

Abstract
Summary: Frozen robust multiarray analysis (fRMA) is a single-array preprocessing algorithm that retains the advantages of multiarray algorithms and removes certain batch effects by downweighting probes that have high between-batch residual variance. Here, we extend the fRMA algorithm to two new microarray platforms—Affymetrix Human Exon and Gene 1.0 ST—by modifying the fRMA probe-level model and extending the frma package to work with oligo ExonFeatureSet and GeneFeatureSet objects.
Availability and implementation: All packages are implemented in R. Source code and binaries are freely available through the Bioconductor project. Convenient links to all software and data packages can be found at http://mnmccall.com/software
Contact: mccallm@gmail.com
The majority of methods for the preprocessing and analysis of microarray gene expression data rely upon the simultaneous analysis of multiple arrays (Hochreiter et al., 2006; Irizarry et al., 2003; Li and Wong, 2001). However, most traditional preprocessing methods struggle with modern microarray applications, such as large meta-analyses, because data preprocessed separately cannot be combined without introducing artifacts (McCall et al., 2010; Ramasamy et al., 2008). Furthermore, clinical applications necessitate the analysis of individual arrays, and datasets that grow incrementally must be preprocessed each time a new array is added.
Frozen robust multiarray analysis (fRMA) (McCall et al., 2010) addressed these challenges by implementing a modified version of the RMA algorithm (Irizarry et al., 2003). Additionally, by modeling probe-specific variances, fRMA showed improved precision of gene expression estimates and reduced susceptibility to batch effects (McCall et al., 2010; McCall and Irizarry, 2011). The fRMA algorithm was initally implemented on two of the most widely used microarray platforms—Affymetrix GeneChip Human Genome U133A and U133 Plus 2.0. Since then, it has been implemented for several other platforms.
In 2007, Affymetrix released two new microarray platforms—Human Exon 1.0 ST (HuEx) and Human Gene 1.0 ST (HuGene). In contrast to previous platforms that targeted the 3′ end of transcripts, these new platforms contain probes for each exon. This design change allowed researchers to assess exon-level expression and detect alternative splicing. However, it also posed a challenge to those who wanted to use these arrays to assess gene expression using the same preprocessing algorithms that were designed for the previous generation of Affymetrix microarrays. Specifically, the majority of preprocessing algorithms assume that each probe within a probeset was designed to measure the expression of the same target transcript; however, when a probeset is composed of probes targeting different exons, this assumption may be violated due to alternating splicing. This is particularly problematic given that splice variants are estimated to occur in 35–59% of genes (Modrek et al., 2002).
By summarizing probes at the exon level, one revalidates the assumption that each probe within a probeset is measuring the same target. This is more feasible for the HuEx platform, which often has four probes per exon, than for the HuGene platform, which contains fewer probes (roughly 35% of exons are targeted by only one probe). However, the small number of probes per probeset limits the ability to generate robust estimates of expression.
To address these limitations and aid researchers seeking to assess gene-level expression using HuEx or HuGene arrays, we have implemented a modified version of the fRMA model for gene-level summarization:
| (1) |
with 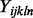 representing the
representing the 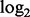 background corrected and normalized intensity of probe j, targeting exon l, of gene n on array i in batch k. Identical to the standard fRMA model,
background corrected and normalized intensity of probe j, targeting exon l, of gene n on array i in batch k. Identical to the standard fRMA model, 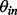 represents the expression of gene n on array i and is the parameter of interest. Here,
represents the expression of gene n on array i and is the parameter of interest. Here, 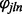 represents the global probe effect for the jth probe targeting the lth exon of gene n, and
represents the global probe effect for the jth probe targeting the lth exon of gene n, and 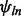 represents the exon effect for the lth exon of gene n. These parameters are both constrained to sum to zero within exon and gene, respectively. Finally,
represents the exon effect for the lth exon of gene n. These parameters are both constrained to sum to zero within exon and gene, respectively. Finally, 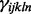 is a random effect representing the batch-specific change in the global probe effect. This model is fit as described in McCall et al. (2010) with an additional step to estimate the exon effects,
is a random effect representing the batch-specific change in the global probe effect. This model is fit as described in McCall et al. (2010) with an additional step to estimate the exon effects, 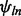 . For a new array, gene-level expression estimates are obtained as robust-weighted averages of the probe- and exon-effect adjusted expression values.
. For a new array, gene-level expression estimates are obtained as robust-weighted averages of the probe- and exon-effect adjusted expression values.
By using a large biologically diverse database of microarrays from a large number of different laboratories spanning several years, the fRMA algorithm is able to differentiate between outliers and probes that show a consistent susceptibility to batch effects. These batchy probes are downweighted during summarization to minimize their effect on expression estimates. HuEx and HuGene arrays add an additional layer of complexity—when summarizing at the gene level, a probe may show high between-batch residual variance due to either batch effects or alternative splicing (Fig. 1). The former should be downweighted, whereas the later may contain highly interesting biological information that could be captured by subsequent analysis of residuals, such as those proposed in Robinson and Speed (2009). For this reason, even when summarizing to the gene level, we weight probes based on their exon-level between-batch residual variance. Unfortunately, this is only feasible for exons targeted by multiple probes. For single-probe exons, it is impossible to assess residual variance at the exon level and, therefore, impossible to distinguish between batch effects and splice variants. For these probes, one must rely on robust summarization methods and post-preprocessing batch-effect correction algorithms such as ComBat (Johnson et al., 2007) or Surrogate Variable Analysis (Leek and Storey, 2007).
Fig. 1.

Residuals for probes targeting one of two exons are shown after fitting a standard RMA model to 100 arrays from 20 different batches (unique experiment/tissue combinations) at both gene (upper panels) and exon levels (lower panels). For both exons, Probe 1 (solid black line) appears to have a strong batch effect (high between-batch residual variance) when assessing probes at the gene level. However, in the case of Exon 96 615 750, the other three probes targeting this exon have nearly the same pattern of residuals across batches. This suggests that the high residual variance may be due to alternative splicing rather than a batch effect. By assessing probes at the exon level (lower panels), one still observes the high between-batch residual variance seen for Probe 1 targeting Exon 96 611 882 (left), but not for the probes targeting Exon 96 615 750 (right). By evaluating probe behavior at the exon level, we are able to distinguish between batch effects and splice variants
The two versions of the fRMA algorithm described earlier are implemented in the frma package and take advantage of the raw data structures implemented in the oligo package (Carvalho and Irizarry, 2010), allowing greater control over the level of summarization. Specifically, this is handled by the target argument passed to the frma function. The frozen parameter vectors for HuEx and HuGene arrays were created using 240 arrays from 48 batches and 1005 arrays from 201 batches, respectively. Here, a batch is defined as a unique tissue type/experiment combination. The frozen parameter vectors are stored in the huex.1.0.st.v2frmavecs and hugene.1.0.st.v1frmavecs annotation packages.
The frmaTools package (McCall and Irizarry, 2011), which allows users to create their own frozen parameter vectors, has also been updated to work with oligo GeneFeatureSet and ExonFeatureSet objects. This allows users to create custom vectors for the HuEx and HuGene platforms and to implement fRMA on other Affymetrix Exon and Gene ST platforms that are not currently supported.
ACKNOWLEDGEMENTS
The authors thank the maintainers of GEO and ArrayExpress for making the data publicly available, Marvin Newhouse and Jiong Yang for helping manage the data and the members of the La Calestienne Meeting, especially Hinrich Gohlmann and Willem Talloen, for their helpful discussions.
Funding: This work was funded by National Institutes of Health (CA009363 to M.N.M.), National Institutes of Health (GM083084, RR021967 and GM103552 to H.A.J.) and partially funded by National Institutes of Health (GM083084, RR021967 and UL1RR025005 to R.A.I.).
Conflict of Interest: none declared.
REFERENCES
- Carvalho B, Irizarry R. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter S, et al. A new summarization method for affymetrix probe level data. Bioinformatics. 2006;22:943–949. doi: 10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- Irizarry R, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Johnson W, et al. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Leek J, Storey J. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong W. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl Acad. Sci. USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M, Irizarry R. Thawing frozen robust multi-array analysis (fRMA) BMC Bioinformatics. 2011;12:369. doi: 10.1186/1471-2105-12-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M, et al. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, et al. A genomic view of alternative splicing. Nat. Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- Ramasamy A, et al. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:e184. doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M, Speed T. Differential splicing using whole-transcript microarrays. BMC Bioinformatics. 2009;10:156. doi: 10.1186/1471-2105-10-156. [DOI] [PMC free article] [PubMed] [Google Scholar]