

Abstract
A candidate gene approach was used to identify levels of nucleotide diversity and to identify genes departing from neutral expectations in coniferous species of the Alpine European forest. Twelve samples were collected from four species that dominate montane and subalpine forests throughout Europe: Abies alba Mill, Larix decidua Mill, Pinus cembra L., and Pinus mugo Turra. A total of 800 genes, originally resequenced in Pinus taeda L., were resequenced across 12 independent trees for each of the four species. Genes were assigned to two categories, candidate and control, defined through homology-based searches to Arabidopsis. Estimates of nucleotide diversity per site varied greatly between polymorphic candidate genes (range: 0.0004–0.1295) and among species (range: 0.0024–0.0082), but were within the previously established ranges for conifers. Tests of neutrality using stringent significance thresholds, performed under the standard neutral model, revealed one to seven outlier loci for each species. Some of these outliers encode proteins that are involved with plant stress responses and form the basis for further evolutionary enquiries.
Keywords: candidate gene, neutrality tests, nucleotide diversity, single nucleotide polymorphisms
Introduction
Subalpine landscapes in the Northern Hemisphere are dominated by coniferous tree species. These ecosystems are sensitive to climate change (Cannone et al. 2008; Theurillat and Guisan 2001). The ability of conifer populations to track climate optima through genetic adaptation will be difficult given the rapidity of climate change (Pautasso 2009), thus raising questions about the relative roles of in situ versus ex situ conservation efforts (Aitken et al. 2008). Genetic diversity is fundamentally important to both approaches, because the continued adaptability, health, and long-term productivity of trees are driven by genetic diversity (Schaberg et al. 2008). Conservation efforts for tree populations, therefore, will benefit greatly from descriptions of genetic diversity. Here, we estimate levels of nucleotide diversity for four dominant tree species in the European forests –Abies alba Mill, Larix decidua Mill, Pinus cembra L., and Pinus mugo Turra – using DNA sequence data from 800 genes. These species are the main components of the subalpine forest ecosystem of the Alps, being found along an altitudinal gradient from 500 to over 2000 m.
Trees growing at alpine and subalpine zones may be under strong abiotic stresses and often display phenotypic responses to stressful environments (Gamache and Payette 2004). In high mountain areas, both species distribution and the forest composition are shaped by climatic conditions (Bonan 2008; Grace 2002). An increase in temperature could shift species ranges to higher elevations, as has been shown for Pinus peuce Griseb. (Meshinev et al. 2000), Fagus sylvatica L. (Peñuelas and Boada 2003), and P. mugo (Camarero et al. 2005). Conversely, climate change has led to a complex shift of species elevational distributions in the Sierra Nevada Mountains of California (Crimmins et al. 2011).
Genetic studies will help to better understand the complexity of adaptation, by demonstrating the role of natural selection in this process. The importance of natural selection has traditionally been demonstrated in trees using common garden experiments (Eveno et al. 2008; González-Martínez et al. 2006). The timing of annual growth (phenology) and response to abiotic stresses (e.g., temperature, moisture) are primary target traits for adaptive studies (Garnier-Géré and Ades 2001; Aitken and Hannerz 2001). However, the genes underlying these quantitative traits, and the segregating polymorphism within these genes, remain unknown. Single nucleotide polymorphisms (SNPs) within candidate genes for complex adaptive traits provide informative markers for studies of natural selection (Neale and Savolainen 2004). Loci with unusually high or low levels of variation (outlier loci) may be affected by selective forces (Luikart et al. 2003) and can be detected by outlier analysis (Kelley et al. 2006). Patterns of diversity and divergence can be predicted for simple null models, so that values observed for sampled genes can be assessed for their consistency to expectations derived from these null models.
The outlier approach has been used in several forest trees, including species of Quercus (Scotti-Saintagne et al. 2004), Pinus (González-Martínez et al. 2006), Pseudotsuga (Eckert et al. 2009), and Picea (Namroud et al. 2008; Holliday et al. 2010), to detect genes showing contrasting patterns of variation. In particular, several studies have focused on specific genes involved in the adaptation to cold (Eckert et al. 2009; Wachowiak et al. 2009) and to drought tolerance (Pyhäjärvi et al. 2007; Eveno et al. 2008; González-Martínez et al. 2008; Grivet et al. 2009, 2011), thus enabling the application of a population genetic approach to the study of adaptation.
The specific aims of this study were (i) to discover SNPs sampled from natural populations of each species; (ii) to estimate nucleotide diversity in the four target species; (iii) to identify among a large set of putative candidate genes those that may be under natural selection; and (iv) to determine whether the genes found to be potentially under selection were common among the four species.
Material and methods
Species
Abies alba is primarily a mountain species, distributed throughout western, central, and southern Europe (Farjon 1990). The natural range for this species is patchy, as a result of several migratory pathways following the end of the last ice age (Liepelt et al. 2009). Larix decidua is naturally distributed in both central and eastern Europe (Rubner 1953). It occurs in the high mountains of central Europe between 1000 and 2200 m, while in Central Alps, it can be found even at higher elevation (Farjon 1990). Pinus cembra and P. mugo are both pioneer species that form pure or mixed stands; above timberline, P. cembra is usually present as solitary individuals, while P. mugo may form pure and dense stands. Pinus mugo is phenotypically variable with a complex classification (Monteleone et al. 2006), mainly due to its high morphological variability in growth habit (Christensen 1987; Wachowiak and Prus-Głowacki 2008). It is native to the mountains in central and southern Europe. Pinus cembra is a glacial relict that has survived in the high European mountains (Höhn et al. 2005). Its fragmented range is mainly due to postglacial competition with Picea abies Karst (Höhn et al. 2009).
Sampling
Sampled trees were selected to cover the natural distribution of each species. Several seeds were collected from one individual at each of the 12 natural populations per species located in European Mountains (Fig. 1; Table S1). For each sampled tree, the latitude and longitude positions were recorded.
Figure 1.
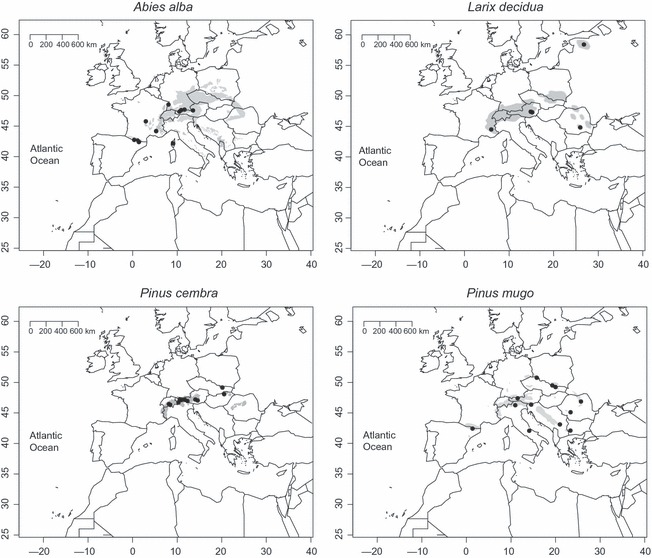
Sampling locations across species distribution in Europe.
Identification of candidate genes
Sequencing primers used in this study resulted from previous research on P. taeda L. (cf. http://dendrome.ucdavis.edu/NealeLab/adept2/), where a set of 7535 primer-pairs was developed for Sanger resequencing (Eckert et al. 2010a), using clustered ESTs (Expressed Sequence Tags). A subset of 800 primer pairs (Data S1) that successfully amplified and resequenced in other species within the Pinaceae (cf. http://dendrome.ucdavis.edu/NealeLab/crsp/) were used in the present study. The process of data generation is outlined in Fig. S1. A total of 698 genes of the 800 attempted had sequence data for at least one species following the removal of duplicate sequences and organellar sequences. To identify the potential protein function of the 698 genes, BLAST, BLASTp, and tBLASTx analyses were performed in the NCBI (http://www.ncbi.nlm.nih.gov/) and TAIR (http://www.arabidopsis.org/) databases, using published sequences of Arabidopsis thaliana. When possible, previous studies on gene expression in other conifers were also taken into account when defining candidate genes (Eveno et al. 2008). From the unique gene data set, a subset of 430 candidate genes was identified based on their biological function in Arabidopsis. The remaining 268 genes were used as genomic controls to contrast with estimates of nucleotide diversity, divergence, and outlier detection in candidate genes (Devlin et al. 2000). After removing low-quality data, the data set consists of 246 candidate genes and 156 control genes in total. The exact number of genes for each species is reported in Fig S1.
DNA isolation and resequencing
After seed germination, DNA was isolated from the haploid megagametophytes using Qiagen Plant Mini kits (Qiagen, Valencia, CA, USA) and stored at −80°C. Each DNA extraction consisted of a single megagametophyte. The DNA was amplified subsequently with RepliG kit (Qiagen) following the manufacturer’s protocol. The amplified product was purified and sequenced directly following standard protocols (Eckert et al. 2010b). The 800 amplicons were resequenced using Sanger sequencing methods by Agencourt Bioscience Corporation (Beverly, MA, USA).
Sequence analysis
A customized pipeline, PineSAP (Wegrzyn et al. 2009), was used to generate sequence alignments and to identify SNPs. A customized Perl script (https://dendrome.ucdavis.edu/TGPlone/research-projects/ace-sap/) was used subsequently to identify sequencing primers in each alignment and to mask bases outside the sequence interval defined by these primers whose signal would be due to mispriming. As megagametophyte samples were not pooled, each DNA sample is derived from a single meiotic event, so it is not expected to see problems associated with Sanger sequencing in diploid tissue (e.g., calling heterozygous SNPs or phasing owing to heterozygous indels). This also allowed rejecting samples that contained potentially paralogous sequence by detecting the presence of secondary peaks in the chromatograms. The quality of base calls, especially those associated with SNPs, was confirmed visually using CONSED (Gordon et al. 1998). The sequence alignments were subsequently aligned with P. taeda, which was used as an outgroup for each species separately using the profile-profile option in MUSCLE (Edgar 2004). Manual adjustments to these alignments were performed using Se-Al version 2.0a9 when necessary (Sequence Alignment Editor version 2.0, Rambaut 1996–2002). All sequences were deposited in GenBank (JQ440374-JQ445205).
Sites within genetic loci were annotated manually by first aligning sequences with the P. taeda ESTs to identify introns. A tBLASTx analysis was subsequently performed against the refseqRNA database for Arabidopsis to identify the putative coding intervals for each gene. Finally, BLASTp analyses against Arabidopsis thaliana gene models used to derive coding intervals were used to verify delineation of coding intervals and frame for each locus. These annotations were successfully performed for 80% of the candidate genes and 50% of the control genes (Data S4).
Nucleotide diversity and divergence
Aligned sequences were analyzed with the DNA Sequence Analysis and Manipulation (DnaSAM) program (Eckert et al. 2010b). Sites with missing data, with indels, or that violated the infinite sites mutational model were not included in the analysis. For each gene, nucleotide diversity per site was estimated with θπ (Tajima 1983; Nei 1987), the average pairwise difference between sequences, and θw (Watterson 1975), which is based on the number of segregating sites. Pairwise divergence (Dxy) was computed between each species and P. taeda, which was represented as a majority rules consensus sequence. Based on the subset of the annotated genes, nucleotide diversity at synonymous and nonsynonymous sites was calculated with the polydNdS program in the analysis package of libsequence C++ library (Thornton 2003). Using gestimator from the same package, divergence per site was calculated for different categories of sites. Differences between species-specific means for diversity and divergence estimates were evaluated through bootstrapping across loci (n = 10 000 replicates). The 95% bootstrap confidence intervals were compared to assess differences between means. All statistical analyses were performed using the boot package in R (R Development Core Team 2007).
Neutrality tests
To search for patterns of diversity that were not consistent with the standard neutral model (SNM), four neutrality statistics were computed for each species: Ewens–Watterson F (Ewens 1972), Tajima’s D (Tajima 1989), Fay and Wu’s normalized H (Fay and Wu 2000), and Kelly’s Zns (Kelly 1997). Each statistic for each species was tested against the SNM model using coalescent simulations (n = 10,000). Coalescent simulations were conducted using the ms program (Hudson 2002) as implemented using DnaSAM to estimate P-values under the SNM.
The multidimensional DHEW test that combines three neutrality tests – Tajima’s D, Fay and Wu’s normalized H, and Ewens–Watterson F (Zeng et al. 2006, 2007) – was applied to find the loci that departed from neutrality. The nominal threshold for the calculation of multidimensional P-values was set at a = 0.0001, from which an adjusted nominal significance level (P*) was estimated from 50 000 coalescent simulations, conditional on the SNM and the observed value of θπ. All multidimensional tests were conducted using DnaSAM. The genes with only one SNP were removed from the analysis to avoid false-positive outliers owing to their low nucleotide diversity.
Results
Nucleotide diversity and divergence in all genes
The average gene fragment length for all species was between 380 bp (±117) and 401 bp (±109) (Table 1), and average number of sequences per gene ranged from three (L. decidua) to six (P. cembra). On average, nearly half of the candidate genes were polymorphic in all species, with the exception of P. mugo, where more than 70% of the genes displayed variation. Among the control genes, P. mugo was also the most polymorphic, but for this set of genes, L. decidua and P. cembra had less than 50% polymorphic genes.
Table 1.
Sequencing summary statistics for the candidate genes and the control genes for each species.
| Candidate | Control | |||||||
|---|---|---|---|---|---|---|---|---|
| Abies alba | Larix decidua | Pinus cembra | Pinus mugo | A. alba | L. decidua | P. cembra | P. mugo | |
| Total genes | 70 | 61 | 171 | 190 | 32 | 35 | 109 | 120 |
| Length mean (bp) | 380 ± 117 | 389 ± 123 | 398 ± 117 | 386 ± 119 | 389 ± 105 | 401 ± 109 | 379 ± 106 | 381 ± 106 |
| No. of samples | 5 ± 3 | 3 ± 1 | 6 ± 3 | 4 ± 2 | 5 ± 3 | 4 ± 2 | 6 ± 3 | 5 ± 2 |
| Polymorphic genes (%) | 54.29 | 50.82 | 45.03 | 73.16 | 56.25 | 37.14 | 34.26 | 73.33 |
| Total no. of SNPs | 197 | 219 | 284 | 900 | 131 | 88 | 90 | 487 |
| No. of SNPs per gene | 3 ± 6 | 4 ± 9 | 2 ± 4 | 5 ± 9 | 4 ± 9 | 3 ± 7 | 1 ± 2 | 4 ± 7 |
| SNP frequency | 135 | 108 | 239 | 81 | 95 | 159 | 459 | 94 |
| Watterson’s θ | 0.0059 0.014 | 0.0077 0.020 | 0.0025 0.007 | 0.0082 0.039 | 0.0064 0.014 | 0.0044 0.012 | 0.0013 0.006 | 0.0069 0.013 |
| Nucleotide diversity (π) | 0.0059 0.0134 | 0.0078 0.020 | 0.0024 0.007 | 0.0081 0.079 | 0.0061 0.013 | 0.0047 0.014 | 0.0013 0.006 | 0.0067 0.013 |
| Divergence | 0.0873 0.0326 | 0.0829 0.031 | 0.0378 0.017 | 0.0157 0.013 | 0.0820 0.039 | 0.0734 0.036 | 0.0373 0.025 | 0.0159 0.012 |
Mean values are reported in boldface type with their standard deviation.
The total number of SNPs ranged from 197 in A. alba to 900 in P. mugo in the candidate genes, while in the control genes, it ranged from 88 in L. decidua to 487 in P. mugo (Table 1). The total number of SNPs per gene ranged from 0 to a high of 56 that was found in L. decidua (Fig. S4), and the average number of SNPs per gene ranged from 2 to 5 (Table 1). However, for the candidate genes, the only significant pairwise difference in the mean number of SNPs was between P. cembra and P. mugo, supported by the bootstrapped 95% confidence intervals (Fig. 2A).
Figure 2.
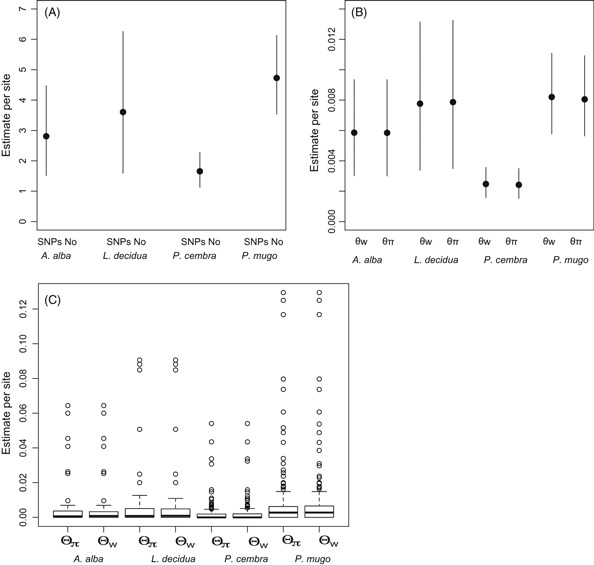
Mean values of (A) number of SNPs and (B) nucleotide diversity (θw, θπ) with the bootstrapped 95% confidence intervals and (C) distribution of nucleotide diversity in candidate genes for the different species. The box shows the upper and lower quartiles, while the line represents the median of the sample.
Among candidate genes, average estimates of Watterson’s θ ranged from 0.0025 in P. cembra to 0.0082 in P. mugo (Table 1). With the exception of A. alba, the average estimates of Watterson’s θ were lower in the control genes than in candidate genes (Table 1). The two average estimates of diversity (θπ vs θw) did not differ within species (Fig. 2B). The distribution of θπ and θw varied widely in the candidate genes (Fig. 2C) and the control genes (Fig. S2A). Nucleotide diversity estimates varied significantly among species, supported by the bootstrapped 95% confidence intervals (Fig. 2B). However, for the candidate genes, the only significant pairwise difference in nucleotide diversity estimates was between P. cembra and P. mugo, supported by the bootstrapped 95% confidence intervals (Fig. 2B).
Estimates of nucleotide divergence for candidate and control genes varied among species (Table 1) but were consistent with their phylogenetic position relative to P. taeda (Gernandt et al. 2008; Eckert and Hall 2006; Willyard et al. 2007). Abies alba showed the highest divergence from P. taeda. According to a conservative fossil calibration (Gernandt et al. 2008), the separation between the two subfamilies (Abietoideas and Pinoidea) occurred around 136 million years ago (mya). A more recent separation within the subfamily Pinoideae determined the formation of genus Larix (around 133 mya). Moreover, the two pines had different estimates of divergence. This result is consistent with their membership in the two subgenera of Pinus; P. cembra belongs to the subgenus Strobus, while both P. mugo and P. taeda belong to subgenus Pinus.
Nucleotide diversity and divergence in the annotated candidate genes
The percentage of silent sites ranged from 35% in L. decidua to 40% in P. cembra (Table 2). The number of segregating sites in all regions ranged from 80 in L. decidua to 574 in P. mugo, whereas the percentage of segregating sites in nonsynonymous regions varied from 29% in P. cembra to 43% in A. alba. In all species, the majority of the SNPs were silent with values ranging from 57% in A. alba to 71% in P. cembra. On average, two nonsynonymous SNPs per gene were found in P. mugo (range: 0–46), one in A. alba (range: 0–20) and 0.5 SNPs per gene in both L. decidua (range: 0–16) and P. cembra (range: 0–12).
Table 2.
Levels of nucleotide polymorphism in the annotated candidate genes
| Species | Parameters | Alla | N-codinga | N-Syna | Syna | All silenta |
|---|---|---|---|---|---|---|
| Abies alba | Sitesb | 23 299 | 4362 | 14 650 | 4005 | 8367 |
| 18.72% | 62.88% | 17.19% | 35.91% | |||
| Segregating sitesb | 146 | 23 | 63 | 60 | 83 | |
| 15.75% | 43.15% | 41.10% | 56.85% | |||
| Watterson’s θc | 0.0050 | 0.0027 | 0.0034 | 0.0097 | ||
| 0.0133 | 0.0129 | 0.0097 | 0.0299 | |||
| Nucleotide diversity (π)c | 0.0050 | 0.0027 | 0.0034 | 0.0097 | ||
| 0.0133 | 0.0013 | 0.0097 | 0.0294 | |||
| Larix decidua | Sitesb | 20 265 | 3571 | 12 862 | 3559 | 7130 |
| 17.62% | 63.47% | 17.56% | 35.18% | |||
| Segregating sitesb | 80 | 14 | 26 | 40 | 54 | |
| 17.50% | 32.50% | 50.00% | 67.50% | |||
| Watterson’s θc | 0.0032 | 0.0008 | 0.0017 | 0.0087 | ||
| 0.0075 | 0.0021 | 0.0058 | 0.0206 | |||
| Nucleotide diversity (π)c | 0.0033 | 0.0009 | 0.0017 | 0.0088 | ||
| 0.0075 | 0.0023 | 0.0058 | 0.0207 | |||
| Pinus cembra | Sitesb | 57 152 | 13 292 | 33 950 | 9343 | 22 635 |
| 23.26% | 59.40% | 16.35% | 39.60% | |||
| Segregating sitesb | 222 | 69 | 64 | 89 | 158 | |
| 31.08% | 28.83% | 40.09% | 71.17% | |||
| Watterson’s θc | 0.0022 | 0.0013 | 0.0010 | 0.0049 | ||
| 0.0057 | 0.0057 | 0.0035 | 0.0131 | |||
| Nucleotide diversity (π)c | 0.0022 | 0.0013 | 0.0010 | 0.0049 | ||
| 0.0057 | 0.0057 | 0.0034 | 0.0131 | |||
| Pinus mugo | Sitesb | 62 260 | 12 541 | 38 447 | 10 693 | 23 234 |
| 20.14% | 61.75% | 17.17% | 37.32% | |||
| Segregating sitesb | 574 | 123 | 234 | 217 | 340 | |
| 21.43% | 40.77% | 37.80% | 59.23% | |||
| Watterson’s θc | 0.0057 | 0.0028 | 0.0034 | 0.0112 | ||
| 0.0129 | 0.0068 | 0.0102 | 0.0261 | |||
| Nucleotide diversity (π)c | 0.0055 | 0.0028 | 0.0033 | 0.0108 | ||
| 0.0128 | 0.0068 | 0.0101 | 0.0258 |
All, all sites; N-coding, noncoding sites; N-Syn, nonsynonymous sites; Syn, synonymous sites; All silent, all silent sites.
Numbers are the total number of sites and the total number of segregating size.
Bold numbers are the average across loci with their standard deviations in regular type.
Average estimates of nucleotide diversity per site did not differ between θπ and θw; they varied among genes (Fig. S3A) and among species with values from θπ = 0.0022 in P. cembra to θπ = 0.0055 in P. mugo (Table 2). However, the only significant pairwise difference in the estimates of θπ for all sites was between P. cembra and P. mugo, supported by the bootstrapped 95% confidence intervals (Fig. 3A). Within each species, average estimates of θπ at noncoding sites were generally lower than the estimates of θπ at nonsynonymous sites, with the exception of P. cembra (Fig. 3A). The ratio of the nonsynonymous to synonymous substitution rate is an indicator of selection. Neutral genes should have a ratio close to one. All polymorphic genes showed a ratio θπa/θπs lower than 1.0 in A. alba and P. cembra, suggesting the presence of purifying selection, while one control gene had a ratio higher than 1.0 in L. decidua. In P. mugo, five candidate genes had ratios greater than one, with values falling in the range between 1.09 and 1.99 (Data S5).
Figure 3.
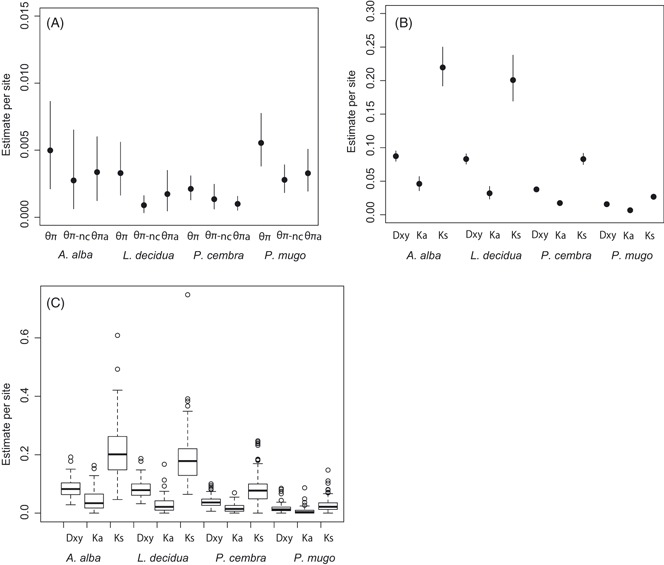
Mean values of (A) π for all sites for the annotated genes (θπ), at noncoding sites (θπ-nc), at nonsynonymous sites (θπa) (B) divergence for all sites (Dxy), divergence at nonsynonymous sites (Ka), divergence at synonymous sites (Ks), with the bootstrapped 95% confidence intervals and (C) distribution of divergence from loblolly pine across site categories in candidate genes. The box shows the upper and lower quartiles, while the line represents the median of the sample.
The estimate of divergence from P. taeda in the annotated candidate genes varied among species supported by the bootstrapped 95% confidence intervals (Fig. 3B). The highest divergence calculated for all sites was found for A. alba and L. decidua, whose values were almost three times higher than what was found in P. cembra. Moreover, the average estimates of divergence varied within species among category of sites (Fig. 3B): divergence at synonymous sites (Ks) was threefold greater than at nonsynonymous sites (Ka). Within each species, estimates of divergence varied among genes (Fig. 3C). In general, estimates of divergence varied more in A. alba (range: 0.01476–0.02093) and L. decidua (range: 0.0063–0.1869) than in the two pines (range: 0.004–0.1622 in P. cembra and 0.0014–0.085 in P. mugo).
The majority of the candidate genes exhibited a Ka/Ks ratio lower than 1.0 in all species, indicating the possible presence of purifying selection. One candidate gene (locus CL3007Contig1) showed a ratio higher than 1.0 in both A. alba and L. decidua. Four candidate genes in P. cembra (range: 1.10–1.83) and eight genes in P. mugo (range: 1.05–11.33) had a ratio greater than 1.0. In the latter species, the ratio ranged from 1.05 to 4.36; however, one gene showed an extremely high ratio (locus 0_4032, Ka/Ks = 11.33) owing to both a lower Ka and a higher Ks compared to average estimates.
The percentage of silent sites in the annotated control genes ranged from 36.86% in P. cembra to 40.39% in P. mugo (Table S2). Average estimates of nucleotide diversity per site did not differ between θπ and θw; they varied among species (Fig. S2) and among genes (Fig. S3B) with values from θπ = 0.0005 in P. cembra to θπ = 0.0112 in P. mugo (Table S2).
Neutrality tests
The results of individual candidate and control gene neutrality tests are presented in Data S3 and S6, and values varied enormously among genes. Average estimates of the neutrality test statistics reflect the differences in nucleotide diversity between candidate and control genes (Table S3). Among the candidate genes, Tajima’s D had a negative value on average in the majority of the species (A. alba = −0.13, P. cembra = −0.02, P. mugo = −0.21), reflecting the excess of low-frequency SNP alleles, indicating population size expansion and/or purifying selection or selective sweep, whereas a positive average Tajima’s D value was found in L. decidua (1.02), indicating a decrease in population size and/or balancing selection. Fay and Wu’s normalized H value was negative in all species (A. alba = −0.13, L. decidu = −0.26, P. cembra = −0.48, P. mugo = −0.28), showing the excess of high-frequency-derived SNP alleles. Estimates of average Tajima’s D were calculated for the control genes, and estimates for A. alba (−0.44) and P. mugo (−0.42) were more than twice that of the candidate genes. Furthermore, the average Fay and Wu’s normalized H estimates in the candidate genes ranged from −0.13 in A. alba to −0.48 in P. cembra. In candidate genes, the average estimates of Ewens–Watterson F ranged from 0.61 in P. mugo to 0.81 in P. cembra. The average estimates of Kelly’s Zns were generally lower in control genes than in the candidate genes, with the exception of P. cembra. In the control genes, Zns ranged from 0.59 in P. mugo to 0.92 in P. cembra and varied from 0.66 in P. mugo to 0.85 in L. decidua and P. cembra.
The critical P-values of the compound test were calculated for each gene in each species by taking into account the number of individuals and its nucleotide diversity, under the SNM assumption (Data S6). The compound DHEW test was significant for 10 candidate genes (Table 3) and eight control genes (Table S4) in just P. cembra and P. mugo. No significant DHEW values were found in A. alba and L. decidua in both gene categories.
Table 3.
List of the outliers from the standard neutral model (SNM) across the candidate genes
| Species | Gene | Putative protein | Sa | PDb | PHb | PEWb | PZnsa | DHEW-P*c | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Pinus cembra | 0_18619 | Protein kinase family protein | 6 | 0.0207 | 0.0257 | 0.8835 | 0.8812 | 0.0622 | |
| 0_2775 | spx domain-containing protein | 2 | 0.118 | 0.0773 | 0.8062 | 1 | 0.2949 | Wang et al. 2004 | |
| 0_8111 | 6-phosphogluconate dehydrogenase | 3 | 0.0798 | 0.0331 | 0.9009 | 1 | 0.1807 | Dal Bosco et al. 2004 | |
| 2_1528 | Reduced epidermal fluorescence 4 | 3 | 0.0491 | 0.0829 | 0.7472 | 0.5466 | 0.2256 | ||
| 2_6731 | E3 ubiquitin complex protein | 3 | 0.0451 | 0.0169 | 0.746 | 0.5456 | 0.2276 | Lozano-Durán et al. 2011 Bentsinka and Koornneef 2008 | |
| CL1659Contig1 | Chloride channel-like protein | 4 | 0.0786 | 0.0533 | 0.9346 | 0.9552 | 0.1050 | ||
| CL1661Contig1 | Acetyl-CoA carboxylase 2 | 3 | 0.0453 | 0.0851 | 0.9167 | 0.8924 | 0.2272 | Li-Beisson et al. 2010 | |
| Pinus mugo | 0_13913 | Exocyst subunit EXO70 family protein | 4 | 0.1146 | 0.0803 | 0.9236 | 0.8508 | 0.0985 | Synek et al. 2006 |
| 2_8627 | Carbon–sulfur lyase | 10 | 0.1131 | <0.0001 | 0.9846 | 1 | 0.0176 | Mikkelsen et al. 2004 | |
| 2_8852 | Galactokinase | 8 | 0.0155 | <0.0001 | 0.9919 | 1 | 0.0291 | Yang et al. 2009 |
The P value of Kelly Zns (PZns) and the number of SNPs per locus (S).
Results of each test (D = Tajima’s D; H = Fay and Wu’s H and EW = Ewens–Watterson’s F) are presented as P-value.
The critical P-values calculated with the compound DHEW test. (Tajima‘s D, Fay and Wu‘s H and Ewens-Watterson's F tests).
Under neutrality, the proportion of outlier loci should be the same between candidate and control genes; under non-neutrality, the proportion of candidate genes is expected to be larger than the proportion of control genes. In P. cembra, 4.09% the candidate genes were outliers and 2.75% the control genes were outliers; thus, there was no difference in the proportion of outliers detected between groups. In P. mugo, there were proportionately more outliers in the control genes (4.17%) than in the candidate genes (1.58%).
In P. cembra, one outlier candidate gene (locus 0_18619) encodes for a kinase family protein. Two loci (0_2775 and CL1659) encode for a transmembrane transport protein, while locus 0_8111 is involved in oxidoreductase activity. One locus (2_6731) is involved in gibberellic acid (GA) signaling. The Arabidopsis homologue to locus 2_1528 is involved in reduced epidermal fluorescence, while the CL1661Contig1 homologue has acetyl-CoA carboxylase activity that takes part in fatty acid biosynthetic process. In P. mugo, two candidate genes are involved in metabolic processes, such as carbon–sulfur lyases (locus 2_8627) and galactokinase protein (locus 2_8852), while locus 0_13913 is a member of the EXO70 gene family protein.
The distinction between candidate and control genes was made using the protein function of the Arabidopsis gene homologue with the aim to distinguish genes potentially involved in cell metabolism from unknown genes. No information were available on those latter genes; therefore, it is not possible to exclude that some control genes may be meaningful in terms of functionality. Moreover, the candidate genes were not defined according to their role in any specific adaptation to the environment, so many of these might be effectively neutral.
Discussion
The overarching goal of this study was to compare and contrast patterns of nucleotide diversity and tests of neutrality in four conifer species of the alpine mountain region of central Europe. These results give an overview of nucleotide diversity in four coniferous species and provide a useful SNP resource that can be applied in landscape genomic studies. In spite of the result that there were no strong and consistent differences in the proportion of outliers detected between candidate and control genes, there were several putative outlier genes that may be related to environmental adaptations. A unique aspect of this study is the comparison of diversity and departure from neutrality among four tree species living in montane ecosystems.
Pinus cembra showed lower diversity than the other tree species
Pinus cembra showed the lowest diversity among the four species, with values falling in the range of species belonging to the subgenus Strobus, such as P. chiapensis (Syring et al. 2007) and P. albicaulis (A. J. Eckert, A. D. Bower, K. D. Jermstad, J. L. Wegrzyn, B. J. Knaus, J. V. Syring and D. B. Neale, unpublished manuscript) and somewhat less than that found in other coniferous species (González-Martínez et al. 2006; Savolainen and Pyhäjärvi 2007). To the contrary, P. mugo showed the highest nucleotide diversity among the four species, with values similar to other species belonging to the subgenus Pinus (Grivet et al. 2009, 2011; Ma et al. 2006; Shiraishi and Shiraishi 2011; Wachowiak et al. 2009; Eveno et al. 2008).
The contrasting patterns of nucleotide diversity of the two pines, growing in similar altitudinal ranges (1200–2300 m for P. cembra and 1000–2000 m for P. mugo), call for an interpretation. This result may be linked to the different demographic histories of the two pines, because P. cembra is characterized by two distinct postglacial refugia in the Carpathians and in the Alps (Höhn et al. 2009), whereas in the Pliocene, the large range of P. mugo was separated into several refugia that are poorly known (Sandoz 1983,Heuertz et al. 2010). Moreover, P. cembra, like P. albicaulis Engelm., has bird-dispersed seeds (Tomback 2005), which may lead to higher levels of inbreeding (Rogers et al. 1999). Low genetic diversity within P. cembra populations in the northern Alps may be due to genetic drift by restricted gene flow (Gugerli et al. 2009). The nonpine species of this study, A. alba and L. decidua, had fairly high estimates of nucleotide diversity compared to two other pines, P. sylvestris (Pyhäjärvi et al. 2007; Palmé et al. 2008) and P. luchuensis (Shiraishi and Shiraishi 2011).
Pinus cembra shows proportionally more outlier loci
The compound DHEW test detected the presence of outlier loci in only P. cembra and P. mugo, although the single-locus tests revealed the possible presence of outlier loci in A. alba and L. decidua as well. These results suggest that selection may have acted more in the two pines than in the other two species, although this interpretation could be confounded by the fact that proportionally more highly conserved genes were tested in A. alba and L. decidua or that P. cembra had the lowest average diversity.
The presence of several genes, especially in P. mugo, showing higher nucleotide diversity at synonymous sites compared to the other site categories is an indication of purifying selection (e.g., Palmé et al. 2009), in accordance with the expectation that in coding regions, most mutations are probably disadvantageous. Moreover, divergence at synonymous sites was three times the value at nonsynonymous sites and up to eight genes per species displayed Ka/Ks ratios greater than one, which may indicate the presence of positive selection (Palmé et al. 2008). In the candidate genes, the negative average estimate of Tajima’s D found in A. alba and in the two pines may indicate the presence of recent demographic events, such as population size expansion or purifying selection or selective sweeps. Moreover, the positive value of Tajima’s D in L. decidua may indicate a decrease in population size and/or balancing selection.
Several loci deviating from neutrality were found in both control and candidate gene sets in P. cembra. Among candidate outlier loci, locus 2_6731 is the most interesting as its homologue encodes for the E3 ubiquitin complex protein, an F-box protein that is involved in GA signaling. Ubiquitination controls most of the hormonal responses in plants and is one of the dominant plant regulatory mechanisms (reviewed in Dreher and Callis 2007; Santner and Estelle 2009). Plant DNA viruses (Geminiviruses) may interfere with several responses regulated by the ubiquitin E3 ligases, making the plant more susceptible to virus infection (Ascencio-Ibáñez et al. 2008; Lozano-Durán et al. 2011). Moreover, GA modulates plant growth and development throughout the whole lifecycle of the plant (Sun 2010). Additionally, two outliers encoded for proteins related with membrane transporters (loci 0_2775, CL1659Contig1). In particular, the Arabidopsis homologue of locus 0_2775 is involved in cellular uptake of inorganic phosphate in the root xylem (Wang et al. 2004). In the same species, one outlier (locus CL1661Contig1) encodes for acetyl-CoA carboxylase, the enzyme that catalyzes the first committed step in fatty acid synthesis (Konishi et al. 1996; Li-Beisson et al. 2010). Acyl lipids constitute the membrane between cell and organelles. These genes may be important for tree fitness, because organelle proteins change in abundance during stress, as an immediate response to abiotic stress (Taylor et al. 2009).
Several outliers were also found in both control and candidate gene sets in P. mugo. Among the candidate gene outliers, locus 0_13913 encodes for a member of EXO70 family protein, which is involved in exocytosis. One member of this gene family (AtEXO70A1) was found to be crucial for polar growth and plant development (Synek et al. 2006). Locus 2_8627 has a catalytic activity for the carbon–sulfur lyase that is involved in glucosinolate biosynthesis (Mikkelsen et al. 2004). Glucosinolates are amino acid-derived natural plant products in Arabidopsis, implicated in plant defense (Halkier and Gershenzon 2006). Locus 2_8852 encodes for a galactokinase that is involved in the synthesis of d-galacturonic acid (d-GalA) polysaccharides (Yang et al. 2009).
To compare our results with other coniferous species and between species in this study, it is important to consider our results in the context of possible biases owing to (i) sequence conservation that determined the number of loci successfully resequenced, (ii) number of trees sampled, (iii) species range covered by the sampling and (iv) the effect of demography that differs from the SNM assumptions. The much lower success in the resequencing in A. alba and L. decidua than in the two pines was a direct effect of sequence conservation with P. taeda from which primers were designed. The unbalanced number of sequences per tree may have affected the estimates of nucleotide diversity, especially in L. decidua, which had the lowest average number of sequences per gene (n = 3 ± 1). In the same species, the low number of reads may have biased outlier detection; nevertheless, the sample number was used in the estimation of the neutrality tests.
Furthermore, the small number of trees sampled, the partial coverage of species ranges, and a nonuniform sample distribution according to species demographic history could all affect estimates of diversity (Städler et al. 2009). Abies alba was sampled mainly in the central-west of Europe; this might bias the estimation of nucleotide diversity, because there is a clear separation into two maternal lineages in A. alba (Liepelt et al. 2009). In P. cembra, more trees were sampled in the Alps than in the Carpathian Mountains. These two areas belonged to two different lineages, with the Carpathian populations being more polymorphic than the populations in the Alps (Belokon et al. 2005; Höhn et al. 2009). For P. mugo, the sampling covered the species range, including the area in which the different varieties (P. mugo s.s. and P. uncinata) overlap. Moreover, for the identification of the outlier loci, the estimation of the P-value in the neutrality tests did not take into account species demography. It should be noted, however, that the compound test is fairly robust to demographic deviations from the SNM (Zeng et al. 2006, 2007). The bias in sequence conservation may also have affected the identification of the outliers from SNM in the studied species, because the percentage of outliers per gene set per species ranged from 4.17% to 1.58% in the two pines, while no outliers were identified in A. alba and L. decidua.
Conclusions
This study is a first step toward trying to understand the molecular basis of adaptation, both lineage-wide and locally, for these alpine conifers. Patterns of nucleotide diversity showed that the two pines sharing the same high altitudinal habitat had contrasting levels of diversity, while the nonpine species had intermediate values. The low nucleotide diversity and the abundance of outlier loci found in P. cembra compared to the two nonpine species and to the other pine P. mugo may suggest that P. cembra may have gone through different demographic events that may have changed the original population size. Therefore, P. cembra might be more susceptible to changing climate, not having sufficient diversity to adapt to changing environment.
This research is an exploratory study on genetic diversity in four forest species that provides a new set of genetic markers. Among the present methods for the SNP discovery in nonmodel species, the candidate gene approach (Sanger sequencing) is widely used and generally requires primers that are specific to the target gene (Garvin et al. 2010). In this research, a set of candidate genes, first developed in P. taeda, was successfully transferred to other coniferous species for SNPs discovery. For example, the candidate gene approach was applied to investigate plant adaptation to drought in P. taeda (González-Martínez et al. 2006). A more recent and powerful approach, although not available at the time of this study, is RNA-Seq technology (Wang et al. 2009). A recent example of RNA-Seq technology applied to a conifer is that in P. contorta (Parchman et al. 2010).
This study is a first step in developing a polymorphism resource for four important nonmodel species of European Alpine forests. Future studies will focus on SNP genotyping across a large geographic area, with the goal of understanding the relationship between tree genotype and environmental factors, such as altitude, temperature, and water availability.
Acknowledgments
The authors would like to thank Santiago González-Martínez for his help in data analysis and all colleagues for their helpful comments. ACE-SAP project was partially funded by the Autonomous Province of Trento (Italy), with the regulation No. 23, June 12, 2008, of the University and Scientific Research Service.
Data Archiving Statement
Raw data sequences are currently available in the following database at the University of California: http://loblolly.ucdavis.edu/bipod/ftp/Sanger_resequencing/. In this database, the files are separated according to species and then locus. The studied species are coded in this way: Abies alba (Abal), Larix decidua (Lade), Pinus cembra (Pice), and Pinus mugo (Pimg). The samples are coded with the Acesap project name. The sequence data underlying the main results of the study were submitted to the GenBank website (http://www.ncbi.nlm.nih.gov/genbank/). These sequences were accepted by GenBank, and their accession number goes from JQ440374 to JQ445205. Sequences shorter than 200 bp were submitted to EMBL website (https://www.ebi.ac.uk/Databases/). The accession number for those sequences goes from HE663538 to HE663608 and from HE681087 to HE681096.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Primer sequences.
Data S2. All studied genes and their blast.
Data S3. DnaSAM results for each gene.
Data S4. Annotation input for each gene.
Data S5. PolyDNdS output and gestimator output.
Data S6. List of outliers for the compoundneutrality test (DHEW-P*).
Figure S1. Flow-chart of the data generation and screening.
{kind=link}
Figure S2. Distribution of nucleotide diversity (θw, θπ) in control gene forthe different species (A) and distribution of divergence from loblolly pine across several site categories (B).
{kind=link}
Figure S3. Distribution of nucleotide diversity (θw, θπ) in candidate genes (A) and control ones (B) across several site types in the four species.
{kind=link}
{kind=link}
Figure S4. Frequency plots of the number of SNP per gene in the candidate genes for each species.
{kind=link}
Table S1. List of the species samples with their geographical location.
Table S2. Estimates of nucleotide diversity in the control genes for several site types.
Table S3. Estimates of the neutrality tests per gene set (candidate versus control) for each species.
Table S4. List of the outliers from the standard neutral model (SNM) across the control genes.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Literature cited
- Aitken SN, Hannerz M. Genecology and gene resource management strategies for conifer cold hardiness. In: Bigras FJ, Columbo SJ, editors. Conifer Cold Hardiness. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001. pp. 23–53. [Google Scholar]
- Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S. Adaptation, migration or extirpation: climate change outcomes for tree populations. Evolutionary Applications. 2008;1(1):95–111. doi: 10.1111/j.1752-4571.2007.00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascencio-Ibáñez J-T, Sozzani R, Lee T-L, Chu T-M, Wolfinger R-D, Cella R, Hanley-Bowdoin L. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during Geminivirus Infection. Plant Physiology. 2008;148:436–454. doi: 10.1104/pp.108.121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belokon MM, Belokon YS, Politov DV, Altukhov YP. Allozyme polymorphism of Swiss stone pine Pinus cembra L. in mountain populations of the Alps and the Eastern Carpathians. Russian Journal of Genetics. 2005;41(11):1268–1280. [PubMed] [Google Scholar]
- Bentsinka L, Koornneef M. 2008. Seed dormancy and germination. The Arabidopsis Book, Number 6.
- Bonan GB. Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science. 2008;320(5882):1444–1449. doi: 10.1126/science.1155121. [DOI] [PubMed] [Google Scholar]
- Camarero JJ, Gutierrez E, Fortin MJ, Ribbens E. Spatial patterns of tree recruitment in a relict population of Pinus uncinata: forest expansion through stratified diffusion. Journal of Biogeography. 2005;32(11):1979–1992. [Google Scholar]
- Cannone N, Diolaiuti G, Guglielmin M, Smiraglia C. Accelerating climate change impacts on Alpine glacier forefields ecosystems in the European Alps. Ecological Applications. 2008;18(3):637–648. doi: 10.1890/07-1188.1. [DOI] [PubMed] [Google Scholar]
- Christensen KL. Taxonomic revision of the Pinus mugo complex and P. rhaeticaP. mugo sylvestris) (Pinaceae) Nordic Journal of Botany. 1987;7(4):383–408. [Google Scholar]
- Crimmins SM, Dobrowski SZ, Greenberg JA, Abatzoglou JT, Mynsberge AR. Changes in climatic water balance drive downhill shift in plant species’ optimum elevations. Science. 2011;331:324–327. doi: 10.1126/science.1199040. [DOI] [PubMed] [Google Scholar]
- Dal Bosco C, Lezhneva L, Biehl A, Leister D, Strotmann H, Wanner G, Meurer J. Inactivation of the chloroplast ATP synthase y subunit results in high non-photochemical fluorescence quenching and altered nuclear gene expression in Arabidopsis thaliana. The Journal of Biological Chemistry. 2004;279(2):1060–1069. doi: 10.1074/jbc.M308435200. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K, Wasserman L. Genomic control for association studies: a semiparametric test to detect excess-haplotype sharing. Biostatistics. 2000;1(4):369–387. doi: 10.1093/biostatistics/1.4.369. [DOI] [PubMed] [Google Scholar]
- Dreher K, Callis J. Ubiquitin, hormones and biotic stress in plants. Annals of Botany. 2007;99:787–822. doi: 10.1093/aob/mcl255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, Hall BD. Phylogeny, historical biogeography, and patterns of diversification for PinusPinaceae): phylogenetic tests of fossil-based hypotheses. Molecular Phylogenetics and Evolution. 2006;40(1):166–182. doi: 10.1016/j.ympev.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Eckert AJ, Wegrzyn JL, Pande B, Jermstad KD, Lee JM, Liechty JD, Tearse BR, et al. Multilocus patterns of nucleotide diversity and divergence reveal positive selection at candidate genes related to cold hardiness in coastal Douglas Fir (Pseudotsuga menziesii var. menziesii. Genetics. 2009;183(1):289–298. doi: 10.1534/genetics.109.103895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, van Heerwaarden J, Wegrzyn JL, Dana Nelson C, Ross-Ibarra J, González-Martínez SC. Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae. Genetics. 2010a;185(3):969–982. doi: 10.1534/genetics.110.115543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, Liechty JD, Tearse BR, Pande B, Neale DB. DnaSAM: software to perform neutrality testing for large datasets with complex null models. Molecular Ecology Resources. 2010b;10(3):542–545. doi: 10.1111/j.1755-0998.2009.02768.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high Throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveno E, Collada C, Guevara MA, Léger V, Soto A, Díaz L, Léger P. Contrasting patterns of selection at Pinus pinaster Ait. Drought stress candidate genes as revealed by genetic differentiation analyses. Molecular Biology and Evolution. 2008;25(2):417–437. doi: 10.1093/molbev/msm272. [DOI] [PubMed] [Google Scholar]
- Ewens WJ. The sampling theory of selectively neutral alleles. Theoretical Population Biology. 1972;3:87–112. doi: 10.1016/0040-5809(72)90035-4. [DOI] [PubMed] [Google Scholar]
- Farjon A. 1990. Pinaceae: drawings and descriptions of the genera Abies, Cedrus, Pseudolarix, Keteleeria, Nothotsuga, Tsuga, Cathaya, Pseudotsuga, Larix and Picea. Regnum Veg. 121. (Pinaceae)
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155(3):1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamache I, Payette S. Height growth response of tree line black spruce to recent climate warming across the forest-tundra of eastern Canada. Journal of Ecology. 2004;35(5):1–845. [Google Scholar]
- Garnier-Géré PH, Ades PK. Environmental surrogates for predicting and conserving adaptive genetic variability in tree species. Conservation Biology. 2001;15:1632–1644. [Google Scholar]
- Garvin MR, Saitoh K, Gharrett AJ. Application of single nucleotide polymorphisms to non-model species: a technical review. Molecular Ecology Resources. 2010;10:915–934. doi: 10.1111/j.1755-0998.2010.02891.x. [DOI] [PubMed] [Google Scholar]
- Gernandt DS, Magallón S, López GG, Flores OZ, Willyard A, Listonk A. Use of simultaneous analyses to guide fossil-based calibrations of Pinaceae phylogeny. International Journal of Plant Sciences. 2008;169(8):1086–1099. [Google Scholar]
- González-Martínez SC, Ersoz E, Brown GR, Wheeler NC, Neale DB. DNA sequence variation and selection of tag single-nucleotide polymorphisms at candidate genes for drought-stress response in Pinus taeda L. Genetics. 2006;172(3):1915–1926. doi: 10.1534/genetics.105.047126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Martínez SC, Huber D, Ersoz E, Davis JM, Neale DB. Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity. 2008;101:19–26. doi: 10.1038/hdy.2008.21. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome research. 1998;8(3):195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Grace J. Impacts of climate change on the tree line. Annals of Botany. 2002;90(4):537–544. doi: 10.1093/aob/mcf222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivet D, Sebastiani F, González-Martínez SG, Vendramin GG. Patterns of polymorphism resulting from long-range colonization in the Mediterranean conifer Aleppo pine. New Phytologist. 2009;184(4):1016–1028. doi: 10.1111/j.1469-8137.2009.03015.x. [DOI] [PubMed] [Google Scholar]
- Grivet D, Sebastiani F, Alía R, Bataillon T, Torre S, Zabal-Aguirre M, Vendramin GG, et al. Molecular footprints of local adaptation in two Mediterranean conifers. Molecular Biology and Evolution. 2011;28(1):101–116. doi: 10.1093/molbev/msq190. [DOI] [PubMed] [Google Scholar]
- Gugerli F, Rüegg M, Vendramin GG. Gradual decline in genetic diversity in Swiss stone pine populations (Pinus cembra) across Switzerland suggests postglacial re-colonization into the Alps from a common eastern glacial refugium. Botanica Helvetica. 2009;119:13–22. [Google Scholar]
- Halkier BA, Gershenzon J. Biology and biochemistry of glucosinolates. Annual Review of Plant Biology. 2006;57:303–333. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- Heuertz M, Teufel J, González-Martínez SC, Soto A, Fady B, Alía R, Vendramin GG. Geography determines genetic relationships between species of mountain pine (Pinus mugo complex) in Western Europe. Journal of Biogeography. 2010;37(3):541–556. [Google Scholar]
- Höhn M, Brán A, Vendramin GG. Genetic analysis of Swiss stone pine populations (Pinus cembra L. subsp. cembra) from the Carpathians using chloroplast microsatellites. Acta Silvatica et Lignaria Hungarica. 2005;1:39–47. [Google Scholar]
- Höhn M, Gugerli F, Abran P, et al. Variation in the chloroplast DNA of Swiss stone pine (Pinus cembra L.) reflects contrasting post-glacial history of populations from the Carpathians and the Alps. Journal of Biogeography. 2009;36(9):1798–1806. [Google Scholar]
- Holliday JA, Ritland M, Aitken SN. Postglacial history of a widespread conifer produces inverse clines in selective neutrality tests. Molecular Ecology. 2010;19:3857–3864. doi: 10.1111/j.1365-294X.2010.04767.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics Applications Note. 2002;18(2):337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Research. 2006;16(8):980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JK. A test of neutrality based on interlocus associations. Genetics. 1997;146(3):1197–1206. doi: 10.1093/genetics/146.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi T, Shinohara K, Yamada K, Sasaki Y. Acetyl- CoA carboxylase in higher plants: most plants other than gramineae have both the prokaryotic and the eukaryotic forms of this enzyme. Plant Cell Physiology. 1996;37:117–122. doi: 10.1093/oxfordjournals.pcp.a028920. [DOI] [PubMed] [Google Scholar]
- Li-Beisson Y, Shorrosh B, Beisson F, et al. 2010. Acyl-lipid metabolism. The Arabidopsis Book 8.
- Liepelt S, Cheddadi R, De-Beaulieu J, et al. Postglacial range expansion and its genetic imprints in Abies alba (Mill.) – a synthesis from palaeobotanic and genetic data. Review of Palaeobotany and Palynology 153(1-2) 2009;13:9–149. [Google Scholar]
- Lozano-Durán R, Rosas-Díaz T, Gusmaroli G, Luna AP, Taconnat L, Deng WX, Bejaranoa ER. Geminiviruses subvert ubiquitination by altering CSN-mediated derubylation of SCF E3 ligase complexes and inhibit jasmonate signaling in Arabidopsis thaliana. The Plant Cell. 2011;23:1014–1032. doi: 10.1105/tpc.110.080267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nature Reviews Genetics. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Ma X-F, Szmidt AE, Wang X-R. Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Molecular Biology and Evolution. 2006;23(4):807–816. doi: 10.1093/molbev/msj100. [DOI] [PubMed] [Google Scholar]
- Meshinev T, Apostolova I, Koleva E. Influence of warming on timberline rising: a case study on Pinus peuce Griseb. in Bulgaria. Phytocoenologia. 2000;30:431–438. [Google Scholar]
- Mikkelsen MD, Naur P, Halkier BA. Arabidopsis mutants in the C–S lyase of glucosinolate biosynthesis establish a critical role for indole-3-acetaldoxime in auxin homeostasis. The Plant Journal. 2004;37:770–777. doi: 10.1111/j.1365-313x.2004.02002.x. [DOI] [PubMed] [Google Scholar]
- Monteleone I, Ferrazzini D, Belletti P. Effectiveness of neutral RAPD markers to detect genetic divergence between the subspecies uncinata and mugo of Pinus mugo Turra. Silva Fennica. 2006;40(3):391–406. [Google Scholar]
- Namroud M–C, Beaulieu J, Juge N, Laroche J, Bousquet J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Molecular Ecology. 2008;17:3599–3613. doi: 10.1111/j.1365-294X.2008.03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale DB, Savolainen O. Association genetics of complex traits in conifers. Trends in Plant Science. 2004;9(7):325–330. doi: 10.1016/j.tplants.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. New York, NY: Columbia University Press; 1987. [Google Scholar]
- Palmé AE, Wright M, Savolainen O. Patterns of divergence among conifer ESTs and polymorphism in Pinus sylvestris identify putative selective sweeps. Molecular Biology and Evolution. 2008;25(12):2567–2577. doi: 10.1093/molbev/msn194. [DOI] [PubMed] [Google Scholar]
- Palmé AE, Pyhäjärvi T, Wachowiak W, Savolainen O. Selection on nuclear genes in a Pinus phylogeny. Molecular Biology and Evolution. 2009;26(4):893–905. doi: 10.1093/molbev/msp010. [DOI] [PubMed] [Google Scholar]
- Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation and marker discovery. BMC Genomics. 2010;11:180–196. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautasso M. Geographical genetics and the conservation of forest trees. Perspectives in Plant Ecology, Evolution and Systematic. 2009;11:157–189. [Google Scholar]
- Peñuelas J, Boada M. A global change-induced biome shift in the Montseny mountains (NE Spain) Global Change Biology. 2003;9(2):131–140. [Google Scholar]
- Pyhäjärvi T, García-Gil MR, Knürr T, Mikkonen M, Wachowiak W, Savolainen O. Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics. 2007;177(3):1713–1724. doi: 10.1534/genetics.107.077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2007. http://www.R-project.org (accessed on 10 March 2011) [Google Scholar]
- Rambaut A. 1996. –2002. Sequence alignment editor v2.0a11. http://tree.bio.ed.ac.uk/software/seal/ (accessed on 14 March 2011)
- Rogers DL, Millar CI, Westfall RD. Fine-scale genetic structure of whitebark pine (Pinus albicaulis): associations with watershed and growth form. Evolution. 1999;53:74–90. doi: 10.1111/j.1558-5646.1999.tb05334.x. [DOI] [PubMed] [Google Scholar]
- Rubner K. Die Pflanzengeographischen Grundlagen des Waldbaues. Radebeul, Berlin: Neumann; 1953. [Google Scholar]
- Sandoz H. Sur la plausibilité de l’installation de refuges pléistocènes du Pin mugho ou Pin pumilo (Pinus mughus Scopoli = Pinus pumilio Haenke) dans la basse vallée de la Durance (Provence occidentale – France) Revue Générale de Botanique. 1983;90:23–41. [Google Scholar]
- Santner A, Estelle M. Recent advances and emerging trends in plant hormone signalling. Nature. 2009;459:1071–1078. doi: 10.1038/nature08122. [DOI] [PubMed] [Google Scholar]
- Savolainen O, Pyhäjärvi T. Genomic diversity in forest trees. Current Opinion Plant Biology. 2007;10:1–6. doi: 10.1016/j.pbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Schaberg PG, DeHayes DH, Hawley GJ, Nijensohn SE. Anthropogenic alterations of genetic diversity within tree populations: implications for forest ecosystem resilience. Forest Ecology and Management. 2008;256(5):855–862. [Google Scholar]
- Scotti-Saintagne C, Mariette S, Porth I, Goicoechea PG, Barreneche T, Bodenes C, Burg K, et al. Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. and Q. petraea (Matt.) Liebl.] Genetics. 2004;168:1615–1626. doi: 10.1534/genetics.104.026849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi S, Shiraishi S. Nucleotide diversities and genetic relationship in the three Japanese pine species; Pinus thunbergiiPinus densiflora, and Pinus luchuensis. Diversity. 2011;3(1):121–135. [Google Scholar]
- Städler T, Haubold B, Merino C, Stephan W, Pfaffelhuber P. The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics. 2009;182(1):205–216. doi: 10.1534/genetics.108.094904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T-P. Gibberellin-GID1-DELLA: a pivotal regulatory module for plant growth and development. Plant Physiology. 2010;154:2567–2570. doi: 10.1104/pp.110.161554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synek L, Schlager N, Eliáš M, Quentin M, Hauser M-T, Žárský V. AtEXO70A1, a member of a family of putative exocyst subunits specifically expanded in land plants, is important for polar growth and plant development. Plant Journal. 2006;48(1):54–72. doi: 10.1111/j.1365-313X.2006.02854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syring J, del Castillo RF, Cronn R, Liston A. Multiple nuclear loci reveal the distinctiveness of the threatened, neotropical Pinus Chiapensis. Systematic Botany. 2007;32(4):703–717. [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NL, Tan Y–F, Jacoby RP, Millar AH. Abiotic environmental stress induced changes in the Arabidopsis thaliana chloroplast, mitochondria and peroxisome proteomes. Journal of proteomics. 2009;72(3):367–378. doi: 10.1016/j.jprot.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Theurillat J, Guisan A. Potential impact of climate change on vegetation in the European alps: a review. Climatic Change. 2001;50:77–109. [Google Scholar]
- Thornton K. Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics. 2003;19(17):2325–2327. doi: 10.1093/bioinformatics/btg316. [DOI] [PubMed] [Google Scholar]
- Tomback DF. The impact of seed dispersal by Clark’s nutcracker on whitebark Pine: multi-scale perspective on a high mountain mutualism. In: Broll G, Keplin B, editors. Mountain Ecosystems: Studies in Treeline Ecology. Berlin, Germany: Springer; 2005. pp. 181–201. [Google Scholar]
- Wachowiak W, Prus-Głowacki W. Hybridisation processes in sympatric populations of pines Pinus sylvestris L., P. mugo Turra and P. uliginosa Neumann. Plant Systematics and Evolution. 2008;271:29–40. [Google Scholar]
- Wachowiak W, Balk PA, Savolainen O. Search for nucleotide diversity patterns of local adaptation in dehydrins and other cold-related candidate genes in Scots pine (Pinus sylvestris L.) Tree Genetics & Genomes. 2009;5(1):117–132. [Google Scholar]
- Wang Y, Ribot C, Rezzonico E, Yves P. Structure and expression profile of the Arabidopsis PHO1 gene family indicates a broad role in inorganic phosphate homeostasis. Plant Physiology. 2004;135:400–411. doi: 10.1104/pp.103.037945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites. Theoretical Population Biology. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Wegrzyn JL, Lee JM, Liechty J, Neale DB. PineSAP – pine alignment and SNP identification pipeline. Bioinformatics. 2009;25(19):2609–2610. doi: 10.1093/bioinformatics/btp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willyard A, Syring J, Gernandt DS, Liston A, R Cronn. Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Molecular Biology and Evolution. 2007;24:90–101. doi: 10.1093/molbev/msl131. [DOI] [PubMed] [Google Scholar]
- Yang T, Bar-Peled L, Gebhart L, Lee SG, Bar-Peled M. Identification of galacturonic acid-1-phosphate kinase, a new member of the GHMP kinase superfamily in plants, and comparison with galactose-1-phosphate kinase. The Journal of Biological Chemistry. 2009;284(32):21526–21535. doi: 10.1074/jbc.M109.014761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K, Fu Y-X, Shi S, Wu C-I. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics. 2006;174(3):1431–1439. doi: 10.1534/genetics.106.061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K, Suhua S, Wu C-I. Compound tests for the detection of hitchhiking under positive selection. Molecular Biology & Evolution. 2007;24:1898–1908. doi: 10.1093/molbev/msm119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.