

Abstract
Recessive osteogenesis imperfecta (OI) is caused by defects in genes whose products interact with type I collagen for modification and/or folding. We identified a Palestinian pedigree with moderate and lethal forms of recessive OI caused by mutations in FKBP10 or PPIB, which encode endoplasmic reticulum resident chaperone/isomerases FKBP65 and CyPB, respectively. In one pedigree branch, both parents carry a deletion in PPIB (c.563_566delACAG), causing lethal type IX OI in their two children. In another branch, a child with moderate type XI OI has a homozygous FKBP10 mutation (c.1271_1272delCCinsA). Proband FKBP10 transcripts are 4% of control and FKBP65 protein is absent from proband cells. Proband collagen electrophoresis reveals slight band broadening, compatible with ≈10% overmodification. Normal chain incorporation, helix folding, and collagen Tm support a minimal general collagen chaperone role for FKBP65. However, there is a dramatic decrease in collagen deposited in culture despite normal collagen secretion. Mass spectrometry reveals absence of hydroxylation of the collagen telopeptide lysine involved in cross-linking, suggesting that FKBP65 is required for lysyl hydroxylase activity or access to type I collagen telopeptide lysines, perhaps through its function as a peptidylprolyl isomerase. Proband collagen to organics ratio in matrix is approximately 30% of normal in Raman spectra. Immunofluorescence shows sparse, disorganized collagen fibrils in proband matrix.
Keywords: osteogenesis imperfecta, Bruck syndrome, FKBP65, FKBP10, PPIB, peptidylprolyl isomerase
Introduction
Osteogenesis imperfecta (OI; MIM#s 166200, 166210, 259420, 166220, 610967, 613982, 610682, 610915, 259440, 613848, 610968, 613849) is a collagen-related disorder, with the more prevalent (≈85%) classical/dominant forms caused by defects in type I col lagen, and the relatively rare (10–15%) recessive forms caused by defects in proteins that interact with collagen for folding or posttranslational modification. OI is characterized by bone fragility and deformity, as well as growth deficiency. Dominant forms of OI are generally described by clinical and radiographic severity using the Sillence classification, with types I–IV OI specifying mild to lethal, progressive deforming, and moderate phenotypes, respectively [Sillence et al., 1979]. In the last 6 years, deficiency of eight separate proteins has been shown to cause recessive OI and OI-like conditions, including components of the collagen prolyl 3-hydroxylation complex (CRTAP, P3H1, CyPB), collagen chaperones (HSP47 and FKBP65), the collagen-interacting anti-angiogenic factor PEDF, the osteoblastic transcription factor osterix, and most recently, the collagen C-propeptide cleavage enzyme BMP1 [Alanay et al., 2010; Barnes et al., 2006, 2010; Becker et al., 2011; Cabral et al., 2007; Christiansen et al., 2010; Lapunzina et al., 2010; Martinez-Glez et al., 2012; Morello et al., 2006; van Dijk et al., 2009].
Two of these proteins, cyclophilin B (also known as peptidylprolyl cis–trans isomerase [PPIase] B, CyPB; encoded by PPIB, NM_000942.4) and FKBP65 (FK506 binding protein, 65 kDa; encoded by FKBP10, NM_021939.3) are immunophilins, members of a family of proteins with PPIase activity. Immunophilins catalyze cis–trans isomerization of peptidylprolyl bonds, important for the proper folding of proteins. PPIases are important for type I collagen folding, as approximately one-sixth of the collagen primary sequence consists of proline residues. Both CyPB and FKBP65 are located in the endoplasmic reticulum (ER), can bind to a gelatin column, and have chaperone activity for collagen in vitro [Alanay et al., 2010; Ishikawa et al., 2008; Patterson et al., 2000; Smith et al., 1995; Zeng et al., 1998]).
CyPB is a ubiquitously expressed cyclophilin, the subset of immunophilins that can be inhibited by cyclosporine A (CsA). Its importance to collagen folding was demonstrated when CsA treatment of cells slowed type I collagen folding; the authors concluded that CyPB catalyzed the rate-limiting step in collagen folding [Steinmann et al., 1991]. In its role as a component of the collagen 3-hydroxylation complex, CyPB is involved in modification of select prolines on types I, II, V, and XI collagen, and acts as a collagen chaperone [Ishikawa et al., 2009; Weis et al., 2010]. Recessive mutations in PPIB show phenotypic variability, ranging from moderately severe to lethal [Barnes et al., 2010; Pyott et al., 2011; van Dijk et al., 2009].
FKBP65 is the largest member of the immunophilin subfamily that binds FK506; it is one of the five FKBPs (FK506 binding proteins) that localize in the ER, with 4 PPIase domains, 2 Ca2+-binding EF hand motifs, and an ER retention sequence at its carboxyl end [Coss et al., 1995]. Tropoelastin was shown to be a ligand for FKBP65 in the secretory pathway over a decade ago [Davis et al., 1998]. FKBP65 also stains strongly in vascular and smooth muscle cells in the developing lung, where elastin fibers are localized [Patterson et al., 2000]. In mice, FKBP65 is highly expressed in developing, but not adult, aorta and lung, as well as brain and kidney, suggesting the existence of other ligands, although skeletal tissues were not examined [Patterson et al., 2000]. The functioning of collagen as an additional FKBP65 ligand was suggested by the binding of FKBP65 to a gelatin-sepharose column, and its enhancement of type III collagen folding [Zeng et al., 1998]. More recently, FKBP65 was shown to act as a chaperone in well-described assays, inhibiting the aggregation of citrate synthase and rhodanese [Ishikawa et al., 2008]. It also delayed fibril formation of type I collagen in vitro. FKBP65 is unique among ER-resident chaperones in that it is rapidly degraded in response to ER stress [Murphy et al., 2011].
Deficiency of FKBP65 in recessive type XI OI was first reported in 2010 in cases of progressive deforming OI from Turkey and Mexico [Alanay et al., 2010]. It quickly became clear that the phenotype of Bruck syndrome type I, with congenital joint contractures as well as osteoporosis, fragile bones, and short stature, overlapped with the phenotype of FKBP10 mutations [Kelley et al., 2011; Setijowati et al., 2012; Shaheen et al., 2010, 2011]. Five independent pedigrees with the same FKBP10 frameshift mutation (c.831_832insC) were reported, three of which included individuals with congenital joint contractures [Alanay et al., 2010; Kelley et al., 2011; Shaheen et al., 2011]. The pathogenesis of OI related to FKBP65 deficiency is still unclear. The type I collagen produced by the previously reported cases of type XI OI has normal helical posttranslational modification and normal α1(I)Pro986 3-hydroxylation, suggesting no significant defect in collagen folding from loss of FKBP65's cis–trans isomerase activity [Alanay et al., 2010; Venturi et al., 2012]. A moderate delay of collagen secretion and the formation of collagen aggregates in one cell line with a FKBP10-null mutation suggested a potential collagen chaperone role [Alanay et al., 2010].
We have identified a homozygous frameshift mutation in FKBP10 in a Palestinian child with moderately severe OI without contractures. Investigation of cells from this proband provides the first demonstration that type I collagen folding kinetics are normal in the absence of FKBP65, but collagen cross-linking and deposition of collagen into matrix are strongly decreased, resulting in matrix with sparse and disorganized collagen fibrils. The decreased collagen deposition is caused by near absence of collagen telopeptide hydroxylation in proband-secreted collagen. Interestingly, the same extended consanguineous pedigree included perinatal lethal recessive OI in another branch, caused by a PPIB mutation.
Materials and Methods
Patient Samples
White blood cell genomic DNA (gDNA) was collected from nine members of an extended Palestinian pedigree, with informed consent. In addition, a skin biopsy was taken at 9 years of age from proband 9, providing primary fibroblasts for preparation of fibroblast gDNA using a Gentra Puregene DNA extraction kit (Qiagen, Valencia, CA) and isolation of total RNA.
Mutation Detection
Initially, probands 1, 2, 5, 6, and 9 leukocyte gDNAs were screened for mutations in two components of the collagen 3-hydroxylation complex, CRTAP (NM_006371.4) and LEPRE1 (NM_022356.3). Then, PPIB was sequenced in the parents of the lethal infants (probands 1 and 2) and also in proband 9. A PPIB mutation was detected in the family with lethal OI (probands 1–4), followed by amplification and HpaII restriction digestion of PPIB exon 5 of all nine family members. This PPIB mutation was confirmed in both parents of the lethal infants but not in the pedigree branch with nonlethal proband 9. In addition, proband 9 complementary DNA (cDNA) was screened for mutations in SP7 (NM_001173467.1) by PCR and sequencing. Although proband 9 did not have contractures or the skeletal deformity previously reported for FKBP10 mutations, FKBP10 transcripts in proband cDNA were measured by real-time (RT)-PCR (see below). Because reduced FKBP10 transcripts were detected, the 10 exons of FKBP10 were amplified and sequenced in proband 9, resulting in identification of a homozygous mutation in exon 8. All nine family members were then screened for this mutation by AvaII restriction digestion. The FKBP10 mutation was confirmed in homozygous configuration in both of her parents and in one sibling; the FKBP10 mutation was confirmed to be absent in the pedigree branch with lethal OI. All PCR products were sequenced on a CEQ2000 DNA Sequencer (Beckman, Fullerton, CA). The cDNA (c.) numbering is in accordance with journal guidelines (www.hgvs.org/mutnomen), with +1 corresponding to the first nucleotide of the ATG start codon. The PPIB and FKBP10 variants were submitted to the OI Variant Database (http://www.LOVD.nl/PPIB or http://www.LOVD.nl/FKBP10.)
RT-PCR
Total RNA was extracted from control and proband 9 primary fibroblasts using TriReagent (Molecular Research Center, Cincinnati, OH); RNA integrity was verified on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). cDNA was reversed transcribed from 5 μg RNA using a High Capacity cDNA Archive Kit (Applied Biosystems/Life Technologies Corporation, Carlsbad, CA). Taqman Gene Expression Assays were used to determine the transcript levels of CRTAP, LEPRE1, PPIB, SERPINH1, and FKBP10. Relative expression of each gene of interest was compared with expression of GAPDH, B2M, and 18s. Expression levels were normalized to control primary fibroblasts.
TOPO Cloning
cDNA was reverse transcribed from proband 9 fibroblast RNA using Oligo(dT) primers. Cells were not treated with emetine prior to RNA collection so that only stable transcripts were analyzed. Proband 9 cDNA was used to amplify exon 6 through the 3′ UTR of FKBP10. This PCR fragment was cloned into a TOPO TA vector (Invitrogen, Life Technologies, Grand Island, NY), and clones were screened by PCR and sequencing. Intron 7 retention was detected in 3/45 clones.
Western Blot
Cell lysates were collected in RIPA buffer supplemented with a protease inhibitor cocktail (Sigma–Aldrich, St. Louis, MO). Proteins were separated on precast 4–15% Ready Gels (Bio-Rad, Hercules, CA), transferred to 0.2 μm nitrocellulose membranes, and blocked with 5% bovine serum albumin plus 1× casein before probing with antibody overnight. Antibodies used were as follows: FKBP65, CRTAP, LEPRE1 MaxPab (Abnova, Taipei, Taiwan), cyclophilin B (Abcam, Cambridge, MA), HSP47 (Stressgen, Enzo Life Sciences, Farmingdale, NY), actin (Sigma-Aldrich), and LF-39 (α1[I] N-propeptide; a generous gift from Dr. Larry Fisher, NIH). Blots were washed, incubated with secondary IR-conjugated antibodies for 1 hr, washed, and visualized on a LI-COR Odyssey infrared imager (LI-COR, Lincoln, NE).
Amino Acid Analysis
Amino acid analysis to quantify 4-hydroxyproline, proline, hydroxylysine, and lysine was performed by high-pressure liquid chromatography (AIBiotech, Richmond, VA). Prolyl 3-hydroxylation and telopeptide lysyl hydroxylation were assessed by ion-trap mass spectrometry, as previously described [Barnes et al., 2006]. Secreted procollagen was precipitated by 1 M (NH4)2 SO4, from which proα-chains were resolved by SDS-PAGE and subjected to in-gel trypsin digestion for analysis of targeted peptides by electrospray mass spectrometry.
Collagen Biochemistry
For type I collagen studies, control and proband 9 fibroblasts were grown to confluence in six-well culture dishes. Steady-state collagen analysis was conducted as previously described [Cabral et al., 2005]. Cells were incubated with 437.5 μCi/ml L-[2,3,4,5–3H] proline for 16–18 hr, prior to collection and ammonium sulfate precipitation, then analyzed by 6% SDS–urea–PAGE.
The chain incorporation assay was performed as described by Chessler et al. (1993). Briefly, procollagen alpha chains were labeled with a pulse of 140 μCi/ml L-[2,3,4,5-3H] proline for 80 min, then chased with DMEM containing 10% serum, 50 μg/ml ascorbic acid, and 10 mM proline. Samples were ethanol precipitated and procollagens were separated by 5.5% SDS–urea–PAGE.
The collagen folding assay was adapted from Steinmann et al. (1991). Cells were pulsed with 1.4 μCi/ml 14C-proline for 15 min to label procollagen chains, then the cell layer was collected every 5 min. Each sample was added to a tube containing a final concentration of 0.2% Triton X-100, 100 μg/ml trypsin, and 250 μg/ml chymotrypsin (Sigma–Aldrich), and digested for 2 min at room temperature. The reaction was stopped by the addition of a final concentration of 1 mg/ml soybean trypsin inhibitor (Sigma–Aldrich). Samples were ethanol precipitated and then run on 6% SDS–urea–PAGE.
For extracellular matrix deposition, control and proband cells were plated at the same cell density. Once cells reached confluence, they were stimulated with 100 μg/ml ascorbic acid every other day for 14 days. After 14 days, fibroblasts were either labeled for 24 hr with 406.25 μCi/ml L-[2,3,4,5-3H]-proline and the media and cell layer were harvested, as described previously [Bateman and Golub, 1994], or used for Raman spectroscopy. The 3H-proline-labeled collagens were sequentially extracted from the cell layer fraction with neutral salt, acetic acid, and pepsin. In brief, newly synthesized collagens were extracted for 24 hr with neutral salt (0.15 M NaCl in 50 mM Tris–HCl, pH 7.5). Then, collagens with acid labile cross-links were extracted for 24 hr with 0.5 M acetic acid. Finally, collagens with mature cross-links were extracted by pepsin digestion (0.1 mg/ml) for 24 hr. All matrix fractions were precipitated with 2 M NaCl. Matrix extracts were run on 6% SDS–urea–PAGE. Samples were loaded on the gel to provide an equal signal by densitometry and then total signal for each fraction was calculated from the volume of sample loaded to achieve an equivalent signal.
Pulse-chase assays were performed as previously described [Forlino et al., 1997]. Collagen secretion from control fibroblasts was compared with secretion from proband 9 fibroblasts and with fibroblasts with mutations in LEPRE1 c.1080+1G>T (homozygous) or COL1A1 c.3523G>A, p.Gly1175Ser (heterozygous), helical designation Gly997Ser. Cells were labeled for 4 hr with 2.5 μCi/ml [14C]-proline and then chased with fresh medium containing 2 mM cold proline. Medium and cell layer procollagens were harvested at the indicated times and digested with pepsin. Collagen secretion rate and absolute collagen concentration were quantified by adding AlexaFluor488-labeled MMP-1 fragments of type I mouse-tail-tendon collagen as an internal standard, followed by Cy5 labeling of the mixture as previously described [Makareeva et al., 2010]. Secretion was measured in the presence of ascorbic phosphate and 0.5% serum. Sample loading for gel electrophoresis was normalized to cell number. Collagen was quantitated by densitometry of autoradiograms and fluorescence on an FLA5000 scanner (Fuji Medical Systems, Tokyo, Japan) and analyzed with Multi-Gauge software supplied with the scanner.
Differential scanning calorimetry (DSC) of collagen solutions in 0.2 M sodium phosphate and 0.5 M glycerol at pH 7.4 was performed from 10 to 55°C in a Nano III DSC instrument (Calorimetry Sciences Corporation, Lindon, UT), as previously described [Makareeva et al., 2008].
Indirect Immunofluorescence Microscopy
For FKBP65, Golgin-97, and type I collagen immunofluorescence staining, fibroblasts were plated on two-well chamber slides (Nalge Nunc International, Rochester, NY), grown for 48 hr and stained. Briefly, for costaining of FKBP65 (BD, Franklin Lakes, NJ)/LF-68 (α1[I] C-telopeptide; a generous gift from Dr. Larry Fisher, NIH) or Golgin-97 (Life Technologies)/LF-68, cells were washed with PBS and then fixed in 4% paraformaldehyde. Fixed cells were washed, permeabilized on ice in 1% goat serum plus 0.2% TritonX-100 for 5 min, washed again, and blocked with 1% BSA in PBS. Cells were incubated with primary antibody for 1 hr, washed in PBS, and incubated with Alexa Fluor-555- or Alexa Fluor-488-conjugated secondary antibodies for 1 hr, then washed extensively. Slides were mounted using mounting media with 4′, 6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA) and coverslips. Stained cells were imaged using a Zeiss LSM 510 Inverted Meta microscope and LSM510 software.
Staining of the extracellular matrix was performed essentially as described [Valli et al., 2011]. Cells were grown to confluence on chamber slides, then treated with DMEM + 10% FBS containing 100 μg/ml ascorbic acid twice a week for 2 weeks. Matrices were washed in PBS, then fixed in 4% paraformaldehyde. The matrix was blocked in 1% BSA in PBS plus 0.02% Tween-20, washed with PBS, and incubated with primary antibody (LF-68; α1[I] C-telopeptide) in 1% BSA/PBS for 1 hr. After washing, the matrix was incubated with secondary antibody for 1–1.5 hr, washed, and examined as above.
Raman Microspectroscopy
Pieces of confluent cell cultures fixed in 1% formaldehyde in PBS for 4 hr at 37°C were placed between 1 and 0.16 mm quartz slides (ESCO Products), hydrated with PBS, and mounted into a Sen-terra confocal Raman microscope (Bruker Optics) equipped with 40×/0.95NA objective, a 50 μm pinhole, and a depolarized laser (532 nm, 14 mW at sample). Autofluorescence images of the pieces were taken under 365 nm light to highlight cell cytoplasm. The excitation laser was focused to 2 × 2 μm areas pointed either at cell cytoplasm (between nuclear edge and cell periphery) or at cell periphery. Raman-scattered light was collected from a 7 μm high focal volume, which encapsulated both cells and their matrix. Raman scattering spectra were recorded with 10–15 cm−1 resolution for 90 s. The spectra were collected from different 140 × 170 μm regions within each culture, averaged over 10 points of each type within each region, and corrected for water and quartz contributions. Matrix collagen to cell organics ratios were evaluated from decomposition of the corrected spectra on spectra of collagen-free cell cytoplasm and of purified collagen normalized to have similar integral intensities of stretching vibrations of all organic CH groups. Decomposition was in the regions of the CH stretching (2,780–3,040 cm−1) or in the amide III region (1,180–1,380 cm−1) where collagen and cells have very distinct spectra. Alternatively, the spectra were decomposed from ratios of integral intensities of baseline-corrected, spectral bands dominated by (hydroxy)proline (Pro-A at 832–893 cm−1 or Pro-B at 901–980 cm−1) to those of stretching (2,810–3,028 cm−1) or bending (1,400–1,511 cm−1) vibrations of the CH groups or amide I (1,620–1,705 cm−1) vibration of proteins. Content of intracellular collagen in these cultures was negligible. Standard errors and P values (two-tail t-test) were calculated for variations between different regions within the control (n = 17) and FKBP10-null (n = 8) cultures.
Results
Case Reports
An extensive Palestinian pedigree was identified with lethal and moderately severe OI in different branches. In one branch of the pedigree, two infants (offspring of probands 1 and 2, who are first cousins) had lethal OI (Fig. 1). The first infant was a girl, born with multiple deformities, who died immediately after birth; no medical records were available. The second infant was a boy, who weighed 2,300 g (<5th percentile), with a length of 40 cm (<5th percentile), a head circumference of 32 cm (2nd percentile), and Apgar scores of 7, 8, and 9 at 1, 5, and 10 min respectively. On newborn exam, there was a wide separation of the cranial sutures, and deformities of all extremities with palpable callus. Skeletal radiographs showed thin cortices and multiple fractures (data not shown). Two subsequent offspring were unaffected.
Figure 1.

Extended Palestinian pedigree with Types IX and XI OI. Pedigree of extended Palestinian family members, including probands 1–9, with a history of lethal OI in one branch of the family (probands 1–4) and moderately severe OI (probands 5–9) in another branch. Proband 2 is a heterozygous carrier of a PPIB c.563_566delACAG mutation in exon 5 of the gene. A HpaII restriction enzyme digest confirms that parents 1 and 2 are both carriers and unaffected siblings 3 and 4 are not carriers of the PPIB mutation. Proband 9 has a homozygous FKBP10 c.1271_1272delCCinsA mutation, leading directly to a PTC and a null allele. This mutation was confirmed to be heterozygous in parents 5 and 6 and unaffected sibling 8 by AvaII restriction enzyme digestion.
In another branch of the pedigree, there was a proband with moderately severe OI (proband 9, Fig. 1). This 8 and 9/12-year-old girl came to medical attention with a history of multiple fractures. Her parents were second cousins. The mother gave a history of an uneventful pregnancy and delivery. The child had a birth weight of 2,500 g (10th percentile) and her length was described as normal. At birth, she had a deformity in her right thigh, associated with a right femoral fracture. There were multiple fractures since birth. After several corrective orthopedic procedures in Germany, she was able to ambulate with a walker. After she attained mobility with a Zimmer frame, she had a left arm fracture, and later walked with the aid of elbow crutches. At 6 years of age, after recurring lower extremity fractures, an intramedullary rodding procedure was performed in Germany. Radiographs taken before surgery (Fig. 2) show well-preserved vertebral height with minimal central compression and gracile lower long bones with minimal bowing.
Figure 2.
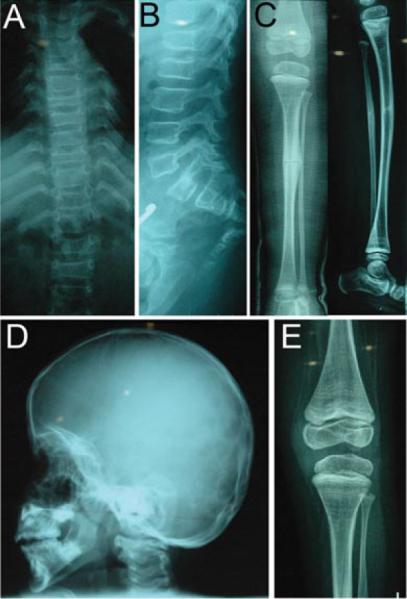
Radiographs of child with FKBP10 mutation. A: Spine radiograph showing minimal curvature. B: Vertebrae are osteoporotic but height is well preserved with minimal central compression. C: Tibiae are osteoporotic with reduced cortical thickness and mild bowing, but are well modeled. D: Skull is undermineralized without wormian bones. E: Knee radiographs reveal open growth plates without popcorn formation, sclerotic bands from multiple cycles of bisphosphonate treatment are evident.
The proband has mild short stature, 120 cm at age 8 and 9/12 years (50th percentile for a 7-year-old girl), when her weight was 29 kg (50th percentile). She had relative macrocephaly, white sclerae, and normal dentition. She does not have limb contractures. She had unspecified surgeries to correct loss of bladder control around age 8 years old. Her intellectual development is normal.
Proband Mutations in FKBP10 and PPIB
Proband 1, 2, 5, 6, and 9 gDNA was screened for recessive mutations in CRTAP and LEPRE1, by PCR amplification and sequencing. In addition, proband 9 was screened for SP7 mutations. The sequencing of PPIB in the parents of the lethal infants (probands 1 and 2) and proband 9 revealed that the parents of the lethal infants are both heterozygous carriers of a PPIB c.563_566delACAG mutation, located in the final exon of the gene (exon 5). Although this mutation leads to a premature termination codon (PTC) six nucleotides downstream (p.Asp188Alafs*6), it is consistent with a lethal OI phenotype in homozygous form, as the transcripts would be expected to escape nonsense-mediated decay. The fragment containing the 4-bp deletion was detectable after HpaII digestion, and confirmed that both parents are carriers of the same mutation, while the unaffected offspring (probands 3 and 4) have two normal alleles; members of the other branch of the pedigree do not carry this mutation. Only gDNA of parents was available for the PPIB mutation, so no further studies could be done.
RNA from proband 9 fibroblasts was used to determine the relative expression of genes causing recessive OI (CRTAP, LEPRE1, PPIB, FKBP10, and SERPINH1) (Table 1). Because proband 9 cells contained reduced levels of FKBP10 transcripts, proband 9 was screened by PCR and sequencing for FKBP10 mutations. Proband 9, with moderately severe OI, was found to be homozygous for an FKBP10 c.1271_1272delCCinsA mutation, located in exon 8, which encodes the 4th PPIase domain. The mutation introduces an AvaII restriction site, which was used to confirm heterozygosity in both parents (probands 5 and 6) and one unaffected sibling (proband 8). This mutation is functionally null, with FKBP10 transcripts at 4% of control cell levels (Table 1), consistent with the frameshift leading directly to a PTC in exon 8 only 12 codons downstream of the mutation (p.Ala424Aspfs*12). In addition, a minor portion (less than one-tenth) of stable proband FKBP10 transcripts has retention of intron 7 (data not shown). Since the mutation is 15 bp into exon 8 and would not directly affect the splice acceptor site, splicing intermediates cannot be ruled out. The transcript with intronic retention has a PTC in the retained intron.
Table 1.
Effect of FKBPW10 Mutation on Gene Expression
| RNA expressiona | versus GAPDH | versus B2M | versus 18s | Geometric mean |
|---|---|---|---|---|
| FKBP10 | 0.06 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.02 | 0.04 |
| SERPINH1 | 2.33 ± 0.08 | 1.72 ± 0.06 | 1.50 ± 0.01 | 1.82 |
| CRTAP | 1.21 ± 0.22 | 0.79 ± 0.14 | 1.22 ± 0.01 | 1.05 |
| LEPRE1 | 1.12 ± 0.06 | 1.63 ± 0.12 | 1.20 ± 0.03 | 1.30 |
| PPIB | 1.62 ± 0.34 | 2.03 ± 0.15 | 1.29 ± 0.05 | 1.62 |
The relative expression of each mRNA was normalized to GAPDH, B2M, or 18s expression and then compared with the relative mRNA expression of a control fibroblast cell line (which was arbitrarily set to 1), n = 2–3, run in triplicate.
FKBP10 Mutation has Minimal Effect on Collagen Folding and Modification
Type I collagen steady-state electrophoresis of proband 9 is minimally abnormal (Fig. 3A), with slight broadening of α1(I) and α2(I) bands. This is consistent with the approximately 10% increase in helical hydroxylysine and hydroxyproline residues in proband collagen (Table 2). This minimal overmodification did not increase collagen Tm, determined by DSC (Fig. 3B). Collagen secretion from proband cultured cells was not significantly delayed in a pulse-chase assay (Fig. 3C). Quantification of collagen secretion rate and absolute collagen concentration using an internal collagen standard confirmed absolute proband collagen secretion was in the normal range (proband 0.87 pg collagen/hr vs. control 0.95 pg collagen/hr) (Fig. 3D). Both the association of proα1(I) chains into trimers (Fig. 3E) and collagen helical folding in a direct folding assay (Fig. 3F) are normal in FKBP10-null cells.
Figure 3.
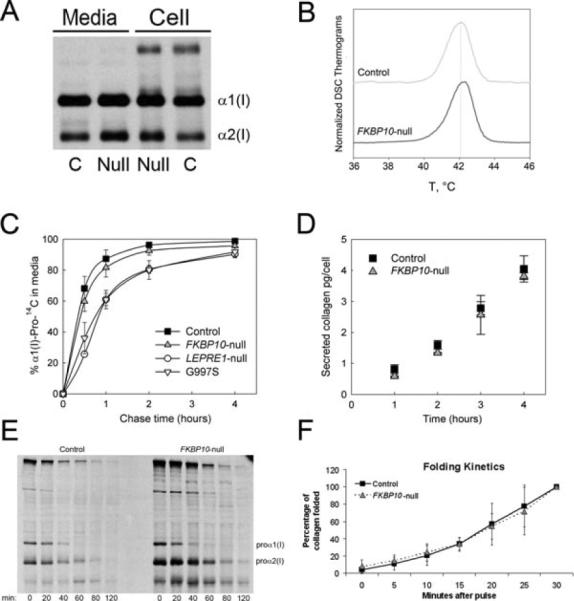
FKBP10-null mutation causes minimal changes in type I collagen. A: Steady-state collagen electrophoretic analysis, showing slight broadening of α1(I) and α2(I) bands of proband, suggestive of minimal delay in collagen folding. B: Thermal stability of type I collagen, measured by differential scanning calorimetry (DSC). Collagen melting temperature (Tm) was normal. C: On pulse-chase assay, secretion of proband collagen from cultured fibroblasts is minimally delayed versus control, and less delayed than collagen with LEPRE1 or a COL1A1 structural mutation (n = 2). D: Collagen secretion rate per cell was similar between control and proband 9 at all time points (n = 2). E: Chain incorporation assay shows free proα1(I) was incorporated into trimers at a similar rate as control. F: Collagen folding rate was measured after digestion with trypsin and chymotrypsin at various timepoints. Proband collagen folded at a normal rate (n = 3).
Table 2.
Effect of the FKBP10 Mutation on Collagen and Collagen Chaperones
| Proband 9 | Control | % Change | |
|---|---|---|---|
| Protein expression | |||
| FKBP65 | <0.01 | 1 | |
| HSP47 | 0.82 ± 0.19 | 1 | |
| COL1A1 | 0.84 ± 0.12 | 1 | |
| CRTAP | 1.12 ± 0.08 | 1 | |
| P3H1 | 1.04 ± 0.09 | 1 | |
| CyPB | 1.17 ± 0.32 | 1 | |
| Hydroxylation (%) | |||
| Hydroxylysine/total lysines | 26.5 | 23.5 | +12.75 |
| Hydroxyproline/total prolines | 47.7 | 44.6 | +7.0 |
| 3-hydroxylation of α(I)Pro986 | 98 | 94–97 | |
| Telopeptide lysyl hydroxylation | <1 | 58–59 |
Values are means ± SD, n = 2–3. The relative levels of protein were normalized to actin, then compared with normal protein levels from a control cell line (arbitrarily set to 1).
Effect of FKBP10 Mutation on Other Collagen Chaperones
The transcript and protein levels of other known collagen chaperones were measured in proband 9 fibroblasts (Tables 1 and 2 and Fig. 4A). Transcript levels of CRTAP and LEPRE1 were normal, while chaperones PPIB and SERPINH1 were slightly elevated (1.62- and 1.82-fold, respectively). Transcripts of SERPINH1 might be elevated in an attempted compensation for the low FKBP10 transcript levels, since HSP47 and FKBP65 have been speculated to form a complex [Kelley et al., 2011]. There are no known interactions of FKBP65 and CyPB to account for the elevation of PPIB transcripts.
Figure 4.

Effects of FKBP10 mutation on collagen and collagen chaperone levels. A: Western blots of FKBP10-null fibroblast lysates confirm the absence of FKBP65 protein, and demonstrate normal levels of type I procollagen (COL1A1) and the collagen chaperones HSP47, CRTAP, P3H1, and CyPB (See also Table 2). B: Immunofluorescence staining of control fibroblasts shows FKBP65 is localized to the ER. Collagen antibody staining revealed a similar pattern in proband and control, with absence of collagen aggregation in the proband samples. C: Immunofluorescence of control fibroblasts shows more structured Golgi (Golgin-97) with increased overlap with type I collagen (COL1A1), as reflected by the yellow color in the overlay panel. Proband fibroblasts showed less overlap of collagen with the Golgi, and the Golgi appeared more fragmented or condensed (see inset). Scale bars on inset = 10 μm.
All chaperones had normal protein levels by Western blot (Table 2). Costaining for FKBP65 and type I collagen did not reveal collagen aggregation in cells (Fig. 4B). Colocalization of type I collagen with Golgin-97, a marker for the trans-Golgi network, appeared less distinct and more fragmented in proband 9 cells compared with control, with less signal overlap (Fig. 4C).
FKBP10 Mutation Decreases Collagen Cross-Linking in Matrix
We investigated the effect of the FKBP10 mutation on deposition of collagen into matrix in culture. After extraction of the matrix, samples were balanced for equal signal, analyzed by densitometry and normalized for sample loading. The proportion of proband collagen in the media fraction is increased 8–10% compared with control. However, there is a decrease in all fractions of proband 9 matrix when compared with control: by 70–95% in the non-cross-linked neutral salt fraction, by 90–95% in the immaturely cross-linked acetic acid fraction, and by >95% in the pepsin-digested fraction, which contains collagen with mature cross-links (Fig. 5A). Furthermore, the cross-linked β-forms of collagen are nearly eliminated from proband matrix, with only a residual amount of β1–2 form in the acetic acid fraction containing collagen with immature cross-links.
Figure 5.

Collagen is sparse and disorganized in extracellular matrix deposited by fibroblasts. A: Collagen content of extracellular matrix deposited by FKBP10-null and control fibroblasts was examined. The collected matrix was extracted into three fractions: neutral salt (NS, newly incorporated collagen without cross-links), acetic acid (AA, immaturely cross-linked collagen), and pepsin digested (P, mature cross-linked collagen), and compared with collagen secreted in the media. Thirty-fold more sample was loaded for the proband to get an equivalent signal in the maturely cross-linked collagen fraction, with a loss of collagen cross-linked β-forms (n = 2). B: The extracellular matrix deposited by cultured control and FKBP10-null fibroblasts was stained with a collagen antibody (LF-68, an α1[I] C-telopeptide antibody) and DAPI, then imaged using a confocal microscope at 20× (left) or 63× (right, z-stacked images). Control matrix has abundant long fibrillar collagen strands, while collagen in proband matrix appears mesh-like, branched, thinner, and more sparse. Matrix from a patient with classical OI has abnormal fibrils, but they are as abundant as control (n = 2, representative panels shown). C: Matrix collagen to cell organics ratio in cultures measured by Raman microspectroscopy at locations where matrix and cell cytoplasm overlap. The ratios were measured in the spectral regions of amide III, CH-stretching or proline A bands. Consistent reduction in collagen of the FKBP10-null culture was also found using proline-B band, CH-bending band of all organic molecules, or amide I band of all proteins. The error bars represent the standard errors. *** P < 0.0005.
To investigate whether the decreased collagen in matrix represented an inability of proband collagen to engage in cross-links, we examined the telopeptide lysyl hydroxylation in secreted proband collagen by mass spectrometry. Less than 1% of telopeptide lysines were hydroxylated in proband collagen vs. about 60% hydroxylation in two independent normal controls (Table 2). Since telopeptide lysines tend to be partially hydroxylated in dermal cell cultures despite not being hydroxylated in mature skin tissue [Eyre et al., 2011], this assay indicates a failure of the telopeptide lysines to be hydroxylated by FKBP10 mutant cells. The decrease in matrix deposition in culture therefore is attributable to abnormal posttranslational telopeptide hydroxylation and cross-linking, rather than to less collagen being available, since proband collagen secretion level was normal (Fig. 3D).
Matrix Deposited by Proband Cells has Sparse and Disorganized Collagen Fibrils
When the matrix deposited by proband 9 cells in long-term culture was stained for collagen fibrils and visualized with z-stacked images, collagen fibrils were barely detectable (~10%) in proband matrix as compared with control or classical OI matrix (Fig. 5B). The fibrils that were detectable were disorganized, compared with the orderly bundles of fibrils in matrix deposited by control cells.
Since fibers thinner than 300 nm are not well detected with immunofluorescence, Raman microspectroscopy was employed to detect the presence of collagen in fibers of all diameters. The ratio of matrix collagen to cell organics in proband cell cultures was estimated to be about one-third that of control matrix from characteristic Raman peaks of collagen versus other organic molecules (Fig. 5C). The collagen to organics ratios were similar at locations where the laser beam passed through matrix and cells at the cell periphery or at the middle of the cell cytoplasm, suggesting that the reduced collagen deposition by FKBP10-null cells was not associated with differences in cell morphology or packing. A proband skin biopsy was not available to determine whether fibrils in tissue were of smaller diameter.
Discussion
The expanding panorama of gene defects causing recessive OI is identifying proteins crucial for normal bone development and providing novel information on the mechanism of OI. We studied an extensive consanguineous pedigree, in which homozygous null mutations in two different genes known to cause recessive OI, FKBP10 and PPIB, were found in two distinct branches of the pedigree. Cultured cells from the FKBP10-null proband were used to provide new insights into the mechanism of this form of recessive OI. Hydroxylation of the collagen α1(I) telopeptide lysine crucial for collagen cross-linking in extracellular matrix was absent in proband secreted collagen. This resulted in dramatically decreased deposition of collagen detected by in vitro collagen deposition assays, immunofluorescence microscopy, and Raman spectroscopy. Type I collagen helical folding was normal by a direct intracellular assay in FKBP10-null cells.
For the proband with the PPIB mutation, we were unable to conduct further studies because no cells were available. However, the PPIB mutation described here (c.563_566delACAG) is very similar to a previously described mutation (c.556_559delAAGA), diagnosed as lethal OI on ultrasound [van Dijk et al., 2009]. The mutation described by van Dijk et al. (2009) caused loss of cyclophilin B protein and a decrease of α1(I)Pro986 3-hydroxylation. Both our mutation and the mutation reported by van Dijk et al. (2009) are in the final exon of PPIB. These mutations could lead to truncated CyPB with dominant-negative effects on the 3-hydroxylation complex, consistent with the lethal phenotype.
The prior reports of FKBP10 defects, which cause recessive OI encompass 11 independent mutations [Alanay et al., 2010; Kelley et al., 2011; Setijowati et al., 2012; Shaheen et al., 2010, 2011; Steinlein et al., 2011; Venturi et al., 2012]. The phenotypic spectrum of these cases includes both deforming OI and Bruck syndrome, a recessive OI with congenital pterygium formation. These conditions were shown to be variable manifestations of defects in the same gene, even by the same mutation. The severity of the OI symptoms has varied from progressive deforming to moderately severe in terms of fractures, bone deformity, and scoliosis; sclerae, hearing, and (usually) teeth [Setijowati et al., 2012] have been normal.
Proband 9's FKBP10 mutation is located in the 4th PPIase domain, only a few codons away from a previously reported compound heterozygous mutation in a young adult with congenital contractures and progressive deforming OI [Kelley et al., 2011]. However, our proband is at the milder end of the FKBP10 clinical spectrum, having moderate OI without contractures. Mutations expected to produce a null allele have been located in each of the four PPI-ase domains. In the two cases with molecular studies, the FKBP10 transcript levels varied from half to absent [Alanay et al., 2010; Venturi et al., 2012]. Also, FKBP65 protein was undetectable in the cells with half normal transcript levels [Venturi et al., 2012]. Cells from our proband had only 4% of the normal FKBP10 transcript level. The alternatively spliced transcripts were predicted to lead to frameshifts, PTCs, and nonsense-mediated decay, consistent with the undetectable FKBP65 protein in proband cells.
Proband 9 collagen biochemistry was minimally affected, with normal secretion and about 10% increase in helical modification, which would not account for the bone phenotype. Collagen trafficking through the Golgi was subtly altered. The Golgi may be providing a level of post-ER quality control; however, the large amount of collagen secretion may saturate the response [Arvan et al., 2002] since ultimately near-normal levels of procollagen are secreted from the cells. Most importantly, we found that collagen deposition into matrix by proband cells was significantly abnormal. Collagen deposition into the immaturely cross-linked fraction was reduced 90–95% despite a normal absolute collagen secretion into media. This included >95% reduction in the proportion of collagen in the maturely cross-linked pepsin-extracted fraction, and almost total absence of cross-linked β-forms. A crucial role for FKBP65 in formation of cross-links in extracellular matrix has previously been speculated for tropoelastin cross-linking and assembly of elastic fibers in matrix [Davis et al., 1998]. For tropoelastin, FKBP65 may be important at cross-linking domains in which lysine pairs are separated and flanked by prolines. The cis–trans configuration of the surrounding prolines could have a steric effect on the lysine residues and their availability for cross-linking. In addition, the overall folding of tropoelastin monomers could be affected, playing a role in the alignment of cross-linking domains. The resulting matrix would be insufficient and disorganized, with an absence of mature cross-links.
The overlap in phenotype between Bruck syndrome caused by mutations in PLOD2 (which encodes lysyl hydroxylase-2, LH2) and FKBP10 mutations [Kelley et al., 2011] suggests that FKBP65 protein may be required for activity of LH2 against collagen telopeptide domains. Since the posttranslational hydroxylation of the telopeptide lysine regulates the chemical quality of cross-linking, we investigated whether collagen telopeptide lysyl hydroxylation was reduced in proband collagen. Mass spectrometry demonstrated less than 1% proband telopeptide lysyl hydroxylation versus 60% in control collagen. Although telopeptide lysines are not normally hydroxylated in type I collagen of mature dermis, they can be hydroxylated in fibroblast cultures. The observed lack of telopeptide hydroxylation, if it occurred in bone collagen in vivo, would result in the absence of the normally occurring cross-links of bone matrix. This might occur if FKBP65 is required in osteoblasts to catalyze the isomerization of certain prolines in LH2 and/or its collagen substrate for the correct folding and activity of LH2 against telopeptide lysines. Alternatively, absence of FKBP65 might directly affect LH2 expression or stability. Altered levels of different cross-linked collagen forms have previously been reported in osteoporotic bone, and inhibition of cross-link formation leads to a decrease in bone biomechanical properties [Bailey et al., 1993; Oxlund et al., 1995, 1996].
In the case of type XI OI reported here, collagen fibrils deposited in matrix were sparse and disorganized, consistent with the defect in cross-linking. Collagen monomers not able to cross-link may simply dissociate from fibers due to a low collagen concentration in media. Conversely, collagen cross-linking may actually catalyze fiber formation, since fibrillogenesis of collagen lacking telopeptides is inefficient [Helseth and Veis, 1981]. In addition, many of the proband fibrils detected on immunofluorescence appeared thinner than normal. Raman spectra, which detect fibrils of all sizes, yielded a proband collagen to organics ratio of about 30% of the control level, suggesting that a substantial proportion of proband fibrils are too thin to be detected by immunofluorescence. The combined matrix insufficiency and altered, disorganized fibril structures would be expected to produce significant bone tissue pathology. Further investigation is required to understand whether the variable appearance of contractures in these patients is related to deficiency of collagen cross-links.
Acknowledgments
We would like to acknowledge the NICHD Microscopy and Imaging Core, in which the confocal microscopy was conducted.
Contract grant sponsors: NICHD Intramural Funding (to J.C.M. and S.L.); NIH grants AR37318 and AR36794; the Burgess Chair endowment.
Footnotes
Note in proof: The FKBP10-null mutation reported in this article (c.1271_1272delCCinsA) was also recently published in a paper about Bruck Syndrome by Puig-Hervás et al. (2012) These authors demonstrated the phenotypic variability of this mutation, since they found congenital contractures as well as severe OI in two unrelated Egyptian families.
Disclosure Statement: The authors declare no conflict of interest.
References
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 2002;3:771–780. doi: 10.1034/j.1600-0854.2002.31102.x. [DOI] [PubMed] [Google Scholar]
- Bailey AJ, Wotton SF, Sims TJ, Thompson PW. Biochemical changes in the collagen of human osteoporotic bone matrix. Connect Tissue Res. 1993;29:119–132. doi: 10.3109/03008209309014239. [DOI] [PubMed] [Google Scholar]
- Barnes AM, Carter EM, Cabral WA, Weis M, Chang W, Makareeva E, Leikin S, Rotimi CN, Eyre DR, Raggio CL, Marini JC. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JF, Golub SB. Deposition and selective degradation of structurally-abnormal type I collagen in a collagen matrix produced by osteogenesis imperfecta fibroblasts in vitro. Matrix Biol. 1994;14:251–262. doi: 10.1016/0945-053x(94)90189-9. [DOI] [PubMed] [Google Scholar]
- Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, de Vries P, Wirth B, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88:362–71. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral WA, Makareeva E, Colige A, Letocha AD, Ty JM, Yeowell HN, Pals G, Leikin S, Marini JC. Mutations near amino end of alpha1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem. 2005;280:19259–19269. doi: 10.1074/jbc.M414698200. [DOI] [PubMed] [Google Scholar]
- Chessler SD, Wallis GA, Byers PH. Mutations in the carboxyl-terminal propeptide of the pro alpha 1(I) chain of type I collagen result in defective chain association and produce lethal osteogenesis imperfecta. J Biol Chem. 1993;268:18218–18225. [PubMed] [Google Scholar]
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coss MC, Winterstein D, Sowder RC, 2nd, Simek SL. Molecular cloning, DNA sequence analysis, and biochemical characterization of a novel 65-kDa FK506-binding protein (FKBP65) J Biol Chem. 1995;270:29336–29341. doi: 10.1074/jbc.270.49.29336. [DOI] [PubMed] [Google Scholar]
- Davis EC, Broekelmann TJ, Ozawa Y, Mecham RP. Identification of tropoelastin as a ligand for the 65-kD FK506-binding protein, FKBP65, in the secretory pathway. J Cell Biol. 1998;140:295–303. doi: 10.1083/jcb.140.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre DR, Weis M, Hudson DM, Wu JJ, Kim L. A novel 3-hydroxyproline (3Hyp)-rich motif marks the triple-helical C terminus of tendon type I collagen. J Biol Chem. 2011;286:7732–7736. doi: 10.1074/jbc.C110.195768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlino A, D'Amato E, Valli M, Camera G, Hopkins E, Marini JC, Cetta G, Coviello DA. Phenotypic comparison of an osteogenesis imperfecta type IV proband with a de novo alpha2(I) Gly922-> Ser substitution in type I collagen and an unrelated patient with an identical mutation. Biochem Mol Med. 1997;62:26–35. doi: 10.1006/bmme.1997.2620. [DOI] [PubMed] [Google Scholar]
- Helseth DL, Jr., Veis A. Collagen self-assembly in vitro. Differentiating specific telopeptide-dependent interactions using selective enzyme modification and the addition of free amino telopeptide. J Biol Chem. 1981;256:7118–7128. [PubMed] [Google Scholar]
- Ishikawa Y, Vranka J, Wirz J, Nagata K, Bachinger HP. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J Biol Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Wirz J, Vranka JA, Nagata K, Bachinger HP. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J Biol Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S, Willaert A, Elcioglu N, Van Maldergem L, Verellen-Dumoulin C, Gillerot Y, Napierala D, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R, Martinez-Glez V, Elhossini R, Amr K, Vilaboa N, Ruiz-Perez VL. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makareeva E, Han S, Vera JC, Sackett DL, Holmbeck K, Phillips CL, Visse R, Nagase H, Leikin S. Carcinomas contain a matrix metalloproteinase-resistant isoform of type I collagen exerting selective support to invasion. Cancer Res. 2010;70:4366–4374. doi: 10.1158/0008-5472.CAN-09-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makareeva E, Mertz EL, Kuznetsova NV, Sutter MB, DeRidder AM, Cabral WA, Barnes AM, McBride DJ, Marini JC, Leikin S. Structural heterogeneity of type I collagen triple helix and its role in osteogenesis imperfecta. J Biol Chem. 2008;283:4787–4798. doi: 10.1074/jbc.M705773200. [DOI] [PubMed] [Google Scholar]
- Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, Tenorio J, Pulido V, Lindert U, Rohrbach M, Eyre D, Giunta C, Lapunzina P, Ruiz-Perez VL. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bachinger HP, Pace JM, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Murphy LA, Ramirez EA, Trinh VT, Herman AM, Anderson VC, Brewster JL. Endoplasmic reticulum stress or mutation of an EF-hand Ca(2+)-binding domain directs the FKBP65rotamase to anERAD-basedproteolysis. Cell StressChaperones. 2011;16:607–619. doi: 10.1007/s12192-011-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxlund H, Barckman M, Ortoft G, Andreassen TT. Reduced concentrations of collagen cross-links are associated with reduced strength of bone. Bone. 1995;17:365S–371S. doi: 10.1016/8756-3282(95)00328-b. [DOI] [PubMed] [Google Scholar]
- Oxlund H, Mosekilde L, Ortoft G. Reduced concentration of collagen reducible cross links in human trabecular bone with respect to age and osteoporosis. Bone. 1996;19:479–484. doi: 10.1016/s8756-3282(96)00283-9. [DOI] [PubMed] [Google Scholar]
- Patterson CE, Schaub T, Coleman EJ, Davis EC. Developmental regulation of FKBP65. An ER-localized extracellular matrix binding-protein. Mol Biol Cell. 2000;11:3925–3935. doi: 10.1091/mbc.11.11.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Hervas MT, Temtamy S, Aglan M, Valencia M, Martinez-Glez V, Ballesta-Martinez MJ, Lopez-Gonzalez V, Ashour AM, Amr K, Pulido V, et al. MUTATIONS in PLOD2 cause autosomal recessive connective tissue disorders within the bruck syndrome-osteogenesis imperfecta phenotypic spectrum. Hum Mutat. 2012 doi: 10.1002/humu.22133. http://dx.doi.org/10.1002/humu.22133. [DOI] [PubMed]
- Pyott SM, Schwarze U, Christiansen HE, Pepin MG, Leistritz DF, Dineen R, Harris C, Burton BK, Angle B, Kim K, Sussman MD, Weis M, et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum Mol Genet. 2011;20:1595–1609. doi: 10.1093/hmg/ddr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setijowati ED, van Dijk FS, Cobben JM, van Rijn RR, Sistermans EA, Faradz SM, Kawiyana S, Pals G. A novel homozygous 5bp deletion in FKBP10 causes clinically Bruck syndrome in an Indonesian patient. Eur J Med Genet. 2012;55:17–21. doi: 10.1016/j.ejmg.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Al-Owain M, Faqeih E, Al-Hashmi N, Awaji A, Al-Zayed Z, Alkuraya FS. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A. 2011;155A:1448–1452. doi: 10.1002/ajmg.a.34025. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Al-Owain M, Sakati N, Alzayed ZS, Alkuraya FS. FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification? Am J Hum Genet. 2010;87:306–307. doi: 10.1016/j.ajhg.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ. Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J Biol Chem. 1995;270:18323–18328. doi: 10.1074/jbc.270.31.18323. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Aichinger E, Trucks H, Sander T. Mutations in FKBP10 can cause a severe form of isolated osteogenesis imperfecta. BMC Med Genet. 2011;12:152. doi: 10.1186/1471-2350-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann B, Bruckner P, Superti-Furga A. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J Biol Chem. 1991;266:1299–1303. [PubMed] [Google Scholar]
- Valli M, Barnes A, Gallanti A, Cabral W, Viglio S, Weis M, Makareeva E, Eyre D, Leikin S, Antoniazzi F, Marini J, Mottes M. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01794.x. doi: 10.1111/j.1399-0004.2011.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MH, Elting MW, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi G, Monti E, Carbonare LD, Corradi M, Gandini A, Valenti MT, Boner A, Antoniazzi F. A novel splicing mutation in FKBP10 causing osteogenesis imperfecta with a possible mineralization defect. Bone. 2012;50:343–349. doi: 10.1016/j.bone.2011.10.023. [DOI] [PubMed] [Google Scholar]
- Weis MA, Hudson DM, Kim L, Scott M, Wu JJ, Eyre DR. Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J Biol Chem. 2010;285:2580–2590. doi: 10.1074/jbc.M109.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B, MacDonald JR, Bann JG, Beck K, Gambee JE, Boswell BA, Bachinger HP. Chicken FK506-binding protein, FKBP65, a member of the FKBP family of peptidylprolyl cis-trans isomerases, is only partially inhibited by FK506. Biochem J. 1998;330:109–114. doi: 10.1042/bj3300109. [DOI] [PMC free article] [PubMed] [Google Scholar]