

Abstract
Bacterial cell division and cell wall synthesis are highly coordinated processes involving multiple proteins. Here, we show that Rv0008c, a novel small membrane protein from Mycobacterium tuberculosis, localizes to the poles and on membranes and shows an overall punctate localization throughout the cell. Furthermore, Rv0008c interacts with two proteins, CrgA and Wag31, implicated in peptidoglycan (PG) synthesis in mycobacteria. Deletion of the Rv0008c homolog in M. smegmatis, MSMEG_0023, caused bulged cell poles, formation of rounded cells, and defects in polar localization of Wag31 and cell wall synthesis, with cell wall synthesis measured by the incorporation of the [14C]N-acetylglucosamine cell wall precursor. The M. smegmatis MSMEG_0023 crgA double mutant strain showed severe defects in growth, viability, cell wall synthesis, cell shape, and the localization of the FtsZ, FtsI, and Wag31 proteins. The double mutant strain also exhibited increased autolytic activity in the presence of detergents. Because CrgA and Wag31 proteins interact with FtsI individually, we believe that regulated cell wall synthesis and cell shape maintenance require the concerted actions of the CrgA, Rv0008c, FtsI, and Wag31 proteins. We propose that, together, CrgA and Rv0008c, renamed CwsA for cell wall synthesis and cell shape protein A, play crucial roles in septal and polar PG synthesis and help coordinate these processes with the FtsZ-ring assembly in mycobacteria.
INTRODUCTION
Since its reemergence in the early 1990s, tuberculosis caused by Mycobacterium tuberculosis remains a leading cause of global morbidity and mortality. With nearly 10 million new cases reported each year and 2 billion people infected, as well as the emergence of extensively drug-resistant and completely drug-resistant strains, sustained interest in improved control measures and tuberculosis research is crucial (4, 40). These reports emphasize the importance of understanding the basic biology of the pathways needed for pathogen proliferation, namely, DNA replication and cell division, and the development of novel strategies for combating M. tuberculosis infection (27).
Cell division in bacteria is accomplished by the coordinated actions of multiple proteins that form a septal ring in the middle of a dividing cell. FtsZ, a cytoskeletal protein and a GTPase, initiates the cytokinesis process by the GTP-dependent midcell assembly of a ring structure called the Z ring (2). Constriction of the Z ring, driven by the GTPase activity of FtsZ, initiates cell division. During various stages of cell division, FtsZ interacts with a number of proteins, and these interactions modulate its midcell localization, membrane tethering, biochemical activities, and influence on the assembly of other septal ring members, including some proteins involved in peptidoglycan (PG) synthesis (reviewed in reference 2). Recent data also suggest that, at least in some bacteria, FtsZ plays a role in guiding cell wall synthesis (1, 37).
In the last decade, significant advancements have been made not only in identifying the players involved in the cell division and cell wall biosynthesis processes in mycobacteria but also in understanding various unique features of these processes. M. tuberculosis FtsZ (FtsZTB) exhibits slow polymerization and weak GTPase activities and is essential for viability (12, 31, 39). FtsZTB interacts with the FtsW, FipA, and CrgA proteins (8, 29, 33). Increased levels of ChiZ and ClpX affect FtsZ assembly and possibly its cell division potential (5, 13). FtsZTB assembly and cell division are also modulated during growth of M. tuberculosis in macrophages (6). Furthermore, M. tuberculosis FtsI interacts with FtsW; FtsZ, FtsW, and FtsI may form a 3-protein complex (9). FtsZTB interacts with and is phosphorylated in vitro by an essential serine/threonine protein kinase, PknA (34). Phosphorylation impairs the GTP-dependent polymerization activity of FtsZTB, which suggests that phosphorylation regulates cell division. More recently, Aldridge et al. (3) showed that cell division in mycobacteria is asymmetric due to unipolar growth and produces daughter cells that differ in growth rate and size.
Limited information on the protein factors involved in PG synthesis in mycobacteria is also available. RipA, an essential cell wall hydrolase and a target of the MtrA response regulator (28), interacts with RpfB, a lytic transglycosylase and one of the five resuscitation-promoting factors in M. tuberculosis. The RipA-RpfB-mediated synergistic hydrolysis of PG is modulated by PBP 1 (16, 17). Wag31, a coiled-coil protein and a homolog of cell shape/cell division protein DivIVA, is essential for mycobacterial viability as well as for septal and polar PG synthesis and localizes at poles and septa (20, 25). Wag31 is phosphorylated by PknA, and the phosphorylated form is competent for polymerization and localization to sites of PG synthesis (19, 20). Our recent studies showed that CrgATB is a FtsZ-interacting protein that localizes at septa, at poles, and on membranes and that it plays an important role in septal PG synthesis and cell shape morphogenesis (29). These studies also revealed that CrgATB interacts with FtsI and FtsQ and that the loss of CrgA results in a severe reduction in the septal and polar localization of FtsI, indicating that CrgA is important for cell wall PG synthesis (29). Here, we report the characterization of another small membrane protein encoded by Rv0008c. This protein was found to interact with CrgA and Wag31, two proteins important for cell wall PG synthesis; hence, the Rv0008c-encoded protein is referred to here as CwsA. Our characterization of cwsA single and cwsA crgA double mutant strains revealed that CwsA, along with CrgA and Wag31, is required for regulated cell division, cell wall synthesis, and the maintenance of cell shape.
MATERIALS AND METHODS
Growth of bacterial strains.
Escherichia coli Top10F′ strain was used for cloning purposes, and recombinant strains were propagated in Luria-Bertani (LB) broth or LB agar containing one or more of the following antibiotics: 50 μg/ml kanamycin (Km), 200 μg/ml hygromycin (Hyg), and 100 μg/ml ampicillin (Amp) (29). M. smegmatis mC2155 and M. tuberculosis H37Rv strains were grown in Middlebrook 7H9 broth containing 0.01% Tween 80 and supplemented with albumin-dextrose and with oleic acid, albumin, dextrose, and sodium chloride, respectively (29). Recombinants were selected on 7H10 agar supplemented with 10 μg/ml kanamycin or 50 μg/ml hygromycin. As needed, gentamicin and zeocin were added at 10 μg/ml. Induction of Pami and Ptet promoters was with 0.02 to 0.2% acetamide and 10 to 100 ng/ml anhydrotetracycline, respectively. Plate and broth cultures were grown at 37°C, and growth of broth cultures was monitored by measuring the absorbance at 600 nm. M. smegmatis Δwag31 Pami::wag31 was maintained in medium containing 0.2% acetamide (20).
Molecular cloning.
Various DNA fragments were generated by PCR with Phusion or Deep Vent DNA polymerase (NE Biolabs) and cloned into appropriate vectors following standard molecular cloning protocols. The cloning vectors used and the recombinant plasmids generated are listed in Table 1, whereas the oligonucleotide primers used for PCR are listed in Table S1 in the supplemental material. The cwsA (Rv0008c) gene was cloned into the pTBSG vector with an N-terminal tobacco etch virus (TEV) protease-cleavable histidine tag (30), and the crgA (Rv0011c) gene was cloned into pET29b with a C-terminal uncleavable 6× histidine tag (22) or into pTBMalE with an N-terminal maltose binding protein (MBP) fusion (30). For controls, the coding region of the M2 protein from influenza A virus was cloned into the pTBSG vector with the N-terminal TEV protease-cleavable histidine tag. For expression in mycobacteria, the cwsA-, crgA-, and wag31-coding regions were cloned under the inducible amidase (Pami) or tetracycline (Ptet) promoter. The wag31-mCherry fusion was expressed from Pami and contained a sequence encoding the STNNNLQ linker between the two coding regions. Construction of ecfp-crgA was described previously (29). The gfp-cwsA fusion construct was created by placing the gfp-coding region upstream of cwsA and a linker encoding NNNLQGS. For bacterial two-hybrid (BACTH) constructs, the coding regions of the various genes were cloned in the BACTH vectors as in-frame fusions to Bordetella pertussis adenylate cyclase (Table 1) (21). All cloned fragments were confirmed by sequencing.
Table 1.
Strains and plasmids used
| Strain or plasmid | Source or description | Reference or source |
|---|---|---|
| E. coli Top10F′ | Invitrogen Inc. | |
| M. tuberculosis H37Rv | Lab stock | |
| M. smegmatis | ||
| mC2155 | Lab stock | |
| ΔcrgA | ΔcrgA Gentr | 29 |
| Δwag31 | Δwag31 Hygr | 20 |
| ΔcwsA | ΔcwsA Gentr | This study |
| DKO | ΔcrgA ΔcwsA Gentr Zeor | This study |
| Plasmid | ||
| pMV306 | Integration proficient E. coli-Mycobacterium shuttle vector, Kmr | 32 |
| pLR56 | E. coli-Mycobacterium shuttle vector, integrating, with tet promoter and tet repressor, Hygr | 13 |
| pLR52 | E. coli-Mycobacterium shuttle vector, replicating, with tet promoter and tet repressor, Hygr | 13 |
| pMG101 | E. coli-Mycobacterium shuttle vector, integrating, with amidase promoter, Hygr | This study |
| pMG103 | E. coli-Mycobacterium shuttle vector, integrating, with amidase promoter, Kmr | 13 |
| pGOAL17 | Carrying 6.1-kb PacI cassette, Kmr | 26 |
| pET-19b | E. coli expression vector allowing fusions to N-terminal 6× histidine tag (10× His), Ampr | Novagen |
| pET29b | E. coli expression vector allowing fusions to C-terminal 6× histidine tag (10× His), Kmr | |
| pTBSG | E. coli expression vector allowing fusions to N-terminal 6× histidine tag (6× His), Ampr | 30 |
| pTBMalE | E. coli expression vector allowing fusions to N-terminal maltose binding protein, Ampr | 30 |
| pKT25 | E. coli expression vector allowing fusions to C terminus of the T25 fragment of cyaA, Kmr | 21 |
| pUT18C | E. coli expression vector allowing fusions to C terminus of the T18 fragment of cyaA, Ampr | 21 |
| pKNT25 | E. coli expression vector allowing fusions to N terminus of the T25 fragment of cyaA, Kmr | 21 |
| pUT18 | E. coli expression vector allowing fusions to N terminus of the T18 fragment of cyaA, Ampr | 21 |
| pTBSG-8c | M. tuberculosis cwsA cloned in pTBSG, Ampr | 30 |
| pET29b-11c | M. tuberculosis crgA cloned in pET29b, Kmr | 22 |
| pTBMalE-11c | M. tuberculosis crgA cloned in pTBMalE, Ampr | 30 |
| pTBSG-M2 | Tobacco etch virus M2-coding region in pTBSG, Ampr | 30 |
| pPP7 | Pami::gfp-cwsA in pJFR19, Hygr | This study |
| pNM9 | CwsAsol (aa 1–98)-coding region in pLR52, Hygr | This study |
| pNM10 | PcwsA::cwsAsmeg in pMV306K, Kmr | This study |
| pPP17 | Pami::gfp-cwsAsol in pMG103, Hygr | This study |
| pPP30 | crgATB-cfp cloned in pLR52 vector, Hygr | 29 |
| pPP48 | crgATB cloned in pUT18C vector, Ampr | 29 |
| pPP53 | crgATB in pKT25 vector, Kmr | 29 |
| pPP46 | cwsATB cloned in pUT18C vector, Ampr | This study |
| pPP47 | cwsATB in pKT25 vector, Kmr | This study |
| pPP64 | cwsAsol in pUT18, Ampr | This study |
| pPP65 | cwsAsol in pKNT25 vector, Kmr | This study |
| pPP79 | Pami::gfp-ftsI in pMG103 | 29 |
| pRD65 | wag31 in pUT18 vector, Ampr | This study |
| pRD57 | wag31 in pKNT25 vector, Kmr | This study |
| pKS34 | Coding region for Wag31 C-terminal aa 61–260 cloned in pUT18, Ampr | This study |
| pKS35 | Coding region for Wag31 C-terminal aa 61–260 cloned in pKNT25, Kmr | This study |
| pEB6 | wag31-mCherry fusion cloned in pMG103, Kmr | This study |
| pEB16 | wag31-mCherry fusion cloned in pMG101, Hygr | This study |
| pJFR79 | ftsZsmeg-gfp in pJAM2, Kmr | 31 |
| pPP105a | MSMEG_0023 (cwsAsmeg) with flanking sequences (417 bp upstream and 600 bp downstream) cloned in PstI-NdeI sites of pGEM-T Easy, Ampr | This study |
| pPP105 | Blunt-ended gentamicin resistance cassette inserted in HincII site (37 bp from start codon) of cwsA-coding region in pPP105a, Ampr Gentr | This study |
| pPP106 | 6.1-kb PacI cassette from pGOAL17 inserted in pPP105, Ampr Gentr Sucr lacZ | This study |
| pPP115 | Blunt-ended zeocin resistance cassette inserted in HincII site (37 bp from start codon) of cwsA-coding region in pPP105a, Ampr Zeor | This study |
| pPP116 | 6.1-kb PacI cassette from pGOAL17 inserted in pPP115, Ampr Gentr Sucr lacZ | This study |
Construction of M. smegmatis cwsA knockout and cwsA crgA DKO strains.
M. smegmatis cwsA knockout and cwsA crgA double-knockout (DKO) strains were created following the two-step recombination protocol (26). To create an M. smegmatis cwsA mutant, the MSMEG_0023-coding region was cloned in the PstI and NdeI sites of the pGEM-T Easy vector, generating pPP105a (Table 1). Next, a gentamicin resistance cassette was inserted at a HincII site 37 nucleotides (nt) downstream of the start codon to create plasmid pPP105 (Table 1). Finally, a 6.1-kb PacI cassette carrying the lacZ, aph, and sacB genes was inserted next to the cwsA-coding region, creating the suicide recombination plasmid pPP106 (Table 1). The pPP106 plasmid was used to create the cwsA mutant by homologous recombination, and the mutant strain was confirmed by Southern hybridization. The cwsA crgA double mutant was created in the crgA mutant background (29) by carrying out homologous recombination protocols with the pPP116 suicidal plasmid (Table 1) that is similar to pPP106, except that it has the cwsA-coding region interrupted by a zeocin resistance cassette. The cwsA crgA double mutant strain was confirmed by PCR and quantitative real-time PCR.
Microscopy.
M. smegmatis and M. tuberculosis cultures were diluted to an A600 of 0.1 and grown for the various indicated time points in the absence or presence of an appropriate inducer (0.02 or 0.2% acetamide or 50 ng/ml anhydrotetracycline). Samples were collected, resuspended in phosphate-buffered saline, and visualized by microscopy. M. tuberculosis cells were always fixed in 4% paraformaldehyde prior to visualization. In some experiments, M. smegmatis cultures were grown to an A600 of ≥2.0 and examined by microscopy. Other strain-specific growth conditions were as detailed in the respective figure legends. For staining of nascent PG, M. smegmatis strains were grown with 1.1 μg/ml vancomycin (BODIPY fluoresceinated vancomycin [Van-FL]-vancomycin, 0.1:1) for 2 h and imaged as described previously (5). A Nikon Eclipse 600 microscope fitted with a ×100 Nikon Plan Fluor oil-immersion objective with a numerical aperture of 1.4 and appropriate filter sets (Chroma) was used for bright-field and fluorescence microscopy. Images were acquired using a Photometrics Coolsnap ES camera and Metamorph (version 6.2) imaging software (Universal Imaging Corporation). All fluorescent images for the various sample sets compared were acquired at identical exposure times and optimized to similar extents with respect to brightness and contrast using Adobe Photoshop CS4. Multiple independent samples were imaged, and data from 2 to 3 independent experiments were used for statistical analysis. At least 100 cells were scored from each experiment. We defined bulgy cells as those with bulged regions (visibly increased cell width) either at the poles or along the cell length.
Protein-protein interaction assays. (i) BACTH assay.
A BACTH system kit from Euromedex was used to investigate the interactions of CwsA with CrgA, Wag31, and other cell division proteins as described previously (13, 29). E. coli BTH101 recombinants containing various combinations of BACTH constructs are listed in Table 1. Initial experiments were carried out on LB agar plates containing kanamycin, ampicillin, 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and confirmed by broth assays as described previously (29). β-Galactosidase activity 5-fold higher than that of the BTH101 control, which carried a single copy of the gene and an empty vector, was considered indicative of an interaction. E. coli BTH101 transformants obtained with pKT25-GCN4 and pUT18C-GCN4 served as positive controls for complementation (Table 1).
(ii) Pulldown assay.
CwsA-CrgA interaction was examined by pulldown assay as described previously (29). E. coli BL21(DE3) cells were used to overproduce His-CrgA or His-CwsA fusion proteins (Table 1). For pulldown assays, the supernatant of solubilized His-CrgA or His-CwsA cell lysate in 40 mM Tris-HCl, pH 8.0, 300 mM NaCl with 3% Empigen detergent (buffer A) was incubated with Ni-nitrilotriacetic acid (NTA) (Qiagen) resin at 4°C. Unbound proteins were removed by washing the beads 3 times with buffer A plus 20 mM imidazole. The beads were equilibrated with a buffer containing 20 mM Tris HCl, pH 8.0, 50 mM NaCl, 0.25% Empigen, and 10% glycerol (buffer B), tag-free CwsA or tag-free CrgA was added to beads, and complexes were allowed to form for 2 h at room temperature. The beads were then washed with buffer B plus 5 mM imidazole, followed by buffer B without imidazole. The His-CrgA-CwsA and His-CwsA-CrgA complexes were eluted with buffer B plus 250 mM imidazole. Eluted proteins were separated in an SDS-polyacrylamide gel and visualized following staining with EZ-blue stain (Sigma-Aldrich). For negative controls, His-CwsA/tag-free M2 pulldown was performed. M2 protein is an acid-activated proton-selective-channel 12-kDa protein from influenza A virus. Expression and purification of M2 were similar to those of CwsA, except that the detergent octyl-glucopyranoside was used during purification of M2. The His-CwsA and tag-free M2 protein pulldown assay was similar to the His-CwsA-CrgA assay.
Immunoblotting.
Wag31 protein was detected in M. smegmatis strains by immunoblotting using protein-specific antibodies as previously described (29). For the loading control, anti-SigA was used to probe parallel blots. Immunoblots were processed using an ECF Western blotting kit (GE Life Sciences, Piscataway, NJ) and scanned using a Bio-Rad molecular imager (FX). As needed, protein levels were determined using the volume analysis function of QuantityOne software.
RNA isolation and quantitative real-time PCR (qRT-PCR).
Total RNA was isolated from actively growing cultures of wild-type (WT), ΔcrgA, ΔcwsA, and DKO strains of M. smegmatis following protocols described previously (14). RNA was reverse transcribed using random hexamers (Invitrogen) and Moloney murine leukemia virus reverse transcriptase (Promega), and the cDNA thus obtained was PCR amplified using gene specific primers (see Table S1 in the supplemental material) in a Bio-Rad iCycler iQ5 real-time PCR detection system and 2× iQ SYBR supermix. The threshold cycle (CT) value of cwsA was normalized to the CT value for sigA, and fold expression was calculated using the formula 2−Δ(ΔCT). Data presented were obtained from 2 independent RNA preparations reverse transcribed and quantified by real-time PCR in triplicate.
Incorporation of 14C-labeled GlcNAc into cell walls.
In vivo labeling of cell walls with 14C labeled N-acetylglucosamine was done essentially as described previously (15). Overnight cultures of M. smegmatis strains grown in GAG-GAS medium (15) were diluted to an A600 of 0.1 and grown to an A600 of 0.5. The actively growing cultures were diluted 5-fold with prewarmed GAG-GAS medium, and a 5-ml culture was set up for each strain. To each culture, 0.25 μl (final concentration, 0.97 μM) of 14C-labeled N-acetyl-d-glucosamine (GlcNAc; GE Life Sciences) plus 100 μM cold GlcNAc (Sigma-Aldrich) was added, and cultures were incubated at 37°C with shaking. A parallel culture without radioactive label was grown as a control. At the indicated times after addition of GlcNAc, 0.5 ml of culture was removed and filtered through a 0.2-μm-pore-size nitrocellulose filter. The filters were washed with 0.5 ml of GAG-GAS medium and air dried, and radioactivity was measured in a liquid scintillation counter. Aliquots from cold cultures for the corresponding time points were used for measuring the A600. The data were normalized to growth.
Measurement of cell wall turnover in WT and mutant M. smegmatis strains.
Following incorporation of 14C-labeled GlcNAc for 6 h (see above), cells were collected by centrifugation, washed twice with prewarmed 7H9 broth, and finally resuspended in 7H9 broth containing 100 μM cold GlcNAc. The cells were then grown with shaking at 37°C, and 0.5 ml of sample was drawn at the various indicated time points. Cells were pelleted and resuspended in 4.0% SDS and boiled for 30 min, and the boiled extract was passed through a 0.22-μm-pore-size filter. The filters were washed once with 3 ml of 0.1 M LiCl and 2 times with 3 ml of water. The filters were air dried and transferred to vials containing scintillation cocktail. The retained radioactivity was measured in a liquid scintillation counter, and the PG turnover at each time point was calculated with respect to that at time zero. Data were collected from three independent experiments, and turnover rates were analyzed by paired Student's t test and one-way analysis of variance (ANOVA), followed by Tukey's multiple-comparison test.
Autolysis assay.
Exponential cultures of M. smegmatis WT and various mutant strains were pelleted by centrifugation and washed once with a solution of 50 mM glycine, pH 8.0, and 0.05% Tween 20. The cell pellet was resuspended in the same buffer to an A600 of 1.0 and incubated at 37°C with shaking. At various indicated time points, the A600 was measured and the data were plotted in the Excel program. The autolysis rate was defined as the time required to achieve a 50% decrease in the A600 (10). Data from three independent experiments were used to calculate autolysis rates and were analyzed using two-tailed paired Student's t test.
Cell wall hydrolysis assays.
Preparations of crude cell walls containing mycolyl arabinogalactan (mAGP) from WT and various mutant M. smegmatis strains were labeled with fluorescein as described previously (5). Known amounts of labeled cell walls were incubated in a buffer containing 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 1 mg/ml lysozyme for 4 h. At the end of incubation, reactions were stopped by adding an equal volume of 4 M lithium chloride, the reaction mixtures were centrifuged, and the fluorescent label released into the supernatant was measured in a Jasco FP6500 spectrofluorimeter as described previously (5). Percent hydrolysis was calculated relative to the input fluorescence counts. Assays were done in triplicate, and average percent hydrolysis was plotted.
RESULTS
CwsA interacts with CrgA.
Our preliminary structural characterization studies of small membrane proteins from M. tuberculosis revealed the possible interaction of CrgATB with another small membrane protein, CwsA. We expanded these initial observations and confirmed with pulldown assays using Ni-NTA affinity chromatography that the recombinant His-CrgA protein was able to pull down tag-free CwsA protein (Fig. 1A, top). In a reverse reaction, tag-free CrgA (or MBP-CrgA) coeluted with His-CwsA (Fig. 1A, middle). In a control assay, His-CwsA did not pull down tag-free M2 protein, a 12-kDa protein from influenza A virus (Fig. 1A, bottom). For further confirmation of CrgA and CwsA interactions, we performed bacterial two-hybrid (BACTH) assays (21) and found that the cwsA and crgA gene products produced as C-terminal fusions to the T25 and T18 domains of adenylate cyclase interacted (Fig. 1B).
Fig 1.
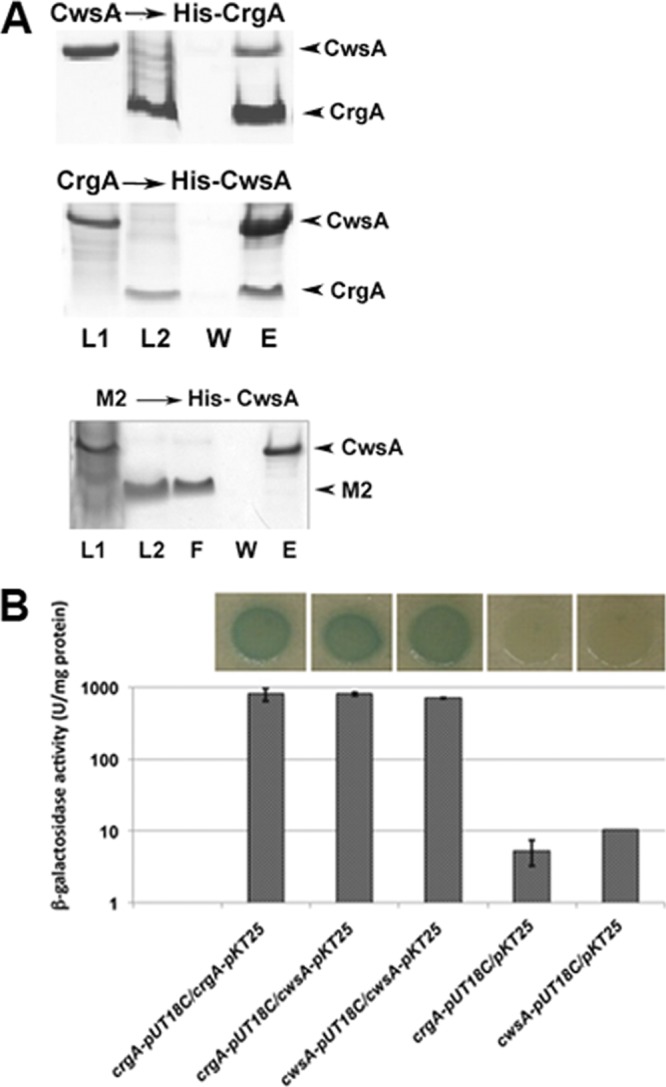
CwsATB interacts with CrgATB. (A) Interaction of CwsA with CrgA by pulldown assay. His-CrgA (top) or His-CwsA (middle) cell lysates were incubated with Ni-NTA (Qiagen) resin at 4°C. Unbound proteins were washed off, the beads were equilibrated with a tag-free CwsA or tag-free CrgA, and complexes were allowed to form for 2 h at room temperature. The His-CrgA-CwsA and His-CwsA-CrgA complexes were eluted, separated in SDS-polyacrylamide gels, and visualized by staining with EZ-blue stain. As a control, His-CwsA bound to Ni-NTA beads was also incubated with influenza A virus M2 protein (bottom) and processed as described above. L1, CwsA lysate; L2, CrgA lysate (middle) or M2 lysate (bottom); F, flow-through fraction; W, wash fraction; E, eluted fraction. (B) Interaction of CwsA with CrgA by BACTH. CrgA and CwsA fusions to T25 and T18 fragments of adenylate cyclase in the BACTH vectors (Table 1) were used to transform E. coli BTH101 and recombinants plated on LB agar supplemented with X-Gal and IPTG. Green-blue colonies, indicative of strong interactions, were subsequently propagated in LB broth (top), and β-galactosidase activity was measured as described in the text (bottom). Values shown are means ± standard deviations from at least 3 independent experiments.
CwsA localizes to poles and midcell sites.
The interactions between CwsA and CrgA suggest a role in cell division/cell wall synthesis. To investigate this issue further, we examined the localization of green fluorescent protein (GFP)-CwsA protein in M. smegmatis. The GFP-CwsA protein fusion showed distinct membrane and polar localization in actively growing M. smegmatis cells (Fig. 2ii and iv). In dividing cells with distinct constriction, midcell and polar localization was visible (Fig. 2ii and iv, arrowheads). Additionally, an all-over-the-cell punctate localization was observed (Fig. 2ii and iv). The GFP-CwsA localization patterns observed in M. tuberculosis were also similar to those observed in M. smegmatis, except that the fluorescent structures were less distinct and the punctate localization patterns were absent (see Fig. S1 in the supplemental material). Presumably, the diffuse and weak fluorescence was due to paraformaldehyde fixation, a safety step used prior to visualization of structures in M. tuberculosis; paraformaldehyde fixation is known to quench the fluorescence intensity of some cell division proteins (28, 29). We also examined GFP-CwsA localization in the M. smegmatis crgA mutant background. Loss of crgA had only a modest effect on GFP-CwsA localization (Fig. 2v and vi). Although punctate localization was less distinct, dispersed fluorescent foci, polar localization, and sharp membrane-bound signals were evident in the mutant cells.
Fig 2.

Cellular localization of CwsA. Exponential cultures of M. smegmatis Pami::gfp-cwsA (i to iv) and M. smegmatis ΔcrgA Pami::gfp-cwsA (v, vi) were induced with 0.2% acetamide for 5 h and visualized by bright-field (i, iii, and v) and fluorescence (ii, iv, and vi) microscopy. Images in panels iii and iv were slightly enlarged to show details of GFP-CwsA localization. White arrows, punctate localization; arrowheads, polar (P) or new pole (midcell) localization.
cwsA deletion leads to defects in cell shape and polar PG synthesis.
CwsATB is a 15.6-kDa membrane protein with a 98-amino-acid (aa) N-terminal soluble end, a 22-aa transmembrane domain, and a short 21-aa extracytoplasmic tail. The CwsA protein homologs in mycobacterial members share a significant degree of homology (see Fig. S2 in the supplemental material). The CwsA proteins of M. tuberculosis and M. smegmatis are ∼80% similar. Therefore, to obtain further insights into the role(s) of CwsA, we created a cwsA mutant (ΔcwsA) strain of M. smegmatis by inserting a gentamicin resistance cassette 37 bp downstream of the cwsA start codon by homologous recombination (Fig. 3A; Table 1). The mutant strain was confirmed by Southern hybridization and qRT-PCR (Fig. 3A, inset, and B). qRT-PCR revealed a complete absence of cwsA expression in the ΔcwsA strain (Fig. 3B). The ΔcwsA strain showed a modest growth defect (Fig. 3C) but exhibited distinct defects in cell shape (Fig. 3D). Nearly 43% of the ΔcwsA cells showed a bulgy phenotype. In contrast, a small proportion of WT cells showed a modestly swollen cell morphology (Fig. 3D; compare panels i and ii). The proportion of bulged cells in the ΔcwsA strain remained nearly unchanged during stationary-phase growth, although a small increase was noted for WT cells (see Fig. S3 in the supplemental material). Unlike the ΔcrgA strain (29), the ΔcwsA strain showed only a modest 2-fold increase in chain-linked cells (data not shown). Expression of either cwsA from its own promoter (PcwsA::cwsAsmeg) or gfp-cwsATB from a heterologous amidase promoter (Pami::gfp-cwsATB) restored the cell shape to the ΔcwsA cells (see Fig. S4 in the supplemental material). These results established that CwsATB can replace the function of CwsAsmeg and, importantly, that the GFP-CwsATB fusion protein is fully functional (see Fig. S4iv and v in the supplemental material).
Fig 3.

Loss of CwsA affects growth and cell shape. (A) Cartoon of 2. 7-kb cwsA (msmeg0023) region of M. smegmatis (top) with an inserted copy of a gentamicin resistance cassette (bottom) (Table 1, pPP105). Black arrow, cwsA-coding region; gray arrows, coding regions of neighboring genes; white bars, intergenic regions; black bar, probe region. Genomic DNA was isolated from WT, two single-crossover colonies (lanes SCO), and one double-crossover colony (DCO), digested with EcoRI, separated by agarose gel electrophoresis, and transferred to a nitrocellulose membrane. The blot was probed with a 32P-labeled MSMEG_0023 fragment probe. The positions of expected bands in WT (2.7 kb) and double-crossover (3.55 kb) lanes are marked. The single-crossover colonies gave a band corresponding to the WT copy (2.7 kb) and another band of ∼2.3 kb (arrowhead) due to insertion of the recombination vector pPP106. (B) qRT-PCR for mutant strains. RNA was isolated from M. smegmatis WT, ΔcwsA, and DKO strains as described in the text. Quantitative real-time PCR was performed for cwsA expression levels, and data were normalized to those for housekeeping gene sigA expression (see Materials and Methods). cwsA expression levels in the two mutant strains are presented with respect to that for WT (for which the level is equal to 1). Mean expression levels ± standard deviations from two independent experiments done in triplicate are shown. (C) Growth curve for cwsA and crgA strains. Growth of M. smegmatis mC2155 (WT), ΔcrgA, ΔcwsA, and DKO strains was monitored for the indicated time points. Briefly, overnight cultures of various strains were diluted to an A600 of 0.1 and grown in 7H9 broth with albumin-dextrose, the A600 was recorded at the indicated time points, and the data were plotted. The experiment was carried out 3 times, and one representative data set is shown. (Inset) Serial dilutions of exponential cultures of indicated strains were prepared, and 5 μl was spotted on 7H10 agar medium plates and grown at 37°C. (D) Morphology of M. smegmatis and various strains. Exponential cultures of WT (i), ΔcwsA (ii), and ΔcwsA Pami::cwsAsol (iii) strains of M. smegmatis were grown in 7H9 broth with albumin-dextrose and imaged by bright-field microscopy. Arrowheads, bulgy cells. Numbers below each panel indicate percent average number of bulgy cells ± standard error from two independent experiments. Each experiment included ≥250 cells from each of the three strains.
Cell wall synthesis occurs at cell poles and midcell sites in mycobacteria, and perturbations in PG synthesis lead to a swollen tip and bulged cell morphology (reviewed in reference 29). This suggests that the cwsA gene product may be involved in cell wall synthesis. To investigate this issue, we stained the WT and ΔcwsA cells with Van-FL to label the sites of nascent PG synthesis. Consistent with our published data, Van-FL staining was detected at the poles and septal sites (Fig. 4A). Distinct midcell and bipolar staining was also evident in constricting cells (Fig. 4Aii, arrow) (5). We also found that the staining was asymmetric, with preferential staining of one pole over the other, as recently reported (3). There was an ∼50% reduction in Van-FL staining in ΔcwsA cells (Fig. 4A, compare panel ii with panel iv). We also found that the bulged regions of the ΔcwsA cells showed faint or no Van-FL staining (Fig. 4Aiv). Since old-growth poles stain preferentially with Van-FL (3), it is likely that the bulged regions in ΔcwsA represent defective growth poles. Together, these results suggest that CwsA, like the Wag31 (20, 25) and CrgA (29) proteins, is important for cell wall synthesis.
Fig 4.
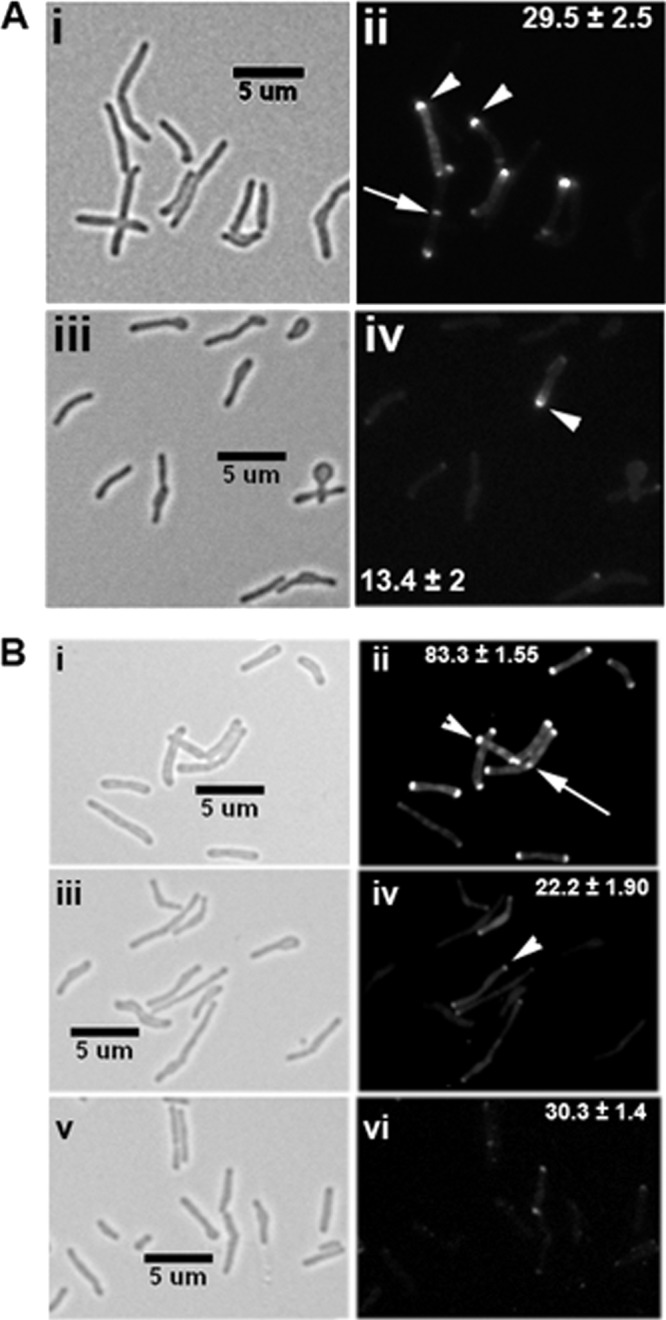
The cwsA mutant is defective for cell wall synthesis and Wag31 localization. (A) Exponential cultures of M. smegmatis WT (i, ii) and ΔcwsA (iii, iv) strains were grown in the presence of Van-FL for 2 h and imaged by bright-field (i, iii) and fluorescence (ii, iv) microscopy. Arrowheads, polar staining; arrow, midcell staining. Polar staining patterns from three independent experiments were scored, and average percentages ± standard errors are given in the respective fluorescent panel for clarity. Each experiment included ≥170 cells from each strain. Midcell staining, 4.14% ± 0.77% for WT and 1.71% ± 0.47% for the ΔcwsA strain. (B) Wag31-mCherry fusion localization in M. smegmatis WT (i, ii), ΔcwsA (iii, iv), and ΔcrgA (v, vi) strains. Exponential cultures of respective recombinant strains expressing Pami::wag31-mCherry were grown in the presence of 0.2% acetamide for 2 h and imaged by bright-field (i, iii, v) and fluorescence (ii, iv, vi) microscopy. Arrow, midcell localization; arrowheads, polar localization. Percent polar localizations from two independent experiments were scored, and averages ± standard errors are given in the respective fluorescent panel. Each experiment included ≥150 cells from each strain. Midcell localization, 7.7% ± 0.9% for WT, 2.6% ± 0.86% for the ΔcwsA strain, and <1% for the ΔcrgA strain.
cwsA deletion compromises Wag31 polar and septal assembly.
Because CwsA interacts with CrgA, we examined the effect of CwsA absence on CrgA localization in cells expressing Ptet::ecfp-crgA by bright-field and fluorescence microscopy, as reported previously (29). A modest 7% increase in the polar localization of the enhanced cyan fluorescent protein (ECFP)-CrgA fusion was noted in the ΔcwsA strain (see Fig. S5 in the supplemental material). Next, we visualized the localization of the Wag31-mCherry fusion protein in the ΔcwsA mutant background. We first confirmed that the Wag31-mCherry fusion protein is functional and served as the sole source for Wag31 in a Δwag31 mutant strain (see Fig. S6 in the supplemental material). Next, consistent with published reports, we found that Wag31-mCherry, like Wag31-GFP or GFP-Wag31, localized at poles and midcell sites (Fig. 4Bii) (20, 25). Interestingly, we found a sharp reduction in the number of ΔcwsA cells showing Wag31-mCherry localization and in the signal intensity of Wag31-mCherry in the mutant cells (Fig. 4B; compare panel ii with panel iv). Immunoblot analyses indicated that Wag31 levels were the same in the ΔcwsA strain as in the WT strain (see Fig. S7 in the supplemental material). We also found that Wag31-mCherry localization was compromised in the ΔcrgA mutant (Fig. 4B; compare panels ii and vi). Together, these data suggest that loss of CwsA modestly increases CrgA localization and that both proteins are required for optimal Wag31 localization.
cwsA interacts with Wag31.
Next, we tested interactions between the Wag31 and CwsA proteins by BACTH assays. Strong interactions of CwsA with Wag31 were noted (Fig. 5); however, no interactions of CwsA with FtsZ, FtsQ, FtsI, and FtsW were noted (see Fig. S8 in the supplemental material). We also did not observe interaction between Wag31 and CrgA (see Fig. S8 in the supplemental material). Together, the localization and interaction data suggest that Wag31 and CwsA interact at the poles and septal sites, the sites of potential PG synthesis. Wag31TB is a soluble protein that contains two coiled-coil domains, present at aa 24 to 66 and aa 123 to 226. The N-terminal amino acids 46NSD48 have been shown to interact with FtsI, and this interaction is reported to protect FtsI from proteolytic cleavage (24). The interactions between Wag31 and CwsA suggest that the soluble N-terminal region of CwsA (CwsAsol) would interact with Wag31. Indeed, BACTH assays showed robust interactions of CwsAsol with Wag31 (Table 1; Fig. 5). Furthermore, the C-terminal 200 aa of Wag31 encompassing coiled-coil domain 2 was sufficient for interaction with CwsA (see Fig. S8 in the supplemental material). Next, we addressed whether CwsAsol is functional in vivo by expressing Ptet::cwsAsol in the presence of 50 ng/ml anhydrotetracycline in the ΔcwsA strain and examined the strain by bright-field microscopy. We found a partial reversal of the bulged cell morphology defect (Fig. 3Diii). We also found that CwsAsol partially restored the Wag31-mCherry localization defect associated with the ΔcwsA strain (Fig. 6A; compare panels ii and iv). Because the phenotype reversal is not complete, it is likely that both membrane and extracellular regions of CwsA are necessary for optimal Wag31 localization. The GFP-CwsAsol fusion protein showed polar and throughout-the-cell punctate localization and, as expected, lacked membrane localization in the WT background (Fig. 6Bii).
Fig 5.
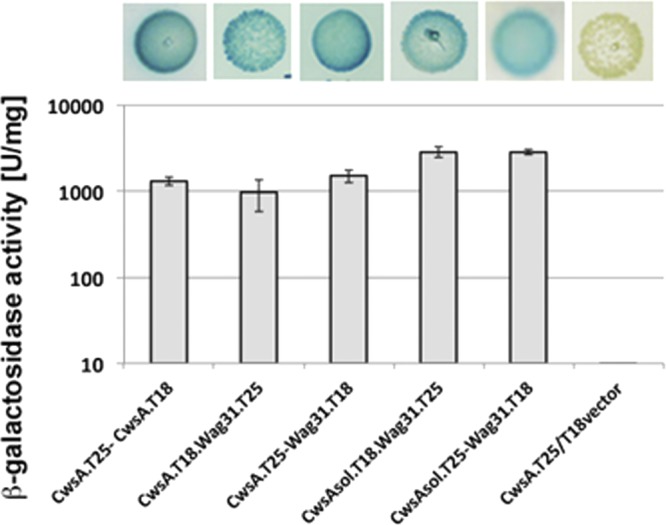
CwsA interacts with Wag31. CwsA, CwsAsol, and Wag31 fusions to T25 and T18 fragments of adenylate cyclase in the BACTH vectors (Table 1) were used in various combinations to transform E. coli BTH101 and recombinants plated on LB agar supplemented with X-Gal and IPTG. Green-blue colonies were subsequently propagated in LB broth (top), and β-galactosidase activity was measured as described in the text (bottom). Values shown are means ± standard deviations from at least 3 independent experiments.
Fig 6.
CwsAsol does not fully complement the defect in the ΔcwsA strain. (A) Exponential cultures of M. smegmatis WT (i, ii) and the ΔcwsA strain complemented with cwsAsol (iii, iv) expressing Pami::wag31-mCherry were induced with 0.2% acetamide for 2 h and imaged by bright-field (i, iii) and fluorescent (ii, iv) microscopy. Arrows, polar localization of Wag31-mCherry. Percent polar localizations from two independent experiments were scored, and averages ± standard errors are given in the respective fluorescent panel. Each experiment included ≥100 cells from each strain. (B) GFP-CwsAsol localization. The M. smegmatis Pami::gfp-cwsAsol strain was grown with acetamide for 5 h and imaged as described in the legend to Fig. 2. (i) Bright-field image; (ii) fluorescent image. Note the polar (arrow) and punctate (arrowhead) patterns of GFP-CwsAsol and absence of membrane localization in panel ii (compare with Fig. 2).
A crgA-cwsA double-deletion mutant shows severe growth, morphology, and cell division defects.
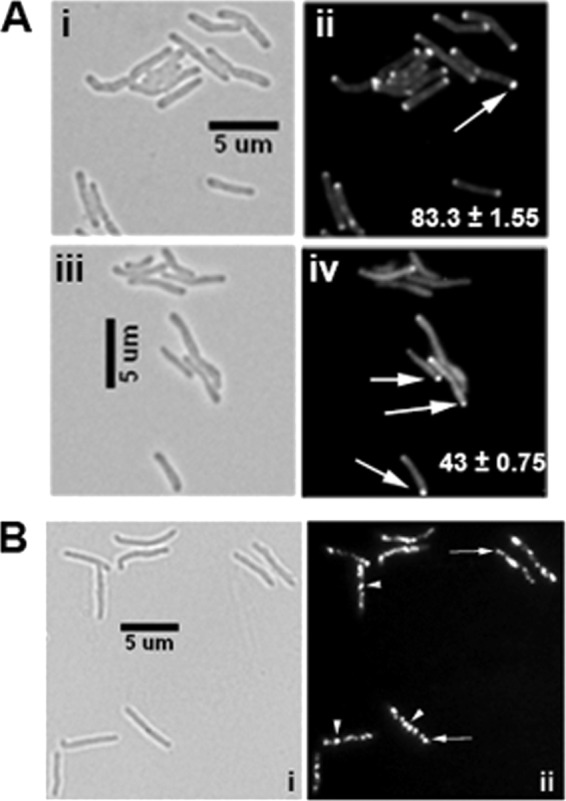
The interactions of CwsA with CrgA and Wag31 (this study) and interactions of CrgA with FtsZ and FtsI (29) suggest an intimate involvement of CwsA and CrgA in cell division and cell wall synthesis in mycobacteria. To gain further insights into the roles of the two proteins, we created a crgA cwsA DKO strain by disrupting the cwsA-coding region in the crgA mutant background (29). A zeocin resistance cassette was inserted 37 nt downstream of the cwsA start codon (Table 1), and the cwsA disruption in the DKO strain was confirmed by PCR (see Fig. S9 in the supplemental material). Furthermore, qRT-PCR showed the absence of cwsA expression in the DKO strain (Fig. 3B). The DKO strain showed severe growth and cell shape defects distinct from the defects observed in either of the single-deletion strains (Fig. 3C and 7A; in Fig. 7A, compare the ΔcrgA strain and DKO panels with WT and with ΔcwsA in Fig. 3D). Two-thirds of the double mutant cells either were bulged or showed abnormal cell widths and/or cell lengths (Fig. 7A). The localization of Wag31-mCherry was substantially compromised in the DKO strain compared to the WT and the single mutants (Fig. 7Bii and iv and 4Biv and vi). We showed earlier that in the absence of CrgA, GFP-FtsI localization is compromised, whereas FtsZ-GFP assembly remains unaffected (29). However, both GFP-FtsI localization (Fig. 7B; compare panels vi and viii) and FtsZ-GFP assembly were severely compromised in the DKO background (Fig. 7C; compare panels ii and iv). The combined loss of crgA and cwsA possibly has a more severe effect on PG synthesis, which in turn affects membrane integrity and the stability of FtsZ rings. Alternatively, the perturbed FtsZ localization could be due to the defective growth phenotype of the DKO strain. Overall, the data presented above suggest an intimate involvement of CrgA and CwsA proteins in cell wall synthesis and morphogenesis in mycobacteria.
Fig 7.

Characterization of crgA cwsA double mutant. (A) Morphology of the crgA cwsA (DKO) strain. M. smegmatis WT, ΔcrgA, and DKO strains were grown to exponential phase and imaged by bright-field microscopy. The numbers in each panel indicate the average percentage of bulgy cells ± standard error from two or three independent experiments. Each experiment included ≥115 cells from each of the three strains. (B) Localization of Wag31-mCherry and GFP-FtsI in the crgA cwsA mutant strain. The M. smegmatis WT (i, ii, v, and vi) and crgA cwsA mutant (iii, iv, vii, and viii) expressing either Pami::wag31-mCherry (i to iv) or Pami::gfp-ftsI (v to viii) were grown with 0.2% acetamide for 2 h and 3 h, respectively, and examined by bright-field and fluorescence microscopy. Arrowheads in panels ii and iv, polar Wag31-mCherry localization; arrow, septal localization. Percent polar localizations from two independent experiments were scored, and averages ± standard errors are given in the respective fluorescent panel. Each experiment included ≥100 cells from each strain. Arrowheads in panel vi, GFP-FtsI localization in WT cells. Note the near complete absence of GFP-FtsI fluorescence localization in the double mutant (viii). (C) Localization of FtsZ-GFP in the crgA cwsA mutant. Exponential cultures of M. smegmatis WT (i, ii) and DKO (iii, iv) strains expressing Pami::ftsZ-gfp were examined by bright-field and fluorescence microscopy. Percent midcell FtsZ-GFP localizations from two independent experiments were scored, and averages ± standard errors are given in the respective fluorescent panel. Each experiment included ≥130 cells from each strain. Arrows, midcell ring. Note the reduced and aberrant (fluorescent spots) localization of FtsZ-GFP in the double mutant (arrowhead in panel iv).
crgA and cwsA are involved in cell wall synthesis.
To obtain direct evidence for a role in cell wall synthesis, we examined the incorporation of 14C-labeled GlcNAc into cell walls of the mutant strains. When grown in the presence of glucose, M. smegmatis has been shown to incorporate the majority of the radiolabeled GlcNAc (93 to 95%) into PG and to incorporate a relatively small amount into the arabinogalactan-linkage unit (15). The incorporation of 14C-labeled GlcNAc into crgA and cwsA mutant strains was reduced compared with that in the WT strain, and the incorporation profiles of the two mutant strains were similar to each other (Fig. 8A). Interestingly, the DKO strain showed a substantially lower 14C-labeled GlcNAc incorporation rate and final yield than the individual single-deletion strains. There was an initial increase in the incorporation, which reached a plateau within 3 h of incubation with 14C-labeled GlcNAc. The similar but reduced incorporation rates and the growth profiles of the single mutant strains suggest that the activities of both CwsA and CrgA are required for PG synthesis, optimal growth, and viability. Next, we examined the loss of 14C-labeled GlcNAc from cell walls after culturing cells in the presence of 14C-labeled GlcNAc for 6 h (∼2 generations) and resuspending the cells in prewarmed broth containing unlabeled GlcNAc. Loss of cwsA had no effect on cell wall turnover (Fig. 8B), whereas the loss of crgA resulted in a modest, statistically significant reduction in turnover. The DKO strain showed higher turnover rates than the WT and single mutants (Fig. 8B). These results suggest that CrgA activity facilitates cell wall turnover, whereas CwsA does not directly contribute to turnover, although the latter is required along with CrgA for optimal cell wall turnover.
Fig 8.

Loss of crgA and cwsA causes severe defects in cell wall synthesis. (A) 14C-labeled GlcNAc incorporation in M. smegmatis WT, ΔcwsA, ΔcrgA, and DKO strains. Exponential cultures were grown with 14C-labeled GlcNAc as described in Materials and Methods. At 1 min, 2 h, 4 h, and 6 h after addition of the label, 0.5 ml of culture was filtered through a 0.2-μm-pore-size nitrocellulose filter and washed, the filters were air dried, and radioactivity was measured in a liquid scintillation counter. Data shown are normalized to the A600 at each given point. Means ± standard deviations from 3 independent experiments are shown. (B) Cell wall turnover in M. smegmatis strains from panel A. Cells were labeled with [14C]GlcNAc for 6 h as described for panel A and pelleted. The labeled cells were resuspended in medium without label and grown at 37°C with shaking. At 0, 3, 4, 5, 6, 8, 10, and 12 h following growth in label-free medium, 0.5 ml of culture was pelleted, boiled in 4.0% SDS for 30 min, and passed through a 0.22-μm-pore-size filter. The filters were processed as described in Materials and Methods, and the retained radioactivity was measured in a liquid scintillation counter. Percent PG turnover at each time point was calculated with respect to that at time zero. Marker labels are as described for panel A. Means ± standard deviations from 3 independent experiments are shown. Statistical analysis of data using paired Student's t test indicated significant differences between WT and the ΔcrgA strain (P = 0.003), WT and DKO (P = 0.005), and WT and the ΔcwsA strain (P = 0.011). One-way ANOVA followed by Tukey's multiple-comparison test revealed significant differences between the ΔcrgA and ΔcwsA strains (P = 0.0035), the ΔcrgA and DKO strains (P = 0.0058), and the ΔcwsA and DKO strains (P = 0.0249). (C) Autolysis of M. smegmatis strains. Various M. smegmatis strains (as described for panel A) were grown to an A600 of 0.5, and cells were pelleted, resuspended in 50 mM glycine, pH 8.0, containing 0.05% Tween 20, and incubated with shaking at 37°C. At time points of 0, 1, 2, 4, 6, 8, 10, 12, 14, and 16 h, aliquots of cells were removed and the A600 was measured. The percentage of the initial optical density (OD) retained was calculated with respect to that at time zero. Data shown are means ± standard deviations from 3 independent experiments. Paired Student's t test showed significant differences in autolysis between the WT and ΔcwsA strains (P = 0.0078), the WT and DKO strains (P = 0.0028), the ΔcwsA and DKO strains (P = 0.0026), and the ΔcrgA and DKO strains (P = 0.0014) but not between the WT and the ΔcwsA strains (P = 0.063). (D) Lysozyme sensitivity. Fluorescein-labeled cell walls were prepared from WT and mutant M. smegmatis strains as described in the text and incubated with 1 mg/ml lysozyme in a buffer containing 50 mM Tris and 1 mM EDTA for 4 h. Reactions were stopped by addition of an equal volume of 4 M LiCl and spun, and the fluorescent label released in the supernatant was quantitated in a Jasco FP6500 fluorimeter. Reactions were done in triplicate, and average percent hydrolysis is shown.
Cell wall synthesis involves a balance between the activities of cell wall synthetic and hydrolytic enzymes (18). Our results showing substantial differences in the cell wall synthesis rates (Fig. 8A) but modest differences in the turnover rates (Fig. 8B) prompted us to examine the autolysis rates of these strains (Fig. 8C). The ΔcwsA and DKO strains showed statistically significantly higher autolysis rates than the WT and ΔcrgA strains; furthermore, the DKO strain showed near complete lysis after approximately 16 h (data not shown). These results suggest that CwsA activity somehow affects autolysis and that CrgA does not directly affect autolysis per se, although the combined activities of CwsA and CrgA appear to be critical for regulation of autolysis.
Given the involvement of the CwsA and CrgA proteins in cell wall synthesis and changes in cell morphology phenotypes, we next examined whether isolated cell walls from the mutant strains show altered access to lysozyme compared to the wild-type strain. The PG of mycobacteria is generally resistant to lysozyme, an enzyme that cleaves glycosidic linkages between sugar residues of PG (11, 23). The cell wall preparations from all mutant strains showed enhanced susceptibility to lysozyme compared with those from the WT strain (Fig. 8D). Presumably, defects in cell wall synthesis in single mutants compromised cell wall integrity and, hence, increased the susceptibility to lysozyme (Fig. 8D). Together, these data support the involvement of the CrgA and CwsA proteins in PG synthesis and cell shape determination.
DISCUSSION
The genetic determinants of septal Z-ring assembly and cell wall synthesis processes in mycobacteria are not completely known. In other rod-shaped bacteria, actin- and tubulin-like cytoskeletal elements direct the cell wall synthesis and cell division processes, respectively (reviewed in references 35 and 38). For example, it is reported that the MreB protein coordinates the cell wall elongation steps, whereas the tubulin-like FtsZ protein initiates the cell division process by promoting the assembly of septasomal components and helps direct septum formation (2, 38). Interactions among proteins involved in the two pathways are believed to be essential for coordinating growth, cell division, cell wall synthesis, and cell shape maintenance (35). M. tuberculosis and other mycobacterial members lack MreB-like proteins but contain FtsZ and its interacting partners, some of which are novel proteins not present in other bacteria (7, 29). Thus, it is an enigma how the cell wall synthesis process occurs and is coordinated with the Z-ring assembly. In the present study, we provide evidence that CwsA is an important protein involved in cell wall synthesis and cell shape maintenance in mycobacteria. The observed membrane, polar, and septal localization patterns of CwsA (Fig. 2), reduction in the rates of 14C-labeled GlcNAc incorporation (Fig. 8A), increased autolysis rates (Fig. 8C), susceptibility of the isolated cell walls to the lysozyme (Fig. 8D), and importantly, defects in cell shape with bulged cell morphology of the ΔcwsA mutant are consistent with the notion that CwsA is required for cell wall synthesis and cell shape maintenance (Fig. 3D). Earlier studies reported that CrgA and Wag31 proteins contribute to some steps of cell wall synthesis and cell shape (19, 29). Thus, CwsA joins the list of the components comprising the mycobacterial cell wall synthesis machinery. It is also likely that CwsA, owing to its interactions with CrgA, is important for coordination of the cell wall synthesis process with the Z-ring assembly (see below).
Our results also showed physical and possibly functional interactions of CwsA with Wag31 and CrgA proteins. One functional consequence of the CwsA interaction with CrgA is to promote Wag31 localization because the Wag31 localization pattern was compromised in the mutants with single mutations of cwsA and crgA (Fig. 4B), although the latter gene product does not interact with Wag31 (see Fig. S8 in the supplemental material). CrgA interacts with FtsI, a Wag31-interacting protein; and FtsI localization is shown to be severely compromised in the ΔcrgA background (29). CrgA also interacts with FtsZ, FtsQ, and possibly other unidentified cell division proteins (29). Thus, the decreased localization of Wag31 in the crgA background could be due to a compromised FtsI-CrgA interaction, which then influences FtsI-Wag31 interactions and, in turn, cell wall synthesis. Therefore, CwsA directly promotes and CrgA indirectly promotes Wag31 localization. We propose that CrgA and CwsA proteins together contribute to the FtsI- and Wag31-mediated cell wall synthesis steps and that a coordinated action of these four proteins is required for optimal cell wall synthesis (Fig. 9). Disruption of CrgA and CwsA interactions could compromise the stability of the PG synthesis network and thereby impact cell wall synthesis.
Fig 9.
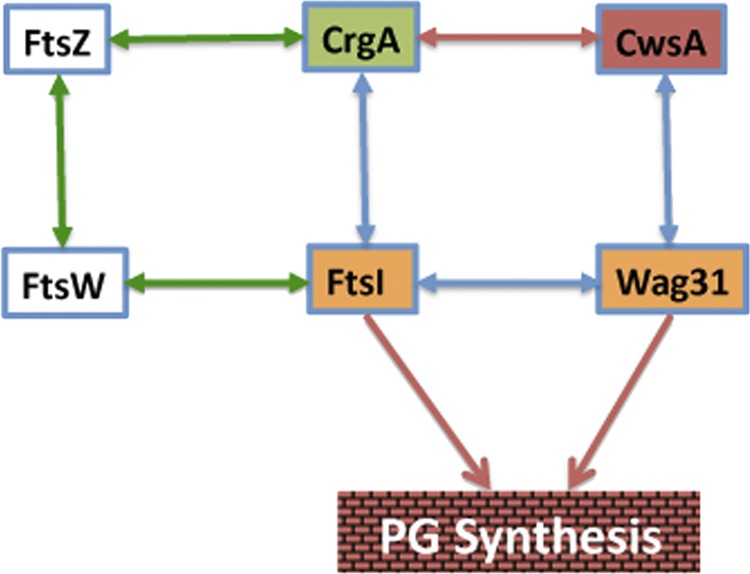
Protein-protein interactions among cell division and cell wall synthesis proteins in mycobacteria. Interactions of CrgA and CwsA with each other and with FtsI and Wag31, respectively, contribute to cell division and cell wall synthesis (Fig. 8). Green and blue arrows indicate interactions of essential (core) proteins with each other and with accessory proteins CrgA and CwsA; red arrow, interaction of CrgA with CwsA.
Characterization of the DKO strain in this study, however, provided clues as to how interactions between CwsA and CrgA lead to a regulated cell wall synthesis and possibly cell shape maintenance in mycobacteria. The DKO strain showed significant defects in growth, viability, PG synthesis, and cell shape morphology, as well as increased autolysis and cell wall turnover compared with either of the mutants with a single cwsA or crgA mutation (Fig. 3 and 8). Furthermore, the localization patterns of Wag31 and FtsI were severely compromised in the DKO background (Fig. 7B). These results support a model involving two interdependent but closely connected routes for PG synthesis and cell shape maintenance (Fig. 9). The presence of two routes for PG synthesis ensures continual PG synthesis even when the functions of CwsA or CrgA proteins are compromised. For example, in the absence of CwsA function, cell wall synthesis and cell division proceed, albeit defectively, via a route involving CrgA, FtsI, and Wag31, whereas in the absence of CrgA, this route could involve the actions of the CwsA, Wag31, and FtsI proteins. We propose that interactions between CwsA and CrgA are critical for tethering the complex together and promoting optimal cell wall synthesis. Finally, it is known that FtsZ and FtsI proteins interact with each other via FtsW (9). Thus, it is also possible that cell wall synthesis in the cwsA crgA DKO strain proceeds in a compromised manner involving the actions of FtsZ, FtsW, FtsI, and Wag31 (Fig. 9).
Close inspection of autolysis and cell wall turnover data revealed that the CrgA and CwsA proteins differentially modulate these processes. For example, while both proteins are required for cell wall synthesis, the absence of crgA only modestly reduced turnover but did not affect autolysis, whereas the absence of cwsA increased autolysis but did not affect turnover. On the other hand, the absence of both proteins increased autolysis and turnover. These results suggest that the underlying mechanisms regulating autolysis and turnover involving CwsA and CrgA and, possibly, their interactions are rather complex and require more detailed investigations. It should be noted that the CrgA localization was modestly enhanced in the cwsA background (see Fig. S5 in the supplemental material). The possibility that elevated levels of CrgA somehow modulate autolysis rates in the cwsA background remains open.
One interesting aspect of our data is that the FtsZ-GFP localization was severely compromised in the cwsA crgA DKO strain (Fig. 7C). It was reported earlier that although CrgA interacts with FtsZ, the absence of CrgA does not affect FtsZ localization (29). It seems likely that CrgA and CwsA function as a complex and help coordinate FtsZ-ring assembly at the septa with downstream cell wall synthesis events. Defective FtsZ septal assembly either due to compromised cell wall synthesis or due to defective growth of DKO might then affect the loading of other divisome components at the septa and therefore productive cell division. This is an not unreasonable assumption, as perturbations to cell wall integrity due to overproduction or loss of the cell wall hydrolase ChiZ have been shown to affect FtsZ-ring stability in M. smegmatis and M. tuberculosis (5, 36). Further studies are required to address how cell wall synthesis coordination with Z-ring assembly takes place in mycobacteria. In summary, the results presented in this study indicate that CwsA and CrgA together with Wag31 and FtsI are members of a multiprotein complex engaged in PG synthesis in mycobacteria. Large multiprotein complexes made up of proteins spanning different cellular compartments, viz., the cytoplasm, inner membrane, and periplasm, are proposed to be involved in various stages of PG synthesis in E. coli, Bacillus subtilis, and other bacteria (reviewed in reference 38). Further studies on the roles of CrgA and CwsA will likely aid our understanding of cell division and cell wall synthesis processes in mycobacteria.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the M. Madiraju and M. Rajagopalan labs for discussions and E. V. P. Pandeeti and S. T. Howard for critical comments, Hau Nguyen (T. A. Cross lab) for providing purified CwsA protein, Tanya Parish for the mCherry plasmid, and Choong-Min Kang for the M. smegmatis wag31 mutant strain.
This work was supported by NIH grants AI48417 (to M.R.), AI084734 (to M.M.), and AI73891 (T.A.C.).
Footnotes
Published ahead of print 21 September 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Aaron M, et al. 2007. The tubulin homologue FtsZ contributes to cell elongation by guiding cell wall precursor synthesis in Caulobacter crescentus. Mol. Microbiol. 64:938–952 [DOI] [PubMed] [Google Scholar]
- 2. Adams DW, Errington J. 2009. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 7:642–653 [DOI] [PubMed] [Google Scholar]
- 3. Aldridge BB, et al. 2012. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrews JR, et al. 2007. Multidrug-resistant and extensively drug-resistant tuberculosis: implications for the HIV epidemic and antiretroviral therapy rollout in South Africa. J. Infect. Dis. 196(Suppl 3):S482–S490 [DOI] [PubMed] [Google Scholar]
- 5. Chauhan A, et al. 2006. Interference of Mycobacterium tuberculosis cell division by Rv2719c, a cell wall hydrolase. Mol. Microbiol. 62:132–147 [DOI] [PubMed] [Google Scholar]
- 6. Chauhan A, et al. 2006. Mycobacterium tuberculosis cells growing in macrophages are filamentous and deficient in FtsZ rings. J. Bacteriol. 188:1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cole ST, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 8. Datta P, Dasgupta A, Bhakta S, Basu J. 2002. Interaction between FtsZ and FtsW of Mycobacterium tuberculosis. J. Biol. Chem. 277:24983–24987 [DOI] [PubMed] [Google Scholar]
- 9. Datta P, et al. 2006. Interaction between FtsW and penicillin-binding protein 3 (PBP3) directs PBP3 to mid-cell, controls cell septation and mediates the formation of a trimeric complex involving FtsZ, FtsW and PBP3 in mycobacteria. Mol. Microbiol. 62:1655–1673 [DOI] [PubMed] [Google Scholar]
- 10. de Jonge BL, de Lencastre H, Tomasz A. 1991. Suppression of autolysis and cell wall turnover in heterogeneous Tn551 mutants of a methicillin-resistant Staphylococcus aureus strain. J. Bacteriol. 173:1105–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dhople AM, Hanks JH. 1980. Effect of cholesterol, lecithin and nucleotides on the in vitro growth of Mycobacterium lepraemurium. Jpn. J. Exp. Med. 50:469–471 [PubMed] [Google Scholar]
- 12. Dziadek J, et al. 2002. Physiological consequences associated with overproduction of Mycobacterium tuberculosis FtsZ in mycobacterial hosts. Microbiology 148:961–971 [DOI] [PubMed] [Google Scholar]
- 13. Dziedzic R, et al. 2010. Mycobacterium tuberculosis ClpX interacts with FtsZ and interferes with FtsZ assembly. PLoS One 5:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fol M, et al. 2006. Modulation of Mycobacterium tuberculosis proliferation by MtrA, an essential two-component response regulator. Mol. Microbiol. 60:643–657 [DOI] [PubMed] [Google Scholar]
- 15. Hancock IC, et al. 2002. Ligation of arabinogalactan to peptidoglycan in the cell wall of Mycobacterium smegmatis requires concomitant synthesis of the two wall polymers. Microbiology 148:3059–3067 [DOI] [PubMed] [Google Scholar]
- 16. Hett EC, Chao MC, Deng LL, Rubin EJ. 2008. A mycobacterial enzyme essential for cell division synergizes with resuscitation-promoting factor. PLoS Pathog. 4:e1000001 doi:10.1371/journal.ppat.1000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hett EC, Chao MC, Rubin EJ. 2010. Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog. 6:e1001020 doi:10.1371/journal.ppat.1001020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holtje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:181–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang CM, et al. 2005. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19:1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang CM, et al. 2008. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology 154:725–735 [DOI] [PubMed] [Google Scholar]
- 21. Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korepanova A, et al. 2005. Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci. 14:148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizuguchi Y, Tokunaga T. 1975. Genetics of Mycobacterium. Nippon Saikingaku Zasshi 30:297–313. (In Japanese.) [PubMed] [Google Scholar]
- 24. Mukherjee P, et al. 2009. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol. Microbiol. 73:103–119 [DOI] [PubMed] [Google Scholar]
- 25. Nguyen L, et al. 2007. Antigen 84, an effector of pleiomorphism in Mycobacterium smegmatis. J. Bacteriol. 189:7896–7910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parish T, Stoker NG. 2000. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146(Pt 8):1969–1975 [DOI] [PubMed] [Google Scholar]
- 27. Payne DJ. 2008. Microbiology. Desperately seeking new antibiotics. Science 321:1644–1645 [DOI] [PubMed] [Google Scholar]
- 28. Plocinska R, et al. 2012. Septal localization of the Mycobacterium tuberculosis MtrB sensor kinase promotes MtrA regulon expression. J. Biol. Chem. 287:23887–23899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plocinski P, et al. 2011. Characterization of CrgA, a new partner of the Mycobacterium tuberculosis peptidoglycan polymerization complexes. J. Bacteriol. 193:3246–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qin H, et al. 2008. Construction of a series of vectors for high throughput cloning and expression screening of membrane proteins from Mycobacterium tuberculosis. BMC Biotechnol. 8:51 doi:10.1186/1472-6750-8-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajagopalan M, et al. 2005. Mutations in the GTP-binding and synergy loop domains of Mycobacterium tuberculosis ftsZ compromise its function in vitro and in vivo. Biochem. Biophys. Res. Commun. 331:1171–1177 [DOI] [PubMed] [Google Scholar]
- 32. Stover CK, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460 [DOI] [PubMed] [Google Scholar]
- 33. Sureka K, et al. 2010. Novel role of phosphorylation-dependent interaction between FtsZ and FipA in mycobacterial cell division. PLoS One 5:e8590 doi:10.1371/journal.pone.0008590 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Thakur M, Chakraborti PK. 2006. GTPase activity of mycobacterial FtsZ is impaired due to its transphosphorylation by the eukaryotic-type Ser/Thr kinase, PknA. J. Biol. Chem. 281:40107–40113 [DOI] [PubMed] [Google Scholar]
- 35. Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10:123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vadrevu IS, et al. 2011. ChiZ levels modulate cell division process in mycobacteria. Tuberculosis (Edinb.) 91(Suppl 1):S128–S135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Varma A, Young KD. 2009. In Escherichia coli, MreB and FtsZ direct the synthesis of lateral cell wall via independent pathways that require PBP 2. J. Bacteriol. 191:3526–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White CL, Gober JW. 2012. MreB: pilot or passenger of cell wall synthesis? Trends Microbiol. 20:74–79 [DOI] [PubMed] [Google Scholar]
- 39. White EL, et al. 2000. Slow polymerization of Mycobacterium tuberculosis FtsZ. J. Bacteriol. 182:4028–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young DB, Perkins MD, Duncan K, Barry CE., III 2008. Confronting the scientific obstacles to global control of tuberculosis. J. Clin. Invest. 118:1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.