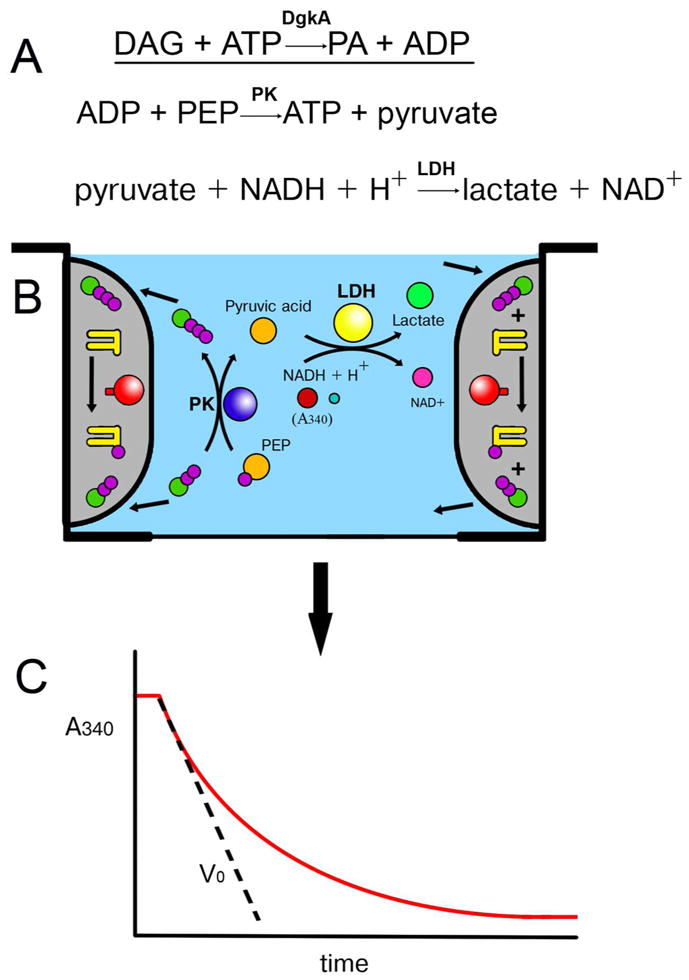
Monitoring the γ-phosphoryl group transfer activity of diacylglycerol kinase (DgkA) reconstituted into the bilayer of the cubic phase by a coupled enzyme assay method (A) in a muli-well plate.62 Protein-laden mesophase (shaded grey in (B)) is positioned on the wall of the well where it remains in place throughout the assay as a consequence of its intrinsic viscosity and stickiness. The mesophase is bathed in buffer (blue) containing the water-soluble ingredients of the coupled assay (ATP, ADP, pyruvate, lactate, phosphoenolpyruvate (PEP), NADH/NAD+, pyruvate kinase (PK) and lactate dehydrogenase (LDH)). Water soluble substrate, ATP, diffuses into the nanoporous mesophase for use by DgkA in synthesizing phosphatidic acid from diacylglycerol both of which reside in and are confined to the bilayer of the mesophase. Water soluble product, ADP, diffuses out of the mesophase into the bathing solution where it is used by the coupled enzyme assay system to regenerate ATP. The coupling process that involves PK and LDH leads to a drop in the concentration of NADH which, in turn, is monitored continuously in situ in a multi-plate reader by a reduction in absorbance at 340 nm of the bathing solution (C). The slope of the progress curve (C) provides a measure of the initial velocity, Vo, as indicated.
Diffusion
Crystallization, regardless of how it happens, requires transport, i.e., the movement of the crystallant from the bulk medium up and into the face of the crystal. If transport is impeded or does not happen, crystallogenesis will suffer. Under in meso conditions, mobility must take place both in the bilayer and in the aqueous channels of the mesophase. When working with colored proteins, such as bacteriorhodopsin, mobility is noticeable with a simple light microscope. As crystals form, flecks of dark purple appear surrounded by a zone of colorless mesophase, whose extent away from the crystal expands with time and crystal growth. This is evidence that the protein is moving from the mesophase reservoir, presumably in the lipid bilayer, to the crystal. Additional and more quantitative evidence that mobility in the bilayer is required for in meso crystallogenesis comes from recent fluorescence recovery after photobleaching measurements performed with labeled bacteriorhodopsin and a GPCR chimera.64 In this case, diffusion and a high fractional recovery of fluorescence in the bleached area correlated with known in meso crystallization conditions.
The diffusion of gramicidin, LHII, and a highly lipophilic dye, Sudan Red, has been used to characterize the transport properties of the cubic phase.60,65, To this end, a bolus of diffusant-loaded cubic phase, the source, was placed in direct contact with a bolus of diffusant-free cubic phase, the sink, at a sharp interface. Transfer of diffusant between the two boluses and subsequent diffusion in the sink was monitored by UV-visible spectroscopy and was shown to occur. In the case of the brightly red colored Sudan Red, transport could be seen with the naked eye. These observations show that the cubic phase supports transport and, because at least two of these diffusants are highly apolar, that diffusion is most likely taking place within the lipid bilayer. The results also highlight the fusogenic nature of the cubic phase, suggesting that the bilayer of one bolus can become continuous with the bilayer of the other bolus with which it makes contact. This means that the bilayer composition of a given bolus can, within limits, be adjusted at will, which has implications for seeding, co-crystallization, and complex formation by a stepwise approach to in meso crystallogenesis.
In the context of in meso crystallogenesis the cubic phase is viewed as a porous molecular sponge consisting of two interpenetrating nanochannels filled with an aqueous medium and coated by a common lipid bilayer. In the preceding paragraphs it was shown that proteins move within the membrane, a requirement for crystallogenesis. Mobility within the aqueous channels is also a prerequisite for crystal growth, at the very least to enable precipitant components to access the interior of the bolus and to trigger nucleation and crystal growth.
Several studies have been performed that support such transport and, for reasons of experimental simplicity, most were done by following release of water-soluble diffusants from a bolus of preloaded cubic phase.62,66–68 The studies show that the diffusion rate was dependent on the size of diffusant molecules as expected, given that the channels within the mesophase have a diameter of approximately 50 Å. Remarkably, transport was observed with apo-ferritin, whose size (~100 Å diameter) far exceeds that of the aqueous channel, suggesting a molecular breathing or peristalsis type of facilitated diffusion4.66 Exquisite control over the rate of movement within the aqueous channels was achieved by adjusting (a) channel dimensions (see Sponge Phase below), (b) the partitioning of the diffusant on or into the lipid bilayer, (c) the electrostatic interaction strength, and (d) histidine-tag displacement. Thus, although the mesophase channels are small and confined - just 15 water molecules wide - they enable simple and well-behaved transport.
In support of this, ultrafast hydration dynamics studies revealed that the channels include a water core with bulk-like dynamics and orientational relaxation properties consistent with transport.70 In contrast, the water at the aqueous/bilayer interface is dynamically rigid. The bilayer is surrounded by a hydrogen-bonded network of water with dynamic relaxations intermediate between those of the interfacial and core water. Taken together these data support the view that the cubic phase behaves as a nanoporous molecular sponge into and out of which water-soluble substances of a wide range of sizes and chemistries can diffuse, which is integral to the in meso crystallization model.
The ability of the cubic phase to act as a nanoporous membrane mimetic for integral membrane enzymes also supports the view that facile transport into and out of the aqueous channels of the mesophase does happen.62 Two lipid metabolizing enzymes were assayed kinetically on the basis of water soluble substrates diffusing in and water soluble products diffusing out of the mesophase bolus in which the enzymes were reconstituted. In one case, the activity measurement was done by coupled assay, in the other a direct assay was performed.
Type I Crystal Packing and the Lamellar Phase
The hypothesis posits that the protein migrates from the bulk mesophase reservoir to the face of the crystal by way of a lamellar conduit.1,43,44 Using a sub-micrometer-sized X-ray beam, the interface between a growing membrane protein crystal and the bulk cubic phase has been examined with micrometer spatial resolution.71 Characteristic diffraction from the lamellar phase was observed at the crystal interface, which supports the proposal that the protein uses a lamellar portal on its way from the bulk mesophase up and into the face of the crystal.
There are two reports based on microscopy that address the in meso growth of membrane protein crystals by way of a lamellar conduit. The first of these involved freeze-fracture electron microscopic (EM) examinations of acetylcholine receptor-α-bungarotoxin complex microcrystals grown from within a lipid mesophase.53 EM images showed highly ordered domains of the complex next to lipid lamellae, consistent with the working hypothesis. In the second study, atomic force microscopy was used to demonstrate the existence of a lamellar conduit between the bacteriorhodopsin crystal and the bulk cubic phase.72
In meso crystallization is predicted to produce Type I crystals (Figures 5 and 6). Here, proteins are arranged in planar sheets that stack one atop the other. Direct protein-protein interactions within the plane of a given layer can be extensive in Type I crystals. Type II crystals2 are commonly encountered when grown by in surfo methods. In this case, a torus of detergent coats the protein where it contacted the apolar region of the biomembrane from which it came originally. As a result, direct contact between the apolar midriff of the protein is much less likely in Type II crystals, and packing density and diffracting power can be low. To date, all membrane proteins that have been crystallized by the in meso method have given rise to Type I crystals (Figure 6) (http://www.mpdb.tcd.ie/),73 consistent with the hypothesis. However, non-lamellar-type packing could be observed at some point with in meso-grown crystals. This might come about by a polymorphic transition in the solid state.44 Presumably, the Type I crystal will form first to be replaced by a more stable polymorph in which the proteins are no longer arranged in distinct lamellae. It is possible too that the transition will occur after the protein concentrates locally in the partially ordered lamellar domain (Figure 5) that subsequently produces crystals.74
TERMINOLOGY
As noted, the cubic phase method is based on the assumption that the protein to be crystallized is initially reconstituted into the lipid bilayer of the cubic phase. However, the phase that feeds the face of the growing crystal is likely to be lamellar, not cubic.43,44,71 Further, while the monoolein/water phase diagram (Figure 2) upon which the method is based shows that the cubic phase is stable under conditions that approximate those used in crystallization, there are other ingredients in the crystallization mix that can destabilize the cubic phase, locally or even totally converting it to another phase. It is for this reason that we sought to determine the identity of the bulk mesophase/s present before and during crystal growth. SAXS measurements were used for purposes of phase identification. For the most part, the prevailing mesophase was found to be of the cubic type. However, this is not always the case and the phases present vary with the concentration of protein (and detergent), and the identity and concentration of the precipitants used.75 Thus, we have found that at high concentrations of added protein – bacteriorhodopsin was the test membrane protein – coexisting lamellar (Lα) and cubic phases form initially. Upon incubation with the Na+/K+ phosphate salt ‘precipitant’, the Lα phase converted to the cubic phase such that crystallization took place from a bulk cubic phase medium.76 In other systems, the cubic phase does not necessarily remain stable throughout the crystal growing period and it can transform with time to a birefringent or a liquid phase, depending on the precipitant used. Increasingly, the sponge phase is identified as the medium in which crystals grow (see below under Sponge Phase).
From this discussion it is apparent that the exclusivity of the cubic phase as the hosting and portal medium is not cast in stone and that other mesophases, or media derived from and reminiscent of them, may play a role. It is for this reason that the less limiting in meso, as opposed to the original in cubo or lipidic cubic phase (LCP) or more recent lipidic sponge phase (LSP) descriptors is preferred and continues to be used by the authors. An equally accurate and acceptable descriptor is the ‘lipidic mesophase crystallization’ or LMC method.
MINIATURIZATION AND HIGH-THROUGHPUT CRYSTALLIZATION
The protocol described above under Experimental Aspects refers to the manual mode of setting up crystallization trials. Accurate and precise delivery of the sticky and viscous protein-laden mesophase in volumes that range from pico- all the way to micro-liters was made possible by use of an inexpensive repeat dispenser in combination with differently sized micro-syringes.77,78 The smaller volumes means that the in meso method works with miniscule quantities of target protein. Thus, extensive crystallization screening can be done with just a few micrograms of valuable membrane protein making the in meso method one of the most efficient in terms of required protein, lipid and precipitant.
The repeat dispenser greatly facilitated the in meso method. However, it was still a manual set-up with limits to the numbers of trials that any one person could comfortably set up at a sitting. The need to automate the process was obvious. With the assistance of A. Peddi and Y. Zheng, engineers at The Ohio State University where the original work was done, we were able to perform a proof-of-principle robotics exercise employing LabView-controlled motorized translation stages operating and supporting a micro-syringe and a crystallization plate. With it, we demonstrated that the viscous mesophase could be dispensed automatically and wells filled in such a way that eventually yielded crystals. This was enough to secure funding for a robot which was custom-designed and built in-house to our specifications (Figure 9).
Figure 9.

Approaches and equipment used to set up in meso crystallization trials since the method was introduced in the mid-nineties. The original method used repeated centrifugation in a fixed angle rotor (A) to effect lipid and protein solution homogenization and cubic phase formation.38 The coupled syringe mixing device49 (B) was introduced in 1998 as a more practical and efficient means to generate and to dispense conveniently nanoliter volumes of protein-laden mesophase for use in in meso crystallization trials. Manual dispensing of the protein-laden mesophase prepared in the coupled syringe mixing device was greatly facilitated by the repeat dispenser (C).77 The x, y and z motions executed in dispensing mesophase manually, as in (C), inspired the building of a prototype robot consisting of a series of motorized orthogonal translation stages connected to a computer under LabView control (D). The success of the prototype in ‘automatically’ setting up crystallization plates in which membrane protein crystals grew was proof-of-concept and enough to secure funding with which to build in-house a custom-designed in meso crystallization robot (E).51 Variations on the original robot shown in (E) are now available commercially.79–83 The instrument shown in the figure comes equipped with a 4-tip liquid handling dispensing arm. An 8-tip version of the instrument is available commercially.
The in meso robot has two arms programmed to move simultaneously over a stationary crystallization plate.79 One arm dispenses the viscous, protein-laden mesophase while the other dispenses precipitant. Typical volumes used are 50 nL mesophase (usually consisting of 20 nL protein solution and 30 nL monoolein) and 800 nL precipitant solution. Custom, 96-well glass sandwich plates were designed which take just 5 minutes to fill using an 8-tip robot. The robot enables the precise and accurate setting up of in meso crystallization trials with very small volumes in high-throughput mode and, if required, under challenging conditions of reduced temperature and controlled lighting. Given the success of the original in meso robot, several such instruments, available through commercial vendors,79–83 are currently in use in labs throughout the world. Variants on the original design, where tip alignment is done automatically and/or where precipitant is handled by disposable tips, are now available commercially.80,83 Another dispenses 96 precipitant solutions simultaneously providing for very rapid plate set up.81 These represent important advances simplifying in meso crystallogenesis and making the method user-friendly. An on-line video of how to set up trials robotically is in preparation (Li, D., Boland, C., Walsh, K., and Caffrey, M. Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. In press)
With the success that the in meso method has had it perhaps is not unexpected to find products appearing on the market in support of this proven crystallogenesis approach. In addition to the in meso robots, these include a number of precipitant screen kits, glass and plastic sandwich plates, and a plate that comes complete with lipid-coated wells. The latter are convenient in that they can be used with a liquid dispensing robot for protein solution delivery first and precipitant post-swelling. Variations on this approach have been reported.63,84,85 It is important to note when using this passive approach that the time required for complete hydration of the lipid and reconstitution ahead of precipitant addition will depend on the specifics of the target protein, the composition of the solution in which it is dissolved, the thickness and identity of hosting lipid, and temperature. These, in turn, can impact on reproducibility. Protracted incubation, with a view to improving reproducibility, may compromise the protein.
MEMBRANE PROTEIN TARGET ISSUES
General
Although the focus of this article is on crystallization, a word about the protein ingredient of the crystal is in order. As noted, suitable starting material is often in short supply. Much effort is currently devoted to increasing the yield of membrane proteins in a crystallizable form. Sources include cellular membranes where the protein calls home. Under the best of circumstances, the membrane will come enriched naturally in the target protein, as in the case of bacteriorhodopsin and the purple membrane. At the other extreme are proteins that are not at all plentiful, and enormous amounts of biomaterials, effort and time must be devoted to procuring mere microgram quantities. The GPCRs and the cystic fibrosis transmembrane conductance regulator (CFTR) are such proteins.86,87 Overexpression in a host organism can be used to boost yield. However, it is not unusual for the membrane protein to compromise or kill the host cell when overproduced. One way around this is to bypass the membrane altogether and to express the protein in vivo as an insoluble cytoplasmic inclusion body88,89 or in vitro in a variety of dispersed states.90 This then requires that the protein be solubilized, often in concentrated solutions of urea or guanidine hydrochloride, with or without detergent, followed by a refolding step in the presence of detergent as a prelude to crystallogenesis. As an alternative, we have proposed using solubilized inclusion bodies for direct in meso crystallization.68 The logic is as follows. The protein, dissolved in a concentrated denaturant (urea, for example) solution, is used to form the cubic phase. Upon incubation with excess refolding buffer, the urea rushes out of the porous mesophase, and the protein begins to refold. Since it is essentially trapped in the narrow aqueous confines of the cubic phase, the protein is only ångströms away from the lipid bilayer and spontaneously reconstitutes into it. The protein-laden mesophase can then be used directly for in meso crystallogenesis. The fact that such proteins never encounter a natural membrane raises questions regarding the fidelity of the structure so determined. With OmpG91,92 and a recent case involving the biofilm alginate transporting AlgE from Pseudomonas aeruginosa, virtually identical structures were obtained with protein isolated from membranes (Tan, J., Li, D., Aragao, D., Pye, V., and Caffrey, M., unpublished) and protein refolded from inclusion bodies.93
When cloning is used, advantage can be taken of the ability to engineer in sequences (affinity tags) and/or fusion proteins that facilitate purification and crystallization, as well as amino acid analogs, such as seleno-methionine for phasing purposes. Fusions that include the green fluorescent protein (GFP) or its homologs can be used for convenient and high-throughput screening of expression, purification and indeed crystallization94,95. For this to work however the GFP tag needs to be cytoplasmic and the tagged protein should be properly folded. Protocols and vectors are available to ensure a cytoplasmic localization.96 Including in the recombinant protein a protease site for optional removal of the tag or fusion protein prior to crystallization is the norm. The recombinant approach also affords the opportunity to modify the target should the native protein prove refractory to crystallization. Such modifications include N- and/or C-terminal as well as internal sequence trimming or extension and removal of undesired post-translational modification sites. Exploring the crystallizability of thermostabilized mutants and of homologues of the target protein from other organisms is a proven strategy with membrane proteins.94,95,97–99 Often, where the perceived business end of the molecule is not in the membrane, the membrane-anchoring part of the protein is removed. This reduces the task to crystallizing a soluble polypeptide whose structure, it is hoped, will faithfully represent that of the intact membrane-associated protein. Neuraminidase is one such example.100–103
This same ‘pruning’ approach can be taken as a last resort in pursuit of at least some structural information on the more complex, multi-domain membrane proteins, such as the CFTR. Domains that are not likely to be buried in the membrane can be expressed separately or excised from the intact protein and used in crystallization trials. However, functional insights gleaned from structural information derived using this ‘divide and conquer’ approach must be evaluated with caution. A bonus is that should diffraction quality crystals of the intact protein be obtained subsequently, the solved soluble domain structure might be used for phasing by molecular replacement.
As with soluble proteins, every effort must be made to ensure that the membrane protein used in crystallization trials is stable and of the highest possible biochemical and conformational purity and homogeneity. Assessments of purity based on electrophoresis, size exclusion chromatography, single particle electron microscopy, analytical ultracentrifugation and light and X-ray scattering can be used to advantage here. More recently, mass spectrometry (MS) has emerged as an important alternative and/or supplement to the more traditional analytical techniques given that it offers picomole sensitivity, high mass accuracy, high-throughput capability and speed, all at a reasonable cost.104–106 Furthermore, mass spectrometers are ubiquitous and most institutions now provide MS facilities and support on a routine service basis.
With regard to target purity a word of caution is in order in that a membrane protein can be ‘too pure’ and, as a result, does not yield crystals of the desired quality. This happens when the protein is purified to such an extent that it is stripped of structurally important lipids and/or cofactors. Thus, working with a less pure preparation is worth trying. Alternatively, lipids can be included in the purification buffers or added back to the protein preparation ahead of crystallization trials. There are several examples in the literature where the right amount and type of added lipid proved critical to the production of structure quality crystals.107–110 In the case of the GPCRs, cholesteryl hemi-succinate (CHS) is commonly included in the buffers used for purification and the hosting mesophase for crystallization is spiked with cholesterol to this same end.7–10,12–20,22,25,26,111–114
GPCRs
In addition to the in meso crystallization technology, novel protein related complimentary strategies have proven crucial to the recent spate of high resolution crystal structures of GPCRs, both in the inactive and active conformations. These involve increasing crystal contact surface area, conformational homogeneity, and thermostability of purified receptors.
Crystal contact enhancement involved the replacement of the third intracellular loop with a modified T4 lysozyme (T4L). Extensive mutagenesis and biochemical studies were performed to show that the flexible third-intracellular loop of the β2-adrenergic receptor (β2AR) could be replaced with the relatively stable, ordered and crystallization prone T4L.22 The resulting β2AR-T4L fusion protein displayed similar pharmacological properties but enhanced stability compared to the wild-type receptor. Further, the fusion construct trafficked to the cell surface and was crystallisable by the in meso and bicelle methods.7,22 This highly effective T4L fusion approach has since been applied to a number of other GPCRs whose crystal structure has been determined by the in meso method.8–10,13–18,20,111–114 A recent variation on this approach leaves the third intracellular loop intact and fuses a thermostabilized apo-cytochrome b562RIL from Escherichia coli to the truncated N-terminus of the receptor to promote receptor stability and in meso crystallogenesis.19 Separately, a rhodopsin-inspired single amino acid (3.41 according to Ballesteros-Weinstein numbering115) mutation in the third transmembrane helix that significantly enhances the functional expression and thermal stability of Class A GPCRs is now standard practice.116 To produce a structure of the fully-active, agonist bound form of β2AR, high affinity nanobodies (camelid antibodies) were generated that, in association with the receptor or the receptor-Gs complex, promoted active state conformational homogeneity, stability of the G protein complex, and crystal contacts that facilitated in meso crytallogenesis.12,25 Receptor stability can be optimized using a convenient, high-throughput fluorescence assay that measures cysteine accessibility upon denaturation of purified receptor.117 An alternative approach uses ligand binding assays to membranes isolated from recombinant E. coli in what is referred to as “conformational thermostabilization”.98,118–122 To date, this method that employs scanning and additive mutagenesis has been used for GPCR structure determination only by the in surfo method of crystal production.11,123–125
SAMPLES WITH LOW PROTEIN CONCENTRATION
The driving force for nucleation is greater the more supersaturated is the system. Thus, a common strategy in crystallization is to work at the highest possible protein concentration to favor nucleation and to lower its concentration subsequently to just above the solubility limit for slow, orderly growth of a few good quality crystals. It is likely that the same principles apply to crystallization in meso where initially, the highest possible protein concentration should be used in support of nucleation. There are at least two issues that must be dealt with in this context that apply to membrane proteins. Firstly, most membrane proteins are prepared and purified in combination with detergents. Thus, the detergent is carried along with the protein into the crystallization mix. It follows then that as the protein concentration increases, the detergent concentration will rise in parallel. This may work against crystallization because high levels of detergent destabilize the hosting mesophase.76,126 Of course, the sensitivity to added detergent will depend, among other things, on the identities of the hosting lipid and detergent. Completely removing the detergent before folding the protein into the crystallization mix is usually not an option because it is commonly required to keep the protein soluble as a mixed micelle. One alternative is to reduce the detergent load to an acceptable level before combining the protein with the hosting lipid. This can be done with BioBeads76,127 or by eluting the protein in a highly concentrated form from an affinity column. Using detergents with low critical micelle concentration values, such as lauryl maltose neopentyl glycol (MNG-DDM) is also worth investigating.
The second issue has to do with raising the concentration of protein in the lipid bilayer of the cubic phase to facilitate nucleation. Two approaches can be tried that are quite different but that achieve the same end. The first exploits the water-carrying capacity of the cubic phase, a property that varies with lipid identity (see Figure 2).128–133 Thus, the reconstituted protein will be more concentrated in the bilayer of the cubic phase prepared with a lipid of high water-carrying capacity than would obtain for a less hydrating lipid. The second approach involves sequential reconstitutions where the protein concentration in the bilayer rises with each round5.
LIMITS TO MESOPHASE COMPATIBILITY
Protein Solution Components
As alluded to above, what happens during in meso crystallization is intimately tied up with lipid mesophase behaviour.44 The working hypothesis for how nucleation comes about begins with the protein reconstituting into the continuous bilayer of the cubic phase (Figure 2). Precipitant is added which triggers local formation of a lamellar phase into which the protein preferentially partitions and concentrates in a process that leads to nucleation and crystal growth (Figure 5). As noted, experimental evidence in support of aspects of this model has been reported.
Experience, built up over several years working with the in meso method, suggests that the mesophase behaviour observed during the course of the crystallization process mimics that of the relevant MAG (where the default MAG is monoolein)/water system (Figure 2). The implication therefore is that the protein solution has minimal effect on the phase behaviour of the hosting lipidic mesophase into which the protein is reconstituted. That solution, along with the target protein, typically includes lipid, detergent, buffers, and salt at a minimum. Other components such as glycerol, sulfydryl reagents, denaturants, etc., are not uncommon. Each of these can impact on phase behaviour and, by extension, on the outcome of a crystallization trial. In the interests of learning about component compatibility, the sensitivity of the monoolein/water cubic phase system to their inclusion has been evaluated. Our findings indicate that the default cubic mesophase is remarkably resilient and retains its phase identity in the presence of a vast array of different additives. These include glycerolipids, cholesterol, free fatty acids, detergents, denaturants, glycerol and sulfydryl reagents, among others.43,60,66–68,75,76,126,134 Of course, for each there is a concentration beyond which the cubic phase is no longer stable. In most cases, these limits have been identified.
Occasionally, the concentration of a protein solution component is not known exactly. Detergent is a case in point. This poses a problem because if there is too much detergent the bulk lamellar phase may form and this will not support crystallization.76,126 It may also be that a new detergent is being used, whose compatibility with the cubic phase is not known. In this case, a small amount of the buffer used to solubilise the protein or the protein solution itself can be used to prepare mesophase. The physical texture (high viscosity), appearance between crossed polarizers, or SAXS behaviour of the mesophase will indicate which phase has been accessed. If, for example, it is a lamellar phase that forms suggesting too much detergent then another purification step where its concentration in the final protein solution is reduced may be enough to solve the problem. We have encountered situations with bacteriorhodopsin where the particular preparation ended up having an excess of detergent. The mesophase first formed was lamellar but when it was used in combination with certain precipitants a transition back to the cubic phase was induced which went on to support crystal growth.76
Crystallization Screen Solution
As noted, in meso crystallization relies upon a bicontinuous mesophase which acts as a reservoir to feed protein into nucleation sites and for crystal growth (Figure 2). The crystallization screening process requires that chemical space be interrogated over wide limits. In the screening process therefore, the protein-laden mesophase is exposed to precipitant solutions that encompass hundreds, perhaps even thousands of different chemical compositions. Screen solution components include buffers that cover a wide buffer type and pH range, polymers, salts, small organics, detergents, apolar solvents, amphiphiles, etc., and all at different concentrations. Each component can potentially destabilize the mesophase. In a separate study using SAXS, we examined the compatibility of the reference monoolein/water cubic phase with various commonly used precipitant screen solutions.75 What we found was hardly surprising. Compatibility was temperature dependent and the usual suspects, that included organic solvents, destroyed the cubic phase rendering these screen solutions effectively useless. A goal of the study was to design screens that were mesophase friendly. However, that goal was never pursued; instead we have opted for the convenience of commercial screen kits mindful of the fact that certain conditions are not relevant. As a result, certain kits are simply not used because they contain too few conditions that are compatible with the cubic phase. Others are used in diluted form. For example, PACT premier (MD1-36, Molecular Dimensions) is used at 50 – 65 % of full-strength.
SPONGE PHASE
During the course of mesophase compatibility studies we noticed that particular screen components caused the cubic phase to ‘swell’ and, under certain conditions, to form what is referred to as the sponge phase. The latter evolves from the cubic phase as a result of the ‘spongifying’ component lowering bilayer interfacial curvature thereby enabling the mesophase to imbibe more lyotrope (aqueous solution). This is revealed in the SAXS pattern where the lattice parameter of the cubic phase rises. Eventually, the mesophase looses order and the low-angle diffraction pattern becomes diffuse. Fortunately, the sponge phase retains its bicontinuity and, as a result, can support in meso crystallogenesis.44,60,135 One advantage of the sponge phase is that its aqueous channels are dilated. Thus, proteins with large extra-membrane domains should be accommodated in and amenable to crystallogenesis from the sponge phase. Further, the reduced interfacial curvature is likely to facilitate more rapid and long range diffusion within the lipid bilayer. Since net movement of protein from the bulk mesophase reservoir to the nucleation and growth sites is a requirement for crystallization this effect alone should contribute to improved crystallization. Interestingly, many of the proteins that have yielded to the in meso method have been crystallized under conditions that favour sponge phase formation (www.mpdb.tcd.ie).73
Reflecting the utility of the sponge phase for in meso crystallogenesis a number of commercial screening kits now include spongifiers such as polyethyleneglycol, Jeffamine, butanediol, 2-methyl-2,4-pentanediol (MPD), and pentaerythritol propoxylate (PPO) among others. Some of these provide a preformed sponge phase to which the protein solution is added directly. We continue to use the original method that involves an active protein reconstitution step with pure lipid where the entire crystallization screen space is available for sampling.
LIPID RATIONAL DESIGN
Low Temperature Crystallogenesis
The MS&FB Group has devoted considerable time and effort to establishing the structure-function rules for rationally designing lipids with specific end uses.1,136–138 One such application concerned the development of a host lipid for use in in meso crystallogenesis at low temperatures. Certain proteins are labile and require handling in the cold. The problem with the in meso method, in the default mode at least, is that it relies on monoolein as the hosting lipid. The cubic phase formed by monoolein is not stable below about 17 °C (Figure 2B)48 and performing crystallization trials in a cold room at 4–6 °C is risky. For this low temperature application therefore a cis-monounsaturated monoacylglycerol, 7.9 MAG, was designed, using the rules referred to above. The target MAG was synthesized and purified in-house and its phase behaviour mapped out using SAXS.136 As designed, it produced the cubic phase stable in the range from 6 °C to 85°C. 7.9 MAG has been used in the crystallization of a number of membrane proteins in the MS&FB Group.
The word ‘risky’ was used in the previous paragraph when referring to low temperature crystallization with monoolein as the hosting lipid. This reflects the fact that it is possible to do successful in meso work with monoolein at 4 °C provided the system undercools. Fortunately, the cubic phase is noted for this capacity48,131 and we perform successful crystallization trials regularly with monoolein in the 4 to 17°C range. As expected, under these metastable conditions occasionally the mesophase converts to the bulk lamellar crystalline (Lc) or solid phase which is no use as far as crystallogenesis is concerned6.
Tailoring Mesophase Microstructure to Match the Target Protein
With regard to in meso crystallization, it is important to appreciate that the microstructure of the phase can change with, among other things, temperature, sample composition (hydration is one example) and lipid identity.48,140 By microstructure is meant the lattice parameter of the phase and how it is constituted. Thus, for example, the lamellar phase consists of planar sheets of lipid bilayers each separated by a layer of water (Figure 2). As temperature, composition, and lipid identity change, the thickness of the lipid bilayer as well as that of the water layer can change. The same holds for the other mesophases, including the bicontinuous phases (Figure 10).
Figure 10.

Temperature-induced changes in the lipid and aqueous channel dimensions of the cubic phase. Temperature dependence of the lipid length in the cubic phase (A) and of fully hydrated, cubic-Pn3m phase water channel radius (B) for three monoacylglycerols. Lipid identity is reported in the N.T notation. Data are from references 128,130 and 131.
Hydrated monoolein in the cubic phase at 20 °C may provide a suitable matrix in which to grow membrane protein crystals. However, dropping temperature to 4 °C, while preserving the cubic phase as a result of metastability, will cause the lattice parameter to change and, along with it, the dimensions of the lipid bilayer (Figure 10A) and the water channels (Figure 10B) of the cubic phase.48,131 It is possible that such changes may no longer ensure retention of protein activity or support crystal growth for a host of reasons. By the same token, it may well provide an even more stabilizing and a better crystal-growing environment. We are currently quantifying the effects that lipid and water compartment sizes of the cubic phase have on the stability and crystallizability of several membrane proteins.
Reference has just been made to the sensitivity of phase microstructure to lipid identity. Support for this statement is based on X-ray diffraction measurements performed on the cubic phase of a homologous series of MAGs (Figure 10).1 The data show expected behavior in that as chain length decreases so too does the thickness of the lipid layer that creates the apolar fabric of the cubic phase, when evaluated at a single temperature (Figure 10A). Less intuitive perhaps is the finding that the aqueous channel diameter drops as chain length increases (Figure 10B). This is consistent with a ‘flattening’ and an attenuating curvature at the polar/apolar interface with the shorter chained lipids.
While lipid identity can be used to tailor phase microstructure, it is possible that the desired microstructure might not be accessible with a single lipid species in the temperature range of interest. In this case, it is possible to fine tune by using mixtures of MAGs with different acyl chain characteristics where the mole ratio is adjusted to set microstructure at the desired intermediate value.1
It is apparent from the data just presented that it is possible to engineer the microstructure of the mesophase over relatively wide limits by manipulating temperature and/or lipid identity and composition. However, it is also important to note that the two metrics of the cubic phase - the polar and apolar compartment dimensions - are not independently adjustable and indeed are tightly coupled, as indicated in Figure 10. Nonetheless, this feature of tunability is an important tool that is available to the crystallographer in search of a suitable lipid matrix in which to grow crystals. Thus, proteins with transmembrane and extramembrane domains that come in a variety of sizes can be accommodated as can those that originate from native membranes with different hydrophobic thicknesses.141
Proteins and Complexes with Large Membrane Footprints and Large Extramembrane Domains
In what follows, two recent examples of rational lipid design for use in crystallizing targets with large footprints in the plane of the membrane and/or extensive extramembrane domains are described. The first refers to cytochrome caa3 oxidase from Thermus thermophilus. This terminal oxidase is a large 120 kDa hetero-trimeric protein with 23 transmembrane helices and a cytochrome c-like domain as a C-terminal extension to one of its subunits. Extensive crystallization trials by traditional vapor diffusion methods failed to produce structure-grade crystals. The in meso method was considered as an appropriate alternative. At the time the study was undertaken, in meso crystallization had generated crystals and a structure of a protein, LHII, whose bulk in the plane of the membrane resembled that expected for caa3. Initial in meso trials with the default lipid, 9.9 MAG or monoolein, failed to produce useful crystals. Anticipating the likelihood that 9.9 MAG would not suit every membrane protein, the lipid synthesis program in the MS&FB Group provided alternative MAGs with which to screen for crystallogenesis. The first of these tested was 7.7 MAG which has an acyl chain 14 carbon atoms long and a cis double bond between carbons 7 and 8. 7.7 MAG had been shown to form a cubic mesophase with a thinner, less highly curved bilayer and with enlarged aqueous channels.133 A thinner bilayer was considered desirable for use with caa3 because it more suitably complimented the hydrophobic thickness predicted for related cytochrome oxidases of known structure. Additionally, the bigger aqueous channels provided by 7.7 MAG were attractive in the context of caa3 with its added extramembrane bulk in the form of a cupredoxin and a tethered cytochrome c-like domain. As expected, 7.7 MAG produced crystals; upon optimization they provided a structure at 2.36 Å.137
The second example is the β2-adrenergic receptor – Gs protein complex. Earlier work had shown that the free receptor produced structures to high resolution in the default lipid, 9.9 MAG, using the in meso method. However, efforts to grow structure grade crystals of the receptor as a complex with its cognate Gs protein in monoolein failed. The Gs protein is itself a large hetero-trimeric complex with a molecular weight of ~80 kDa. It binds to the exposed intracellular surface of the receptor and adds considerable extramembrane bulk to the target. In this particular instance the Gs protein had bound to it a camelid antibody or nanobody (15 kDa) and T4L (19 kDa) was fused to the N-terminus of the receptor. Both contributed additional extramembrane heft to the complex. Given that the cubic phase prepared with monoolein alone has aqueous channels in which the water-soluble domains must reside that are only 50 Å in diameter,131 failure to crystallize in monoolein did not come as a surprise. 7.7 MAG, with its significantly larger aqueous channels, was immediately identified as a suitable alternative hosting lipid and, with some limited optimizations, it generated diffraction quality crystals and a structure of the complex.12 Interestingly, the precipitant used for the production of final crystals included PEG 400, a known spongifier, and crystals were harvested from what appeared to be a sponge phase. It seems likely therefore that the short chain MAG and the spongifier worked hand in hand to generate a bicontinuous medium that accommodated unrestricted diffusion and that facilitated crystallization of the complex with its extensive extramembrane domain. Future in meso crystallization trials with targets of this sort will undoubtedly benefit from the use of short chain MAGs in concert with sponge phase inducing precipitants. Commercial crystallization kits that include such materials are likely forthcoming. It is important to note that in all of the aforementioned GPCR work, the hosting MAG was doped with cholesterol (see following section).
LIPID SCREENING
Host Lipid
The original lipid used for in meso crystallogenesis was monoolein. It was recognized from the outset that this one lipid may not work with all target membrane proteins. The rationale was that these come from a variety of native membranes which differ in lipid composition, bilayer thickness, surface charge and packing density, fluidity and polarity profile, intrinsic curvature, etc. Thus, having a range of MAGs that differed in acyl chain characteristics available for screening was deemed important. Using principles of rational design a number of suitable MAGs were identified with the requirement that they form the inverse cubic phase at or close to 20 °C under conditions of full hydration. Several lipids meeting these specifications have been synthesised and characterized in-house. They now constitute an invaluable hosting lipid screen in the MS&FB Group. With a number of membrane proteins, that include β-barrels, α-helical proteins and complexes, and an integral peptide antibiotic, crystals have been grown by the in meso method using these alternative hosting MAGs.12,74,133,136,138 In a number of instances, monoolein either failed to produce crystals or the crystals it did produce were not of diffraction quality. It was only when MAGs from the hosting lipid screen were used that structure grade crystals were obtained (Figure 4). Some of these novel MAGs are available commercially.142
Additive Lipid
Early on in the development of the in meso method it was recognized that monoolein, as the lipid used to create the hosting mesophase, is not a typical membrane lipid.1,134 The sense was that it might be recognized as foreign by certain target proteins and trigger a destabilization reaction. One solution considered was to employ a naturally occurring membrane lipid that forms the requisite cubic phase under crystallization conditions. Unfortunately, none was available. An alternative was to use the default MAG, monoolein, as the hosting lipid and to supplement it with typical membrane lipids with a view to creating a more native like environment. Accordingly, the carrying capacity of the monoolein cubic phase for a number of different lipids was established using SAXS.134 This amounted to about 20 mol% in the case of phosphatidylcholine, phosphatidylethanolamine, and cholesterol with lesser amounts of phosphatidylserine and cardiolipin being accommodated. With time, other lipids will need to be included in in meso crystallization trials. The carrying capacity of the cubic phase for such lipids can be evaluated by SAXS, as noted,134 or less quantitatively but more simply and immediately by evaluating texture and optical clarity and by polarized light microscopy.45 The strategy of using additive lipids has proven particularly useful with GPCR targets where cholesterol augmentation of the cubic phase was critical to the production of diffraction quality crystals and was seen in the final solved structure (Figure 11).7–10,12–20,22,25,26,111–114 Given the success of this approach pre-prepared lipid mixtures for use in in meso crystallization trials are available commercially.143
Figure 11.
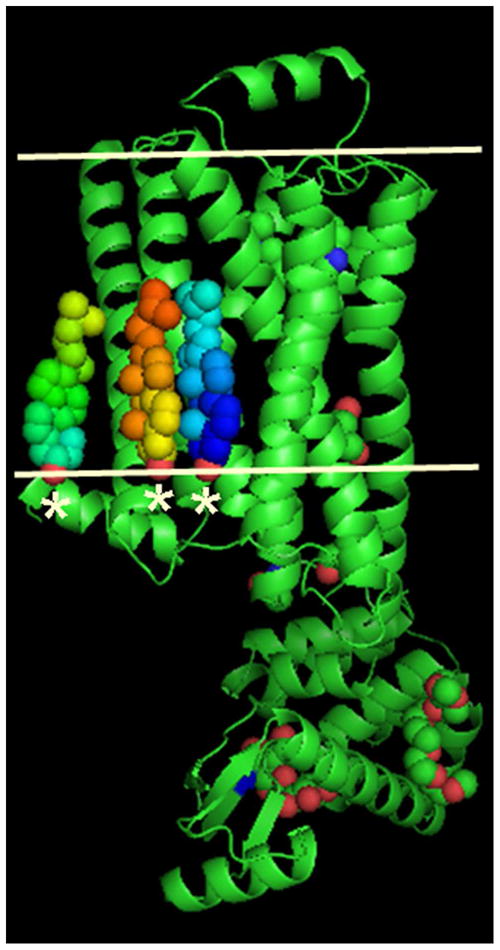
Structure of the β2-adrenoreceptor solved with crystals grown by the in meso method using monoolein doped with cholesterol as an additive lipid. Cholesterol molecules (space filling model, asterisks) are part of the crystal structure. White horizontal lines mark the approximate location of the membrane/aqueous interface with respect to the receptor (green model, PDB code 2RH1).
CRYSTAL STRUCTURES
The in meso method accounts for 103 records in the Protein Data Bank (PDB, www.pdb.org) that relate to integral membrane proteins and peptides (Tables 1 and 2; www.mpdb.tcd.ie).73 This corresponds to about 10% of published membrane protein structures and represents at least eight distinct classes of membrane proteins (Figure 12). With successes that include bacterial and eukaryotic rhodopsins, sensory rhodopsin II/transducer complex, LHII, photosynthetic reaction centres, cytochrome oxidases, β-barrels, GPCRs, a GPCR-Gs complex, an exchanger, and an integral membrane peptide the method has a convincing record of versatility and range. Each of these membrane protein types represents families the members of which are also candidates for in meso crystallogenesis. The GPCR family is the latest case in point; it has approximately eight hundred distinct GPCRs and close to twenty cognate G proteins coded for in the human genome alone. The in meso method therefore, in combination with the necessary protein engineering and receptor stabilization strategies, is poised to contribute to the generation of GPCR and receptor-G protein complex structures on an ‘industrialized’ scale. Evidence in support of this statement is the recent spate of GPCR structures courtesy, in part, of in meso crystallogenesis (Table 2). This past year alone has witnessed at least eight such deposited structures in the PDB. It is with some confidence therefore that we can look forward to successes with other membrane protein families.
Figure 12.

A gallery of membrane protein structures solved using crystals grown by the lipidic cubic phase or in meso method. A single representative structure within a membrane protein class is shown along with diffraction resolution and hyperlinked PDB identifier. The figure at the bottom of each panel refers to the number of record entries in the PDB for that particular membrane protein class. Thus, within the peptide class there are 3 records for gramicidin D at different resolutions. Within the β-barrel class there are 7 entries, 1 each for BtuB, OpcA, Intimin, and Invasin, and 3 for OmpF. Within the G protein-coupled receptor class there are 26 records: 1 for the β2-adrenoreceptor-Gs complex, 7 for the β2-adrenorecptor, 3 for the A2a-adenosine receptor, 5 for the CXCR4 receptor, 2 for the sphingosine 1-phosphate subtype 1 receptor, and 1 each for the D3 dopamine receptor, the H1 histamine receptor, the M2 muscarinic receptor, the M3. muscarinic receptor, the δ-opioid receptor, the κ-opioid receptor, the μ-opioid receptor, and the nociceptin receptor. Within the non-GPCR rhodopsin class there are 53 records with 38 for bacteriorhodopsin, 3 for halorhodopsin, 6 for sensory rhodopsin II, 3 for the sensory rhodopsin II/transducer complex, and 1 each for sensory rhodopsin from Nostoc sp. Pcc 7120 and for rhodopin from Acetabularia acetabulum. Within the light harvesting complex class there is a single entry for LHII. Within the photosynthetic reaction center class there are 5 and 4 entries for the reaction centers from Blastochloris viridis and Rhodobacter sphaeroides, respectivly. Within the cytochrome oxidase class there are 2 entries for ba3 and 1 for caa3. Within the exchanger group there is a single entry for the Na+-Ca2+ exchanger. As of June 2012, the total count for in meso structures in the PDB is 103. Hyperlinks: 2Y5M; 2GUF; 3SN6; 1M0K; 2FKW; 2WJN; 2YEV; 3V5U
The further development of the in meso crystallogenesis approach is an important goal for the MS&FB Group. Recently, this has focused on examining the utility of the method with small membrane proteins. A separate analysis performed using a model cubic phase under somewhat limiting conditions indicated that suitable targets would need to include a minimum of five transmembrane helices.144 Our experience with the sponge phase variant of the cubic phase suggested otherwise. Accordingly, the utility of the method with a ‘mini-protein,’ the penta-decapeptide antibiotic, linear gramicidin, was examined. It worked remarkably well providing a structure for the intertwined conformation of the antibiotic with a resolution of 1.08 Å.74,145 Regardless of the chain length of the hosting MAG used the antibiotic grew crystals in the intertwined conformation. A word of caution is in order here. The physiological relevance of the latter, intertwined form has been questioned and the issue was considered in detail by Hoefer et al.74 Several mechanistic proposals for how this, as opposed to the head-to-head conformation crystallized in meso have been presented.74,146 Regardless, the result obtained with linear gramicidin highlights the utility of the method with proteins having small transmembrane domains which abound in nature.146
THE MEMBRANE PROTEIN DATA BANK
Details regarding the structure and function of integral, anchored and peripheral membrane proteins are available online in a convenient and searchable database, the Membrane Protein Data Bank (MPDB, www.mpdb.tcd.ie).73 Records in the MPDB are obtained from the PDB. However, the former limits itself to entries for membrane proteins. Statistical analyses on the contents of the database can be performed conveniently and viewed directly online. Examples include detergents or pH or temperature used for membrane protein structure work, number of structures published annually by method, and so on.
PROSPECTS
The in meso method burst on the scene a decade and a half ago. It was received with great anticipation for what it would deliver; perhaps it was to be the panacea. However, output in the early years was limited to naturally abundant, bacterial α-helical proteins bedecked with stabilizing and highly colored prosthetic groups (www.mpdb.tcd.ie).73 The perceived restricted range, coupled to the challenges associated with handling the sticky and viscous cubic mesophase, meant that subsequent interest in the method waned. This was countered to some degree with the introduction of the in meso robot, a growing understanding for how the method worked at a molecular level, and a continued demonstration of the method’s general applicability. However, interest in the method has rocketed of late with the success it has had in the GPCR field (Table 2).7–10,12–20,22,25,26,111–114
Improvements are needed of course if the method is to become routine. Critically, the specialized materials and supplies upon which the method relies must be made more generally available and the method itself must be made user-friendly. New and improved in meso robots available on the market are tackling the user-friendliness issue. Workshops that involve hands-on demonstrations contribute to making the method more accessible. The author has been active in this area for several years now with recent workshops in Ireland,147 Mexico148, Hawaii,149 Australia150, and China.151 In the past year alone, over 200 students were trained in the practicalities and finer elements of in meso crystallogenesis and related topics at various locations worldwide. Online video demonstrations covering practical aspects of the method are available and in preparation (Li, D., Boland, C., Aragao, D., Walsh, K., and Caffrey, M. Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. In review; Li, D., Boland, C., Walsh, K., and Caffrey, M. Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. In press).46
Developments are needed in the area of crystal identification. Optical clarity is of the highest quality with the glass sandwich plates currently in use and this provides for ready detection of colourless, micrometre-sized crystals in normal light and between crossed polarizers. Detection by UV fluorescence is particularly powerful and convenient for tryptophan containing proteins. Fluorescence labelling52,53 is also a route worth considering for the sensitive detection of early hits. Second-order non-linear optical imaging of chiral crystals (SONICC) is a novel approach introduced by G. Simpson. It has been shown to sensitively and selectively detect membrane protein crystals growing in meso.152
Recovering crystals from the mesophase for data collection is a non-trivial undertaking (Li, D., Boland, C., Aragao, D., Walsh, K., and Caffrey, M. Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. In review).45 This is especially true when harvesting is done directly from glass sandwich plates. Typically, a glass cutter is used to open the well and to expose the mesophase. Teasing out and harvesting the crystal for immediate cryo-cooling is most conveniently done with a cryo-loop. This is a slow, pains-taking and cumbersome process especially if it must be done in a cold room and/or in subdued light. This whole area of harvesting calls out for innovation to include automation.
Data collection at the synchrotron is not exactly straightforward either. Given that in meso-grown crystals tend to be small, a mini-synchrotron X-ray beam is required. Oftentimes, the crystal of interest is hidden from view in a bolus of mesophase on the cryo-loop. This means that locating the crystal and centring it requires rounds of diffraction rastering with a beam of progressively smaller size.153,154 This same approach is used to advantage in finding the best diffracting part of a crystal. Locating crystals and centering based on X-ray fluorescence from heavy atoms in the sample is in development (Aragao, D., Becker, M., Li, D., Hilgart, M., Lyons, J., Yoder, D., Stepanov, S., Fischetti, R., and M. Caffrey, unpublished). Effective and efficient rastering is recognized now as an important feature of the latest MX beamlines at synchrotron facilities worldwide and steady improvements in the rastering process are being made. In situ screening and data collection as well as dynamic focusing for improved signal-to-noise are other areas under investigation. The wherewithal to screen and to collect data efficiently over the internet and without the need for travel to the synchrotron is eagerly anticipated.
The X-ray free electron laser (XFEL) is a tantalizing new technology for MX of membrane proteins.155,156 Here, femtosecond-long pulses at 1012–13 X-rays/pulse strike a flowing stream or extruded bolus micrometers in diameter that ports equally sized or smaller crystals into and through the X-ray beam. Each pulse, delivered at 120 Hz, has enough energy to raise the temperature of the striken crystal to beyond that of the sun’s core. But because the pulse is of such short duration, diffraction from the ‘native state’ can occur, and be collected subsequently, before the crystal is converted to a plasma. Both the cubic and sponge mesophases157 are being investigated as media for transporting crystals into the XFEL beam with the prospect of being able to collect structure-quality diffraction data using material that might otherwise be abandoned as microcrystalline or mistakenly identified as precipitated protein. The distinct possibility exists that such microcrystals are of superior diffraction quality to their more voluminous counterparts requiring shorter times to grow and less material to produce a structure. Additional XFEL sources dedicated to such measurements will be needed.
The structures solved using in meso-grown crystals have, until very recently,158–160 relied on molecular replacement for phasing7. Increasingly, experimental phasing will be required. In our hands, with poorly diffracting crystals, this is proving to be a challenge. Several targets have been tackled using seleno-methionine labelling and pre-labelling, co-crystallization and soaking with heavy atoms with only limited success. Problems derive in part from a low anomalous signal-to-noise due to a combination of background low- and wide-angle scatter from adhering mesophase and the need to work with small and sometimes poorly diffracting, radiation-sensitive crystals. As often as not, data must be collected in angular wedges on different parts of a single crystal or on multiple crystals and merging data satisfactorily is a challenge. This part of the in meso pipeline is in need of work.
Finally, the method should begin to be used with really small and with big proteins and complexes. The sponge phase,60 with its open aqueous channels and flatter bilayer, should prove particularly useful in this regard. Using it in combination with novel hosting and additive lipid screens74,134,138 will go a long way toward producing crystals and ultimately high resolution structures where interactions that are integral to human health are revealed.
Acknowledgments
There are many who contributed to this work and most are from the MS&FB Group, both past and present members. To all we extend our warmest thanks and appreciation.
Funding
This work was supported in part by grants from Science Foundation Ireland (07/IN.1/B1836), FP7 COST Action CM0902, Marie Curie Actions (PIEF-GA-2009-254103) and the National Institutes Health (GM75915, P50GM073210, U54GM094599).
Abbreviations
- CNCbl
cyanocobalamin
- CHS
cholesteryl hemi-succinate
- EM
electron microscopy
- FI
fluid isotropic phase
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- HII
inverted hexagonal phase
- Lα
lamellar liquid crystal phase
- Lc
lamellar crystal phase
- LCP
lipidic cubic phase
- LDH
lactate dehydrogenase
- LH II
light harvesting complex II
- LMC
lipidic mesophase crystallization
- LSP
lipidic sponge phase
- MAG
monoacylglycerol
- MNG
maltose neopentyl glycol
- MPD
2-methyl-2,4-pentanediol
- MPDB
membrane protein data bank
- MS
mass spectrometry
- MS&FB
Membrane Structural and Functional Biology
- MX
macromolecular X-ray crystallography
- PDB
protein data bank
- PEP
phosphoneol pyruvate
- PK
pyruvate kinase
- PPO
pentaerythritol propoxylate
- SAXS
small-angle X-ray scattering
- SONICC
second-order non-linear optical imaging of chiral crystals
- T4L
T4 lysozyme
- UV
ultra-violet
- XFEL
X-ray free electron laser
Footnotes
The bulk of the in meso crystallogenesis performed to date employs MAGs containing cis-monounsaturated fatty acids. A shorthand system for describing the chemical constitution of these MAGs makes use of the N.T notation introduced previously.47 This is based on a rather simplistic view of the MAG molecule as an object consisting of a head, a neck, and a tail with the latter two joined by a trunk. The head is the glycerol head group. It is in ester linkage to the neck corresponding to that part of the acyl chain extending from its carboxyl carbon to the first carbon of the olefin. The trunk is the cis-double bond. The tail extends from the second carbon of the olefin to the chain’s methyl terminus. In the N.T MAG notation, N and T correspond to the number of carbon atoms in the neck and tail, respectively. The total number of carbon atoms in the chain is the sum of N and T. Thus, 11.7 MAG represents monovaccenin, a MAG with a fatty acyl chain 18 carbon atoms long where the cis-double bond resides between carbon atoms 11 and 12. It is an olefinic isomer of 9.9 MAG also known as monoolein.
Should too much protein solution be employed the mesophase will become cloudy and opaque. If possible, this should be avoided as it can create problems recognizing small crystals commonly encountered as initial hits. If the cloudy, two-phase system does form it can still be used for successful crystallization provided the mesophase is of the bicontinuous (non-birefringent) type.45
For comparison, crystals of the β2AR-Gs complex 0.25 mm long formed in meso in 2–3 days.12 This corresponds to an average growth rate of ~5 μm/h.
This is consistent with the observation that transducin, a large hetero-trimeric G protein, can diffuse from solution into a rhodopsin-loaded cubic phase sample and form a functional complex.69
The membrane protein preferentially partitions from the aqueous solution into the bilayer of the cubic phase. If the reconstitution step is repeated using a single mesophase bolus and with a series of solutions of protein at low concentration, the protein load in the mesophase will increase with each reconstitution leaving excess aqueous solution depleted of protein. This protein-depleted solution is usually removed before the next round of reconstitution commences.
Sugar-phytane lipids form the fully hydrated cubic phase in the 10 – 70 °C range.139 These may well find application for in meso crystallization at reduced temperatures. In our hands, we have found these lipids difficult to handle. They require very robust mixing especially below 50 °C.
The recent successes in using experimental phasing for structure determination have been with rhodopsin II from Chlamydomonas reinhardtii (3UG9; mercury-MAD), the Na+-Ca2+ exchanger from Methanocaldococcus jannaschii (3V5U; samarium-SAD), β-barrels from E. coli (4E1S; selenomethionine-SAD) and Yersinia pseudotuberculosis (4E1T; selenomethionine-SAD) and with a membrane kinase from E. coli (Li, D., Lyons, J., Pye, V., Aragao, D., and Caffrey, M. In preparation; selenomethionine-SAD).
References
- 1.Caffrey M. Membrane protein crystallization. J Struct Biol. 2003;142:108–132. doi: 10.1016/s1047-8477(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 2.Michel H. Crystallization of membrane-proteins. Trends Biochem Sci. 1983;8:56–59. [Google Scholar]
- 3.Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T, Shimono K, Kikukawa T, Hato M, Shinya N, Kim SY, Kimura-Someya T, Shirouzu M, Tamogami J, Miyauchi S, Jung KH, Kamo N, Yokoyama S. Crystal structure of the eukaryotic light-driven proton-pumping rhodopsin, Acetabularia rhodopsin II, from marine alga. J Mol Biol. 2011;411:986–998. doi: 10.1016/j.jmb.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Lahiri S, Brehs M, Olschewski D, Becker CFW. Total chemical synthesis of an integral membrane enzyme: Diacylglycerol kinase from Escherichia coli. Angewandte Chemie-International Edition. 2011;50:3988–3992. doi: 10.1002/anie.201006686. [DOI] [PubMed] [Google Scholar]
- 6.Deisenhofer J, Michel H. The photosynthetic reaction center from the purple bacterium Rhodopseudomonas-viridis. Embo Journal. 1989;8:2149–2170. doi: 10.1002/j.1460-2075.1989.tb08338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien EYT, Liu W, Zhao QA, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu BL, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen SGF, DeVree BT, Zou YZ, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H-1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTiC. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prive GG, Kaback HR. Engineering the lac permease for purification and crystallization. J Bioenerg Biomembr. 1996;28:29–34. [PubMed] [Google Scholar]
- 22.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 23.Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, Stevens RC. Fusion partner toolchest for the stabilization and crystallization of g protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunte C, Michel H. Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 2002;12:503–508. doi: 10.1016/s0959-440x(02)00354-8. [DOI] [PubMed] [Google Scholar]
- 25.Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 27.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sennhauser G, Grutter MG. Chaperone-assisted crystallography with DARPins. Structure. 2008;16:1443–1453. doi: 10.1016/j.str.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Ostermeier C, Iwata S, Ludwig B, Michel H. Fv fragment mediated crystallization of the membrane-protein bacterial cytochrome c oxidase. Nat Struct Biol. 1995;2:842–846. doi: 10.1038/nsb1095-842. [DOI] [PubMed] [Google Scholar]
- 30.Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 33.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 35.Uysal S, Vasquez V, Tereshko V, Esaki K, Fellouse FA, Sidhu SS, Koide S, Perozo E, Kossiakoff A. Crystal structure of full-length KcsA in its closed conformation. Proc Natl Acad Sci U S A. 2009;106:6644–6649. doi: 10.1073/pnas.0810663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, Kusano-Arai O, Weyand S, Shimamura T, Nomura N, Cameron AD, Kobayashi T, Hamakubo T, Iwata S, Murata T. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landau EM, Rosenbusch JP. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci U S A. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caffrey M. Crystallizing membrane proteins for structure determination: Use of lipidic mesophases. Annual Review of Biophysics. 2009;38:29–51. doi: 10.1146/annurev.biophys.050708.133655. [DOI] [PubMed] [Google Scholar]
- 40.Caffrey M. Crystallizing membrane proteins for structure-function studies using lipidic mesophases. Biochem Soc Trans. 2011;39:725–732. doi: 10.1042/BST0390725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faham S, Bowie JU. Bicelle crystallization a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 42.Takeda K, Sato H, Hino T, Kono M, Fukuda K, Sakurai I, Okada T, Kouyama T. A novel three-dimensional crystal of bacteriorhodopsin obtained by successive fusion of the vesicular assemblies. J Mol Biol. 1998;283:463–474. doi: 10.1006/jmbi.1998.2103. [DOI] [PubMed] [Google Scholar]
- 43.Caffrey M. A lipid’s eye view of membrane protein crystallization in mesophases. Curr Opin Struct Biol. 2000;10:486–497. doi: 10.1016/s0959-440x(00)00119-6. [DOI] [PubMed] [Google Scholar]
- 44.Caffrey M. On the mechanism of membrane protein crystallization in lipidic mesophases. Crystal Growth & Design. 2008;8:4244–4254. doi: 10.1021/cg101384p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caffrey M, Porter C. Crystallizing membrane proteins for structure determination using lipidic mesophases. J Vis Exp. 2010:e1712. doi: 10.3791/1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misquitta Y, Caffrey M. Rational design of lipid molecular structure: A case study involving the c19: 1c10 monoacylglycerol. Biophys J. 2001;81:1047–1058. doi: 10.1016/S0006-3495(01)75762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu H, Caffrey M. The phase diagram of the monoolein/water system: Metastability and equilibrium aspects. Biomaterials. 2000;21:223–234. doi: 10.1016/s0142-9612(99)00126-x. [DOI] [PubMed] [Google Scholar]
- 49.Chen AH, Hummel B, Qiu H, Caffrey M. A simple mechanical mixer for small viscous lipid-containing samples. Chem Phys Lipids. 1998;95:11–21. doi: 10.1016/s0009-3084(98)00060-7. [DOI] [PubMed] [Google Scholar]
- 50.Cherezov V, Caffrey M. Nano-volume plates with excellent optical properties for fast, inexpensive crystallization screening of membrane proteins. Journal of Applied Crystallography. 2003;36:1372–1377. [Google Scholar]
- 51.Cherezov V, Peddi A, Muthusubramaniam L, Zheng YF, Caffrey M. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallographica Section D-Biological Crystallography. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- 52.Forsythe E, Achari A, Pusey ML. Trace fluorescent labeling for high-throughput crystallography. Acta Crystallographica Section D-Biological Crystallography. 2006;62:339–346. doi: 10.1107/S0907444906000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paas Y, Cartaud J, Recouvreur M, Grailhe R, Dufresne V, Pebay-Peyroula E, Landau EM, Changeux JP. Electron microscopic evidence for nucleation and growth of 3D acetylcholine receptor microcrystals in structured lipid-detergent matrices. Proc Natl Acad Sci U S A. 2003;100:11309–11314. doi: 10.1073/pnas.1834451100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nollert P, Qiu H, Caffrey M, Rosenbusch JP, Landau EM. Molecular mechanism for the crystallization of bacteriorhodopsin in lipidic cubic phases. FEBS Lett. 2001;504:179–186. doi: 10.1016/s0014-5793(01)02747-8. [DOI] [PubMed] [Google Scholar]
- 55.Khelashvili G, Mondal S, Caffrey M, Weinstein H. Quantitative comparison of GPCR interactions with the lipid bilayer of the cubic and lamellar mesophases. Biophys J. 2012;102:467a–468a. [Google Scholar]
- 56.McPherson A. Crystallization of biological macromolecules. Cold Spring Harbor Laboratory Press; New York: 1999. [Google Scholar]
- 57.Cherezov V, Yamashita E, Liu W, Zhalnina M, Cramer WA, Caffrey M. In meso structure of the cobalamin transporter, BtuB, at 1.95 angstrom resolution. J Mol Biol. 2006;364:716–734. doi: 10.1016/j.jmb.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cherezov V, Liu W, Derrick JP, Luan B, Aksimentiev A, Katritch V, Caffrey M. In meso crystal structure and docking simulations suggest an alternative proteoglycan binding site in the OpcA outer membrane adhesin. Proteins-Structure Function and Bioinformatics. 2008;71:24–34. doi: 10.1002/prot.21841. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, Caffrey M. Gramicidin structure and disposition in highly curved membranes. J Struct Biol. 2005;150:23–40. doi: 10.1016/j.jsb.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 60.Cherezov V, Clogston J, Papiz MZ, Caffrey M. Room to move: Crystallizing membrane proteins in swollen lipidic mesophases. J Mol Biol. 2006;357:1605–1618. doi: 10.1016/j.jmb.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 61.Hochkoeppler A, Landau EM, Venturoli G, Zannoni D, Feick R, Luisi PL. Photochemistry of a photosynthetic reaction-center immobilized in lipidic cubic phases. Biotechnol Bioeng. 1995;46:93–98. doi: 10.1002/bit.260460202. [DOI] [PubMed] [Google Scholar]
- 62.Li D, Caffrey M. Lipid cubic phase as a membrane mimetic for integral membrane protein enzymes. Proc Natl Acad Sci U S A. 2011;108:8639–8644. doi: 10.1073/pnas.1101815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Darmanin C, Conn CE, Newman J, Mulet X, Seabrook SA, Liang YL, Hawley A, Kirby N, Varghese JN, Drummond CJ. High-throughput production and structural characterization of libraries of self-assembly lipidic cubic phase materials. Acs Combinatorial Science. 2012;14:247–252. doi: 10.1021/co2001718. [DOI] [PubMed] [Google Scholar]
- 64.Cherezov V, Liu J, Griffith M, Hanson MA, Stevens RC. LCP-FRAP assay for pre-screening membrane proteins for in meso crystallization. Crystal Growth & Design. 2008;8:4307–4315. doi: 10.1021/cg800778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clogston J. PhD thesis. The Ohio State University; Columbus, OH: 2005. Applications of the lipidic cubic phase: From controlled release and uptake to in meso crystallization of membrane proteins. [Google Scholar]
- 66.Clogston J, Caffrey M. Controlling release from the lipidic cubic phase. Amino acids, peptides, proteins and nucleic acids. J Control Release. 2005;107:97–111. doi: 10.1016/j.jconrel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 67.Clogston J, Craciun G, Hart DJ, Caffrey M. Controlling release from the lipidic cubic phase by selective alkylation. J Control Release. 2005;102:441–461. doi: 10.1016/j.jconrel.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 68.Liu W, Caffrey M. Interactions of tryptophan, tryptophan peptides, and tryptophan alkyl esters at curved membrane interfaces. Biochemistry. 2006;45:11713–11726. doi: 10.1021/bi0608414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Navarro J, Landau EM, Fahmy K. Receptor-dependent G-protein activation in lipidic cubic phase. Biopolymers. 2002;67:167–177. doi: 10.1002/bip.10066. [DOI] [PubMed] [Google Scholar]
- 70.Kim J, Lu WY, Qiu WH, Wang LJ, Caffrey M, Zhong DP. Ultrafast hydration dynamics in the lipidic cubic phase: Discrete water structures in nanochannels. Journal of Physical Chemistry B. 2006;110:21994–22000. doi: 10.1021/jp062806c. [DOI] [PubMed] [Google Scholar]
- 71.Cherezov V, Caffrey M. Membrane protein crystallization in lipidic mesophases. A mechanism study using X-ray microdiffraction. Faraday Discussions. 2007;136:195–212. doi: 10.1039/b618173b. [DOI] [PubMed] [Google Scholar]
- 72.Qutub Y, Reviakine I, Maxwell C, Navarro J, Landau EM, Vekilov PG. Crystallization of transmembrane proteins in cubo: Mechanisms of crystal growth and defect formation. J Mol Biol. 2004;343:1243–1254. doi: 10.1016/j.jmb.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 73.Raman P, Cherezov V, Caffrey M. The membrane protein data bank. Cellular and Molecular Life Sciences. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hofer N, Aragao D, Lyons JA, Caffrey M. Membrane protein crystallization in lipidic mesophases. Hosting lipid effects on the crystallization and structure of a transmembrane peptide. Crystal Growth & Design. 2011;11:1182–1192. doi: 10.1021/cg101384p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherezov V, Fersi H, Caffrey M. Crystallization screens: Compatibility with the lipidic cubic phase for in meso crystallization of membrane proteins. Biophys J. 2001;81:225–242. doi: 10.1016/S0006-3495(01)75694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Misquitta Y, Caffrey M. Detergents destabilize the cubic phase of monoolein: Implications for membrane protein crystallization. Biophys J. 2003;85:3084–3096. doi: 10.1016/S0006-3495(03)74727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cherezov V, Caffrey M. A simple and inexpensive nanoliter-volume dispenser for highly viscous materials used in membrane protein crystallization. Journal of Applied Crystallography. 2005;38:398–400. [Google Scholar]
- 78.Cherezov V, Caffrey M. Picolitre-scale crystallization of membrane proteins. Journal of Applied Crystallography. 2006;39:604–606. [Google Scholar]
- 79.Anachem/Gilson Flexus crystals IMP Robot. ( www.gilsonuk.com)
- 80.Formulatrix NT8 LCP Robot. www.formulatrix.com.
- 81.Gryphon LCP Robot. ( www.artrobbins.com)
- 82.Zinsser Analytics ProCrys Meso Robot. ( www.zinsser-analytic.com)
- 83.TTP Labtech Mosquito LCP. ( www.ttplabtech.com)
- 84.Kubicek J, Schlesinger R, Baeken C, Büldt G, Schäfer F, JL Controlled in meso phase crystallization - a method for the structural investigation of membrane proteins. PLoS ONE. 2012;7:e35458. doi: 10.1371/journal.pone.0035458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wallace E, Dranow D, Laible PD, Christensen J, Nollert P. Monoolein lipid phases as incorporation and enrichment materials for membrane protein crystallization. PLoS ONE. 2011;6:e24488. doi: 10.1371/journal.pone.0024488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tate CG, Stevens RC. Growth and excitement in membrane protein structural biology. Curr Opin Struct Biol. 2010;20:399–400. doi: 10.1016/j.sbi.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 87.Hanrahan JW, Gentzsch M, Riordan JR. The cystic fibrosis transmembrane conductance regulator (ABCC7) In: Holland B, Higgins CF, Kuchler K, Cole SPC, editors. ABC proteins: from bacteria to man. Elsevier Sci. Ltd; New York: 2003. pp. 589–618. [Google Scholar]
- 88.Buchanan SK. Beta-barrel proteins from bacterial outer membranes: Structure, function and refolding. Curr Opin Struct Biol. 1999;9:455–461. doi: 10.1016/S0959-440X(99)80064-5. [DOI] [PubMed] [Google Scholar]
- 89.Padan E, Hunte C, Reilander H. Production and purification of recombinant membrne proteins. In: Hunte C, Jagow GV, Schagger H, editors. Membrane protein purification and crystallization A practical guide. Academic Press; San Diego: 2003. pp. 55–78. [Google Scholar]
- 90.Schwarz D, Junge F, Durst F, Frolich N, Schneider B, Reckel S, Sobhanifar S, Dotsch V, Bernhard F. Preparative scale expression of membrane proteins in Escherichia coli-based continuous exchange cell-free systems. Nature Protocols. 2007;2:2945–2957. doi: 10.1038/nprot.2007.426. [DOI] [PubMed] [Google Scholar]
- 91.Yildiz O, Vinothkumar KR, Goswami P, Kuhlbrandt W. Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. Embo Journal. 2006;25:3702–3713. doi: 10.1038/sj.emboj.7601237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Subbarao GV, van den Berg B. Crystal structure of the monomeric porin OmpG. J Mol Biol. 2006;360:750–759. doi: 10.1016/j.jmb.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 93.Whitney JC, Hay ID, Li CH, Eckford PDW, Robinson H, Amaya MF, Wood LF, Ohman DE, Bear CE, Rehm BH, Howell PL. Structural basis for alginate secretion across the bacterial outer membrane. Proc Natl Acad Sci U S A. 2011;108:13083–13088. doi: 10.1073/pnas.1104984108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 95.Drew D, Lerch M, Kunji E, Slotboom DJ, de Gier JW. Optimization of membrane protein overexpression and purification using GFP fusions. Nat Meth. 2006;3:303–313. doi: 10.1038/nmeth0406-303. [DOI] [PubMed] [Google Scholar]
- 96.Hsieh JM, Besserer GM, Madej MG, Bui HQ, Kwon S, Abramson J. Bridging the gap: A GFP-based strategy for overexpression and purification of membrane proteins with intra and extracellular C-termini. Protein Sci. 2010;19:868–880. doi: 10.1002/pro.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bass RB, Strop P, Barclay M, Rees DC. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 2002;298:1582–1587. doi: 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- 98.Miller JL, Tate CG. Engineering an ultra-thermostable beta1-adrenoceptor. J Mol Biol. 2011;413:628–638. doi: 10.1016/j.jmb.2011.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou YF, Bowie JU. Building a thermostable membrane protein. J Biol Chem. 2000;275:6975–6979. doi: 10.1074/jbc.275.10.6975. [DOI] [PubMed] [Google Scholar]
- 100.Ely LK, Fischer S, Garcia KC. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol. 2009;10:1245–1251. doi: 10.1038/ni.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.He XL, Dukkipati A, Garcia KC. Structural determinants of natriuretic peptide receptor specificity and degeneracy. J Mol Biol. 2006;361:698–714. doi: 10.1016/j.jmb.2006.06.060. [DOI] [PubMed] [Google Scholar]
- 102.Laver WG, Bischofberger N, Webster RG. Disarming flu viruses. Sci Am. 1999;280:78–87. doi: 10.1038/scientificamerican0199-78. [DOI] [PubMed] [Google Scholar]
- 103.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG, Robinson CV. Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nat Meth. 2009;6:585–587. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cohen SL, Chait BT. Mass spectrometry as a tool for protein crystallography. Annu Rev Biophys Biomol Struct. 2001;30:67–85. doi: 10.1146/annurev.biophys.30.1.67. [DOI] [PubMed] [Google Scholar]
- 106.Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D, Robinson CV. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guan L, Smirnova IN, Verner G, Nagamoni S, Kaback HR. Manipulating phospholipids for crystallization of a membrane transport protein. Proc Natl Acad Sci U S A. 2006;103:1723–1726. doi: 10.1073/pnas.0510922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hunte C, Richers S. Lipids and membrane protein structures. Curr Opin Struct Biol. 2008;18:406–411. doi: 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 109.Zhang HM, Kurisu G, Smith JL, Cramer WA. A defined protein-detergent-lipid complex for crystallization of integral membrane proteins: The cytochrome b6f complex of oxygenic photosynthesis. Proc Natl Acad Sci U S A. 2003;100:5160–5163. doi: 10.1073/pnas.0931431100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Siderophore-mediated iron transport: Crystal structure of FhuA with bound lipopolysaccharide. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 111.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 Å structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi HJ, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Structure and function of an irreversible agonist-beta2 adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by x-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 116.Roth CB, Hanson MA, Stevens RC. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of glu122(3.41), a critical residue in GPCR structure. J Mol Biol. 2008;376:1305–1319. doi: 10.1016/j.jmb.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alexandrov AI, Mileni M, Chien EYT, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 118.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta 1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Magnani F, Shibata Y, Serrano-Vega MJ, Tate CG. Co-evolving stability and conformational homogeneity of the human adenosine A2A receptor. Proc Natl Acad Sci U S A. 2008;105:10744–10749. doi: 10.1073/pnas.0804396105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shibata Y, White JF, Serrano-Vega MJ, Magnani F, Aloia AL, Grisshammer R, Tate CG. Thermostabilization of the neurotensin receptor NTS1. J Mol Biol. 2009;390:262–277. doi: 10.1016/j.jmb.2009.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lebon G, Bennett K, Jazayeri A, Tate CG. Thermostabilisation of an agonist-bound conformation of the human adenosine A2A) receptor. J Mol Biol. 2011;409:298–310. doi: 10.1016/j.jmb.2011.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Warne T, Serrano-Vega MJ, Tate CG, Schertler GFX. Development and crystallization of a minimal thermostabilised G protein-coupled receptor. Protein Expr Purif. 2009;65:204–213. doi: 10.1016/j.pep.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 123.Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dore AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a beta1-adrenergic g-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ai X, Caffrey M. Membrane protein crystallization in lipidic mesophases: Detergent effects. Biophys J. 2000;79:394–405. doi: 10.1016/S0006-3495(00)76301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rigaud JL, Mosser G, Lacapere JJ, Olofsson A, Levy D, Ranck JL. Bio-beads: An efficient strategy for two-dimensional crystallization of membrane proteins. J Struct Biol. 1997;118:226–235. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- 128.Briggs J. PhD thesis. The Ohio State University; Columbus, OH: 1994. The phase behavior of hydrated monoacylglycerols and the design of an X-ray compatible scanning calorimeter. [Google Scholar]
- 129.Briggs J, Caffrey M. The temperature-composition phase diagram and mesophase structure characterization of monopentadecenoin in water. Biophys J. 1994;67:1594–1602. doi: 10.1016/S0006-3495(94)80632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Briggs J, Caffrey M. The temperature-composition phase diagram of monomyristolein in water: Equilibrium and metastability aspects. Biophys J. 1994;66:573–587. doi: 10.1016/s0006-3495(94)80847-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Briggs J, Chung H, Caffrey M. The temperature-composition phase diagram and mesophase structure characterization of the monoolein/water system. Journal De Physique Ii. 1996;6:723–751. [Google Scholar]
- 132.Qiu H, Caffrey M. Phase behavior of the monoerucin/water system. Chem Phys Lipids. 1999;100:55–79. doi: 10.1016/s0009-3084(99)00040-7. [DOI] [PubMed] [Google Scholar]
- 133.Misquitta LV, Misquitta Y, Cherezov V, Slattery O, Mohan JM, Hart D, Zhalnina M, Cramer WA, Caffrey M. Membrane protein crystallization in lipidic mesophases with tailored bilayers. Structure. 2004;12:2113–2124. doi: 10.1016/j.str.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 134.Cherezov V, Clogston J, Misquitta Y, Abdel-Gawad W, Caffrey M. Membrane protein crystallization in meso: Lipid type-tailoring of the cubic phase. Biophys J. 2002;83:3393–3407. doi: 10.1016/S0006-3495(02)75339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wohri AB, Johansson LC, Wadsten-Hindrichsen P, Wahlgren WY, Fischer G, Horsefield R, Katona G, Nyblom M, Oberg F, Young G, Cogdell RJ, Fraser NJ, Engstrom S, Neutze R. A lipidic-sponge phase screen for membrane protein crystallization. Structure. 2008;16:1003–1009. doi: 10.1016/j.str.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 136.Misquitta Y, Cherezov V, Havas F, Patterson S, Mohan JM, Wells AJ, Hart DJ, Caffrey M. Rational design of lipid for membrane protein crystallization. J Struct Biol. 2004;148:169–175. doi: 10.1016/j.jsb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 137.Lyons JA, Aragao D, Slattery O, Pisliakov AV, Soulimane T, Caffrey M. Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature. 2012 doi: 10.1038/nature11182. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li D, Lee J, Caffrey M. Crystallizing membrane proteins in lipidic mesophases. A host lipid screen. Cryst Growth Des. 2011;11:530–537. doi: 10.1021/cg101378s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hato M, Minamikawa H, Salkar RA, Matsutani S. Phase behavior of phytanyl-chained akylglycoside/water systems. Progr Colloid Polym Sci. 2004;123:56–60. [Google Scholar]
- 140.Luzzati V. Biological membranes, physical facts and function. Academic Press; New York: 1968. [Google Scholar]
- 141.Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 2010;142:158–169. doi: 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Avanti Polar Lipids. ( www.avantilipids.com); Sigma Aldrich. ( www.sigmaaldrich.com); Nu-Chek Prep. ( www.nu-chekprep.com)
- 143.Qiagen. ( www.qiagen.com)
- 144.Grabe M, Neu J, Oster G, Nollert P. Protein interactions and membrane geometry. Biophys J. 2003;84:854–868. doi: 10.1016/S0006-3495(03)74904-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hofer N, Aragao D, Caffrey M. Crystallizing transmembrane peptides in lipidic mesophases. Biophys J. 2010;99:L23–L25. doi: 10.1016/j.bpj.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10:159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pye VE, Aragao D, Lyons JA, Caffrey M. Overview of the 13th international conference on the crystallization of biological macromolecules. Crystal Growth & Design. 2011;11:4723–4730. [Google Scholar]
- 148.ICCBM12. 12th international conference on biological macromolecules (ICCBM12); Mexico: Cancun; 2008. www.iccbm12.com.mx. [Google Scholar]
- 149.GPCR Workshop. Maui; Hawaii, USA: 2011. ( www.gpcrworkshop.com) [Google Scholar]
- 150.Australian Course in Macromolecular Crystallisation. Melbourne, Australia: 2012. ( http://crystal.csiro.au/en/Events/%7E/media/8A1B891319B14FBA93D36416C63A8165.ashx) [Google Scholar]
- 151.GPCR Workshop. Tsinghua University; Beijing, China: 2012. ( www.cls.edu.cn/english/Academicactivities/notices/index1076.shtml) [Google Scholar]
- 152.Kissick DJ, Gualtieri EJ, Simpson GJ, Cherezov V. Nonlinear optical imaging of integral membrane protein crystals in lipidic mesophases. Anal Chem. 2010;82:491–497. doi: 10.1021/ac902139w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Aishima J, Owen RL, Axford D, Shepherd E, Winter G, Levik K, Gibbons P, Ashton A, Evans G. High-speed crystal detection and characterization using a fast-readout detector. Acta Crystallographica Section D-Biological Crystallography. 2010;66:1032–1035. doi: 10.1107/S0907444910028192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hilgart MC, Sanishvili R, Ogata CM, Becker M, Venugopalan N, Stepanov S, Makarov O, Smith JL, Fischetti RF. Automated sample-scanning methods for radiation damage mitigation and diffraction-based centering of macromolecular crystals. Journal of Synchrotron Radiation. 2011;18:717–722. doi: 10.1107/S0909049511029918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FR, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Homke A, Reich C, Pietschner D, Struder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kuhnel KU, Messerschmidt M, Bozek JD, Hau-Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jonsson O, Svenda M, Stern S, Nass K, Andritschke R, Schroter CD, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TR, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B, Spence JC. Femtosecond x-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fromme P, Spence JC. Femtosecond nanocrystallography using x-ray lasers for membrane protein structure determination. Curr Opin Struct Biol. 2011;21:509–516. doi: 10.1016/j.sbi.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Johansson LC, Arnlund D, White TA, Katona G, Deponte DP, Weierstall U, Doak RB, Shoeman RL, Lomb L, Malmerberg E, Davidsson J, Nass K, Liang M, Andreasson J, Aquila A, Bajt S, Barthelmess M, Barty A, Bogan MJ, Bostedt C, Bozek JD, Caleman C, Coffee R, Coppola N, Ekeberg T, Epp SW, Erk B, Fleckenstein H, Foucar L, Graafsma H, Gumprecht L, Hajdu J, Hampton CY, Hartmann R, Hartmann A, Hauser G, Hirsemann H, Holl P, Hunter MS, Kassemeyer S, Kimmel N, Kirian RA, Maia FR, Marchesini S, Martin AV, Reich C, Rolles D, Rudek B, Rudenko A, Schlichting I, Schulz J, Seibert MM, Sierra RG, Soltau H, Starodub D, Stellato F, Stern S, Struder L, Timneanu N, Ullrich J, Wahlgren WY, Wang X, Weidenspointner G, Wunderer C, Fromme P, Chapman HN, Spence JC, Neutze R. Lipidic phase membrane protein serial femtosecond crystallography. Nat Methods. 2012;9:263–265. doi: 10.1038/nmeth.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fairman JW, Dautin N, Wojtowicz D, Liu W, Noinaj N, Barnard1 TJ, Udho5 E, Przytycka TM, Cherezov V, Buchanan SK. Crystal structures of the outer membrane domain of intimin and invasin from enterohemorrhagic E. coli and enteropathogenic Y. pseudotuberculosis. Structure. 2012 doi: 10.1016/j.str.2012.04.011. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kato HE, Zhang F, Yizhar O, Ramakrishnan C, Nishizawa T, Hirata K, Ito J, Aita Y, Tsukazaki T, Hayashi S, Hegemann P, Maturana AD, Ishitani R, Deisseroth K, Nureki O. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482:369–374. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liao J, Li H, Zeng WZ, Sauer DB, Belmares R, Jiang YX. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science. 2012;335:686–690. doi: 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]