

1. Introduction
Acyl-homoserine lactones are small neutral lipid molecules that bacteria in the Proteobacteria group use to sense and signal their cell density. This signaling process, known as quorum sensing, activates differentiation to diverse community oriented lifestyles within the bacterial population. Since the first description of autoinduction 40 years ago 1 and quorum sensing systems in the light producing gram-negative bacterium Vibrio fischeri 2,3, which is a symbiont of the Hawaiian bobtail squid Euprymna scolopes, much has been learned about the molecular mechanism of signaling in these and many related bacteria (reviewed in 4 and Chapters 5-25 in 5). The primary signals used by gram negative bacteria to sense cell density are acyl-homoserine lactone (AHL) molecules, which are composed of a homoserine lactone ring (HSL) with an acyl chain (Figure 1). The acyl-chain length generally varies from C4 to C18 6,7 and this can be modified by a 3-oxo substituent, or in some cases, a 3-hydroxy substituent, a terminal methyl branch, or varied degrees of unsaturation 8. AHLs are synthesized by enzymes known as the AHL synthases 7,9. Once produced, the AHLs diffuse in and out of the cell by passive 10 as well as active transport mechanisms 11. The concentration of AHL eventually reaches a sufficiently high concentration at a given threshold cell number or bacterial “quorum”, and it is then recognized by a receptor protein, which is the second component of the system. The AHL-responsive receptors include a wide variety of transcriptional regulators called “R proteins”, such as LuxR or LasR, which are DNA-binding transcription factors 4,12 and a small family of sensor kinases related to LuxN 13. The binding of the AHL by most of the characterized R-proteins initiates the activation and repression of target genes, and in some cases AHL binding leads to target gene derepression 14-18.
Figure 1.
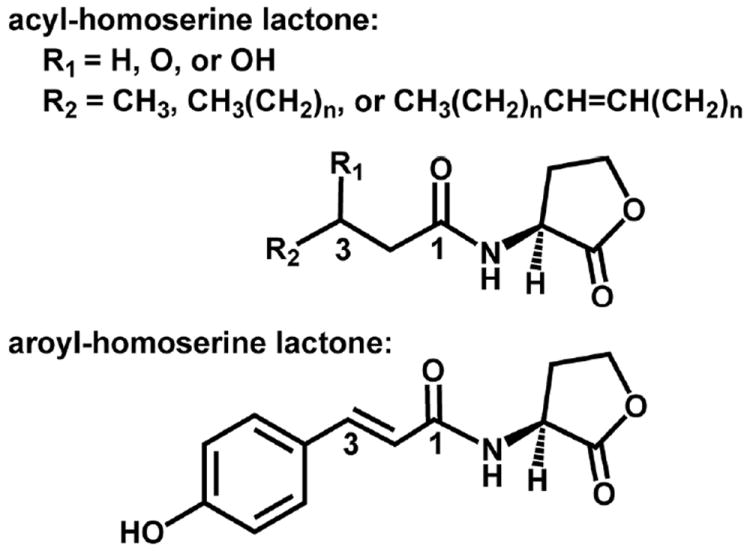
Chemical structure of homoserine lactone-based bacterial signaling molecules. The acyl-homoserine lactones (AHLs) found in Proteobacteria vary by substitution at the C3 position (R1) and the length and unsaturation at the C1 position indicated by R2. Shown also is the structure of the first aroyl-homoserine lactone, para-coumaroyl-HSL (pC-HSL).
The focus of this review article is the structural basis for quorum sensing mediated by AHL signals in gram-negative bacteria (reviewed in 19-21). The structural data in these systems comes from a relatively select number of bacterial species that are illustrated in Figure 2.
Figure 2.
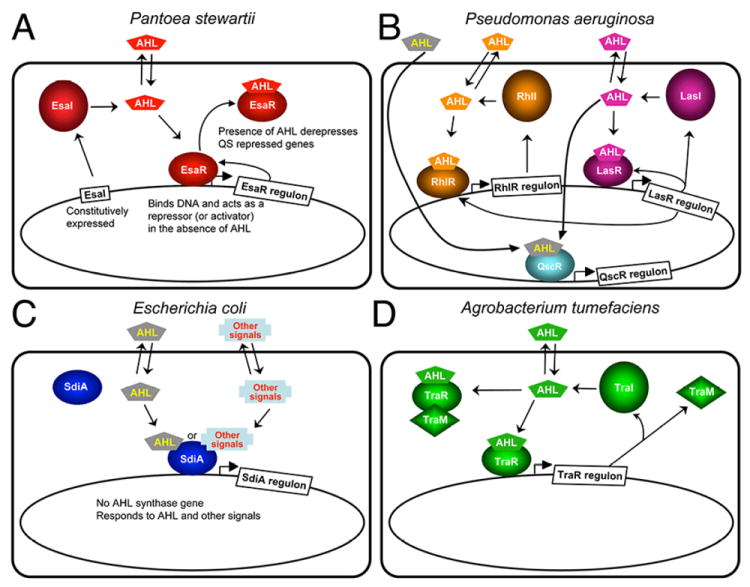
Quorum-sensing systems for which there is structural information. (A) Pantoea stewartii. EsaI constitutively produces 3-oxo-C6-HSL. EsaR binds to the AHL and is derepressed by this binding, which leads to expression of quorum sensing regulated genes in the EsaR regulon and a feedback loop regulating EsaR expression. (B) Pseudomonas aeruginosa. LasI and RhlI produce 3-oxo-C12-HSL and C4-HSL, respectively. The AHLs bind to the cognate R proteins LasR and RhlR, respectively, and activate or repress transcription of the genes in the Rhl and Las regulons. In addition, there is an orphan receptor QscR that has a regulon that overlaps with LasR. QscR can bind to AHLs made by LasI as well as exogenous AHLs that Pseudomonas does not make. The Las system is as at the top of the hierarchy and several feedback loops are found in this quorum sensing system. (C) Escherichia coli. E. coli does not have an AHL synthase, but it does have a LuxR homolog SdiA, which can bind to AHLs and regulate gene expression in response to exogenous AHLs. (D) Agrobacterium tumefaciens. TraI synthesizes 3-oxo-C8-HSL, which binds to TraR, leading to activation of TraR-controlled genes.
2. Homoserine Lactone Signaling Molecules
2.1 Acylhomoserine lactones
The types of AHLs produced and the ways in which AHL production is regulated varies greatly among bacterial species 22. Some species such as Pantoea stewartii, or Agrobacterium tumefaciens, produce mainly a single type of AHL, 3-oxo-C6-HSL23 for Pantoea stewartii or 3-oxo-C8-HSL24 for Agrobacterium tumefaciens, respectively. Such organisms typically possess a single AHL synthase gene, and the AHL synthase enzyme is fairly specific for production of a single AHL species 23,25,26. An increasing number of bacterial genome sequences have revealed the presence of multiple AHL synthase genes and a variety of bioassay studies have revealed some very complex mixtures of AHLs produced in bacteria including species of Burkholderia and Silicibacter 27-29. Burkholderia mallei, for example, has two AHL synthase genes, BmaI1 and BmaI3, which produce C8-HSL and 3-hydroxy-C8-HSL respectively 30,31. Thus, the synthesis of AHLs is controlled by the presence and levels of different enzymes, which also have different specificities for AHL production.
The production of AHL signals is regulated in different ways in a variety of organisms 22. In some organisms, such as Pantoea stewartii, the AHL synthase gene is constitutively expressed (Figure 2A) 23,32. In P. aeruginosa (Figure 2B), LasI, which makes primarily 3-oxo-C12-HSL, is produced first, and as quorum sensing progresses or population density increases, the dominant AHL synthase becomes RhlI, which makes C4-HSL 33-35. In addition, the expression levels of the AHL synthases can be regulated in complex, but as yet incompletely understood ways 36,37. For example, in P. aeruginosa the expression of the RhlI enzyme is under the positive control of the LasR transcriptional regulator, which requires 3-oxo-C12-HSL, the product of LasI (Figure 2B). Furthermore, LasR also regulates LasI expression, which forms a positive feedback loop that rapidly amplifies the amount of LasI to increase the amount of AHLs produced early in the quorum-sensing process 38. One consequence of the production of multiple distinct types of AHLs is that their cognate R-proteins regulate distinct sets of genes that are required at different times during biofilm formation and virulence factor production. Other purposes of this complex pattern of signal production remain unclear, because there are still quorum-sensing functions as well as AHL receptors that remain to be identified. Thus, the synthesis of AHLs is modulated both quantitatively by the concentration of enzymes and substrates and qualitatively by the presence of different enzymes with different specificities. For some organisms, such as E. coli, genes encoding the AHL synthase have not been identified, yet an AHL receptor, SdiA in E. coli, has been found to interact with, notably, a range of ligands including AHLs, and these AHLs are presumably produced by other organisms (Figure 2C) 39. In A. tumefaciens, expression of the AHL synthase TraI is up-regulated by the 3-oxo-C8-HSL-bound TraR transcriptional regulator (Figure 2D), creating a positive feedback mechanism to amplify production of TraI and therefore 3-oxo-C8-HSL, which in turn activates TraR. Interestingly, expression of an antiactivator called TraM (Section 5.2) is also under positive control of TraR: TraM inhibits TraR function by direct formation of the TraR-TraM anti-activation complex. Such a negative feedback control mechanism might prevent cells from prematurely engaging in energy-expensive conjugal transfer at low cell density, a quorum sensing regulated process in A. fumefaciens, and may have evolved to avoid a prolonged quorum-sensing phase or to dampen down TraR activation 40-42.
AHL signaling can be terminated actively or passively, because AHLs are susceptible to degradation by both chemical and enzymatic means 2. AHLs are hydrolyzed rapidly under conditions of alkaline pH 43,44. This sensitivity to hydrolysis of the AHL signal can be exploited by plants that respond to infection by producing alkaline substances at the site of infection 43,45. There is also evidence in Yersinia species that temperature modulates AHL availability in the cellular environment 46,47. AHLs are also susceptible to enzymatic degradation by hydrolysis of the lactone ring and deacylation. Both acylases 48-50 and lactonases have been discovered in a variety of species 51,52. Indeed, the importance of these systems for recycling the AHL signal into products of greater utility to the cell has been demonstrated 53. Furthermore, the utility of these enzymes as AHL-degrading quorum quenchers as a therapeutic or agricultural agents is promising 48, but the role of these enzymes in bacterial virulence is not completely clear 52. Therefore, the levels and the types of AHLs in the cellular environment are also regulated by different conditions of pH and temperature and the presence of degradation enzymes.
2.2 Aroylhomoserine lactones
The most recent discovery of a homoserine lactone-based signaling molecule comes from the plant symbiont Rhodopseudomonas palustris 54. In this organism, an aroyl-homoserine lactone molecule, p-coumaroyl-HSL (pC-HSL) (Figure 1), is produced within the bacterium by using environmental p-coumarate rather than fatty acids from its primary cellular pools of lipids. Plants produce p-coumarate for use in biosynthesis of lignin polymers and flavonoids 55. As such, the precursors to the substrate, which is needed in the bacterial cell for the bacterial pC-HSL synthase to make this specialized pC-HSL signal, are intermediates in plant biosynthetic pathways. It would not be surprising to see other HSL-based signals identified in the future for specialized bacterial signaling programs that could occur in the vicinity of hosts that provide appropriate substrates for AHL synthase-type enzymes 56.
3. Structural Basis of AHL Synthesis
3.1 AHL Synthase Primary Structure
Production of AHLs has been demonstrated in more than 90 species, and three different types of enzymes are now known to synthesize AHLs in vivo. These enzymes include those in the LuxI, HdtS, and LuxM families (reviewed in 20,57). The AHL synthases typified by the LuxI enzyme, the archetype of the class, were first discovered in the lux operon in Vibrio fischeri 58. Hence these enzymes have been assigned a name based on the species of origin or based on the phenotype or systems that they regulate and are frequently referred to as I-proteins. For example, LasI was named for its role in the induction of the elastase virulence factors in Pseudomonas aeruginosa 59. The LuxI-type AHL synthases catalyze the formation of the AHL from the substrates S-adenosyl-L-methionine (SAM) and acyl-acyl carrier protein (acyl-ACP) 60 (Figure 3). Enzymatic synthesis of AHLs using purified substrates for TraI, (from A. tumefaciens) verified that both SAM and acyl-ACP are substrates for AHL synthesis in vitro 61. This finding was confirmed in vivo with studies of TraI 62 and in vitro with Pseudomonas aeruginosa RhlI 63.
Figure 3.
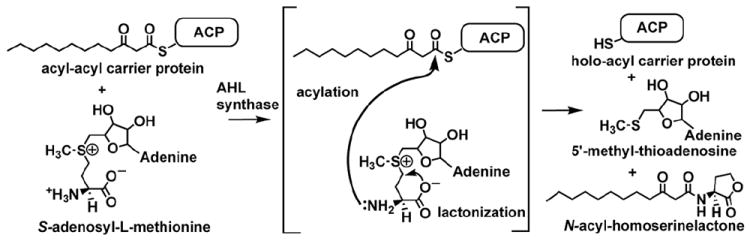
Schematic diagram of the reaction performed by AHL synthase enzymes. AHL synthases catalyze the formation of AHLs from S-adenosyl-L-methionine (SAM) and acyl-ACP by acylation of SAM and lactonization of the methionine moiety to give in addition to the AHL, holo-ACP, and 5’-methylthioadenosine (5’MTA) products 62,63.
Only a few species of γ proteobacteria have representatives of the LuxM family of enzymes. luxM was identified as a gene in Vibrio harveyi that was required for production of specific AHLs 64. In Vibrio fischeri and other related Vibrio species, the AinS and VanM enzymes were also found to produce AHLs 65,66. The VanM and AinS enzymes each have sequence similarity to LuxM. Although, these enzymes do not appear to be related to any other enzyme family, enzymatic studies in vitro show that they use SAM and acyl-ACP as well as acyl-Coenzyme A (acyl-CoA) as substrates 67. These substrate requirements are similar to those of the LuxI-type AHL synthases, with the exception of the utilization of acyl-CoA.
The HdtS enzyme was first identified in Pseudomonas fluorescens F113, where it was shown to produce N-(3-OH-7-cis-tetradecenoyl)-HSL, as well as C6-HSL and C10-HSL as a recombinant protein exogenously expressed in E. coli 68. An HdtS homolog has also been suggested to be responsible for the synthesis of C6-HSL, C8-HSL, and C10-HSL in the Nitrosomonas europaea strain Schmidt 69, although other Nitrosomonas europaea strains appear to carry a distant LuxI-type AHL synthase gene in the genomes. Interestingly, HdtS is a member of the lysophosphatidic acid acyltransferase family 68 and appears to be required for correct acylation of lysophosphatidic acid in Pseudomonas fluorescens 70. HdtS is capable of using both acyl-CoA and acyl-ACP as substrates for acylation of lysophosphatidic acid, and its homologues are ubiquitous among bacteria and eukaryotes 71. The enzymatic mechanism of HdtS-catalyzed AHL synthesis remains uncharacterized. Furthermore, the features of the HdtS enzymes which are required for AHL production are unknown, but there are homologues in organisms that do not produce AHLs. Therefore, this bacterial enzyme family appears to have two functions: acylation of lysophosphatidic acid and AHL synthesis.
3.2 Lux-I type AHL Synthases
The LuxI-type enzymes are by far the most widespread and best understood of the AHL synthases. Identifiable homologues have been found in more than 150 species of α, β, and γ classes of Proteobacteria. There is great diversity among the AHL synthases in their production of AHLs (Table 1), and they cluster into several ill-defined groups based on sequence and phylogenic relationships 72 and likely structural relatedness. These enzymes are composed of a single domain of approximately 205 amino acid residues in length. The sequence conservation between any two members of the AHL synthase family is typically greater than 20% identity / 40% similarity (Figure 4) 25. Interestingly, the Pseudomonas aeruginosa enzyme LasI, a member of the γ class of Proteobacteria is more similar in sequence to enzymes from the α and β classes (≥30% identity / ≥50% similarity) than to EsaI, which is also from the γ class. Despite these differences, there is a simple sequence signature of absolutely conserved amino acid residues that defines the LuxI-type AHL synthase family 73,74. These conserved sequence blocks and conserved amino acid residues are apparent in an alignment of four selected AHL synthases as well as the aroyl-HSL synthase RpaI shown in Figure 4.
Table 1.
AHL signals and bacteria.
| AHL synthase | Species | Signal | Function | Reference |
|---|---|---|---|---|
| 3-oxo-AHLs | ||||
| EsaI | Pantoea sterwartii | 3-oxo-C6-HSL | plant pathogen | 80 |
| LuxI | Vibrio fischeri | 3-oxo-C6-HSL | marine symbiont | 1 |
| YspI | Yersinia pestis | 3-oxo-C6/C8-HSL | Plague causative agent | 66 |
| TraI | Agrobacterium tumefaciens | 3-oxo-C8-HSL | Crown gall causative agent | 81 |
| VanI | Vibrio anguillarum | 3-oxo-C10-HSL | fish pathogen | 82 |
| LasI | Pseudomonas aeruginosa | 3-oxo-C12-HSL | opportunistic pathogen | 83 |
| SinI | Sinorhizobium meliloti | 3-oxo-C14-HSL | nitrogen fixing symbiont | 8 |
| 3-oxo-9-cis-C16-HSL | ||||
| 3-hydroxy-AHLs | ||||
| Xenorhabdus nematophilus | 3-hydroxy-C4-HSL | nematode symbiont | 84 | |
| PhzI | Pseudomonas fluorescens | 3-hydroxy-C6/C8-HSL | phenazine producer | 52 |
| CinI | Rhizobium leguminosarum | 3-hydroxy-7-cis-C14-HSL | nitrogen fixing symbiont | 85 |
| Unsubstituted AHLs | ||||
| RhlI | Pseudomonas aeruginosa | C4-HSL | opportunistic pathogen | 4 |
| CviI | Chromobacterium violaceum | C6-HSL | antibiotic producer | 76 |
| CepI | Burkholderia cepacia | C8-HSL | soil bacterium | 77 |
| BmaI1 | Burkholderia mallei | C8/C10-HSL | Glanders causative agent | 78 |
| CerI | Rhodobacter sphaeroides | 7-cis-C14-HSL | photosynthetic bacterium | 79 |
| SinI | Sinorhizobium melioti | 9-cis-C16-HSL | nitrogen fixing symbiont | 8 |
| C18-HSL |
Figure 4.
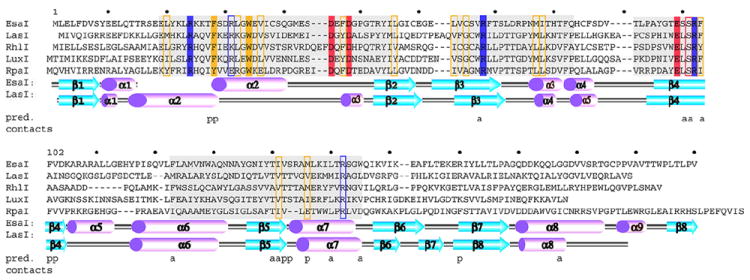
Primary structure of the AHL synthases and the pC-HSL synthase RpaI. A sequence and topology diagram shows the sequences of several AHL synthases mapped to the structural descriptions of the AHL synthases LasI and EsaI. The grey shaded regions are the most conserved sequence blocks within the AHL synthase family. The eight conserved residues of the AHL synthase family are highlighted in solid colors, and those that are similar among AHL synthases are boxed. Below the sequences are shown the alpha helices (magenta) and beta strands (cyan) observed in the structures of EsaI and LasI. The lettering ‘a’ and ‘p’ indicate the amino acid residues proposed to interact with the acyl chain or phosphopantetheine moieties of acyl-ACP, respectively.
The most conserved part of the LuxI-type AHL synthase is the N-terminal region (amino acid residues 1-100). Within this region, there are eight invariant residues, including Arg24, Phe28, Trp34, Asp45, Asp48, Arg68, Glu97, and Arg100, (using the numbering based on the Pantoea stewartii enzyme EsaI (Figure 4)), which are crucial for enzymatic activity 25,73,74. Of these Arg24, Asp45, Asp48, Arg68, Glu97, Ser99, and Arg100 were shown to be essential for AHL synthesis based on mutagenesis analysis of LuxI, RhlI, and EsaI enzymes 25,73,74. The high degree of sequence conservation in this region suggested a role in catalysis and in the binding of the common substrate SAM 74. In contrast, the C-terminal region of the enzyme is less conserved overall, and appears to be less important for activity of RhlI compared to LuxI, for reasons that are still not clear 73,74. However, this region is involved in recognition of the most variable part of the acyl-ACP substrate, the acyl chain, which explains the higher degree of variability in this region. Thus, the entire LuxI-type AHL synthase family share similar structures and mechanisms of AHL synthesis, but what is expected to differ is the manner by which they recognize the varying substrate acyl-ACP.
3.3 Three-dimensional Structure
X-ray crystallographic structural analyses of two of the LuxI-type AHL synthases provided the foundation of the current molecular understanding of AHL synthesis 25,75-77. EsaI produces a 3-oxo-C6-HSL and LasI produces a significantly longer AHL, 3-oxo-C12-HSL, which both contribute to the quorum-sensing regulation of pathogenicity in their respective organisms 23,59. EsaI (Figures 2A and 5A) and LasI (Figure 2B and 5B) are quite divergent AHL synthases, with 28% identity / 44% similarity. Their structures show remarkably similar core elements, but there are some notable differences in the N-terminal region of the enzyme.
Figure 5.
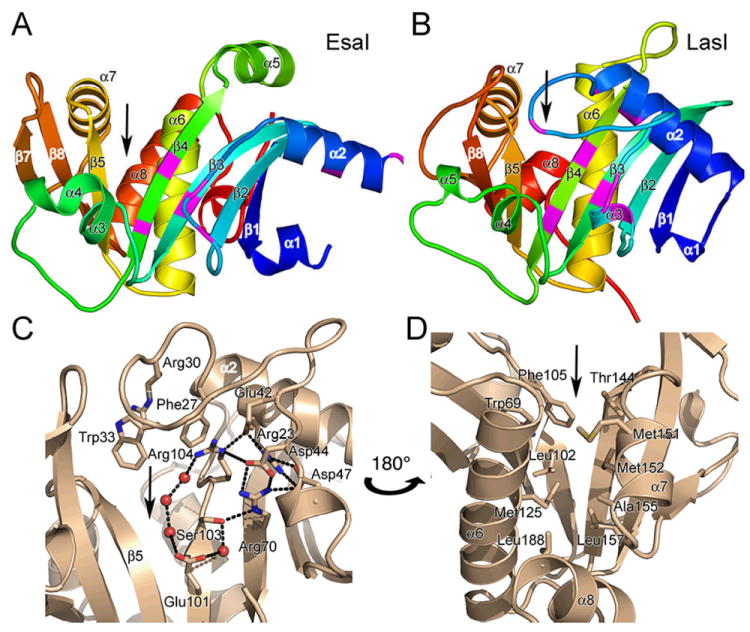
Structure of the AHL synthases EsaI and LasI. (A) Ribbon diagrams depict the backbone structures of EsaI (A) and LasI (B). The structure of the EsaI enzyme (PDB ID 1kzf) from Pantoea stewartii was determined from the native sequence. The structure of the LasI enzyme (PDB ID 1ro5) from Pseudomonas aeruginosa was determined from an active form that had been engineered to improve solubility and crystallization properties. The rainbow coloring is blue to red from the N-terminus to the C-terminus and the major secondary structure elements are labeled. The most conserved residues are indicated with magenta coloring. (C) Close-up view of the LasI active site residues, showing the conserved residues and the electrostatic cluster. Well-ordered water molecules in the active site are shown as red spheres and putative hydrogen bonds are shown as dotted black lines. (D) Close-up view of the LasI acyl-chain binding pocket. The view is rotated approximately 180° about the vertical axis of the page relative to (C) and shows the residues lining the putative acyl-chain binding pocket. In each panel, the active site V-shaped cleft is indicated with a black arrow.
The AHL synthase fold is a mixed α–β–α sandwich with a V-shaped cleft and a deep cavity/tunnel (Figure 5A and B). The predominantly anti-parallel β-sheet has a prominent V-shaped cleft (indicated by arrows in the figure) between the parallel β-strands β4 and β5. A β-bulge between residues 99 and 100 in β4 (EsaI numbering) also appears to be a unique and conserved feature of the AHL synthases (Figure 6C and D) 25,75. There is a structurally conserved core, composed of 74 residues within the beta strands β1, β2, β3, β4, β5, β6, and β8, and α helices α3 and α4, which has a root mean squared deviation (r.m.s.d.) between LasI and EsaI of less than 1 Å 75. Helices α6 and α7 are also conserved, but occupy slightly different positions in the structures. Portions of the relatively unstructured loop between strands β3 and β4 are also conserved. Interestingly, only three of the conserved AHL synthase signature residues, Arg68, Glu97, and Arg100, are found in this structurally conserved core (Figure 4 and 5). The V-shaped cleft is the active site of the enzyme, and its vertex consists of the entry to a hydrophobic cavity that is the site of acyl chain binding 25,26,75.
Figure 6.
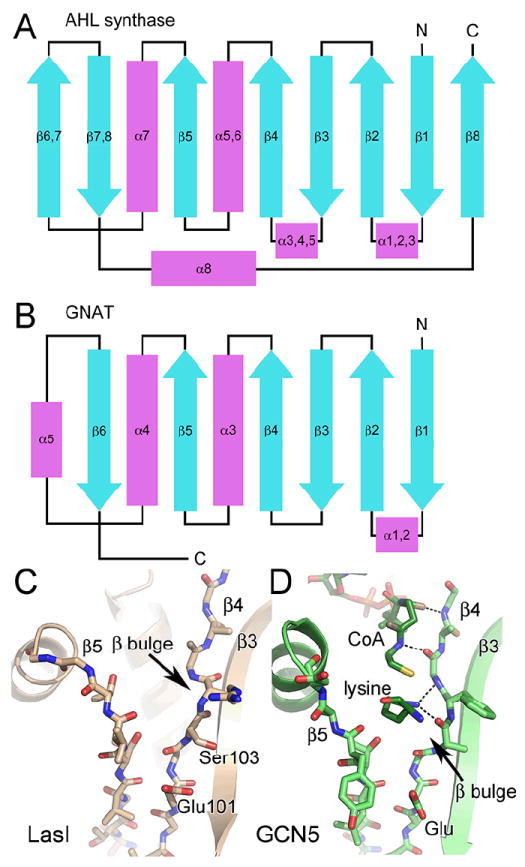
Similarity of AHL synthases to GNAT enzymes. Topology diagrams are shown for the AHL synthase (A) and GNAT (B) families. The alpha helices are in magenta and the beta strands are in cyan. The active site clefts of LasI (C) and Gcn5 (PDB ID 1qsn) (D) show common and different features. Well-ordered water molecules in the active site are shown as red spheres and putative hydrogen bonds are shown as dotted black lines.
Adjacent to the V-shaped cleft is a unique electrostatic cluster in the core of the N-terminal region of EsaI and LasI (Figure 5C). This cluster contains a complex network of ion pairs formed by the side chains from six of the eight conserved residues in the AHL synthase family (Figure 4 and 5C). Mutagenesis analysis of LuxI, RhlI, and EsaI enzymes demonstrated that most of the residues in this cluster, (Arg24, Asp45, Asp48, Arg68, Glu97, Ser99, and Arg100 (in EsaI numbering; Arg30, Asp44, Asp47, Arg70, Glu101, Ser103, and Arg104 in LasI numbering), are essential for AHL synthesis 25,73,74. LasI Ser103, which is conserved as either serine or threonine in all known LuxI-like AHL synthases, lies at the center of this cluster and interacts directly with Arg70 and a bridging water molecule bound to Glu101. This cluster appears to contribute to the stability of interactions between N-terminal helices and the front surface of the β-sheet, but may also perform a catalytic function.
To date there are no structures of acyl-ACP bound to any enzyme, and thus the binding site of the acyl chain, the phosphopantethiene moiety, and protein core of ACP within the AHL synthase had to be interpreted from structural analysis of other enzymes and the locations of the conserved residues. Fortunately, the structure of the highly conserved core domain of the AHL synthases is shared with other enzymes, such as the GCN5-related histone N-acetyltransferases (GNATs) 25,78 and amino-acid acyltransferases 79 (and reviewed in 80). The fold of the enzyme families is similar (Figure 6 A and B), and the main β-strand platform, which forms the active site of the GNATs and the AHL synthases superimpose with an r.m.s.d of approximately 1 Å. This highlights the remarkable structural similarity of the AHL synthases and the GNATs in light of their completely unrelated primary sequences 80 (Figure 6 C and D). A third structurally related protein FeeM, which is an acyl-ACP-dependent amino acid acyltransferase, has features of both the AHL synthases and the GNATs 79. In both the acyl-ACP and acetyl-CoA substrates used by these enzymes, the terminal thiol of phosphopantetheine forms a thioester bond to either a variable length acyl chain or an acetyl group. Holo- and acyl-ACP carry phosphopantetheine via a phosphodiester bond to the hydroxyl oxygen atom of Ser36. In acetyl-CoA, however, phosphopantetheine forms a pyrophosphate linkage to the 5’ phosphate of adenosine 3’5’-diphosphate. The common phosphopantetheine portion of acetyl-CoA in the GCN5-acetyl-CoA complex 81 occupies the central catalytic cleft that is highly conserved structurally with EsaI and LasI (Figure 6 D). This close structural relationship of the AHL synthases with the GNATs and amino-acid acyltransferases has provided great insight into how the AHL synthases may bind phosphopantetheine and carry out acylation reactions.
Based on the analysis of the GNATs and FeeM together with the AHL synthases, the LasI structure (Figure 5B and C) suggested a role for the remaining conserved AHL synthase signature residues. The loop between α2 and α3 that contains the conserved residues Trp33 and Phe27 (LasI numbering) positions them directly in front of the active site. These residues may be conserved for the purpose of binding SAM, perhaps by helping position the SAM amine nitrogen for N-acylation at the C1 position of acyl-ACP. The aromatic side chains would allow a stacked ring sandwich type of binding with the adenine of SAM, as is commonly observed in SAM binding enzymes. The N-terminal region of EsaI exists in a more open conformation compared to LasI, but the conserved residues of EsaI must be brought into proximity of the active site for catalysis to occur. That this region of the AHL synthase varies greatly in structure between EsaI and LasI may be attributed to crystal packing forces, but it also indicates the degree of potential flexibility in this region of the enzyme, and suggests that conformational changes take place when the substrates bind to the AHL synthase in the course of the reaction.
3.4 Specificity in Signal Production
A wide variety of AHL signals are produced in nature (Table 1). They vary greatly in acyl-chain length 82, from the C4-HSL produced by P. aeruginosa RhlI 83, up to C18-HSLs produced by Sinorhizobium meliloti SinI 6 (Table 1). Most recently, AHLs with methyl-branched acyl chains have been discovered in Aeromonas culicicola and AHLs with multiple double bonds in the acyl chain have been found in Jannaschia helgolandensis 8. AHLs produced by different bacterial species also vary in the degree of oxidation at the AHL C3 position. The preference for unsubstituted-, 3-oxo-, or 3-hydroxy-acyl-ACPs is thought to be due to the intrinsic selectivity of the AHL synthase for a particular subset of a pool of available acyl-ACP substrates, which are produced by the reductive-condensation cycle for fatty acid biosynthesis. For example the AHL synthase RhlI produces an unsubstituted, C4-HSL, whereas LasI, produces predominantly 3-oxo-C12-HSL, at the same time in vivo from the same cellular pool of acyl-ACPs.
The preference of AHL synthases for 3-oxo-substituted acyl-ACP substrates appears to be due to hydrogen bonding interactions between the C3 carbonyl of 3-oxo-hexanoyl-ACP and residues in the acyl-chain binding site 25,75. In EsaI and LasI, two hydrogen bonds are predicted to form between the enzyme and the 3-oxo group of acyl-ACP. One of these occurs from a threonine Oγ1, at amino acid position 140 in EsaI and 144 in LasI (Figures 4, 5 and 7) and the other is from the main-chain carbonyl group of the adjacent residue 141 or 145. For enzymes that are known to produce 3-oxo-AHLs, the residue at this position is typically a threonine (Figure 4). Substitution of this threonine to alanine or other amino acids in EsaI and LasI, respectively, increased the amount of the unsubstituted AHL produced relative to 3-oxo-AHL, an indication of some lost degree of specificity for the oxo-group 34. Notably, the enzymes that make 3-hydroxy-AHLs or unsubstituted-AHLs have either a glycine or alanine, or occasionally a serine at that position 84. Proof of the direct interaction of residues at these special positions with the acyl-phosphopantetheine moiety awaits further structural analysis.
Figure 7.
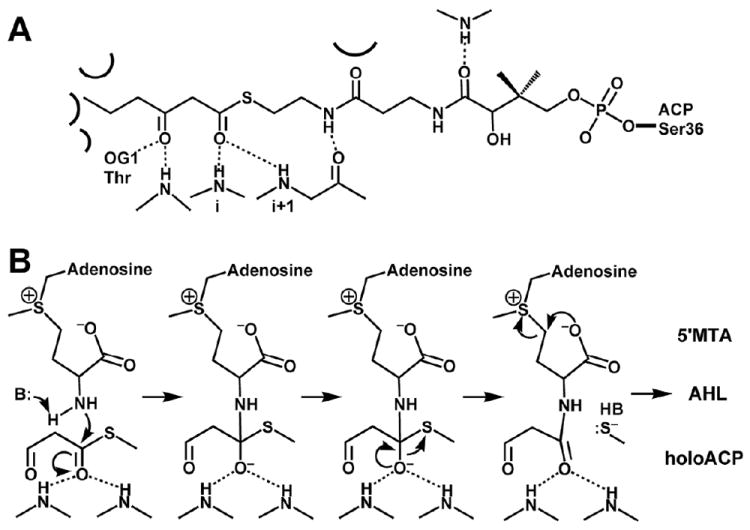
Schematic diagram of the proposed AHL synthesis mechanism. (A) The interactions predicted to form between acyl-phosphopantetheine bound in the active site of a generic AHL synthase are depicted as dotted lines for hydrogen bonds and curved lines for other types of interactions, such as van der Waals contacts. (B) The proposed mechanism of acylation involves a direct nucleophilic attack by the amine of SAM on the C1 position of the acyl chain, which will eventually release holo-ACP. The mechanism of lactonization is a direct nucleophilic attack on the Cγ position of SAM by the carboxylate oxygen atom, which will produce the AHL and release the product 5’MTA.
The structural studies of EsaI provided the first explanation for the selectivity of AHL synthases for acyl-ACPs with short acyl-chain lengths. EsaI produces 3-oxo-C6-HSL, which has a relatively short acyl chain and species with longer acyl chains are not observed for this enzyme in vitro or in vivo 23,25. The structure of EsaI revealed that the acyl-chain binding pocket is an enclosed cavity, composed of a shell of residues directly surrounding the cavity, including Ser98, Met126, Thr140, Val142, Met146, and Leu176 25. Numerous other residues within the protein core that contact these cavity residues but not the hexanoyl chain, also direct the size and shape of the cavity through hydrophobic packing (Figure 5 and 7) 25. The importance of these two layers of residues was demonstrated by Palva and coworkers, who identified mutations in the gene encoding ExpI(SCC1) of Erwinia carotovora that increased the acyl-chain lengths of the AHLs produced in vivo 26. These mutations occurred at one position that lines the acyl-chain binding pocket and one that is in the next layer of residues that would not directly contact the acyl chain 25. Thus acyl-chain length restriction plays a key role in regulating the enzyme’s ability to produce AHLs with relatively short acyl chains.
The LasI structure provided a less clear explanation for the selectivity of AHL synthases for acyl-ACPs with long acyl chains. The LasI acyl-chain binding pocket appeared as an elongated tunnel through the enzyme that is formed by hydrophobic residues (Trp69, Leu102, Phe 105, Met125, Thr144, Met151, Met152, Ala155, Leu157, and Leu188 mainly on helices α6, α7, and α8) at similar positions in the enzyme as those in the EsaI pocket (Figures 4 and 5D) 75. Although the same residue positions are involved in formation of both acyl-chain binding sites, the different sizes of the hydrophobic side chains and slight changes in the orientations of helices α6 and α8 also contribute to differences between the structures of the closed pocket in EsaI versus the open tunnel observed in LasI. Many of the residues in EsaI that occlude the pocket are larger than those in the same position in LasI, which limits the acyl chain size to a C6 acyl chain. Interestingly for LasI, the tunnel places no steric restriction on the acyl-chain length of acyl-ACPs that could bind to LasI. However, there is still a rather narrow distribution of AHLs produced by LasI in vivo 34, which raises the question of how LasI restricts the population of AHLs produced to those with the appropriate length of acyl chain.
Sequence analysis of the AHL synthase family has failed to reveal a robust correlation between sequence composition and acyl-chain length. Although, for the AHL synthases that produce AHLs with short acyl chains, computational analyses have found weak correlations between the identity of amino acids at certain positions in the acyl-chain binding pocket and the length of acyl chain length 85. The EsaI and LasI structures instead suggest that in order to accommodate and create a preference for acyl-ACPs with different acyl-chain lengths, particularly for long acyl-chain lengths, more complex sequence and tertiary structure differences occur in different AHL synthases. Such large changes in helix orientations and tertiary packing are still very difficult to predict from primary sequence.
3.5 Mechanism of Signal Production
Early studies in the Greenberg, Winans, and Cronan labs revealed the substrates for the reaction and general features of the reaction mechanism 9,61,62. For in vitro studies with the enzymes LuxI and TraI 61, the primary acyl chain donor was found to be acyl-ACP, whereas the lactone ring was found to originate in the methionine side chain of S-adenosyl-L-methionine (SAM) 9,61. Studies with LuxI in vivo confirmed these findings and proved the identity of the physiologically relevant substrates illustrated in Figure 3 62. Subsequent enzymatic studies with RhlI examined substrate specificity and patterns of enzyme inhibition 63. These indicated a bi ter sequential ordered reaction, with SAM binding first to the enzyme, followed by acyl-ACP. The binding of acyl-ACP was suggested to trigger the N-acylation reaction, in the form of a nucleophilic attack on the acyl carbonyl carbon (acyl-ACP) by the amine of SAM to form butyryl-SAM to be followed by the lactonization of SAM and release of holo-ACP prior to 5’-methylthioadenosine (MTA). Butyryl-SAM was a good substrate and inhibitor of the reaction, which supported the formation of acyl-SAM as an intermediate and suggested that the acylation reaction preceded lactonization 63. Thus, the reaction mechanism of AHL synthesis has two main steps: acylation and lactonization (Figure 3).
Structural studies of the Lux-I type AHL synthases have helped to clarify the initial steps in the mechanism of AHL synthesis. The model of the acyl-phosphopantetheine of acyl-ACP in the active sites of EsaI and LasI places the acyl chain C1 carbonyl oxygen within hydrogen bonding distance of the backbone amides of residues 100 and 101 that form an unusual β-bulge in the V-shaped cleft (Figure 6 and 7). The location of the residues proposed to interact with SAM, also suggest that the SAM binding site is nearer the exterior of the enzyme than the acyl-ACP binding site. Together these results contrast those described above and rather support a bi-ter sequential ordered reaction mechanism where acyl-ACP binds prior to SAM 25.
The current proposed mechanism of N-acylation by AHL synthase is similar to the GNATs, which involves proton abstraction from the substrate amine to be acylated by an activated water molecule 86,87 (Figure 7). The GNAT and AHL synthase structures have a water molecule bound to a conserved glutamic acid residue (Glu97 in EsaI, Glu 101 in LasI or similar residue in the GNATs) (Figure 5C). By analogy to the GNAT family of enzymes, this water molecule would act as a base to abstract a proton from the amine of SAM making it a better nucleophile for direct attack on the C1 atom of the acyl chain. In support of this, the acyl O1 carbonyl oxygen that will form an oxyanion during the acylation reaction could be stabilized by the β-bulge in β5 (Figure 6C and D), as has been observed for numerous GNAT enzymes (reviewed in 88). There is a second possible N-acylation mechanism that is used by the histone acetyltransferase Esa1 (not be confused with the AHL synthase EsaI), which would proceed via a thioacyl covalent intermediate 89. However, no cysteine residues are present in the AHL synthase active site 25 or found to be important for catalysis 73,74, which ruled out this long-standing hypothesis that AHL-synthesis might proceed through a covalent thio-acyl-enzyme intermediate through an active-site cysteine 61.
The second part of the reaction mechanism is the cyclization of the methionine moiety of SAM, which gives rise to the lactone portion of the AHL and occurs through a lactonization reaction. There are multiple chemical mechanisms by which lactonization could take place. Tipton and coworkers examined the mechanism of lactonization of SAM using deuterium incorporation with the RhlI enzyme 90. In their study, no deuterons were taken up into the product when the reaction was conducted in deuterated buffer. This observation ruled out indirect lactonization mechanisms, such as elimination, which would proceed through an intermediate such as N-butyrylvinylglycine. Therefore, the lactonization reaction most likely proceeds through direct nucleophilic attack on the Cγ of SAM by the carboxylate oxygen, via an SN2 chemical reaction 90. For the SN2 reaction to occur (illustrated in Figure 7), the methionine moiety must be in a near cyclic conformation (<± 20°). Under transient-state kinetic methods, the Tipton group verified the formation of a single reaction intermediate, acyl-SAM 91. This study further demonstrated that the reaction catalyzed by RhlI proceeds via acylation of SAM, followed by lactonization. They further suggested that the acylation step in some way activates the lactonization reaction, but it remains unknown which residues contribute to this specific conformation that likely results in the activation process.
4. Mechanisms of AHL Perception by the LuxR-type Proteins
4.1. Overview of the AHL-responsive transcriptional regulators
AHL signaling molecules are perceived by two types of receptors. LuxR 58,92 and its cognate AHL (3-oxo-C6-HSL) 2 control the quorum-sensing mediated bioluminescence in Vibrio fischeri 1,93. The cytoplasmic LuxR-type proteins direct transcription of genes by forming direct contact with DNA sequences near the promoters of target genes, and this DNA binding activity is regulated by AHLs (for reviews, see 19,94). AHLs can also mediate genetic behaviors via a two-component phosphorelay mechanism: in this mechanism, the AHLs are detected by the membrane-bound sensor kinases such as LuxN and LuxQ of V. harveyi 13, and they control the phosphorylation activity of the response regulators leading to differential gene expression. This section of the review focuses on structure-function aspects of the LuxR-type AHL receptors and their regulatory impact on transcription of target genes. We will not cover structural aspects of the two-component sensor kinases such as LuxQ/LuxN, as these largely share attributes of this general protein family.
LuxR-type AHL receptors are approximately 250 amino acid residues in length folded into two functional modules. The N-terminal domain (NTD) contains the AHL-binding site, and the C-terminal domain (CTD) possesses the DNA-binding activity. LuxR proteins and those related to FixJ/NarL are assigned to the same subclass of transcription factors based on sequence alignment of their CTDs 19. When in isolation, the two domains may independently perform their respective functions; when integrated in a full-length polypeptide, the two domains are linked via a conformationally flexible linker, and AHL binding to the NTD modulates the DNA binding function of the CTD. The LuxR-type proteins can be transcription activators: the presence of AHL leads to binding of LuxR-type proteins to the DNA targets at positions that promote transcriptional activation of the controlled genes. The LuxR-type positive regulators include LuxR 3,58,93,95 (Vibrio fischeri, in bio-luminescence), LasR/RhlR 96-99 (Pseudomonas aeruginosa, virulence factor expression and biofilm formation) (Figure 2B), and TraR 100,101 (Agrobacterium tumefaciens, oncogenic Ti plasmid replication and conjugal transfer) (Figure 2D). Among the few LuxR-type AHL-inactivated transcription factors, EsaR 23 (Pantoea stewartii, in exopolysaccharide production) (Figure 2A) has been characterized at the biochemical level. In direct contrast to the LuxR-type activators, EsaR binds to the DNA target of the promoter region in the absence of AHLs, and when this binding site is at an appropriate position, EsaR can repress the controlled genes 18. The presence of the AHL ligands relieves gene repression, presumably by altering the DNA binding of EsaR. Indeed, the presence of AHL decreases the apparent association of EsaR with DNA 18, but the precise derepression mechanism in vivo for different target genes may be more complex. EsaR can also act as an AHL regulated transcriptional activator, where the presence of AHL antagonizes its activation ability 16,102.
Biochemical and structural studies of the LuxR-type proteins had been hampered by protein solubility 95,103. A major breakthrough in production of native, soluble LuxR-type proteins was made when it was found that inclusion of cognate AHLs in the bacterial culture during elevated expression of the LuxR-type protein TraR, from Agrobacterium tumefaciens, greatly enhanced the stability of the proteins, and yielded a substantial amount of soluble protein 104. This strategy has now proved successful to obtain soluble native LuxR-type activators including LuxR 105, SdiA 106, and LasR NTD 107. For EsaR that functions in the absence of AHLs, these ligands are not required for production of the soluble form 18.
4.2 Structural basis for AHL binding to LuxR-type proteins
Currently, structures of the AHL-binding NTDs of TraR (TraRAt from A. tumefaciens 108,109, and TraRNGR from Rhizobium sp. strain NGR234 110), SdiA (E. coli) 106, and LasR (P. aeruginosa) 107 have been reported. The NTD adopts a GAF/PAS fold with an α-β-α tri-layer topology. One α-helical layer along with the concave surface of the β-sheet (arranged in β2-β1-β5-β4-β3) forms an enclosed pocket for AHL binding. The extensive and intimate interactions between AHLs and their LuxR proteins lead to complete burial of AHLs within the protein matrix and may explain the proposed structural role of AHLs in folding of the LuxR proteins.
In the structures of TraR and LasR, the polar homoserine lactone, which is the common structural moiety in AHLs, is coordinated by hydrogen bonding with several conserved residues: the ring keto group with Trp (Trp57 of TraRAt, Trp59 of TraRNGR, and Trp60 of LasR), the 1-keto group with Tyr (Tyr53 of TraRAt, Tyr55 of TraRNGR, and Tyr56 of LasR), and the amide group with Asp (Asp70 of TraRAt, Asp72 of TraRNGR, and Asp73 of LasR), the charged residue that is unusually buried within the protein matrix. Although the NMR structure of SdiA 106 does not allow precise structural analysis on the AHL-SdiA interaction, the three conserved residues (Trp67, Tyr63, and Asp80) surround the homoserine lactone moiety. The acyl chains of the three bound AHLs are aligned parallel to the β-sheets (Figure 8A). The acyl chain of 3-oxo-C8-HSL is surrounded by residues including Ala38, Leu40, Thr51, Tyr53, Trp57, Tyr61, Phe62, Val72, Val73, Trp85, Met 127 and Thr129 of TraRAt, or Ala40, Leu42, His53, Tyr55, Trp59, Tyr63, Leu64, Val74, Leu75, Trp87, Met129, and Thr131 of TraRNGR. The LasR residues interacting with the acyl chain of 3-oxo-C12-HSL comprise of Leu36, Gly38, Leu39, Leu40, Tyr47, Ala50, Ile52, Tyr64, Ala70, Val76, Cys79, L125, Gly126, and Ala127. Although conformation of the bound C8-HSL within SdiA is highly dynamic, the hydrocarbon chain is found within a pocket formed by residues of Leu44, Cys45, Phe59, Thr61, Tyr63, Trp67, Val68, Tyr71, Asp80, Val82, Leu106, and Leu115. Notably, the AHL-contacting residues are largely nonpolar in all of the structures, consistent with the hydrophobic character of the acyl chain. However, as shown in Figure 8A, the structurally similar acyl chains of 3-oxo-C8-HSL and 3-oxo-C12-HSL adopt strikingly different orientations when bound to their cognate LuxR proteins. The conformation deviation begins at the 3-oxo position. The polar 3-keto group, a common modification in the acyl chain of AHLs, is sequestered by hydrogen-bonding to the polar region (the side chain of Thr129 and the main chain of Ala38 in TraRAt, or the side chain of Thr131 and the main chain of Ala40 in TraRNGR, and NE1 and NH1 of Arg61 in LasR via a bound water molecule) of the mostly hydrophobic acyl-chain binding tunnel. These discrete modes of coordination at the 3-oxo position direct distinct orientations of the rest of the hydrocarbon chains, resulting in different conformations between the bound 3-oxo-C8-HSL and 3-oxo-C12-HSL. The acyl-chain binding tunnels in the three LuxR-type proteins are all located on the interface between the α-helical layer (mainly α4 and α6, numbered according to the TraRAt structure 109) and the β sheet (β1, β2, and β5 strands), but they are formed of different residues and orient the acyl chain in different directions. It appears that the hydrophobic acyl chains of AHLs may exploit the large nonpolar α-β interface to adopt different conformations. Thus, the LuxR-type proteins accommodate various conformations of AHLs, with different side-chain interactions, while maintaining the same overall structural fold. Such structural observations may explain why some LuxR-type proteins, such as TraR, can bind different AHLs when overexpressed. In addition, the observation of the hydrophobic acyl portion of AHLs sandwiched between the α-β structure may suggest the role of AHLs in promoting folding of the LuxR-type proteins by latching and stabilizing the α-β bi-layer of the α-β-α GAF/PAS fold.
Figure 8.
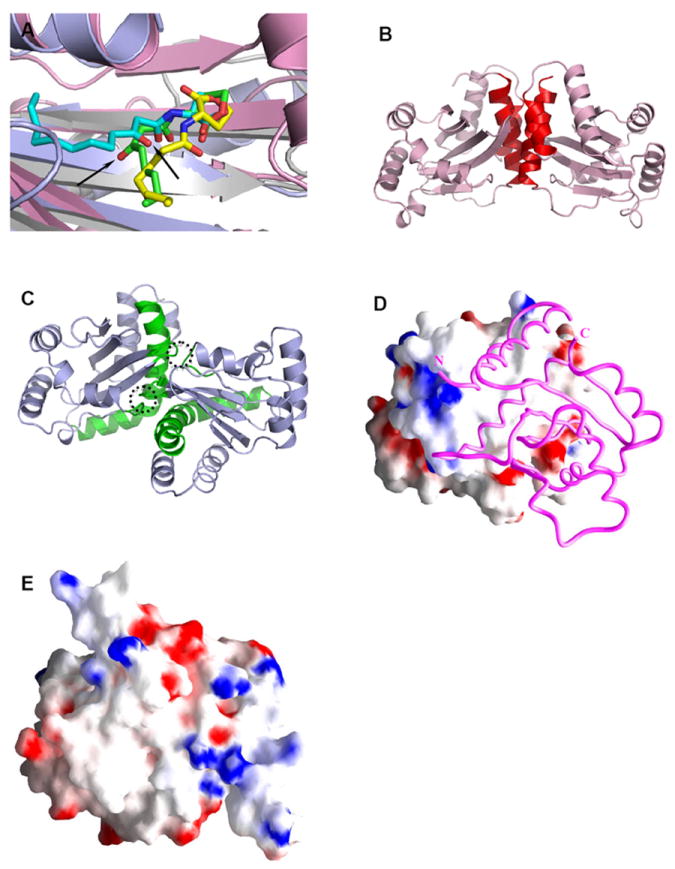
Structural comparison of the LuxR-type proteins. A) Conformations of AHLs in the AHL-binding pockets: 3-oxo-C8-HSL (green) in TraRAt (pink, PDB ID: 1L3L); 3-oxo-C12-HSL (cyan) in LasR (light blue, PDB ID: 2UV0); C8-HSL (yellow) in SdiA (gray, PDB ID: 2AVX). For clarity, structural elements (helices and loops) that are located on the top of the bound AHLs are removed. The different orientations of the 3-oxo group in 3-oxo-C8-HSL and 3-oxo-C12-HSL that lead to drastic conformations of the acyl chains are highlighted with arrows. B) and C) Distinct dimeric assembly of TraR and LasR shown in the same view. Structure of dimeric TraRAt NTD (pink) is overlaid with that of LasR NTD (light blue) by superimposing one (left) NTD of each dimer. The main helices that are located in the TraRAt dimer interface are largely parallel and highlighted in red, and those in LasR are almost perpendicular and in green. The dimer contacts provided by loops of α4-β3 and β4-β5 in LasR are highlighted in dotted circles. D) and E) Surface electrostatic potentials of TraRAt and SdiA, respectively. Surface of one TraRAt NTD is shown, the other NTD is shown as a Cα trace in magenta. The view of SdiA corresponds to that with the surface presentation of TraRAt monomer. Surface with positive potentials is colored in blue, and with negative potentials in red.
4.3 Structural Basis for the Oligomeric States of the LuxR-type Proteins
For the LuxR-type activators, binding of AHLs results in dimerization of the LuxR-type proteins 111-113, which up-regulates their DNA binding activity. Both the full-length TraR (TraRAt and TraRNGR) and the N-terminal domain of LasR are homodimeric when purified in solution in the presence of AHL, and maintain this state when crystallized. The homodimeric interface of both TraR and LasR mainly consists of the first and last helices of this domain, however, the orientations of two monomers in TraR and LasR are surprisingly different. Two main dimerization helices in TraR are arranged more or less parallel to each other, providing an extensive interaction interface (Figure 8B); in contrast, the dimerization helices in LasR are nearly perpendicular. In LasR, additional intermolecular interactions are formed by the α4-β3 and β4-β5 loops from both monomers (Figure 8C): such interactions are mostly absent in the TraR dimer interface. Due to these distinct configurations of the dimer interfaces, the dimeric TraR and LasR structures do not superimpose. Given that the interfaces of dimerization and AHL binding are completely separated and that the AHL binding site is entirely buried, it is unlikely that differences in dimer assembly contribute to AHL discrimination between TraR and LasR. However, they may affect positioning their two DNA binding CTDs, thereby influencing the distribution of recognition motifs in their respective target DNA sequences.
Both polar and nonpolar contacts are found along the NTD dimer interface of TraR and of LasR. For TraR, hydrophobic interactions cluster around both N- and C-termini of α6 (TraRAt: Val146, Ala150 and Ile118; Ala12, Ala157 and Phe161; TraRNGR: the nonpolar portion of Lys148, Gln152 and Val120; Leu14, Tyr159 and Ile163,), and hydrogen bonding and salt bridge interactions are found between these two clusters. For LasR, there are two hydrophobic patches along the dimer interface: one formed by Pro149, Trp152, and Met153 of α6 and the other by Val83, Leu84, Pro85, and Phe87 of β3. Buried within the hydrophobic surface is the charged residue Asp156, which forms hydrogen bonds to Trp152 and Lys155 of the other LasR monomer. The dimer interface in both TraR and LasR is mainly hydrophobic (e.g., Figure 8D for TraRAt).
Interestingly, the SdiA NTD bound with C8-HSL may exist as monomer in solution 106. Unlike LasR and TraR, structural comparisons show that regions in SdiA, which correspond to the hydrophobic areas on the dimer interface of TraR and LasR that were just described, are polar. For example, SdiA Arg159 and Arg167 are structurally correlated with the hydrophobic TraRAt Gly153 and Phe161 (or TraRNGR Met155 and Ile163). Thus, the relative polar surface of SdiA corresponding to the dimer interface of TraR and LasR (Figure 8E) may not need to be sequestered by oligomerization. It is noted, however, that the monomeric SdiA is prone to aggregation, and a non-physiological condition (pH 4.2 and 0.2 M urea) was used in the NMR studies 106.
The presence of the NTDs is sufficient for the LuxR-type proteins to dimerize, but the CTDs may also contribute to dimerization. In the structure of TraRAt-DNA, two α13 helices of the CTD interact with each other (Figure 9A). However, the involvement of CTD in TraR dimerization is likely dictated by the arrangement of their binding sites in DNA and is not required for dimeric TraR formation, as the full-length TraR forms a heterodimer with the natural dominant-negative variant TrlR 114-116, which is missing the CTD (see Section 5.2). Similarly, in complex with the anti-activator protein TraM, the two TraR CTDs are not in direct contact, and their orientations are different from those in the TraR-DNA structure (see Section 5.3) 110. The ability of the flexible CTD to assume various conformations supports the idea that the CTDs are structurally independent from each other and that, although they might augment dimerization, they are not required for TraR dimerization.
Figure 9.
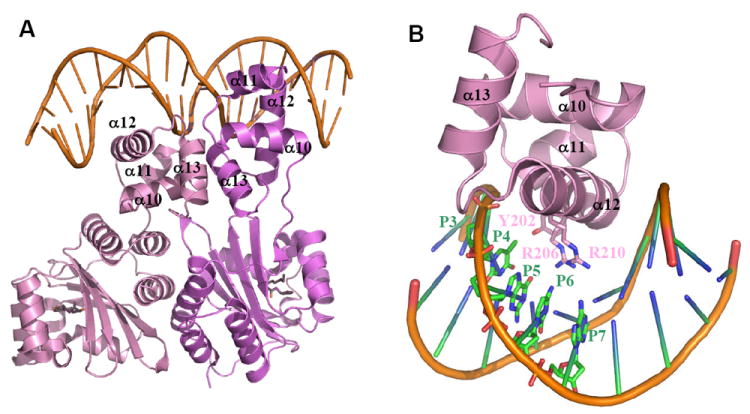
A) The overall structure of the TraRAt-DNA complex (PDB ID: 1L3L). Two TraR monomers (purple and pink) adopt an asymmetric relationship when bound to DNA. The DNA recognition helix α12 projects into the DNA major groove. The α13 helices from each of the two subunits are in contact due to binding of CTDs to the adjacent DNA major grooves. 3-oxo-C8-HSL is shown in green, and DNA in orange. B) Decoding the DNA sequence by TraRAt α12. Side chains of three TraR residues (Tyr202, Arg206 and Arg211) that interact with bases at P3-P7 positions are shown. For clarity, only one CTD and one half-site tra box is shown.
5. Mechanisms of Regulating DNA Activity of the LuxR-type Proteins
5.1 Structural Basis for DNA binding by the LuxR-type Proteins
The C-terminal portion, ~ 1/3 of the full length, of the LuxR-type proteins folds into a functional domain, which when over-expressed may lead to constitutive activation of transcription 117. This CTD contains a predicted helix-turn-helix motif, the prominent DNA binding signature found in many transcription factors. The classic DNA targets of the LuxR-type activators are located around – 41.5 from the transcriptional start site of the first controlled gene 4, 118-120, and those of the LuxR-type AHL-inactivated regulators overlap with the -10 promoter element 18,23,121. These DNA sequences of ~ 20-bp, termed lux boxes, are typically imperfect palindromes. Binding of TraRAt with a DNA fragment of ~250 bp containing the lux - like (tra) box has been reported with a dissociation constant Kd of 10 nM 104.
While no structures of the isolated CTD are available, structures of the CTD in the full-length TraR have been determined in the DNA-bound form (from A. tumefaciens) 108,109 and in the anti-activation complex (from Rhizobium sp. NGR234, see Section 5.3) 110. The CTDs in these different contexts superimpose well with an r.m.s. deviation of 1.1 Å, indicating that structural fold of the CTD is conserved and that the CTD functions as a rigid body. The TraR CTD is comprised of four helices (α10 through α13), and helices of α11 and α12 form the HTH motif. The orientation of the DNA recognition helix α12 is determined by two structural elements. First, a cluster of hydrophobic interactions anchors the beginning residues of α12 (Val205 and Leu209 of TraRAt, or Val207 and Leu211 of TraRNGR) against those from the scaffold helix α11 (Met191, Ile194 and Ala195 of TraRAt, or Ala193 and Thr196 and Ala197 of TraRNGR), the turn (Val200 of TraRAt, or Ile202 of TraRNGR), and α10 (Tyr181 and Ile185 of TraRAt, or Cys183 and Ala187 of TraRNGR). Second, two salt bridges between Glu178 of TraRAt or Glu180 of TraRNGR (α10) and the α12 C-terminal Arg215 of TraRAt or Lys217 of TraRNGR further orient α12 in a direction optimal for DNA binding. The salt bridges are conserved among many HTH-containing proteins, and may contribute to the additional stability of the protein fold.
In the structure of the TraRAt-DNA complex (Figure 9A), the CTD serves as the DNA reading head interacting with the tra box in a sequence-specific manner. Of the 9-bp half-site of the tra box (A1T2G3T4G5C6A7G8A9), bases at five positions (P3-P7) make specific contacts via hydrogen bonds with TraRAt residues from α12 (Figure 9B). Side chains of Arg206 and Arg210 insert into the DNA major groove and hydrogen bond with bases at P4-P7, together decoding the DNA sequence. Tyr202 uses a H2O to form a hydrogen bond with the base at P3. In addition to these sequence specific contacts, TraRAt residues also contact the their sugar moieties and phosphate backbones of the DNA. Subsequent mutational studies showed that most bases in the tra box are important for TraRAt binding and transcriptional activity 122. Bases at positions of P4-P6 confer the specificity for TraRAt function, whereas bases at the center of the tra box (P8 and P9), although not in direct contact with TraRAt, along with that at P7 may function through ‘indirect readout’ likely influencing bending and structural flexibility of the DNA when interacting with TraRAt (below). The modest overall contacts between TraRAt and the base at P3 may account for smaller mutational effects at this position compared to those at P4-P6. Similar mutagenesis analysis has been reported to study the effect of DNA sequence in the lux box on the transcription activation by LuxR 123.
In forming the TraRAt-DNA complex, both TraRAt and the tra box DNA undergo binding induced conformational changes. To present the DNA recognition helices α12 to the two consecutive DNA major grooves formed by the palindromic tra box, the DNA reading heads are projected into different orientations with respect to their NTDs by a 12-residue peptide linker, giving rise to one compact and one extended conformations of TraRAt (Figure 9A). The TraRAt-bound DNA is bent ~30° towards TraRAt, and such DNA bending has been shown to be functionally relevant using permuted DNA fragments including the tra box sequence 109.
5.2 Anti-activators and Structures of Anti-activators
TraR, of the large Rhizobiaceae family, is the only LuxR-type protein known so far whose transcriptional activity is also regulated through protein-protein interaction with anti-activators. TrlR of A. tumefaciens has been shown to antagonize the regulatory function of TraR 114-116. The N-terminal 181-residue portion of TrlR shares greater than 80% identity with that of TraR, while the remaining 31-residue portion is unrelated to any other known proteins. TrlR is considered a C-terminal truncation of TraR, due to a frameshift mutation, because an insertion of a thymine nucleotide between nucleotides 452 and 453 of trlR leads to a gene product that is the same size (234 a.a.) as TraR with 90% identity. As with TraR, TrlR requires AHL to maintain solubility when over-expressed, and binds AHL with 1:1 molar ratio. As expected, TrlR can interact with TraR, presumably via their conserved dimerization NTDs, but this TrlR-TraR heterodimer, with only one CTD per TrlR-TraR dimer, does not bind the tra box containing DNA fragment. Therefore, TrlR inhibits TraR function because it prevents the formation of the DNA-binding competent TraR homodimer by engaging TraR in the formation of an inactive TrlR-TraR complex.
The expression of the other anti-activator found in A. tumefaciens called TraM is positively controlled by TraR, which creates a negative feedback for TraR 40,124. Direct TraR-TraM interaction was first suggested by yeast two-hybrid analysis 125 and subsequently demonstrated by far-western analysis 41. Gel shift assays further showed that TraM sequesters TraR from binding to DNA, presumably via forming the TraR-TraM complex, thereby inhibiting TraR transcriptional activity.
Homologues of TraM have also been identified in several species within the Rhizobiaceae family, including Sinorhizobium meliloti 126, Rhizobium leguminosarum 127, and Rhizobium sp. NGR234 127. Most genetic, biochemical and structural studies have been conducted using TraM proteins from several species of A. tumefaciens (e.g., R10 and C58), while TraM of Rhizobium sp. NGR234 was utilized in the structural study of the anti-activation TraR-TraM complex. All TraM proteins studied so far bind tightly to their respective TraR proteins with dissociation constants (Kd) in 1-50 nM range, and in a 1:1 binding stoichiometry 110,128-132.
The TraM proteins are approximately 100-residue in length, and exist as either homodimers or monomers. TraM of A. tumefaciens (TraMAt) exists as homodimer, and each TraMAt monomer consists of two long (α2 and α4 of ~ 40 Å) anti-parallel α-helices (~27 a.a. each), flanked by two short α-helices (α1 and α3) 128,130,131. In forming a homodimer, two helices from either monomer, one long (α4) and one short (α1), further wrap around to form an intermolecular helical bundle. The dimer interface is extensive (>1000 Å2) and is mainly hydrophobic (Figure 10A); the importance of sequestering this hydrophobic surface is supported by mutational analysis in which substitutions of one hydrophobic residue on the surface to a charged Asp result in insoluble TraM proteins, whereas conservative mutations to Ala do not affect protein solubility 130. Residues whose substitutions to Ala show defects in either TraRAt binding or inhibition of TraRAt activity 125,129 are largely located on both sides of the exposed surface, suggesting that dimeric TraMAt is the functional state when forming a complex with TraRAt. An initial model invoking dissociation of dimeric TraM and TraR to form mixed heterodimers was suggested by crosslinking analysis 130, but has subsequently been shown to be incorrect. We and others have subsequently found that dimeric TraM interacts directly with TraR to form a hetero-octameric (TraRAt)4-(TraMAt)4 complex 128,131,132. To demonstrate directly that the dimeric TraMAt does not dissociate in interacting with TraRAt, Gly23 (~ 2.5 Å away from the cognate Cys71 of the other monomer) was mutated to Cys to generate a non-dissociable, inter-molecularly disulfide bonded dimeric TraMAt 133. Gly23Cys TraMAt in size exclusion chromatography migrated as a molecular species similar to the wild type protein, and as expected, SDS-PAGE analysis showed that Gly23Cys TraMAt is a dimer in the absence of reducing agent DTT and a monomer in the presence of DTT. Notably, the oxidized Gly23Cys TraMAt maintains similar activity (95%) to the wild type in preventing TraRAt from binding to DNA as assayed by gel shift assays. Therefore, these new results indicate that TraMAt maintains the dimer state and does not dissociate in complex with TraRAt.
Figure 10.
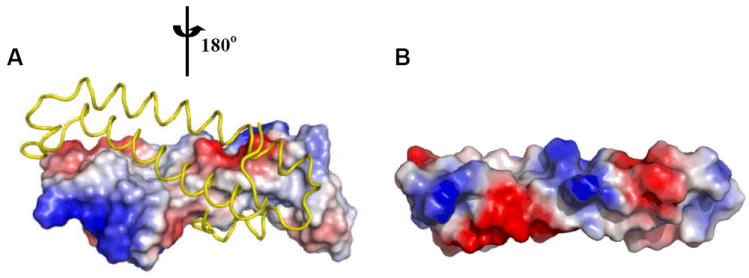
Surface potentials of TraMAt (PDB ID: 1RFY) and TraMNGR (PDB ID: 2Q0O) (from the anti-activation complex). For dimeric TraMAt, surface of one monomer is shown, the other monomer is shown as a Cα trace in yellow. Surface with positive potentials is colored in blue, and with negative potentials in red. TraMNGR is shown in the same view as the surface presentation of TraMAt monomer.
In contrast to the dimeric state of TraMAt, the functional form of TraM of Rhizobium sp. NGR234 (TraMNGR) is monomeric 110. While structure of the isolated TraMNGR has not been determined, structure of the TraMNGR-TraRNGR anti-activation complex may explain the difference in the oligomeric state of TraMNGR and TraMAt. TraMNGR preserves the two-helix coil structure found in one TraMAt monomer: the two core structures superimpose with an r.m.s.d. of 1.8 Å using the Cα atoms from 58 residues. The major structural deviations are found in the two regions that flank the core structure. This structural alignment reveals that the hydrophobic residues that are sequestered within the dimer interface of TraMAt (Leu14, Leu17, Leu20, Ile70 and Ile77) correspond to polar or charged residues (Asn13, Lys16, Arg19, Glu73, and Lys79) in TraMNGR. Thus, the hydrophilic surface of TraMNGR (Figure 10B) may account for its stability as a monomer.
5.3 Structural Basis for Anti-activation
Early genetic and biochemical studies identified residues in TraRAt and TraMAt that are important in TraR-TraM complex formation 41,125,129,132. Leu174, Pro176, Leu182, Gly188, Thr190, Ala195, Met213, and Arg215 of TraRAt CTD are required for efficient TraRAt-TraMAt interaction; however, most of these residues are located not in the DNA recognition helix α12, but instead on α10, suggesting that a direct binding competition between DNA and TraM may not completely account for the anti-activation function of TraM. Many TraMAt residues including those located in the C-terminal tail were found to be important for TraMAt inhibitory function. Interestingly, the Gln82A TraMAt mutant, albeit with high affinity for TraRAt, was defective in inactivating TraRAt. Based on the proposed mechanism that TraM dissociates the TraR-DNA complex via a meta-stable intermediate ((TraMAt)2-(TraR At)2-DNA), it was proposed that the Gln82A TraMAt mutant is not effective in converting the ternary nucleoprotein intermediate to disengage TraRAt completely from the DNA, resulting in deficient inhibition of TraRAt function.
The anti-activation TraRAt-TraMAt complex is a hetero-octamer of 150 kDa, consisting of two dimeric TraMAt and two dimeric of TraRAt 128,131,132; the complex did not produce high quality diffracting crystals 133. The TraRNGR-TraMNGR complex, however, is a hetero-tetramer of 80 kDa, half the size of the octameric TraRAt-TraMAt. When aligned with the identified TraR-TraM systems of different Rhizobecia species, TraRNGR shares 23.5 % amino acid identity with TraRAt, whereas a relatively low identity of 21% exists between TraMNGR and TraMAt, consistent with the divergence in oligomeric states for the two TraM proteins (see Section 5.2). In the crystal structure, the two TraRNGR monomers in TraRNGR-TraMNGR complex adopt open and closed conformations. In both forms, TraRNGR CTD interacts with TraMNGR and in the same manner; in the closed form, TraRNGR NTD also contacts TraMNGR. The open form is involved in crystal contacts, thus it is very likely that both TraRNGR monomers in TraMNGR-TraRNGR adopt the closed form in solution, that is, TraMNGR interacts with both the CTD and NTD of TraRNGR (Figure 11A). Existence of the open form again highlights the capacity of the linker to adopt different conformations (as in TraR-DNA structure, see Section 5.1), and suggests a less significant role of TraRNGR NTD in interaction with TraMNGR. This suggestion is consistent with early genetic and biochemical findings that the TraRAt CTD contributes dominantly to interaction with TraMAt 41.
Figure 11.
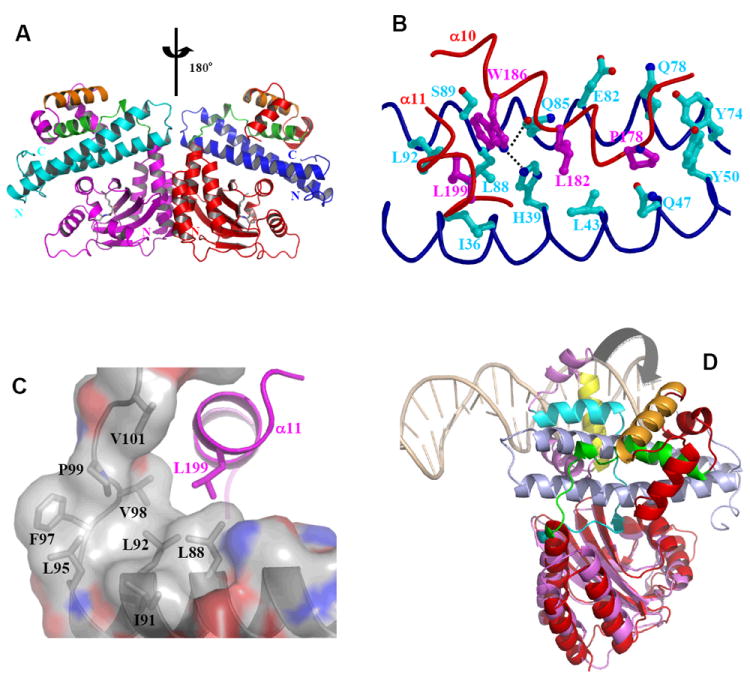
Structure and function of anti-activation by TraM. A) Model of symmetric (TraRNGR-TraMNGR)2 in solution. The model was generated by applying the C2 rotational symmetry of the NTDs to the closed form of dimeric TraRNGR-TraMNGR. The repositioned TraRNGR-TraMNGR pair is colored in red and blue, and the original pair is in magenta and cyan. The main TraM-binding site, α10 and the linker, is in green, and the DNA binding helix, α12, is in orange. The bound 3-oxo-C8-HSL is shown in ball-and-stick representation. B) The interaction between TraRNGR α10 with TraMNGR (PDB ID: 2Q0O). TraMNGR residues (cyan) that are in contact with Pro178, Leu182 and Trp186 (magenta) of TraRNGR α10 are shown. Leu199 from α11 is also shown. The dotted lines denote hydrogen bonds between TraRNGR Trp186 and TraMNGR His39 and Gln85. C) The interaction of TraRNGR L199 with the hydrophobic C-terminal tail of TraMNGR. Surface is colored according to the underlying atoms: red for oxygen, blue for nitrogen, and gray for carbon. Side chains of the hydrophobic TraMNGR residues (gray), along with that of L199 (magenta), are shown. D) Structural comparison of TraRAt-DNA with TraRNGR-TraMNGR. The NTDs of the both structures are superimposed, but for clarity only one monomer of TraRNGR (in red) and TraRAt (in purple, the extended form) from each structure is shown. The large domain movement is highlighted with an arrow. DNA is displayed as a double coil and is light orange, and the DNA-binding helix α12 is yellow in TraRAt-DNA complex and orange in TraRNGR-TraMNGR complex. TraMNGR is light blue, and the TraM-binding site (α10 and the linker) is cyan in TraRAt-DNA and green in TraRNGR-TraMNGR.
The TraRNGR-TraMNGR interface is largely hydrophobic, and the interactions can be separated into two clusters based on TraRNGR structure. First, side chains of P178, L182 and W186 of α10 interact with TraMNGR residues from both long helices as shown in Figure 11B (Ile36, His39, Leu43, Gln47, Tyr50 of one helix, and Tyr74, Gln78, Glu82, Gln85, Leu88, Ser89 and Leu92 of the other helix). In particular, TraRNGR Trp186 forms two pairs of intermolecular H-bonds with TraMNGR His39 and Gln85, and as a result, Trp186 is totally buried by both polar and hydrophobic TraMNGR residues. The functional significance of Trp186 is underscored by its absolute conservation among the LuxR-type proteins. The role of α10 in TraRNGR-TraMNGR interaction revealed in the structure is consistent with early mutational results in A. tumefaciens (above); however, the derived specific residues from those studies (except Pro178) are largely involved in structural organization and positioning of α10. Second, the C-terminus of α11 of TraRNGR, mainly Leu199, is engaged in extensive van der Waals interactions with the C-terminal hydrophobic region, consisting of Leu88, Ile91, Leu92, Leu95, Phe97, Val98, Pro99 and Val101, of TraMNGR (Figure 11C). Mutagenesis studies supported the role of TraMAt C-terminus in inhibiting TraRAt function 129. Structures of this hydrophobic C-terminal tail are superimposable between TraMNGR and TraMAt and are expected to be largely conserved among TraM proteins given the high hydrophobicity in their sequences. Thus, the TraRNGR residues that contact TraMNGR are primarily in α10 and α11, as well as the linker and C-terminal end, but not in the DNA-binding site α12, which is consistent with genetic analysis 132, which indicated separate TraM and DNA-binding sites on TraR.
To understand the structural basis for antagonism of TraR DNA binding activity by TraM, the structure of the activation complex TraRAt-DNA was compared with that of the anti-activation complex TraRNGR-TraMNGR. Superposition of the TraR NTDs shows that the relative orientations of the CTDs to their respective NTDs are drastically different in the two complexes (Figure 11D). In the activation complex, the two TraR CTDs are in contact through the formation of a coiled-coil structure mediated by the two α13 helices; such a conformation orients the two DNA recognition helices (α12) in register with the two consecutive major grooves of the target DNA (Section 5.1). In contrast, the CTDs of the anti-activation complex are distant from each other in a conformation incompatible with simultaneous binding to the closely spaced palindromic sites on DNA. Anti-activators have been shown to occupy the DNA-binding site on their target activators, thereby directly blocking DNA binding 134-136, or to interfere with oligomerization of the activators into their active form (e.g. TrlR-TraR inactive heterodimer 116, mentioned earlier) 137. Here, TraM employs an allosteric approach, by binding to TraR tightly, precluding the domain movements required for the CTDs to interact productively with the palindromic DNA sequence of the tra box.
To disengage TraR from TraR-DNA complex to form TraR-TraM complex, TraM may capture TraR, as it dissociates from DNA as part of the normal dynamic equilibrium, to form the stable anti-activation complex. Alternatively, TraM may play an active role in competing TraR off its DNA binding site. Given that one of the TraM-binding sites (α10 and the linker) in the TraR2-DNA structure is inaccessible (Figure 9A), a stepwise mechanism has been proposed to expose this buried TraM-binding site so as to dissociate TraR-DNA complex 110. This model assumes that the higher affinity of TraR for TraM than for DNA allows binding of one TraM molecule to the accessible TraM-binding site in the extended TraR form to displace this TraR monomer from one half-site on the tra box. The structural flexibility of the resulting TraR2-DNA-TraM intermediate may expose the TraM-binding site buried in the compact TraR monomer so that it can interact with a second TraM, disengaging this TraR monomer from the second half-site of the tra box. In support of this model, a ternary nucleoprotein complex, has been reported in A. tumefaciens when incubating TraMAt with the TraRAt-DNA complex 132.
6. Conclusions and Perspectives
Although the phenotypes controlled by the AHL-dependent quorum sensing system are quite diverse and unique to each species, the key features of AHL synthesis are relatively conserved among numerous Proteobacteria species. The structural and mechanistic analyses described here have provided a basis for a preliminary understanding of the synthesis and detection of AHL signals. They have revealed features of the system that are important for correct AHL-dependent signaling, which is important because many natural and synthetic mechanisms that inhibit or misregulate quorum sensing can provide broad-spectrum control of particular bacterial diseases 32,50,51,138. Thus, the mechanisms by which the metabolic and environmental perturbations alter the ‘language’ spoken by a particular bacterium have broad and important biological consequences.
Current structural studies have provided a chemical basis for how AHLs interact with the LuxR-type activators, offered general insights into AHL specificity of the LuxR-type proteins, and supported the role of AHLs as a folding scaffold for the LuxR-type activators. The structural importance of AHLs in folding of the cognate LuxR-type protein suggests they function as a folding switch to regulate the DNA binding activity of the LuxR-type activators, which in turn leads to transcriptional regulation of target genes. Structural studies of TraR-DNA complex have revealed the atomic details that orchestrate the interactions between the LuxR-type proteins and the target DNA and the principles that contribute to DNA recognition by the LuxR-type proteins. Structural comparison between the anti-activation TraR-TraM complex and the activation TraR-DNA complex has uncovered a unique mechanism to antagonize the DNA binding function. Such a mechanism of allosteric inhibition may be broadly utilized by anti-activators for transcription regulation in a variety of biological processes. With such achievements, it is expected that structural studies will continue to contribute to the mechanistic understanding of AHL signaling and regulation via the LuxR-type regulators, and resolve discrepancies between structural observations and biochemical functions. Included in the areas of interest are as follows:
Mechanism of substrate selectivity of AHL synthases
Current structural analyses of AHL synthases have been limited to the apo-enzymes, because no structures of AHL synthases bound to any substrate are available. The binding sites for the substrates have been inferred from other analogous enzymes 86-88 and experimental studies using mutagenesis followed by functional analysis 25,26,75. Thus, the understanding of the mechanism by which AHL synthases select acyl-ACPs with acyl chains of the appropriate length is still rudimentary. For example, many questions remain on this point. For an AHL synthase that produces long-chain AHLs, such as LasI (3-oxo-C12-HSL), it is unclear how the acyl-chain length is controlled, since there is no apparent restriction on how large an acyl chain can fit in the enzyme 34. Alternatively, how do AHL synthases that have a more restrictive acyl-chain binding pocket, such as RhlI and EsaI obtain most of their binding energy? How does the ACP protein interaction with the AHL synthase influence AHL production in these systems? It is very likely that the binding energy derived from different parts of the substrate influences this selectivity. Very little is known about how SAM interacts with the enzymes and participates in catalysis. Quantitative binding studies with sets of AHL synthases and substrates as well as structures of AHL synthases bound to substrates will begin to answer these questions.
Regulatory mechanism of the LuxR-type AHL-inactivated transcription factors
The most unique feature of this LuxR-type subfamily, represented by EsaR, is that they are soluble in the absence of AHLs and are active for DNA binding 18, contrasting with the essential role of AHLs in folding of the LuxR-type activators (Section 4.2). For 3-oxo-C6-HSL to bind EsaR, the AHL-binding site in EsaR is very likely solvent accessible; if so, it would be interesting to see how the hydrophobic acyl-chain of 3-oxo-C6-HSL is sequestered. Alternatively, the accessible AHL-binding site in EsaR may become closed after 3-oxo-C6-HSL binding, implying conformational changes in EsaR triggered by AHL binding. The AHL-binding induced structural changes in EsaR NTD may influence the conformation of the DNA-binding CTD, thereby regulating the interaction of CTD with DNA. This model of regulation is appealing, because unlike the LuxR-type activators whose DNA-binding function is correlated with protein stability or the AHL-assisted protein folding, EsaR is stable and functional without AHLs, and remains stable structurally in the presence of AHLs. Given the relative structural independence between the NTD and the CTD as observed in TraR, it would be interesting to see how the AHL association in the NTD is allosterically transmitted to the CTD and how the CTD responds resulting in a decreased affinity for DNA. Structural studies of apo-EsaR, EsaR bound with DNA and EsaR with the cognate AHL will unravel the allosteric function of AHLs in regulating DNA binding activity of this subfamily of the LuxR-type proteins.
Molecular steps of regulating DNA binding of the LuxR-type proteins by anti-activators
Although the reported existence of a TraM-TraR-DNA ternary complex in A. tumefaciens supports a stepwise mechanism for TraM to disengage TraR from DNA to form the anti-activation TraR-TraM complex, the transient nature of this nucleoprotein complex prevents further structural characterizations. Alternatively, mutants of TraR or TraM, which are defective after the first TraM binding may stabilize the nucleoprotein intermediate for structural studies. For example, mutations of residues on the TraRNGR-TraMNGR interface or on the TraRNGR linker region may trap the ternary nucleoprotein complex in a stable form. In addition to verifying the pathway of the inhibition, these studies will reveal information on how binding of the first TraM triggers domain movement in TraR and the structural dynamics of TraR-DNA. Given that residues of TraR and TraM involved in the TraRNGR-TraMNGR interface are largely conserved (Section 5.3), the molecular events outlining TraMNGR-mediated dissociation of TraRNGR-DNA should be applicable to the Rhizobiaceae species. However, it is intriguing that TraMNGR did not inhibit TraRAt in an in vivo assay 139. One explanation is that not all the interacting residues contribute equally to the interaction and those that are important may have unique stereochemistry that confers the specificity. Thus, mutational analysis on the interfacing residues from both Rhizobium sp. NGR234 and A. tumefaciens may offer insights into why TraR and TraM of two systems do not react, and whether there exist other anti-regulators in quorum sensing systems.
Acknowledgments
We are very grateful to current and past contributors to this work, including Drs. Susanne Beck von Bodman, John Cronan, Ty Gould, Robert Murphy, Herbert Schweizer, Dale Val, and Bill Watson, as well as Mr. Paul Kirwan, Mr. Tim Minogue, Mrs. Linda Farb, and other members of the Churchill laboratory; Drs. Guozhou Chen, Chao Wang, Clay Fuqua, Lian-Hui Zhang, Mr. Zhida Zheng, Mr. James Malenkos, Mrs. Valena Fiscus, and members of the Chen laboratory. We appreciate the support from the National Science Foundation (MCB-0821220 to M.E.A.C. and MCB-0416447 to L.C.).
Biographies

Mair Churchill was born in England and has lived in the USA since 1963. She received a B.A. in Chemistry from Swarthmore College, and a Ph.D. in Chemistry at the Johns Hopkins University for the solution structural analysis of DNA in the laboratory of Professor Thomas D. Tullius. She returned to England as an American Cancer Society Fellow with Sir Aaron Klug at the Medical Research Council Laboratory of Molecular Biology focusing on structural aspects of DNA recognition by protein motifs in chromosomal proteins. After a Rockefeller Academic Exchange Fellowship in crystallography with Professor Ian Wilson at the Scripps Research Institute, she became Assistant Professor at the University of Illinois at Champaign Urbana. In 1998, she moved to the University of Colorado School of Medicine, where she currently is Professor of Pharmacology.

Lingling Chen was born and raised in Xiamen, Fujian province of China. She received her B.S. in Physical Chemistry from Xiamen University, and continued there for her graduate study in Catalysis. She went to Stanford University to study protein structure and protein folding using small angle x-ray scattering (SAXS) in the laboratory of Keith Hodgson, and obtained Ph.D. in Chemistry in 1996. Dr. Chen received her postdoctoral training in Protein Crystallography in the laboratory of the late Paul Sigler at Yale University as a Helen Hay Whitney Fellow. She became Assistant Professor in Biology Department of Indiana University in 2001, and is now Associate Professor of Molecular and Cellular Biochemistry at Indiana University.
References
- 1.Nealson KH, Platt T, Hastings JW. J Bacteriol. 1970;104:313. doi: 10.1128/jb.104.1.313-322.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. Biochemistry. 1981;20:2444. doi: 10.1021/bi00512a013. [DOI] [PubMed] [Google Scholar]
- 3.Nealson KH. Arch Microbiol. 1977;112:73. doi: 10.1007/BF00446657. [DOI] [PubMed] [Google Scholar]
- 4.Fuqua C, Winans SC. J Bacteriol. 1996;178:435. doi: 10.1128/jb.178.2.435-440.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chemical communication among Bacteria. American Microbiology Association Press; Washington, DC: 2008. [Google Scholar]
- 6.Marketon MM, Gronquist MR, Eberhard A, Gonzalez JE. J Bacteriol. 2002;184:5686. doi: 10.1128/JB.184.20.5686-5695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuqua C, Eberhard A. In: Cell-Cell Communication in Bacteria. Dunny G, Winans SC, editors. American Microbiology Association Press; 1999. [Google Scholar]
- 8.Thiel V, Kunze B, Verma P, Wagner-Dobler I, Schulz S. Chembiochem. 2009;10:1861. doi: 10.1002/cbic.200900126. [DOI] [PubMed] [Google Scholar]
- 9.Schaefer AL, Val DL, Hanzelka BL, Cronan JE, Jr, Greenberg EP. Proc Natl Acad Sci U S A. 1996;93:9505. doi: 10.1073/pnas.93.18.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan HB, Greenberg EP. J Bacteriol. 1985;163:121. doi: 10.1128/jb.163.3.1210-1214.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearson JP, Van Delden C, Iglewski BH. J Bacteriol. 1999;181:1203. doi: 10.1128/jb.181.4.1203-1210.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens AM, Greenberg EP. In: Cell-Cell Communication in Bacteria. Dunny G, Winans SC, editors. American Microbiology Association Press; 1999. [Google Scholar]
- 13.Freeman JA, Lilley BN, Bassler BL. Mol Microbiol. 2000;35:139. doi: 10.1046/j.1365-2958.2000.01684.x. [DOI] [PubMed] [Google Scholar]
- 14.Shadel GS, Baldwin TO. J Bacteriol. 1991;173:568. doi: 10.1128/jb.173.2.568-574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. J Bacteriol. 2003;185:2080. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Bodman SB, Ball JK, Faini MA, Herrera CM, Minogue TD, Urbanowski ML, Stevens AM. J Bacteriol. 2003;185:7001. doi: 10.1128/JB.185.23.7001-7007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuster M, Lostroh CP, Ogi T, Greenberg EP. J Bacteriol. 2003;185:2066. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minogue TD, Wehland-von Trebra M, Bernhard F, von Bodman SB. Mol Microbiol. 2002;44:1625. doi: 10.1046/j.1365-2958.2002.02987.x. [DOI] [PubMed] [Google Scholar]
- 19.Fuqua C, Parsek MR, Greenberg EP. Annu Rev Genet. 2001;35:439. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- 20.Waters CM, Bassler BL. Annu Rev Cell Dev Biol. 2005;21:319. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 21.Whitehead NA, Barnard AM, Slater H, Simpson NJ, Salmond GP. FEMS Microbiol Rev. 2001;25:365. doi: 10.1111/j.1574-6976.2001.tb00583.x. [DOI] [PubMed] [Google Scholar]
- 22.Eberl L. Int J Med Microbiol. 2006;296:103. doi: 10.1016/j.ijmm.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 23.Beck von Bodman S, Farrand SK. J Bacteriol. 1995;177:5000. doi: 10.1128/jb.177.17.5000-5008.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Murphy PJ, Kerr A, Tate ME. Nature. 1993;362:446. doi: 10.1038/362446a0. [DOI] [PubMed] [Google Scholar]
- 25.Watson WT, Minogue TD, Val DL, Beck von Bodman S, Churchill MEA. Mol Cell. 2002;9:685. doi: 10.1016/s1097-2765(02)00480-x. [DOI] [PubMed] [Google Scholar]
- 26.Brader G, Sjoblom S, Hyytiainen H, Sims-Huopaniemi K, Palva ET. J Biol Chem. 2005;280:10403. doi: 10.1074/jbc.M408603200. [DOI] [PubMed] [Google Scholar]
- 27.Lutter E, Lewenza S, Dennis JJ, Visser MB, Sokol PA. Infect Immun. 2001;69:4661. doi: 10.1128/IAI.69.7.4661-4666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohamed NM, Cicirelli EM, Kan J, Chen F, Fuqua C, Hill RT. Environ Microbiol. 2008;10:75. doi: 10.1111/j.1462-2920.2007.01431.x. [DOI] [PubMed] [Google Scholar]
- 29.Gotschlich A, Huber B, Geisenberger O, Togl A, Steidle A, Riedel K, Hill P, Tummler B, Vandamme P, Middleton B, Camara M, Williams P, Hardman A, Eberl L. Syst Appl Microbiol. 2001;24:1. doi: 10.1078/0723-2020-00013. [DOI] [PubMed] [Google Scholar]
- 30.Duerkop BA, Ulrich RL, Greenberg EP. J Bacteriol. 2007;189:5034. doi: 10.1128/JB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duerkop BA, Herman JP, Ulrich RL, Churchill ME, Greenberg EP. J Bacteriol. 2008;190:5137. doi: 10.1128/JB.00246-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck von Bodman S, Majerczak DR, Coplin DL. Proc Natl Acad Sci U S A. 1998;95:7687. doi: 10.1073/pnas.95.13.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Kievit TR, Kakai Y, Register JK, Pesci EC, Iglewski BH. FEMS Microbiol Lett. 2002;212:101. doi: 10.1016/s0378-1097(02)00735-8. [DOI] [PubMed] [Google Scholar]
- 34.Gould TA, Herman J, Krank J, Murphy RC, Churchill MEA. J Bacteriol. 2006;188:773. doi: 10.1128/JB.188.2.773-783.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Kievit TR, Gillis R, Marx S, Brown C, Iglewski BH. Appl Environ Microbiol. 2001;67:1865. doi: 10.1128/AEM.67.4.1865-1873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brencic A, Winans SC. Microbiol Mol Biol Rev. 2005;69:155. doi: 10.1128/MMBR.69.1.155-194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuster M, Peter Greenberg E. Int J Med Microbiol. 2006;296:73. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 38.Seed CP, Passador L, Iglewski BH. J Bacteriol. 1995;177:654. doi: 10.1128/jb.177.3.654-659.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michael B, Smith JN, Swift S, Heffron F, Ahmer BM. J Bacteriol. 2001;183:5733. doi: 10.1128/JB.183.19.5733-5742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang I, Cook DM, Farrand SK. J Bacteriol. 1995;177:449. doi: 10.1128/jb.177.2.449-458.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo ZQ, Qin Y, Farrand SK. J Biol Chem. 2000;275:7713. doi: 10.1074/jbc.275.11.7713. [DOI] [PubMed] [Google Scholar]
- 42.Piper KR, Farrand SK. J Bacteriol. 2000;182:1080. doi: 10.1128/jb.182.4.1080-1088.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byers JT, Lucas C, Salmond GP, Welch M. J Bacteriol. 2002;184:1163. doi: 10.1128/jb.184.4.1163-1171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yates EA, Philipp B, Buckley C, Atkinson S, Chhabra SR, Sockett RE, Goldner M, Dessaux Y, Camara M, Smith H, Williams P. Infect Immun. 2002;70:5635. doi: 10.1128/IAI.70.10.5635-5646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nachin L, Barras F. Mol Plant Microbe Interact. 2000;13:882. doi: 10.1094/MPMI.2000.13.8.882. [DOI] [PubMed] [Google Scholar]
- 46.Atkinson S, Throup JP, Stewart G, Williams P. Mol Microbiol. 1999;33:1267. doi: 10.1046/j.1365-2958.1999.01578.x. [DOI] [PubMed] [Google Scholar]
- 47.Kirwan JP, Gould TA, Schweizer HP, Bearden SW, Murphy RC, Churchill ME. J Bacteriol. 2006;188:784. doi: 10.1128/JB.188.2.784-788.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sio CF, Otten LG, Cool RH, Diggle SP, Braun PG, Bos R, Daykin M, Camara M, Williams P, Quax WJ. Infect Immun. 2006;74:1673. doi: 10.1128/IAI.74.3.1673-1682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang JJ, Petersen A, Whiteley M, Leadbetter JR. Appl Environ Microbiol. 2006;72:1190. doi: 10.1128/AEM.72.2.1190-1197.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leadbetter JR, Greenberg EP. J Bacteriol. 2000;182:6921. doi: 10.1128/jb.182.24.6921-6926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong Y-H, Wang L-H, Xu J-L, Zhang H-B, Zhang X-F, Zhang L-H. Nature. 2001;411:813. doi: 10.1038/35081101. [DOI] [PubMed] [Google Scholar]
- 52.Roche DM, Byers JT, Smith DS, Glansdorp FG, Spring DR, Welch M. Microbiology. 2004;150:2023. doi: 10.1099/mic.0.26977-0. [DOI] [PubMed] [Google Scholar]
- 53.Huang JJ, Han JI, Zhang LH, Leadbetter JR. Appl Environ Microbiol. 2003;69:5941. doi: 10.1128/AEM.69.10.5941-5949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaefer AL, Greenberg EP, Oliver CM, Oda Y, Huang JJ, Bittan-Banin G, Peres CM, Schmidt S, Juhaszova K, Sufrin JR, Harwood CS. Nature. 2008;454:595. doi: 10.1038/nature07088. [DOI] [PubMed] [Google Scholar]
- 55.Boerjan W, Ralph J, Baucher M. Annu Rev Plant Biol. 2003;54:519. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 56.Cooley M, Chhabra SR, Williams P. Chem Biol. 2008;15:1141. doi: 10.1016/j.chembiol.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Churchill ME, Herman JP. In: Chemical Communication Among Bacteria. Winans SC, Bassler BL, editors. American Microbiology Association; Washington D.C.: 2008. [Google Scholar]
- 58.Engebrecht J, Silverman M. Proc Natl Acad Sci U S A. 1984;81:4154. doi: 10.1073/pnas.81.13.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearson JP, Passador L, Iglewski BH, Greenberg EP. Proc Natl Acad Sci U S A. 1995;92:1490. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanzelka BL, Greenberg EP. J Bacteriol. 1996;178:5291. doi: 10.1128/jb.178.17.5291-5294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moré MI, Finger LD, Stryker JL, Fuqua C, Eberhard A, Winans SC. Science. 1996;272:1655. doi: 10.1126/science.272.5268.1655. [DOI] [PubMed] [Google Scholar]
- 62.Val DL, Cronan JEJ. J Bacteriol. 1998;180:2644. doi: 10.1128/jb.180.10.2644-2651.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parsek MR, Val DL, Hanzelka BL, Cronan JEJ, Greenberg EP. Proc Natl Acad Sci U S A. 1999;96:4360. doi: 10.1073/pnas.96.8.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bassler BL, Wright M, Showalter RE, Silverman MR. Mol Microbiol. 1993;9:773. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- 65.Gilson L, Kuo A, Dunlap PV. J Bacteriol. 1995;177:6946. doi: 10.1128/jb.177.23.6946-6951.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milton DL, Chalker VJ, Kirke D, Hardman A, Camara M, Williams P. J Bacteriol. 2001;183:3537. doi: 10.1128/JB.183.12.3537-3547.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hanzelka BL, Parsek MR, Val DL, Dunlap PV, Cronan JEJ, Greenberg EP. J Bacteriol. 1999;181:5766. doi: 10.1128/jb.181.18.5766-5770.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laue BE, Jiang Y, Chhabra SR, Jacob S, Stewart GS, Hardman A, Downie JA, O’Gara F, Williams P. Microbiology. 2000;146(Pt 10):2469. doi: 10.1099/00221287-146-10-2469. [DOI] [PubMed] [Google Scholar]
- 69.Burton EO, Read HW, Pellitteri MC, Hickey WJ. Appl Environ Microbiol. 2005;71:4906. doi: 10.1128/AEM.71.8.4906-4909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cullinane M, Baysse C, Morrissey JP, O’Gara F. Microbiol. 2005;151:3071. doi: 10.1099/mic.0.27958-0. [DOI] [PubMed] [Google Scholar]
- 71.Shih GC, Kahler CM, Swartley JS, Rahman MM, Coleman J, Carlson RW, Stephens DS. Mol Microbiol. 1999;32:942. doi: 10.1046/j.1365-2958.1999.01404.x. [DOI] [PubMed] [Google Scholar]
- 72.Lerat E, Moran NA. Mol Biol Evol. 2004;21:903. doi: 10.1093/molbev/msh097. [DOI] [PubMed] [Google Scholar]
- 73.Parsek MR, Schaefer AL, Greenberg EP. Mol Microbiol. 1997;26:301. doi: 10.1046/j.1365-2958.1997.5741935.x. [DOI] [PubMed] [Google Scholar]
- 74.Hanzelka BL, Stevens AM, Parsek MR, Crone TJ, Greenberg EP. J Bacteriol. 1997;179:4882. doi: 10.1128/jb.179.15.4882-4887.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gould TA, Schweizer HP, Churchill MEA. Mol Micro. 2004;53:1135. doi: 10.1111/j.1365-2958.2004.04211.x. [DOI] [PubMed] [Google Scholar]
- 76.Watson WT, Murphy FV, Gould TA, Jambeck P, Val DL, Cronan JE, Beck von Bodman S, Churchill MEA. Acta Crystallogr. 2001;D57:1945. doi: 10.1107/s0907444901014512. [DOI] [PubMed] [Google Scholar]
- 77.Gould TA, Watson WT, Choi SH, Schweizer HP, Churchill MEA. Acta Crystallogr. 2004;D60:518. doi: 10.1107/S0907444903028300. [DOI] [PubMed] [Google Scholar]
- 78.Clements A, Marmorstein R. Methods Enzymol. 2003;371:545. doi: 10.1016/S0076-6879(03)71041-6. [DOI] [PubMed] [Google Scholar]
- 79.Van Wagoner RM, Clardy J. Structure. 2006;14:1425. doi: 10.1016/j.str.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 80.Churchill MEA. Structure. 2006;14:1342. doi: 10.1016/j.str.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 81.Rojas JR, Trievel RC, Zhou J, Mo Y, Berger SL, Allis CD, Marmorstein R. Nature. 1999;401:93. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- 82.Shaw PD, Ping G, Daly SL, Cha C, Cronan JEJ, Rinehart KL, Farrand SK. Proc Natl Acad Sci U S A. 1997;94:6036. doi: 10.1073/pnas.94.12.6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pearson JP, Pesci EC, Iglewski BH. J Bacteriol. 1997;179:5756. doi: 10.1128/jb.179.18.5756-5767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khan SR, Mavrodi DV, Jog GJ, Suga H, Thomashow LS, Farrand SK. J Bacteriol. 2005;187:6517. doi: 10.1128/JB.187.18.6517-6527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chakrabarti S, Sowdhamini R. Protein Eng. 2003;16:271. doi: 10.1093/proeng/gzg031. [DOI] [PubMed] [Google Scholar]
- 86.Hickman AB, Namboodiri MA, Klein DC, Dyda F. Cell. 1999;97:361. doi: 10.1016/s0092-8674(00)80745-x. [DOI] [PubMed] [Google Scholar]
- 87.Tanner KG, Trievel RC, Huo M-H, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. J Biol Chem. 1999;274:18157. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 88.Berndsen CE, Denu JM. Curr Opin Struct Biol. 2008;18:682. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yan Y, Harper S, Speicher DW, Marmorstein R. Nat Struct Biol. 2002;9:862. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 90.Raychaudhuri A, Jerga A, Tipton PA. Biochem. 2005;44:2974. doi: 10.1021/bi048005m. [DOI] [PubMed] [Google Scholar]
- 91.Raychaudhuri A, Tullock A, Tipton PA. Biochemistry. 2008;47:2893. doi: 10.1021/bi702009n. [DOI] [PubMed] [Google Scholar]
- 92.Engebrecht J, Nealson K, Silverman M. Cell. 1983;32:773. doi: 10.1016/0092-8674(83)90063-6. [DOI] [PubMed] [Google Scholar]
- 93.Eberhard A. J Bacteriol. 1972;109:1101. doi: 10.1128/jb.109.3.1101-1105.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fuqua WC, Winans SC, Greenberg EP. J Bacteriol. 1994;176:269. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaplan HB, Greenberg EP. Proc Natl Acad Sci U S A. 1987;84:6639. doi: 10.1073/pnas.84.19.6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gambello MJ, Iglewski BH. J Bacteriol. 1991;173:3000. doi: 10.1128/jb.173.9.3000-3009.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH. Science. 1993;260:1127. doi: 10.1126/science.8493556. [DOI] [PubMed] [Google Scholar]
- 98.Ochsner UA, Koch AK, Fiechter A, Reiser J. J Bacteriol. 1994;176:2044. doi: 10.1128/jb.176.7.2044-2054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Latifi A, Winson MK, Foglino M, Bycroft BW, Stewart GS, Lazdunski A, Williams P. Mol Microbiol. 1995;17:333. doi: 10.1111/j.1365-2958.1995.mmi_17020333.x. [DOI] [PubMed] [Google Scholar]
- 100.Fuqua WC, Winans SC. J Bacteriol. 1994;176:2796. doi: 10.1128/jb.176.10.2796-2806.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piper KR, Beck von Bodman S, Farrand SK. Nature. 1993;362:448. doi: 10.1038/362448a0. [DOI] [PubMed] [Google Scholar]
- 102.Schu DJ, Carlier AL, Jamison KP, von Bodman S, Stevens AM. J Bacteriol. 2009;191:7402. doi: 10.1128/JB.00994-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stevens AM, Dolan KM, Greenberg EP. Proc Natl Acad Sci U S A. 1994;91:12619. doi: 10.1073/pnas.91.26.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu J, Winans SC. Proc Natl Acad Sci U S A. 1999;96:4832. doi: 10.1073/pnas.96.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Urbanowski ML, Lostroh CP, Greenberg EP. J Bacteriol. 2004;186:631. doi: 10.1128/JB.186.3.631-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yao Y, Martinez-Yamout MA, Dickerson TJ, Brogan AP, Wright PE, Dyson HJ. J Mol Biol. 2006;355:262. doi: 10.1016/j.jmb.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 107.Bottomley MJ, Muraglia E, Bazzo R, Carfi A. J Biol Chem. 2007;282:13592. doi: 10.1074/jbc.M700556200. [DOI] [PubMed] [Google Scholar]
- 108.Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Marco SD. EMBO J. 2002;21:4393. doi: 10.1093/emboj/cdf459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang RG, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. Nature. 2002;417:971. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- 110.Chen G, Jeffrey PD, Fuqua C, Shi Y, Chen L. Proc Natl Acad Sci U S A. 2007;104:16474. doi: 10.1073/pnas.0704843104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kiratisin P, Tucker KD, Passador L. J Bacteriol. 2002;184:4912. doi: 10.1128/JB.184.17.4912-4919.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Luo ZQ, Smyth AJ, Gao P, Qin Y, Farrand SK. J Biol Chem. 2003;278:13173. doi: 10.1074/jbc.M210035200. [DOI] [PubMed] [Google Scholar]
- 113.Lamb JR, Patel H, Montminy T, Wagner VE, Iglewski BH. J Bacteriol. 2003;185:7129. doi: 10.1128/JB.185.24.7129-7139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oger P, Kim KS, Sackett RL, Piper KR, Farrand SK. Mol Microbiol. 1998;27:277. doi: 10.1046/j.1365-2958.1998.00671.x. [DOI] [PubMed] [Google Scholar]
- 115.Zhu J, Winans SC. Mol Microbiol. 1998;27:289. doi: 10.1046/j.1365-2958.1998.00672.x. [DOI] [PubMed] [Google Scholar]
- 116.Chai Y, Zhu J, Winans SC. Mol Microbiol. 2001;40:414. doi: 10.1046/j.1365-2958.2001.02385.x. [DOI] [PubMed] [Google Scholar]
- 117.Choi SH, Greenberg EP. Proc Natl Acad Sci U S A. 1991;88:11115. doi: 10.1073/pnas.88.24.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Devine JH, Shadel GS, Baldwin TO. Proc Natl Acad Sci U S A. 1989;86:5688. doi: 10.1073/pnas.86.15.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Egland KA, Greenberg EP. Mol Microbiol. 1999;31:1197. doi: 10.1046/j.1365-2958.1999.01261.x. [DOI] [PubMed] [Google Scholar]
- 120.Whiteley M, Greenberg EP. J Bacteriol. 2001;183:5529. doi: 10.1128/JB.183.19.5529-5534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carlier AL, von Bodman SB. J Bacteriol. 2006;188:4581. doi: 10.1128/JB.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.White CE, Winans SC. Mol Microbiol. 2007;64:245. doi: 10.1111/j.1365-2958.2007.05647.x. [DOI] [PubMed] [Google Scholar]
- 123.Antunes LC, Ferreira RB, Lostroh CP, Greenberg EP. J Bacteriol. 2008;190:4392. doi: 10.1128/JB.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fuqua C, Burbea M, Winans SC. J Bacteriol. 1995;177:1367. doi: 10.1128/jb.177.5.1367-1373.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hwang I, Smyth AJ, Luo ZQ, Farrand SK. Mol Microbiol. 1999;34:282. doi: 10.1046/j.1365-2958.1999.01595.x. [DOI] [PubMed] [Google Scholar]
- 126.Marketon MM, Gonzalez JE. J Bacteriol. 2002;184:3466. doi: 10.1128/JB.184.13.3466-3475.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Danino VE, Wilkinson A, Edwards A, Downie JA. Mol Microbiol. 2003;50:511. doi: 10.1046/j.1365-2958.2003.03699.x. [DOI] [PubMed] [Google Scholar]
- 128.Vannini A, Volpari C, Di Marco S. J Biol Chem. 2004;279:24291. doi: 10.1074/jbc.M401855200. [DOI] [PubMed] [Google Scholar]
- 129.Swiderska A, Berndtson AK, Cha MR, Li L, Beaudoin GM, 3rd, Zhu J, Fuqua C. J Biol Chem. 2001;276:49449. doi: 10.1074/jbc.M107881200. [DOI] [PubMed] [Google Scholar]
- 130.Chen G, Malenkos JW, Cha MR, Fuqua C, Chen L. Mol Microbiol. 2004;52:1641. doi: 10.1111/j.1365-2958.2004.04110.x. [DOI] [PubMed] [Google Scholar]
- 131.Chen G, Wang C, Fuqua C, Zhang LH, Chen L. J Bacteriol. 2006;188:8244. doi: 10.1128/JB.00954-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qin Y, Su S, Farrand SK. J Biol Chem. 2007;282:19979. doi: 10.1074/jbc.M703332200. [DOI] [PubMed] [Google Scholar]
- 133.Chen G. Indiana University; 2007. [Google Scholar]
- 134.Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA. Cell. 1995;82:701. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 135.Liu D, Ishima R, Tong KI, Bagby S, Kokubo T, Muhandiram DR, Kay LE, Nakatani Y, Ikura M. Cell. 1998;94:573. doi: 10.1016/s0092-8674(00)81599-8. [DOI] [PubMed] [Google Scholar]
- 136.Navarro-Aviles G, Jimenez MA, Perez-Marin MC, Gonzalez C, Rico M, Murillo FJ, Elias-Arnanz M, Padmanabhan S. Mol Microbiol. 2007;63:980. doi: 10.1111/j.1365-2958.2006.05567.x. [DOI] [PubMed] [Google Scholar]
- 137.Masuda S, Bauer CE. Cell. 2002;110:613. doi: 10.1016/s0092-8674(02)00876-0. [DOI] [PubMed] [Google Scholar]
- 138.Smith KM, Bu Y, Suga H. Chem Biol. 2003;10:81. doi: 10.1016/s1074-5521(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 139.He X, Chang W, Pierce DL, Seib LO, Wagner J, Fuqua C. J Bacteriol. 2003;185:809. doi: 10.1128/JB.185.3.809-822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]