

Summary
The promiscuous activity of protein phosphatase one (PP1) is controlled in the cell by associated proteins termed regulatory or targeting subunits. Using biochemical and proteomic approaches we demonstrate that the autosomal recessive nonsyndromic hearing loss gene, taperin (C9orf75), encodes a protein that preferentially docks the alpha isoform of PP1. Taperin associates with PP1 through a classic ‘RVxF’ motif and suppresses the general phosphatase activity of the enzyme. The steady-state localization of taperin is predominantly nuclear, however we demonstrate here that the protein can shuttle between the nucleus and cytoplasm and that it is found complexed to PP1 in both of these cellular compartments. Although originally identified as a hearing loss gene, Western blot analyses with taperin-specific antibodies revealed that the protein is widely expressed across mammalian tissues as multiple splice variants. Taperin is a recent proteome addition appearing during the vertebrate lineage with the PP1 binding site embedded within the most conserved region of the protein. Taperin also shares an ancestral relationship with the cytosolic actin binding protein phostensin, another PP1 interacting partner. Quantitative Stable Isotope Labeling by Amino acids in Culture (SILAC)-based mass spectrometry was employed to uncover additional taperin binding partners, and revealed an interaction with the DNA damage response proteins Ku70, Ku80, PARP and topoisomerases I and IIα. Consistent with this, we demonstrate the active recruitment of taperin to sites of DNA damage. This makes taperin a new addition to the family of PP1 targeting subunits involved in the DNA damage repair pathway.
Keywords: Nonsyndromic deafness, Nucleus, Protein phosphatase one (PP1), Protein phosphorylation, Phostensin, DNA damage
Introduction
High throughput sequencing is accelerating the rate of discovery of disease-linked loci, yet many of these uncovered genes have no known function. One of the necessary future goals of these studies will be understanding the biochemistry, cell biology and function of uncharacterized disease genes. The most common hereditary deafness is autosomal recessive nonsyndromic hearing loss (ARNSHL) with about 60 loci (DFNB) known, including DFNB79, which contains 113 genes within the 3.84 Mb linkage region. Two recent studies using high throughput sequencing of individuals from two separate families identified the mutated gene in this region to be C9orf75 (Li et al., 2010; Rehman et al., 2010). C9orf75 is expressed in multiple tissues, including the cochlea. Immunolocalization places the C9orf75 gene product predominantly in the taper regions of the inner ear hair cells and C9orf75 has thus been given the name taperin (Rehman et al., 2010). Taperin function, however, remains enigmatic.
The specific addition of a phosphate group to a protein is recognized as the most common means of covalent modification to regulate function (Cohen, 2002). Recent mass spectrometry-based investigations have suggested that at least 70% of all human proteins are phosphorylated, and most at multiple sites (Olsen et al., 2010). The phospho-status of any protein is governed by the activities of both protein kinases and phosphatases. The complement of human protein kinases and phosphatases has revealed over 500 and 150 genes, respectively. This apparent dichotomy in total kinase versus phosphatase catalytic subunits is explained by their differing regulatory mechanisms. During the evolution of several protein phosphatase classes the catalytic subunit remained a lone entity and has been recruited to dephosphorylate novel substrates through association with new regulatory subunits (Moorhead et al., 2009; Shi, 2009; Virshup and Shenolikar, 2009). This is best exemplified by protein phosphatase one (PP1), which in humans exists as 3 isoforms (α, β, γ). To date, over 200 proteins have been recognized as PP1 interacting proteins that localize the phosphatase to specific locations in the cell and modulate its activity toward selected substrates (Hendrickx et al., 2009; Moorhead et al., 2008). Regulatory subunits often display a preference for one catalytic subunit isoform, yet the underlying mechanism of specificity remains to be elucidated. Moreover, it is thought that several hundred more human PP1 interacting proteins have yet to be discovered (Hendrickx et al., 2009). In general, misregulated protein phosphorylation is considered a causative agent for numerous human diseases. The identification and functional elucidation of the PP1 interactome is hereby steadily gaining importance because their selective targeting of PP1 substrates, often combined with their preference for a particular isoform, provides more specific targets for the pharmaceutical industry.
Using the independent approaches of SILAC-based quantitative proteomics (Trinkle-Mulcahy et al., 2006) and displacement affinity chromatography (Moorhead et al., 2008) to define the cellular PP1 interactome, we have uncovered the association of PP1 with taperin. Our work characterizes the interaction of taperin with human PP1α and establishes that taperin can shuttle between the nucleus and cytoplasm but remains almost exclusively nuclear. Taperin is expressed as multiple splice variants, and bioinformatic analyses indicate that taperin appeared during the vertebrate lineage, strictly maintains the PP1 docking function across vertebrate species and has an ancestral relationship with the PP1- and actin-binding protein, phostensin.
Results
Taperin (C9orf75) is a nuclear PP1α binding protein expressed as multiple isoforms
We have previously shown the merits of i) SILAC-based quantitative proteomics on the interactome of GFP-PP1 immunoprecipitations (Trinkle-Mulcahy et al., 2006; Trinkle-Mulcahy et al., 2008) and ii) peptide displacement affinity chromatography (Moorhead et al., 2008) for the unbiased identification of nuclear PP1 regulatory subunits. The latter method relies on the binding of microcystin sensitive phosphatases to a matrix coupled with the toxin and the specific release of PP1 associated proteins by incubation with excess peptide encompassing an RVXF motif, the canonical and primary PP1 interaction region within most regulatory subunits (Moorhead et al., 2007). Initial data from the ensuing mass spectrometry defined nuclear PP1 interactomes that showed partial overlap between both approaches, with one common candidate being c9orf75/taperin (Moorhead et al., 2008; Trinkle-Mulcahy et al., 2006). Considering the recent identification of taperin as a protein mutated in patients with non-syndromic, hereditary deafness, we investigated this interaction in more detail.
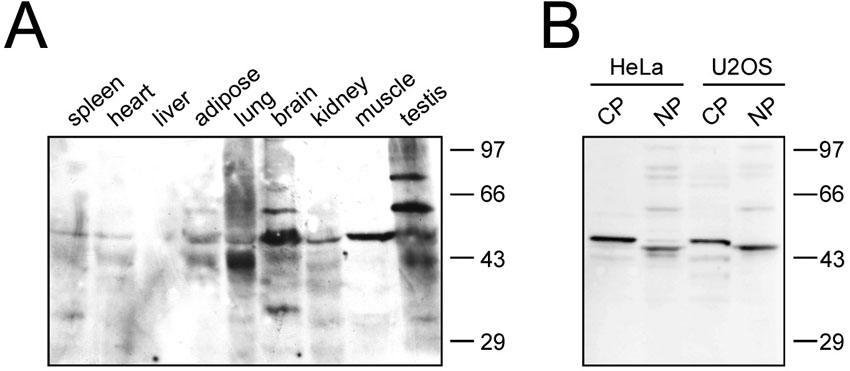
Four human taperin isoforms, generated by alternative splicing events, have been described thus far. The canonical isoform 1 is 711 amino acids long (predicted mass of 75.6 kDa), while the remaining 3 all miss the initial 306 N-terminal amino acids with predicted masses of 44.1, 47.2 and 44.2 kDa, respectively (see supplementary material Fig. S1 for isoform sequence alignments). Isoform 1 is supported by its alignment with mouse sequences and transcript evidence, yet at the time of our study no actual protein based observations supported this. Sequence analysis further showed that all taperin isoforms possessed one potential PP1 docking motif (KISF) between amino acids 577–580 in isoform 1 and amino acids 271–274 in isoforms 2–4. We therefore cloned and expressed isoform 2 (44.2 kDa) as a His-tagged fusion protein for recombinant protein studies and polyclonal antibody production (supplementary material Fig. S2). Antibodies generated against this isoform should ensure recognition of all versions of taperin. The resulting, affinity purified antibodies decorated two major (∼44 and 47kDa) and several minor bands in HeLa and U2OS extracts (supplementary material Fig. S3B, S4B). Further Western blot analyses of various rat tissues identified multiple immunoreactive bands, predominantly around 47 kDa but also a ∼75 kDa version in testis (supplementary material Fig. S4A), a mass reminiscent of the predicted isoform 1 size, and several weaker signals of ∼95 kDa. Interestingly, Western blot analyses mainly recognized ∼44–47 kDa sized taperin isoforms, yet our mass spectrometry results derived from HeLa cells not only identified peptides shared between all four isoforms, but also peptides unique for isoform 1 (supplementary material Fig. S1), thereby providing support for the presence of isoform 1 in certain cells and/or tissues.
Next, we focused on the interaction between PP1 and taperin. We incubated nuclear HeLa extracts with a microcystin matrix to enrich phosphoprotein phosphatases and performed peptide displacement with a RARA peptide (scrambled PP1 interaction motif), a RVRW peptide (GKKRVRWADLE) and finally by sodium thiocyanate (SCN) to selectively elute PP1 binding proteins. Eluted proteins were separated by SDS-PAGE and probed with a polyclonal antibody against taperin (Fig. 1A). Complete displacement with the RVRW peptide suggests association with PP1 occurs primarily through the PP1 dock site (KISF) within taperin. Interestingly, peptide-released taperin ran on SDS-PAGE at an apparent mass of ∼95 kDa (Fig. 1A), a size observed as a minor band in Western blotting (supplementary material Fig. S4). We corroborated these results by immunoprecipitation of endogenous taperin from HeLa cell extracts and revealed that taperin preferentially binds PP1α over PP1γ, but does not bind PP1β (Fig. 1B). To exclude the possibility that PP1 associates with taperin through another protein, we used recombinant DIG-labeled PP1α and PP1γ to perform overlay (Far-Western) experiments with taperin. We again demonstrated a strong preference for recombinant taperin to bind PP1α over PP1γ (Fig. 1C) with the association being completely abolished if the putative PP1 dock site, KISF, is mutated to KASA. Finally, in vitro pulldowns with recombinant taperin and PP1α show a robust interaction, again abolished when the putative PP1 binding KISF motif is mutated (Fig. 1D), establishing this as the primary PP1 dock site on taperin. These results are in line with our quantitative proteomics approach, and identify C9orf75/taperin as a nuclear PP1 interacting protein with a preference for PP1α over PP1γ (supplementary material Fig. S5).
Fig. 1. Taperin (c9orf75) preferentially associates with PP1α.

(A) HeLa cell nuclear extract was incubated with microcystin-Sepharose matrix, washed extensively and sequentially eluted with first the RARA peptide, followed by the RVRW peptide to specifically elute PP1 binding complexes, and finally a 3 M NaSCN step to elute any remaining protein. Eluates were then subjected to SDS-PAGE, transferred to nitrocellulose and a Western blot performed using affinity purified taperin antibodies (2 µg/mL). Endogenous taperin was immunoprecipitated (B) from a HeLa cell extract and blotted with the PP1 isoform specific antibody shown on the right. Control immunoprecipitation was with an equivalent amount of pre-immune IgG. Antibody specificity is shown by running 10 ng, 10 ng and 2 ng of recombinant purified PP1α, β and γ respectively, and as shown in supplementary material Fig. S12 and S13. (C) for far-Western blots, proteins were subjected to SDS-PAGE, transferred to nitrocellulose and overlaid with recombinant digoxigenin-coupled PP1α or PP1γ. Proteins are wild type [KISF] or mutated [KASA] 6His-taperin and 30 µg of rat nuclear extract as control. To ensure equal loading membranes were stripped and re-probed with anti-taperin (lower panels). For pulldowns (D), recombinant PP1α was mixed with Ni-NTA beads alone (control) or with recombinant wild type (KISF) or mutated (KASA) 6His-taperin. After release from the beads with SDS sample buffer, proteins were subjected to Western blot analysis with anti-taperin or anti-PP1 antibodies, as indicated. (E) the protein phosphatase activity of PP1α was monitored when mixed with an equal molar amount of taperin in the presence of increasing amounts of either RVRW peptide (—□—; GKKRVRWADLE) or RARA peptide (—♦—; GKKRARAADLE). Protein phosphatase activity was measured using phosphorylase a as the substrate. Data points are mean ± SD for n = 3.
Taperin inhibits PP1 activity
Using glycogen phosphorylase a as a substrate, we found that taperin is a strong inhibitor of PP1α activity (Fig. 1E). In support of an interaction between PP1 and taperin via its KISF sequence we were able to relieve the inhibition of PP1α activity by titrating in an RVRW peptide, whose high affinity for PP1 disrupted the association of PP1 with the KISF interaction site on taperin (Moorhead et al., 2008). Titrating in a scrambled (RARA) peptide where the key PP1 interaction residues were mutated to alanine had no effect on PP1 activity supporting the idea of KISF as the primary PP1 binding region on taperin.
Taperin is a soluble and predominantely nucleoplasmic protein
Immunostaining of permeabilized, fixed HeLa cells with taperin antibodies revealed that taperin is a predominately nuclear protein that does not accumulate within nucleoli (Fig. 2A). This was further confirmed by transiently expressing GFP-taperin in HeLa (Fig. 2B) and U2OS (Fig. 2C) followed by live cell imaging.
Fig. 2. Taperin is predominantly nucleoplasmic in vivo.
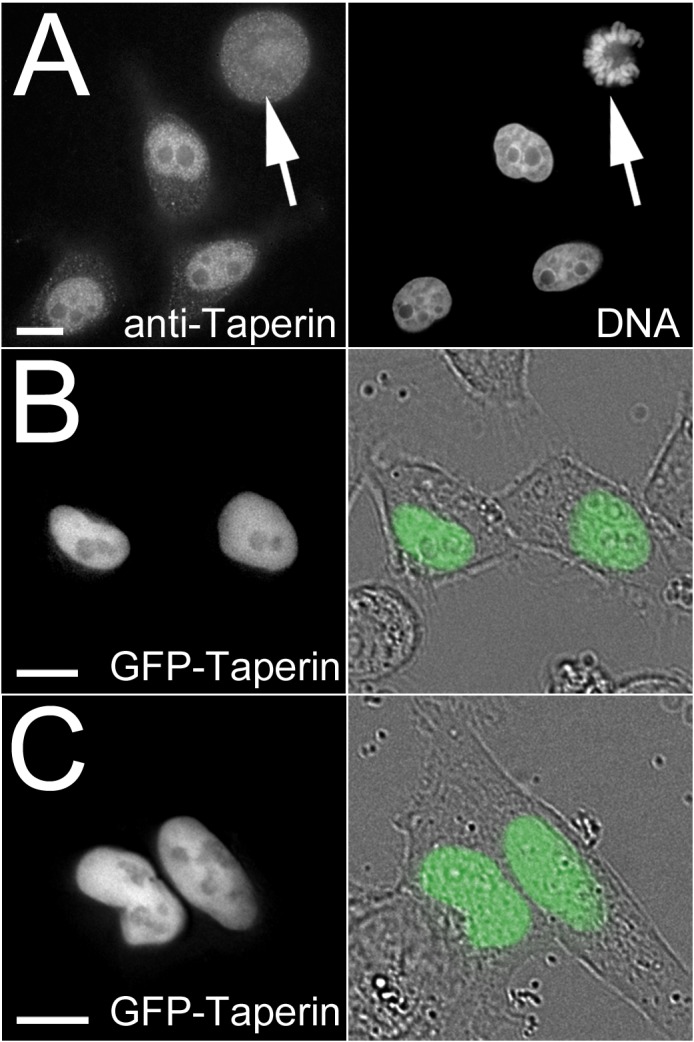
(A) Affinity purified taperin antibodies (5 µg/mL) were used to localize endogenous protein and DNA was stained with Hoechst in PFA-fixed HeLa cells (top panel), revealing a predominant nucleoplasmic localization in interphase and diffuse staining with no apparent accumulations in metaphase (arrow). Taperin is also predominantly nucleoplasmic when transiently expressed as a GFP-tagged fusion protein in both HeLa (B) and U2OS (C) cells. In these two panels the GFP images are shown superimposed on differential interference contrast (DIC) images to the right. Scale bars are 5 µM. Antibody controls are shown in supplementary material Fig. S3.
Our SILAC-based proteomic experiments and Western blot analyses suggested that taperin could associate with PP1 in both nucleoplasm and cytoplasm (supplementary material Fig. S4B, S5) whereas immunofluorescence (Fig. 2) suggests a clear enrichment of taperin in the nucleoplasm. We have occasionally observed this for other soluble, shuttling nuclear proteins, suggesting it may be a fractionation artifact. Alternatively, natural cytoplasmic re-localization may occur only under specific cellular conditions. The ability of taperin to shuttle between the nucleus and cytoplasm was demonstrated using both heterokaryon and fluorescence loss in photobleaching (FLIP) experiments (Fig. 3). In the heterokaryon approach, human cells expressing nucleoplasm accumulated GFP-taperin are fused to non-transfected mouse cells in the presence of cycloheximide to preclude expression of new GFP-taperin. DAPI staining reveals the origin of each nucleus (mouse nuclei show a characteristic pattern of large heterochromatin foci). We observed a fraction of GFP-taperin in the mouse nucleoplasm following fusion, indicating that it migrated from the human nuclei in the heterokaryon (Fig. 3A). This observation was supported by FLIP experiments in which we repeatedly photobleached a region in the cytoplasm in U2OS cells expressing either GFP or GFP-taperin (Fig. 3B–D). We followed its impact by measuring GFP-intensity of a nucleoplasmic area. The bulk of the nucleoplasmic GFP signal is lost from the cell after 35 bleach cycles, while a similar loss of the nucleoplasmic GFP-taperin signal requires 60 bleach cycles. In addition to confirming that taperin shuttles, it also establishes that it is not quite as dynamic as free GFP, an observation supported by FRAP experiments that showed a rapid, albeit slower than GFP, recovery rate for photobleached nucleoplasmic GFP-taperin (data not shown). Taken together, taperin can be regarded as a predominantly nucleoplasmic protein that has the ability to shuttle out of the nucleus.
Fig. 3. Taperin can shuttle between the nucleus and cytosol.

(A) In the heterokaryon approach depicted in the diagram, HeLa cells transiently expressing GFP-taperin (green) were mixed with non-transfected SW3T3 mouse cells. Cytoplasmic membranes were then fused by treatment with 50% PEG for 90 seconds and allowed to recover for 3 hours in media containing cycloheximide. DNA was then stained with Hoechst 33342 (blue) and cells imaged live to assess distribution of GFP-taperin between the original HeLa (arrowhead) and mouse (arrow) nuclei. The dashed line indicates the cell membrane. Scale bars are 5 µM. (B) Diagram depicting the FLIP experiment carried out to measure inter-compartmental dynamics of GFP-taperin, in which a cytoplasmic pool of the fusion protein is repeatedly bleached while the intensity of the nucleoplasmic pool is monitored over time. (C) Graph demonstrating the rapid loss of nucleoplasmic GFP (blue diamonds) when a cytoplasmic pool is bleached. The % maximum fluorescence intensity (normalized for photobleaching due to acquisition) is plotted versus the number of bleach events (each 100% laser power for a 0.05 sec duration). GFP-taperin (red squares) shows slower dynamics than free GFP, but a significant fraction of the nucleoplasmic pool is eventually bleached, indicating shuttling to the cytoplasm. (D) Sample dataset for each FLIP experiment. The lightning bolt indicates the photobleached region while the hashed circle shows the region of interest (ROI) that was monitored within the nucleus in each cell. To normalize for photobleaching caused by acquisition, a ROI was monitored in a neighboring cell. The number of bleach events is indicated in the bottom right corner. Each experiment was repeated 3 times. Scale bars are 5 µM.
In vivo interaction between taperin and PP1
The in vivo interaction of PP1 with taperin was confirmed using Bimolecular Fluorescence Complementation (BiFC) and co-transfecting U2OS cells with fragments of the EYFP protein fused to either PP1γ or PP1 targeting subunits, namely NIPP1, a well-established nuclear PP1 binding protein which serves as control, taperin or mutated taperin (data not shown). The direct vicinity (<10nm) of CYFP-PP1 constructs with either NYFP-NIPP1 or NYFP-taperin formed a competent fluorophore, emitting a clear nuclear EYFP signal (Fig. 4). Mutation of the taperin PP1 docking motif KISF to KASA completely abolished the signal (data not shown). The majority of the taperin-PP1 YFP signal is nucleoplasmic, yet a small fraction can be observed in the cytoplasm. This again suggests that taperin shuttles between these two cellular compartments.
Fig. 4. Bimolecular fluorescence complementation (BiFC) demonstrates the in vivo interaction of taperin and PP1.

The diagram depicts the design of the BiFC experiment, in which PP1γ was fused to an 84 amino acid C-terminal fragment of EYFP (pC/EYFP-PP1) and a targeting subunit fused to a 154 amino acid N-terminal fragment of EYFP (pN-EYFP-targeting subunit). If the proteins interact directly the fragments will complement each other to form a fluorescent EYFP molecule that can be excited with 513 nm light to emit light at 528 nm. As a positive control, U2OS cells were transfected with pNYFP-NIPP1 and pCYFP-PP1γ to demonstrate the nucleoplasmic interaction of these two proteins (green). When pNYFP-taperin was co-transfected with CYFP-PP1γ, a signal was detected throughout the cell (green), indicating interaction of the two proteins in both the nucleus and the cytoplasm. DNA is stained with Hoechst 33342 (blue). No signal was detected when NYFP-taperinKASA was co-transfected with CYFP-PP1γ (data not shown). Scale bars are 5 µM.
It has been shown that over-expression of PP1 regulatory subunits can cause a relocation of PP1 itself, which functions as a strong indicator of their in vivo interaction (Trinkle-Mulcahy et al., 2001). To support the BiFC experiments we studied PP1 relocalization in HeLa cells stably expressing low levels of either EGFP-PP1γ or EGFP-PP1α and transiently expressing either wild type (mCherry-taperin) or mutated (mCherry-taperinKASA) taperin. As shown in Fig. 5A, over-expression of taperin recruits most of the nuclear PP1γ, including the nucleolar pool (arrow), to the nucleoplasm of the cell where taperin resides. Mutation of the PP1 binding site from KISF to KASA abolishes this relocalization of PP1γ (Fig. 5C). As PP1α is already nucleoplasmic, it is difficult to assess whether over-expressed taperin recruited additional PP1α to its localization site (Fig. 5B). Over-expressed taperin remains complexed with PP1 during mitosis, as shown by the failure of EYFP-PP1γ to be recruited to metaphase kinetochores in the presence of excess ECFP-taperin (supplementary material Fig. S6A). Conversely, overexpression of the KASA mutant of taperin has no obvious effect on mitotic PP1γ localization (supplementary material Fig. S6B).
Fig. 5. In vivo relocalization of PP1 by exogenously over-expressed taperin.

(A) Transient overexpression of mCherry-tagged taperin (red) in HeLaEGFP-PP1γ cells relocalizes PP1γ (green) from nucleoli (arrow) and other nuclear sites of accumulation to the characteristic nucleoplasmic localization pattern of taperin itself. (B) As PP1α (green) exhibits a similar nucleoplasmic localization pattern to that of taperin, overexpression of mCherry-taperin (red) in HeLaEGFP-PP1α cells does not lead to an easily observable difference. (C) Over-expression of the non-PP1 binding mutant mCherry-taperinKASA does not lead to a relocalization of PP1γ in interphase HeLaEGFP-PP1γ cells. (D) Heterokaryon experiment in which HeLa cells transiently over-expressing mCherry-taperin (red) were fused to HeLaEGFP-PP1γ cells (green) in the presence of cycloheximide and imaged live 3 hours post-fusion. DNA was stained with Hoechst 33342 (blue). The dashed line indicates the cell membrane. The arrowhead indicates a nucleus containing both fusion proteins, in which PP1 is relocalized out of nucleoli (arrow) by taperin. (E) A similar experiment was carried out with the non-PP1 binding mutant mCherry-taperinKASA (red), which does not relocalize PP1 out of nucleoli (arrow) in nuclei containing both fusion proteins (arrowhead). Scale bars are 5 µM.
These data are further supported by heterokaryon experiments in which mCherry-taperin is introduced into cells to dynamically relocalize EGFP-PP1γ that has already been targeted to its normal intranuclear sites of action. In brief, HeLa cells transiently expressing mCherry-taperin are fused to EGFP-PP1γ expressing cells in the presence of cycloheximide to inhibit new protein translation. When mCherry-taperin enters the nuclei of cells already containing EGFP-PP1γ, it overrides the normal localization pattern by competing PP1 away from other nuclear targeting subunits, resulting in recruitment of the majority of nuclear PP1γ to the nucleoplasm (Fig. 5D). This also highlights the high affinity of the taperin-PP1 interaction. Recruitment does not occur when the PP1 dock site in taperin is mutated to KASA, with PP1γ maintaining its normal nuclear distribution (Fig. 5E).
Additional taperin interaction partners
To assess taperin interaction partners, we carried out a SILAC-based quantitative immunoprecipitation of taperin from nuclear extracts of HeLa cells transiently expressing ECFP-taperin. Mass spectrometry and quantitation of heavy: light amino acid ratios confirmed once again the co-precipitation of PP1 with taperin but also identified the classic DNA damage proteins Ku70, Ku80, PARP1, TOPOI and TOPOIIα (supplementary material Table S1). These were the only proteins identified with more than one peptide and ratios >1, suggesting they are bona fide taperin interactors. To confirm this observation we transiently expressed either GFP alone or GFP-taperin in HeLa cells, immunoprecipitated both from whole cell extracts using the high affinity GFP-Trap®_A reagent and probed Western blots of the eluted complexes with antibodies specific to these putative interaction partners. As shown in Fig. 6, each of these proteins is indeed enriched with GFP-taperin compared to GFP alone. It should be noted that the weak Ku70/80 bands detected in the control IP are not surprising, as these abundant proteins are known “bead contaminants” that can bind non-specifically to affinity matrices (Trinkle-Mulcahy et al., 2008).
Fig. 6. Taperin associates with topoisomerases I and IIα, PARP and Ku.
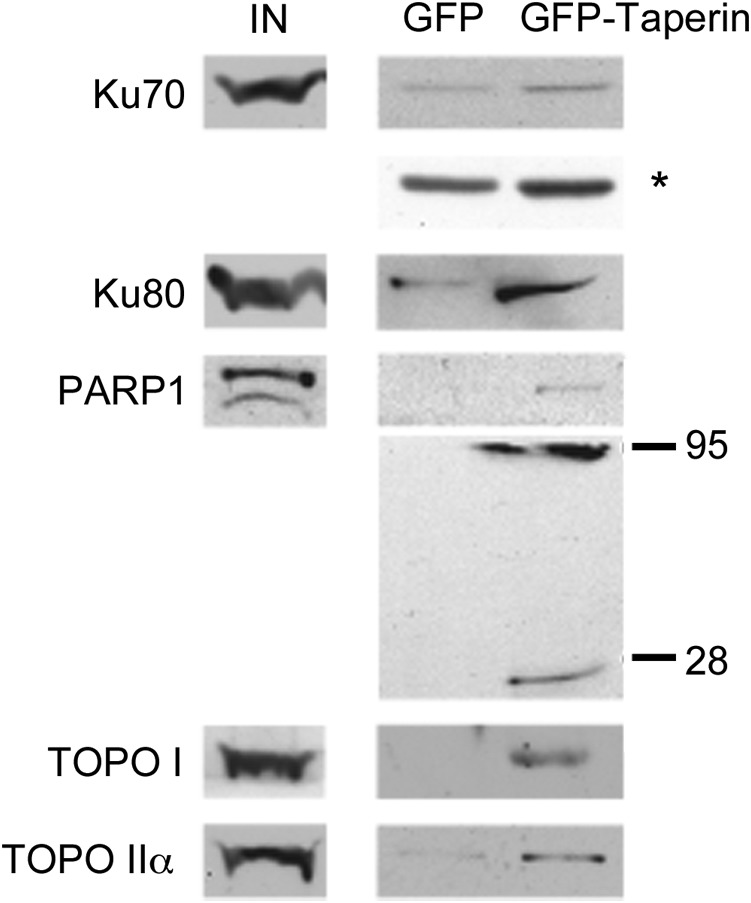
Putative taperin interaction partners identified in a quantitative proteomic screen (supplementary material Table S1) were validated by co-IP and Western blot analysis. Whole cell extracts were prepared from HeLa cells transiently over-expressing either GFP-taperin or GFP alone and subjected to pulldown using the high affinity GFP-Trap_A®. Following wash steps all proteins were eluted from the beads, separated by SDS-PAGE along with input lanes from crude extracts, transferred to nitrocellulose and probed with commercial antibodies raised against Ku70, Ku80, PARP1, TOPOI and TOPOIIα, as indicated. Each protein migrated at its expected molecular mass. PARP1 runs at 116 kDa, but after cleavage is know to have two dominant degradation products of 89 and 24 kDa as shown here. The asterisk (*) indicates a longer exposure of the IP lanes in the Ku70 Western blot.
Taperin accumulates at sites of DNA damage
Given that taperin appears to interact with several proteins known to play a role in the DNA damage response, we sought to determine if taperin is recruited to sites of DNA damage. Several other PP1 regulatory subunits have recently been identified as regulators of the DNA damage response, including GADD34, RepoMan and PNUTS (Kuntziger et al., 2011). We used U2OS cells, transiently expressing GFP alone (as a negative control), PNUTS-GFP (as a positive control), GFP-taperin (WT) or GFP-taperinKASA. Cells were pre-sensitized to double strand breaks by staining with Hoechst 33342 (see Fig. 7A for experimental design) and DNA lesions were induced by UV laser micro-irradiation of discrete regions in the nucleus, similar to the work of Landsverk et al. (Landsverk et al., 2010) with PNUTS-GFP. As expected, GFP alone shows no accumulation at damage foci (data not shown), whereas PNUTS-GFP readily accumulates at these sites (Fig. 7D). GFP-taperin displays an even faster recruitment (Fig. 7B) to DNA damage foci than PNUTS-GFP (Fig. 7D) but not to the same signal intensity (Fig. 7E). The PP1 docking mutant (KASA) of taperin showed very similar kinetics to the PP1 binding version (Fig. 7C,E), suggesting that taperin recruitment may occur prior to and/or independent of PP1 binding. Interestingly, sequence analysis of taperin revealed 2 RGG motifs that may facilitate nucleic acid binding (supplementary material Fig. S7).
Fig. 7. Taperin is recruited to sites of DNA damage in vivo.

(A) Design of the experiment used to assess recruitment to discrete DNA lesions induced by UV laser microirradiation in pre-sensitized (by staining with Hoechst 33342) live cells. Following irradiation of a specific region of interest (ROI) the fluorescence intensity of GFP was then monitored in this same ROI over time. (B) Time-lapse imaging of GFP-taperin demonstrating recruitment of the fusion protein to the site of irradiation (arrow) over a 150 sec period. A pre-irradiation image was taken for comparison, and the first post-irradiation image was collected 0.002 sec after the laser fired. (C) The non-PP1 binding mutant GFP-taperinKASA was subjected to the same treatment and demonstrated similar kinetics of recruitment to sites of UV-induced DNA damage. (D) As a positive control, the recruitment of PNUTS-GFP to DNA lesions was monitored over the same time scale. (E) The average % maximum fluorescence intensity ± SE was plotted versus time for GFP-Taperin (•, n = 10), GFP-TaperinKASA (×, n = 10) and PNUTS-GFP (▴, n = 5). Data were normalized for photobleaching due to acquisition. The dashed line indicates the original fluorescence intensity within the ROI prior to DNA damage. Scale bars are 5 µm.
Taperin is a vertebrate specific protein
The human taperin sequences (isoforms 1–3) were used as a query in a series of pBLAST and tBLASTn searches to identify and collect homologous sequences. Taperin sequences were only found in vertebrates, but within those, mammals, birds and fish are present with no amphibian or reptile sequences represented (supplementary material Fig. S8). Within the taperin sequences, highest conservation is found in the C-terminal half where the PP1 docking motif (KISF) lies in a highly conserved stretch.
Relationship to phostensin
Database searching with the taperin primary sequence revealed phostensin, a previously characterized PP1 interacting protein, as the only sequence with homology with taperin. In turn, we used phostensin sequences as bait in a series of pBLAST and tBLASTn queries, which identified taperin and phostensin homologues. Once again, these are only found in vertebrates with examples in placental and marsupial mammals, but not in monotremes.
Direct alignment of phostensin and taperin reveals some degree of similarity along their entire length (supplementary material Fig. S7). Only a short domain in phostensin is highly related to taperin, and this contains the recognized PP1 interaction region of both proteins. The KISF PP1 dock site is a less frequent, functional version of the common RVXF motif and is found in both taperin and phostensin, with the residues bracketing this site also being well conserved. The sequences surrounding the PP1 docking motif were highly conserved throughout all BLAST derived taperin and phostensin sequences. We therefore used this region to develop a phylogenetic tree, to deduce the evolutionary relationship between both proteins (supplementary material Fig. S8). Both taperin and phostensin sequences align with similar sequences throughout vertebrate organisms, suggesting that these proteins may have originated from a common ancestral protein early in vertebrate evolution.
Previously, taperin exon 1 has been proposed as a mutational hotspot, prone to internal re-arrangements. We suspected that such events could also be observed in the amino acid sequence. Furthermore, considering the relationship between taperin and phostensin, we questioned whether phostensin could possess similar properties. To test the hypothesis of internal re-arrangements throughout evolution for each protein and to assess similarity between both, we generated “HHBlits” to perform “self against self” and “taperin against phostensin” dotmatcher comparisons. HHBlits use P.s.i.-Blast to collect a series of related sequences, derives a Hidden Markov Model (HMM) for the query and its hits, and compares it to a pre-computed database of HMMs formed by sets of related database sequences. Dotmatcher analyses provide a visual representation of the similarity between 2 sequences. Dotplot analyses of self against self comparisons show that not only taperin but also phostensin possess internal rearrangements, exemplified by the offset distribution of short fragments from the diagonal (supplementary material Fig. S9). Moreover, a dotplot of phostensin (613 amino acids) against taperin (711 amino acid version; supplementary material Fig. S9) shows a main diagonal at the N- and C-termini, suggestive of direct similarity, but also several offset diagonals within the interior of the molecules. Thus, taperin and phostensin do share homology outside the highly conserved domain but most likely also have a complex history of differential internal rearrangement from an ancestral sequence to bring about the pattern of internal repeats now observable.
We previously identified phostensin (KIAA1949) in our SILAC-based cellular interactome screen as a PP1α interacting protein (Trinkle-Mulcahy et al., 2006). Here we confirm this interaction in both cytoplasm and nucleoplasm PP1 interactome screens (supplementary material Fig. S5). Phostensin has been described as a 165 amino acid protein (Kao et al., 2007), but recent, database-annotated versions predict the existence of multiple longer versions, up to 613 amino acids. Indeed, mass spectrometry data from our PP1α pulldown identified phostensin peptides spread across the entire predicted 613 amino acid protein (supplementary material Fig. S10). We expressed full-length phostensin as a GFP-tagged fusion protein in HeLa cells and saw an exclusively cytosolic localization pattern with a strong enrichment at the plasma membrane (supplementary material Fig. S11). We also noted significant co-localization with mCherry-actin at both the cell periphery and at stress fibers, which supports a suggested role for phostensin in regulation of actin dynamics (Lai et al., 2009). The cytosolic localization of phostensin is in sharp contrast to taperin, which is a predominantly nucleoplasmic protein. Finally, co-expression of full-length ECFP-phostensin with either EYFP-PP1α or –γ does not cause obvious relocalization of the phosphatase, suggesting that the interaction with PP1 is weaker than that observed for taperin and PP1 (data not shown) or, association may rely on specific cellular conditions.
Discussion
The recent increase in genome and high throughput sequencing has resulted in extensive genetic mapping, including the identification of numerous loci affected within hereditary diseases. Unfortunately, the biological function of the proteins they encode in both healthy and diseased cells remains largely unknown. Non-syndromic deafness, encompassing autosomal dominant (DFNA) or recessive (DFNB) and X-linked (DFNX) gene mutations is a prime example with the identification of 49 linked genes in the last decade (www.hereditaryhearingloss.org), yet a near-complete lack of functional protein studies.
Here we present the first functional study on taperin/c9orf75, mutations of which in exon 1 at the DFNB79 locus lead to non-syndromic autosomal recessive deafness. We have identified taperin as a bona fide protein phosphatase 1 (PP1) interactor, both in vivo and in vitro. Taperin shows a clear preference to dock the alpha isoform of PP1 through its ‘RVXF’ motif (KISF). Deafness-associated taperin mutations documented thus far lead to either missense amino acid stretches followed by a stop codon or to the introduction of a premature stop codon, thought to result in a truncated protein (amino acids 1–412). As a result, none of these forms of taperin contain the PP1 docking motif identified here, and indeed may even act as dominant-negative mutants, competing the full-length protein (and hence taperin-PP1 complexes) away from important cellular substrates. Further studies are required to investigate the substrate(s) of taperin-PP1 in human cells/tissues.
Taperin is highly expressed in the taper region of stereocilia (Rehman et al., 2010) but as we demonstrate here the protein is present in most tissues/cell types. This ubiquitous expression is in line with previous observations for other nonsyndromic deafness genes (Rehman et al., 2010) (and references within). Furthermore, taperin is expressed as multiple splice variants, with variation between tissues. In accordance with previous transcript results, we observed a dominant protein isoform of ∼44 kDa, reminiscent of isoforms 2–4. We also detected a major immunoreactive band at approximately 75 kDa in testis, in line with the predicted mass of isoform 1. In HeLa cell extracts, we identified a predominant 44 kDa isoform by Western blot analyses, while mass spectrometry data from PP1-interacting taperin recognized both the long (isoform 1) and shorter variants (isoforms 2–4), suggesting all isoforms are present and capable of binding PP1 in vivo.
We show that taperin is a predominantly nucleoplasmic protein, which can interact with DNA damage response proteins and accumulate to sites of DNA damage upon UV-irradiation. A role for PP1 in DNA damage repair has been documented previously (Kuntziger et al., 2011). Thus, we can envision a model whereby a nuclear PP1-taperin complex may play a role in the repair of DNA strand breaks which would become disfunctional when taperin no longer binds PP1, due to genetic modifications at the DFNB79 locus.
Taperin sequences are only found in vertebrates, which coincides with vertebrate stereocilia (sensory hair cells) development, currently present at otoliths in amphibia and fish, the cochlear duct in birds and the organ of Corti within the mammalian coiled cochlea (Tanimoto et al., 2011). This distribution supports a key role for taperin in vertebrate hearing, in particular the function of stereocilia.
PP1 may in fact be involved in the biological function of several proteins associated with hereditary non-syndromic deafness. We investigated the protein sequences derived from affected loci for the presence of putative PP1 docking motifs. As of August 2011 the hereditary hearing loss website lists 25 proteins under non-syndromic autosomal dominant (DFNA) and 41 proteins under recessive (DFNB) deafness. The least stringent PP1 docking motif covers 90% of all PP1 interacting motifs and is present in approximately 25% of the human proteome (Hendrickx et al., 2009). This percentage is however significantly higher in both the dominant and recessive protein datasets (56.5% and 57.5%, respectively). Similarly, one out of the two currently listed X chromosome-linked nonsyndromic mixed deafness proteins (POU3F4) also contains a putative PP1 docking motif. Thus, we observed a significantly higher number of PP1 docking motifs within the proteins linked with non-syndromic hereditary deafness. We further observed deafness inducing sequence variants with a detrimental impact on these putative PP1 interaction motifs for 2 proteins within the dominant dataset (GJB2, WFS1), 1 protein in the recessive dataset (GJB2) and as stated, 1 protein in the X-linked dataset (POU3F4). Mutations within the GJB2 (Gap junction beta 2 protein or connexin-26) putative PP1 binding motif influence the same amino acid (R143) but the final amino acid is different between the dominant (Q) and recessive (W) variants. The putative PP1-docking motif (HRVTW) within WFS1 (wolframin) is a highly likely motif as it adheres to the low and the high stringency predictors (Hendrickx et al., 2009). Interestingly, mutation of the essential tryptophan (W) within the motif, most likely eliminating its functionality, is documented for patients with Wolfram syndrome, a rare but severe disorder leading not only to deafness but also to diabetes and additional neurological symptoms (Bespalova et al., 2001; Khanim et al., 2001).
Phostensin was identified as a PP1 binding protein both in our initial nuclear PP1 interactome screen (Trinkle-Mulcahy et al., 2006) and by a yeast two-hybrid screen (Kao et al., 2007). It co-localizes with F-actin in HeLa and MCDK cells and, similar to taperin, is expressed in a number of tissues. Initial work on phostensin detected a 165-residue polypeptide, which binds the pointed end of actin filaments and controls actin dynamics (Lai et al., 2009). We confirm by mass spectrometry the more recent gene prediction programs and define the full length protein at 613 amino acids. Moreover, our phylogenetic tree suggests that, similar to taperin, phostensin is present only in vertebrates and has no direct sequence relatives except for taperin. Furthermore, both proteins carry an identical, but much less common, PP1 docking motif (KISF), implying the potential for serine phosphorylation within the docking motif and consequently PP1 dissociation from phostensin. At present, we cannot deduce a functional reason for these observations due to our limited knowledge on the role of phostensin, except for its recently observed down-regulation in multiple tumors and breast cancer cell lines (Su et al., 2010).
Summarized, this functional study of taperin has brought first insights in the biological role of this protein. Moreover, based on our observations, PP1 may be a common factor in non-syndromic hereditary deafness that merits a number of follow-up studies. Mouse models have been used successfully to study hereditary deafness and could be developed to investigate the impact of PP1 in stereocilia and tissues affected in Wolfram syndrome patients. Although PP1 has long been associated with a number of diseases, this study may lead to the first identification of the underlying causes.
Materials and Methods
General methods
Protein phosphatase assays were performed as in (Ulke-Lemee et al., 2007). All 3 human PP1 isoforms were expressed in E. coli as full-length proteins without tags. PP1γ was purified as in (Douglas et al., 2001), while PP1α and β isoforms were cloned into pK233 vector, expressed as in (Tran et al., 2004) and purified on microcystin-Sepharose (Moorhead et al., 1994). Samples of rat tissues were obtained as described in (Ulke-Lemee et al., 2007).
Cloning, expression and purification of taperin (C9orf75)
Taperin was amplified from an expressed sequence tag (IMAGE clone 4992533; Open Biosystems corresponding to isoform 2 at Uniprot–oligonucleotide sequences available on request) and inserted into the mammalian expresssion vector pECFP-C1 (Clontech), confirmed by DNA sequencing, and further subcloned into pEGFP and mCherry vectors (Clontech). When indicated, the PP1 docking site (KISF) in taperin constructs was mutated (KASA) (Quickchange mutagenesis kit, Stratagene).
For bacterial expression of taperin, the pECFP-taperin construct was used for amplification and the product ligated into the pRSET-A vector (Invitrogen). The resulting 6His-Taperin (KISF or KASA) expression constructs were transformed into Rosetta gami pLysS E. coli, grown to A600 = 0.5 at 37°C and expression induced for 5.5 hr at 37°C (0.5 mM IPTG). Cells were harvested, re-suspended in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 15 mM imidazole, 0.5 mM PMSF, 0.5 mM benzamidine, 5 µg/mL leupeptin, 1 µg/mL pepstatin) and snap frozen in liquid nitrogen. Cells were lysed by two passes through a French Press (SIM Aminco) at 1000 p.s.i. and the crude extract clarified by centrifugation (45 mins, 35000 r.p.m.). The crude sample was batch-loaded onto a 3 mL pre-equilibrated Ni-NTA column (end-over-end, 1 h, 4°C). The column was washed (50 mM Tris pH 7.5, 1.0 M NaCl, 35 mM imidazole, 0.5 mM PMSF, 0.5 mM benzamidine, 0.1% Tween-20), and bound proteins eluted (50 mM Tris pH 7.5, 150 mM NaCl, 300 mM imidazole, 0.5 mM PMSF, 0.5 mM benzamidine). The eluate was dialyzed into buffer A (25 mM Tris pH 7.5, 0.25 mM EDTA, 0.25 mM EGTA, 5% (v/v) glycerol, 0.1% (v/v) β-ME, 0.5 mM PMSF, 0.5 mM benzamidine) before loading on a Mono Q HR 5/5 column. Protein was eluted using a linear salt gradient in buffer A from 100 mM to 500 mM NaCl over 20 mL with 0.5 mL fractions. Protein was visualized on 10% SDS-PAGE and fractions containing the recombinant ∼44 kDa taperin protein were pooled and dialyzed against buffer B (15 mM HEPES pH 7.0, 0.25 mM EDTA, 0.25 mM EGTA, 5% (v/v) glycerol, 0.1% (v/v) β-ME, 0.5 mM PMSF, 0.5 mM benzamidine). Protein was loaded onto a 1 mL Mono S HR 5/5 column and eluted with a linear salt gradient in buffer B from 180 mM to 500 mM NaCl over 20 mL. Peak fractions were dialyzed into PBS, concentrated and stored at −20°C in 50% (v/v) glycerol. A typical yield was 0.1 mg from 4 L of cells. The resulting peak fraction was run on SDS-PAGE, stained with Colloidal-Coomassie blue and the bands marked on supplementary material Fig. S2 excised, digested with trypsin as in (Tran et al., 2004) and identified by MALDI-TOF MS at the Southern Alberta Mass Spectrometry (SAMS) Centre for Proteomics at the University of Calgary, confirming the purification of taperin.
Displacement affinity chromatography to identify nuclear PP1 interactors
Nuclei were purified from HeLa cell suspension cultures and proteins extracted and chromatographed on a microcystin affinity matrix as in (Moorhead et al., 2008). Proteins were eluted from the matrix by sequential elution with the peptides GKKRARAADLE (‘RARA’), GKKRVRWADLE (‘RVRW’), and finally, 3M sodium thiocyanate. Samples were dialyzed, concentrated to an equal volume and run on SDS-PAGE (Moorhead et al., 2008).
SILAC-based mass spectrometry to define PP1 or taperin interactomes
For PP1, quantitative immunoprecipitations were performed using nuclear and cytoplasmic extracts from SILAC-media grown HeLa cell lines stably expressing low levels of GFP-PP1α or GFP-PP1γ with the unmodified parental cell line as control and the GFP-Trap®_A reagent (Chromotek) for affinity binding, as described in (Trinkle-Mulcahy et al., 2006; Trinkle-Mulcahy et al., 2008). To quantitatively define the taperin-interactome, a ECFP-taperin construct or, as a control, ECFP alone, were transiently expressed in Hela cells, with both cultures grown in SILAC media. Nuclear extracts were prepared and affinity purifications performed with a monoclonal anti-GFP antibody (Roche) covalently conjugated to Protein G Sepharose as described (Trinkle-Mulcahy et al., 2006). For direct comparison, affinity and control matrices were combined for protein elution and all subsequent steps, including separation by 1D SDS-PAGE (to reduce complexity) and identification of eluted proteins by mass spectrometry. Quantitation of the heavy:light arginines and lysines in each peptide yields a "H:L or SILAC Ratio", a measure for distinction between non-specific binding of the protein to the affinity matrix (ratio ∼ 1:1) or specific enrichment (ratio>1). Finally, we compared the enrichment and relative abundance of PP1-interacting proteins (taperin and phostensin) directly against each other and against the PP1 isoforms (α, γ) by plotting each average log SILAC Ratios versus their summed peptide intensities, normalized for molecular weight.
Recombinant protein interaction assays
Far-Western analyses were performed as described in (Chamousset et al., 2010) using recombinant DIG-labeled PP1α and γ against recombinant wild type (KISF) and mutated (KASA) 6His-taperin, and a rat nuclear extract prepared as in (Tran et al., 2004). For recombinant protein-interaction and pull down studies, proteins (PP1α + WT, PP1α+KASA, and PP1α alone as a control) were mixed with a 2-fold molar excess of PP1α: taperin in 200 µl reaction buffer (25 mM Tris pH 7.5, 10 mM imidazole pH 7.5, 150 mM NaCl, 5% (v/v) glycerol) and incubated for 15 min at room temperature. Next, protein complexes were incubated with 10 µL of Ni2+-NTA beads (end over end, 1 h, 4°C) followed by 3 washes with 200 µL of dilution buffer plus 300 mM NaCl. Matrix bound protein was eluted with SDS-cocktail and separated on SDS-PAGE.
Bioinformatic analysis
The human taperin (isoforms 1–3 according to uniprotKB) or phostensin (isoforms 1–2 according to uniprotKB) sequences were used as queries in a series of pBLAST and tBLASTn searches to identify and collect homologous sequences. Alignments and tree generation were performed using ClustalW and Genedoc as in (Chamousset et al., 2010).
Antibody production and purification
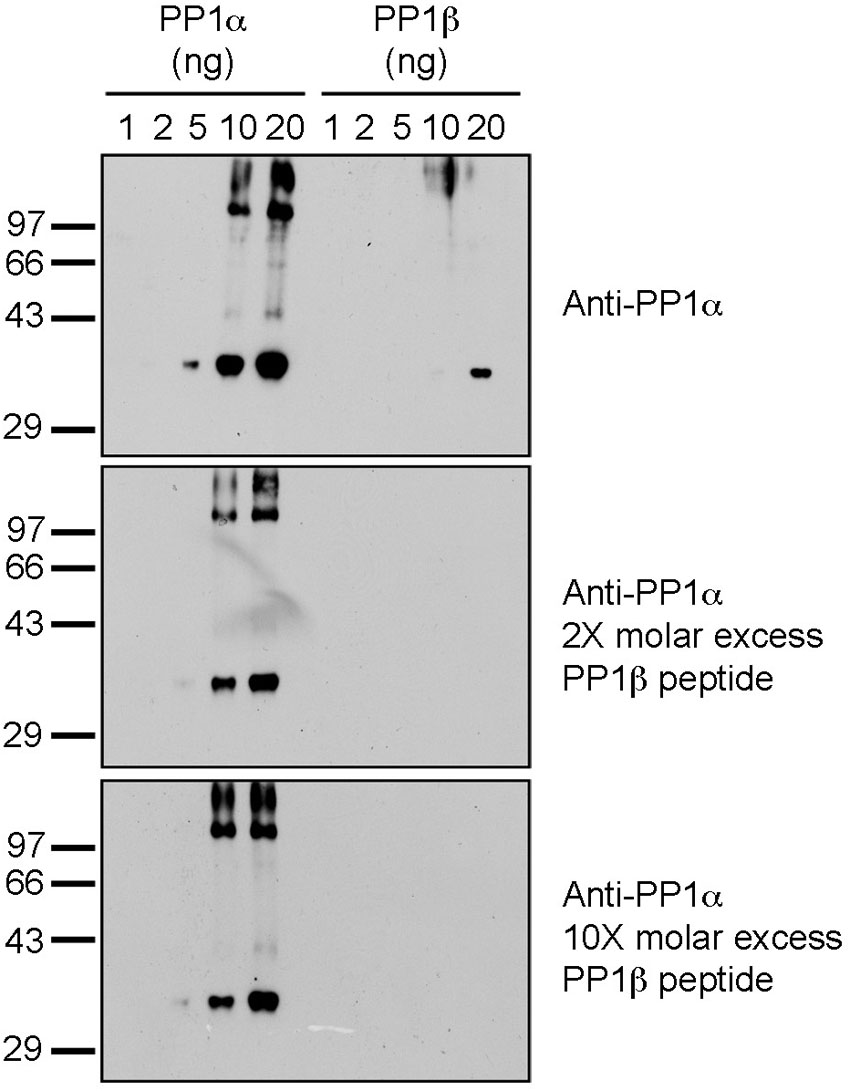
We, and others, have previously noted the unreliability and non-specificity of commercially available PP1 isoform-specific antibodies (Lesage et al., 2004; Trinkle-Mulcahy et al., 2006). To address this issue, we aligned the human PP1α, β and γ amino acid sequences and selected C-terminal peptides unique to each isoform to generate and validate our own isoform-specific antibodies. The following peptides for PP1α (DKNKGKYGQFSGLNPGGR), β (SEKKAKYQYGGLNSGR) and γ (KTPPRGMITKQAKK) were synthesized, coupled to KLH and BSA and used to generate antibodies in rabbits as described in (Smith et al., 2004). For PP1α an additional K (underlined) was added to the N-terminus to aid coupling to carrier proteins. Peptides (1 mg) were coupled to 1 mL of CH-Sepharose and used to affinity purify isoform specific antibodies (Smith et al., 2004). We used 1 µg/mL affinity pure antibodies to test their specificity, first against the original peptides, dotted to a nitrocellulose membrane (supplementary material Fig. S12A) and second, against each recombinant PP1 isoform and a HeLa crude extract, separated on SDS-PAGE and transferred to nitrocellulose membranes (supplementary material Fig. S12B). The PP1α specific antibody weakly detected PP1β, but this signal could be blocked by including a 5-fold molar excess of the PP1β peptide immunogen in the affinity purified PP1α antibody solution (supplementary material Fig. S13). This PP1β peptide was included in all subsequent PP1α blots.
For taperin, the purified recombinant protein (supplementary material Fig. S2) was used as antigen and antibodies made in rabbits (Chamousset et al., 2010). The taperin antibody was affinity purified by running recombinant protein on SDS-PAGE, transfer to a membrane and the taperin band excised. Taperin antibodies were bound and then released from this strip as described in (Chamousset et al., 2010). Pre-immune serum antibodies were purified on Protein-A Sepharose.
Taperin interaction partners
To validate putative taperin interactors identified in the quantitative proteomic screen described above, antibodies were obtained for the top hits (proteins most highly enriched in the FP-taperin IP) and Western blots performed on pulldowns of GFP-taperin and GFP alone transiently expressed in HeLa cells.
All fluorescence imaging was performed using a DeltaVision CORE widefield deconvolution microscope (Applied Precision) fitted with a WeatherStation environmental chamber to maintain temperature at 37°C, a CoolSNAP HQ2 charge-coupled device camera (Roper Scientific) and a quantifiable laser module (QLM; Applied Precision) with 405-nm and 488-nm lasers. Cells were imaged using a 60× NA 1.4 Plan-Apochromat objective (Olympus) and the appropriate filter sets (Chroma Technology Corp.), with 20 optical sections of 0.5 µm each acquired.
Fixed cell imaging
Cells were paraformaldehyde (PFA) fixed on glass coverslips and permeabilized with Triton X-100 as in (Chamousset et al., 2010). Endogenous taperin was stained using affinity purified anti-taperin (detected by Dy549-conjugated anti-mouse secondary antibodies, Jackson Laboratories), DNA was stained using Hoechst 33342 (Sigma) and cells were mounted in FluorSave mounting media (Calbiochem). SoftWorX software (Applied Precision) was used for both acquisition and deconvolution. For Bimolecular Fluorescence Complementation (BiFC) assays, 2 constructs were created. First, full length taperin and mutant taperin (KASA) were subcloned into pNYFP-C1, producing a fusion construct of the N-terminal part of YFP (aa 1–154) followed by taperin. Second, we fused PP1γ to the C-terminal part of YFP (aa 155–238), creating a pCYFP-PP1γ construct. Both constructs were co-transfected into cells, similar to the approach, originally described by Kerppola et al. (Hu et al., 2002). As a positive control, pCYFP-PP1γ was co-transfected with pNYFP-NIPP1, a known high affinity PP1 binding partner (Trinkle-Mulcahy et al., 1999). EYFP fluorescence was assessed as described above.
Live Cell Imaging
Fluorescence Recovery After Photobleaching (FRAP) and Fluorescence Loss in Photobleaching (FLIP) experiments were carried out with HeLa and U2OS cells transiently expressing free GFP, GFP-taperin or mutated GFP-taperin (KASA) as described in (Trinkle-Mulcahy et al., 2007). To monitor the recruitment of GFP-taperin and PNUTS-GFP (Helga Landsverk, Oslo University Hospital) to sites of DNA damage, cells transiently expressing these constructs were pre-sensitized by staining with Hoechst 33342 for 30 min, and then a brief pulse of UV irradiation (0.02 sec, 405 nm, 10% laser power) was applied to a discrete region of the nucleus to induce localized single and double strand DNA breaks.
Heterokaryon experiments
Human U2OS cells transiently expressing GFP-taperin were trypsinized to lift them off the dish, mixed with non-transfected mouse SW3T3 cells and allowed to settle back onto glass coverslips in a 6 cm dish. After 3 hours the cells were rinsed in PBS and covered with a thin layer of 50% PEG solution (Sigma) with gentle rocking for 90 seconds to allow fusion. Cycloheximide was included in the media prior to and following fusion, to prevent translation of new GFP-taperin. The PEG solution was removed by repeated rinses with media and cells allowed to recover for 2 hours. Cells were then fixed, stained with DAPI and mounted for imaging as described above.
Supplementary Material
Acknowledgments
The authors thank David Kerk for assistance with Dotplot analysis and George Templeton for assistance with DNA damage Western blots. Research was supported by NSERC (GM and LTM), Alberta Innovates (GM), Alberta Cancer Board (GM) and the Terry Fox Foundation (LTM).
References
- Bespalova I. N., Van Camp G., Bom S. J., Brown D. J., Cryns K., DeWan A. T., Erson A. E., Flothmann K., Kunst H. P., Kurnool P. et al. (2001). Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum. Mol. Genet. 10, 2501–2508 10.1093/hmg/10.22.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamousset D., De Wever V., Moorhead G. B., Chen Y., Boisvert F. M., Lamond A. I., Trinkle-Mulcahy L. (2010). RRP1B targets PP1 to mammalian cell nucleoli and is associated with Pre-60S ribosomal subunits. Mol. Biol. Cell 21, 4212–4226 10.1091/mbc.E10-04-0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. (2002). The origins of protein phosphorylation. Nat. Cell Biol. 4, E127–E130 10.1038/ncb0502-e127 [DOI] [PubMed] [Google Scholar]
- Douglas P., Moorhead G. B., Ye R., Lees-Miller S. P. (2001). Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem 276, 18992–18998 10.1074/jbc.M011703200 [DOI] [PubMed] [Google Scholar]
- Hendrickx A., Beullens M., Ceulemans H., Den Abt T., Van Eynde A., Nicolaescu E., Lesage B., Bollen M. (2009). Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol. 16, 365–371 10.1016/j.chembiol.2009.02.012 [DOI] [PubMed] [Google Scholar]
- Hu C. D., Chinenov Y., Kerppola T. K. (2002). Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 10.1016/S1097-2765(02)00496-3 [DOI] [PubMed] [Google Scholar]
- Kao S. C., Chen C. Y., Wang S. L., Yang J. J., Hung W. C., Chen Y. C., Lai N. S., Liu H. T., Huang H. L., Chen H. C. et al. (2007). Identification of phostensin, a PP1 F-actin cytoskeleton targeting subunit. Biochem. Biophys. Res. Commun. 356, 594–598 10.1016/j.bbrc.2007.03.026 [DOI] [PubMed] [Google Scholar]
- Khanim F., Kirk J., Latif F., Barrett T. G. (2001). WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum. Mut. 17, 357–367 10.1002/humu.1110 [DOI] [PubMed] [Google Scholar]
- Kuntziger T., Landsverk H. B., Collas P., Syljuasen R. G. (2011). Protein phosphatase 1 regulators in DNA damage signaling. Cell Cycle 10, 1356–1362 10.4161/cc.10.9.15442 [DOI] [PubMed] [Google Scholar]
- Lai N. S., Wang T. F., Wang S. L., Chen C. Y., Yen J. Y., Huang H. L., Li C., Huang K. Y., Liu S. Q., Lin T. H. et al. (2009). Phostensin caps to the pointed end of actin filaments and modulates actin dynamics. Biochem. Biophys. Res. Commun. 387, 676–681 10.1016/j.bbrc.2009.07.086 [DOI] [PubMed] [Google Scholar]
- Landsverk H. B., Mora-Bermudez F., Landsverk O. J., Hasvold G., Naderi S., Bakke O., Ellenberg J., Collas P., Syljuasen R. G., Kuntziger T. (2010). The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. 11, 868–875 10.1038/embor.2010.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage B., Beullens M., Nuytten M., Van Eynde A., Keppens S., Himpens B., Bollen M. (2004). Interactor-mediated nuclear translocation and retention of protein phosphatase-1. J. Biol. Chem. 279, 55978–55984 10.1074/jbc.M411911200 [DOI] [PubMed] [Google Scholar]
- Li Y., Pohl E., Boulouiz R., Schraders M., Nurnberg G., Charif M., Admiraal R. J., von Ameln S., Baessmann I., Kandil M. et al. (2010). Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am. J. Hum. Genet. 86, 479–484 10.1016/j.ajhg.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead G., MacKintosh R. W., Morrice N., Gallagher T., MacKintosh C. (1994). Purification of type 1 protein (serine/threonine) phosphatases by microcystin-Sepharose affinity chromatography. FEBS Lett 356, 46–50 10.1016/0014-5793(94)01232-6 [DOI] [PubMed] [Google Scholar]
- Moorhead G. B., Trinkle-Mulcahy L., Ulke-Lemee A. (2007). Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 8, 234–244 10.1038/nrm2126 [DOI] [PubMed] [Google Scholar]
- Moorhead G. B., Trinkle-Mulcahy L., Nimick M., De Wever V., Campbell D. G., Gourlay R., Lam Y. W., Lamond A. I. (2008). Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochem. 9, 28 10.1186/1471-2091-9-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead G. B., De Wever V., Templeton G., Kerk D. (2009). Evolution of protein phosphatases in plants and animals. Biochem. J. 417, 401–409 10.1042/BJ20081986 [DOI] [PubMed] [Google Scholar]
- Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A. et al. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 10.1126/scisignal.2000475 [DOI] [PubMed] [Google Scholar]
- Rehman A. U., Morell R. J., Belyantseva I. A., Khan S. Y., Boger E. T., Shahzad M., Ahmed Z. M., Riazuddin S., Khan S. N., Friedman T. B. (2010). Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 86, 378–388 10.1016/j.ajhg.2010.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Smith C. S., Morrice N. A., Moorhead G. B. (2004). Lack of evidence for phosphorylation of Arabidopsis thaliana PII: implications for plastid carbon and nitrogen signaling. Biochim. Biophys. Acta 1699, 145–154 10.1016/j.bbapap.2004.02.009 [DOI] [PubMed] [Google Scholar]
- Su Y. A., Yang J., Tao L., Nguyen H., He P. (2010). Undetectable and decreased expression of KIAA1949 (Phostensin) encoded on chromosome 6p21.33 in human breast cancers revealed by transcriptome analysis. J. Cancer 1, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto M., Ota Y., Inoue M., Oda Y. (2011). Origin of inner ear hair cells: morphological and functional differentiation from ciliary cells into hair cells in zebrafish inner ear. J. Neurosci. 31, 3784–3794 10.1523/JNEUROSCI.5554-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H. T., Ulke A., Morrice N., Johannes C. J., Moorhead G. B. (2004). Proteomic characterization of protein phosphatase complexes of the mammalian nucleus. Mol. Cell Proteomics 3, 257–265 10.1074/mcp.M300115-MCP200 [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Ajuh P., Prescott A., Claverie-Martin F., Cohen S., Lamond A. I., Cohen P. (1999). Nuclear organisation of NIPP1, a regulatory subunit of protein phosphatase 1 that associates with pre-mRNA splicing factors. J. Cell Sci. 112 Pt 2, 157–168 [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Sleeman J. E., Lamond A. I. (2001). Dynamic targeting of protein phosphatase 1 within the nuclei of living mammalian cells. J. Cell Sci. 114, 4219–4228 [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Andersen J., Lam Y. W., Moorhead G., Mann M., Lamond A. I. (2006). Repo-Man recruits PP1 gamma to chromatin and is essential for cell viability. J. Cell Biol. 172, 679–692 10.1083/jcb.200508154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Chusainow J., Lam Y. W., Swift S., Lamond A. (2007). Visualization of intracellular PP1 targeting through transiently and stably expressed fluorescent protein fusions. Methods Mol. Biol. 365, 133–154 [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Boulon S., Lam Y. W., Urcia R., Boisvert F. M., Vandermoere F., Morrice N. A., Swift S., Rothbauer U., Leonhardt H. et al. (2008). Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183, 223–239 10.1083/jcb.200805092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulke-Lemee A., Trinkle-Mulcahy L., Chaulk S., Bernstein N. K., Morrice N., Glover M., Lamond A. I., Moorhead G. B. (2007). The nuclear PP1 interacting protein ZAP3 (ZAP) is a putative nucleoside kinase that complexes with SAM68, CIA, NF110/45, and HNRNP-G. Biochim. Biophys. Acta 1774, 1339–1350 [DOI] [PubMed] [Google Scholar]
- Virshup D. M., Shenolikar S. (2009). From promiscuity to precision: protein phosphatases get a makeover. Mol. Cell 33, 537–545 10.1016/j.molcel.2009.02.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}