

Abstract
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that binds to the antioxidant response element, a cis-acting regulatory element that increases expression of detoxifying enzymes and antioxidant proteins. Kelch-like ECH associating protein 1 (Keap1) protein is a negative regulator of Nrf2. Previous work has shown that genetic overexpression of Nrf2 is protective in vitro and in vivo. To modulate the Nrf2-ARE system without overexpressing Nrf2, we used short interfering RNA (siRNA) directed against Keap1. Keap1 siRNA administration in primary astrocytes increased the levels of Nrf2-ARE driven genes and protected against oxidative stress. Moreover, Keap1 siRNA resulted in a persistent upregulation of the Nrf2-ARE pathway and protection against oxidative stress in primary astrocytes. Keap1 siRNA injected into the striatum was also modestly protective against MPTP-induced dopaminergic terminal damage. These data indicate that activation of endogenous intracellular levels of Nrf2 is sufficient to protect in models of oxidative stress and Parkinson's disease.
Background
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcriptional activator of the antioxidant response element, a cis-acting regulatory element in the promoter region of many genes that encode phase II detoxifying enzymes and antioxidants (Itoh et al., 1997, Rushmore et al., 1991). The kelch-like ECH associating protein 1 (Keap1) protein is a negative regulator of Nrf2. Under unstressed conditions, Keap1 binds Nrf2 in the cytoplasm, preventing translocation to the nucleus (Itoh et al., 1999) while functioning as an adaptor protein to E3 ligase to promote the rapid degradation of Nrf2 by the ubiquitin proteasome system (Cullinan et al., 2004, Itoh et al., 2003, McMahon et al., 2003, Zhang and Hannink, 2003, Zhang et al., 2004). Keap1 mediates Nrf2 sequestration and degradation by assembling an E3 ubiquitin ligase with Cul3 and Rbx1 with the BTB domain in Keap1 via the Neh2 domain in Nrf2 to promote the poly-ubiquitization and degradation of Nrf2 (Cullinan et al., 2004, Kobayashi et al., 2004, Zhang et al., 2004). This process requires cysteine 151 on the Keap1 protein (Zhang et al., 2004), which, along with other cysteine residues in Keap1, acts as an electrophilic sensor for the cell. In the presence of electrophiles or stress, Keap1 binding and degradation of Nrf2 is interrupted, possibly through a switch of ubiquitization from Nrf2 to Keap1 (Hong et al., 2005, Zhang et al., 2005), allowing for the intracellular stabilization and nuclear translocation of Nrf2 (Itoh et al., 2003). Thus, a variety of mechanisms can be used to stabilize and activate the Nrf2-ARE pathway in the cell, including protease inhibitors (Itoh et al., 2003), the presence of electrophiles (Itoh et al., 2003, McMahon et al., 2003), the inhibition of Keap1 (Cullinan et al., 2004, Itoh et al., 2003), or the inhibition of other proteins in the Cul3-E3 ligase complex (Cullinan et al., 2004)
It is known that direct modulation of Nrf2 levels, through a genetic knockout of Nrf2 (Chan et al., 1996), reviewed by (Kensler et al., 2006, Motohashi and Yamamoto, 2004) or cell-specific overexpression of Nrf2 (Chen et al., 2009, Schafer et al., 2010, Vargas et al., 2008) can affect the cellular response to oxidative stress. However, much of this work has been performed via genetic modification of Nrf2 levels, resulting in models that do not identify the maximal activation that could occur via the endogenous Nrf2 levels. One could test this by instead modulating the levels of Keap1 without overexpressing exogenous Nrf2. Unfortunately, global knockouts of Keap1 are lethal (Wakabayashi et al., 2003); however, primary cultures derived from Keap1 knockout mice do show protection against oxidative stress (Satoh et al., 2009). To investigate an alternative therapeutic means of modulating the Nrf2-ARE system rather than overexpressing Nrf2, we investigated the activation of the Nrf2-ARE pathway by treatment with short interfering RNA (siRNA) directed against Keap1. Cellular introduction of siRNA, a 21-mer double stranded RNA, has been shown to specifically reduce homologous mRNA levels through the actions of several enzymes (as reviewed in (Agrawal et al., 2003)). Although previous work has indicated that Keap1 siRNA knockdown can result in upregulation of ARE dependent genes in cells from peripheral organs (Cao et al., 2005, Devling et al., 2005, Gan et al., 2010, Singh et al., 2006, Tanigawa et al., 2007), no work has yet looked in depth at the effect of Keap1 siRNA in the brain or in primary neuronal/astrocytic cultures. Here, we characterize the effect of Keap1 siRNA administration in primary astrocytes and their ability to protect neurons from oxidative stress-induced cell death. In conjunction with these in vitro studies, in vivo studies evaluating the ability of striatal injection of Keap1 siRNA to protect against MPTP-induced dopaminergic neuron damage/loss was examined.
1. Materials and Methods
1.1. Primary culture and dosing
For primary astrocyte culture preparation, one litter of P1 mouse pups expressing the ARE-hPAP reporter as reported in (Johnson et al., 2002) were anesthetized with isofluorane and decapitated. The skull and meninges were removed with forceps and scissors and the cortices were removed and pooled in 10 mL ice-cold Ca2+ and Mg2+ free HBSS. Tissue was minced and centrifuged, trypsinized in 0.05% trypsin without EDTA in HBSS and heated in a shaking water bath at 37°C for 10 minutes. Following this, cells were washed 3 times with 5 mL ice-cold CEMEM medium, consisting of minimum essential media (MEM) with Earle's Salts (Life Technologies, Carlsbad, CA) and glutamine, 1% penicillin/streptomycin, and 10% each of 55°C heat inactivated fetal bovine serum and horse serum (Atlanta Biologicals, Inc., Lawrenceville, GA), triturated to a single cell suspension, filtered through a 70 μm cell strainer (BD Bioscience, San Jose, CA), and seeded at a density of 3x104 cells/cm2. Cells were maintained in CEMEM; the medium was changed 24 hours after culture and every 3 days following. Experiments were performed after the cells were confluent. A similar preparation was performed for mixed neuron and astrocyte and neuronally enriched cultures. Embryonic day 15 (E15) embryos were prepared as discussed above and plated on poly-D-lysine at a density of 3×105 cells/cm2. The medium was changed to Neurobasal medium (Life Technologies) supplemented with B27 with antioxidants and 2 mM glutamine (NBM) after 2 days (for mixed culture; 30-50% astrocytes and 50-70% neurons) or 45 minutes (for neuronally enriched culture; 95-99% neurons). Nrf2 knockout (KO) cultures were prepared from embryos of transgenic mice as described in (Chan et al., 1996). Cells were maintained in a tri-gas incubator with O2 and CO2 both held at 5% and N2 at 90%.
After astrocytes were confluent, siRNA was complexed with Lipofectamine™ 2000 (Life Technologies) according to manufacturer's instructions and cells were dosed at a final concentration of 50 nM. Mouse nontargeting siRNA (siNT) or siRNA specific for the mouse Keap1 via the NCBI reference sequence NM_016679 (siKeap1) was obtained from Dharmacon/Thermo Fisher Scientific (Lafayette, CO). Cells were incubated with siRNA for 24 hours followed by a medium change into fresh CEMEM and maintenance for the times indicated. All time points begin at t=0 when the siRNA complexes or compounds were administered to the cells. As a positive control, tert-butyl hydroquinone (tBHQ) was used at a final concentration of 50μM in 0.05% EtOH, with a 0.05% EtOH vehicle used as a control. Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Lafayette, CO).
For co-plating experiments, astrocytes were treated with siRNA as indicated above in 6 well plates, then left for the times indicated after dosing. Astrocytes were then lifted with accutase (Millipore, Billerica, MA), washed and centrifuged twice with fresh medium, resuspended in NBM, counted, diluted out to the numbers indicated, and replated through a ½ medium change onto the neuronally enriched culture. Cells were left for 2 days after co-plating and then the medium was changed to NBM minus antioxidants (AO) for 24 hours in conjunction with tert-butyl hydroperoxide (tBOOH) treatment.
1.2. In vitro assays
The hPAP activity assay protocol was taken from (Johnson et al., 2002, Kraft et al., 2007). Whole cell extracts were prepared by lysing cells in TMNC lysate buffer (50mM TRIS pH 7.5, 5mM MgCl2, 100mM NaCl2, 1% CHAPS detergent), followed by a freeze-thaw at -20°C. 75μL of diethanolamine (DEA) at 200 mM, pH 9.5-10 was added to the bottom of white-bottomed 96 well plates, to which 25μL of cell lysate was added. The plates were sealed and heated in a water bath at 65°C for 30 minutes to inactivate endogenous phosphatase activity. Following this, the substrate mix solution was added (0.8mM CSPD, 2× Emerald (both from Applied Biosystems/Life Technologies), 5mM MgCl2 in DEA buffer) and plates were incubated in the dark for 10 minutes and the luminescent signal was read on an Orion microplate luminometer (Berthold Detection Systems, Pforzheim, Germany) with one second integration. The resulting signal was subtracted from signal from hPAP negative control cultures.
Cell viability was assayed using the tetrazolium salt 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) (Promega, Madison, WI) as a substrate following the manufacturer's protocols. To challenge neurons, a complete medium change into NBM minus AO medium with a free radical generator such as tert-butyl hydroperoxide (tBOOH) was applied at the concentrations indicated. Cells were incubated for 24 hours followed by analysis via the MTS assay. Initial experiments were conducted using the lactate dehydrogenase (LDH) cytotoxicity assay to validate the MTS data. There was a good correlation between the MTS and LDH assay thus only the MTS data is presented. The protocol for astrocyte viability was similar, except that astrocytes were maintained in CEMEM throughout the experiment.
For mRNA analysis, total RNA was isolated from primary astrocyte cultures in 6 well plates, or after homogenization of mouse striatum with Trizol® reagent (Gibco/Life Technologies) following the manufacturer's instructions. RNA quality was evaluated using an Agilent 2100 Bioanlyzer with the RNA 6000 NanoAssay kit (both from Agilent, Santa Clara, CA) and only samples with RNA integrity number (RIN) values of ≥ 8.0 were analyzed. RNA was converted to cDNA using the Reverse Transcription System from Promega following the manufacturer's protocol. Relative cDNA levels were assayed using the LightCycler 480 system by Roche (Basel, Switzerland) with primers for NQO1, GCLC, GCLM, Nrf2, Keap1, hPAP and β -actin as previously described (Chen et al., 2009, Gan et al., 2010, Johnson et al., 2010, Vargas et al., 2008). Primer sequences were as follows: bactin 5′ CAT GAA GAT CCT GAC CGA GCG TG 3′, 5′ TCT GCT GGA AGG TGG ACA GTG AGG 3′; Keap1 5′ AAG GAC CTT GTG GAA GAC CA 3′, 5′ CCC TGT CCA CTG GAA TTG AT 3′, Nrf2 5′ TTC TTT CAG CAG CAT CCT CTC CAC 3′, 5′ ACA GCC TTC AAT AGT CCC GTC CAG 3′, GCLM 5′ GCC ACC AGA TTT GAC TGC CTT TG 3′, 5′ TGC TCT TCA CGA TGA CCG AGT ACC 3′, GCLC 5′ ACA TCT ACC ACG CAG TCA AGG ACC 3′, 5′ CTC AAG AAC ATC GCC TCC ATT CAG 3′, NQO1 5′ GCG AGA AGA GCC CTG ATT GTA CTG 3′, 5′ TCT CAA ACC AGC CTT TCA GAA TGG 3′; hPAP 5′ TTG CCC CAA ATC TCA ACT TC 3′, 5′ TGG GTA GCT GGG ACT ACA GG 3′. Relative cDNA levels were compared to β-actin as an internal control.
For histochemical staining of hPAP activity, heat-inactivated cells or tissue sections were stained for hPAP (antioxidant response element activation) using 1 mg/mL NBT and 1 mg/mL X-phosphate (5-bromo-4-chloro-3-indoyl-phosphate (BCIP); (EMD Chemicals, Gibbstown, NJ) and incubation at 37°C for 30-45 minutes. Immunohistochemical staining for astrocytes was performed using an antibody to glial fibrillary acidic protein (Millipore) at 1/1000. Tyrosine hydroxylase (TH) for dopaminergic neurons was performed using a primary antibody from Millipore at 1/1000. Secondary detection was performed with fluorescently labeled antibodies or conjugated with an avidin-biotin-horseradish peroxidase complex and DAB (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. The Hoescht stain was used to identify nuclear DNA.
1.3. In vivo experiments
All animal work was done in accordance with the University of Wisconsin Madison animal care and use protocols. Intrastriatal injection of siKeap1 was performed on ARE-hPAP mice (Johnson et al., 2002) Mice were anesthetized with 4% isoflurane and maintained at 1.5%. Their skulls were immobilized in a stereotactic injection unit (Stoelting, Wood Dale, IL), eyes were swabbed with aquatears, and the scalp was swabbed with triadine as an antiseptic. The scalp was opened with a razor, a hole was drilled in the skull at AP +0.5 and ML +/- 2.1, and the needle of a syringe preloaded with siKeap1 (20 μM in PBS) or tBHQ (50 mM in 10% EtOH) was lowered 3.3 mm into the brain. One μL of solution was injected with a syringe and 32 gauge needle (Hamilton, Reno NV) at a rate of 0.2 μL/min, the needle was left in place for 5 minutes and then slowly removed over the next 5 minutes. The contralateral hemisphere was injected with control siNT (20 μM in PBS) or vehicle (10% EtOH) in the same manner. The wound was closed using staples and the animal was allowed to recover under a heat lamp.
For the MPTP experiments the mice were injected bilaterally with either siNT or siKeap1 four days prior to initiating MPTP treatment. MPTP was administered to cohorts of 8 week old mice (8-10 per group) intraperitoneally once a day for 5 days with 30mg/kg MPTP in PBS, or an equal volume of PBS as vehicle. After the 5 day injection period, animals were allowed to recover for an additional 7 days before sacrificing and collecting tissues. Animals were kept quarantined after exposure to MPTP.
At the end of the experiment, animals were over-anesthetized with CO2 and transcardially perfused with 10 mL PBS. For histological analysis, mice were subsequently perfused with 10 mL of 4% paraformaldehyde (Electron Microscopy Services, Hatfield, PA), then the brains were removed and the brains were incubated in 4% paraformaldehyde for 2 hours, followed by PBS overnight, and sunk in 30% sucrose followed by freezing and sectioning on a cryostat. For western blots or dopamine metabolite analysis, brains were removed following the PBS perfusion, placed in a rodent brain matrix (ASI Instruments, Warren, MI) to generate a 2mm slice of the brain containing the striatum, the striatum was dissected from the slice, snap frozen in liquid N2, and stored at -80°C. Samples for western blot were prepared and run as previously described (Gan et al., 2010). Briefly, samples were sonicated in RIPA buffer (50mM TRIS-HCl, pH 7.4, 150mM NaCl, 1% NP-40, 0.5% SDS, and Complete Protease Inhibitor from Roche) and protein content was quantified using the BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins (20 μg) were separated on a 10% sodium dodecyl sulfate polyacrylamide gel and transferred to a PVDF membrane. Membranes were blocked with 5% BSA in Tris-buffered saline-Tween 20 and incubated with primary antibodies to tyrosine hydroxylase (Millipore, 1/1000) or to β-actin (Abcam, 1/10,000) overnight, followed by washing and incubation with a secondary antibody labeled with horseradish peroxidase and visualization with Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Samples for HPLC were snap frozen, sonicated in 0.2N perchloric acid, and run on a Shimadzu HPLC (Shimadzu, Kyoto Japan). Dopamine metabolites were detected with an Eicom detector (Eicom, Kyoto, Japan) with an applied potential of 750 mV vs Ag/AgCl electrode according to manufacturer's instructions.
1.4. Statistics
All data reflect at least three independent determinations and significance was determined by using either a unpaired Student's t-test (p<0.05) or one-way ANOVA (p<0.05) followed by a Newman-Keuls posthoc analysis to determine statistically significant paired comparisons (p<0.05) depending on the experimental design.
2. Results
2.1. Keap1 knockdown activates Nrf2 in astrocytes
Primary astrocyte cultures derived from ARE-hPAP reporter mice (Johnson et al., 2002) treated with siKeap1 showed a significant reduction in Keap1 mRNA levels (∼80%) at 1, 2, 4 and 7 days relative to siNT, with no corresponding change in Nrf2 mRNA (Table 1). The effect of siKeap1 administration was compared to tBHQ, a known chemical activator of the Nrf2-ARE pathway. tBHQ treatment actually increased Keap1 mRNA after 1 day, but not after 2, 4 and 7 days. In addition, tBHQ significantly increased Nrf2 mRNA levels at 2 days, but not 1, 4 or 7 days (Table 1). For both siKeap1 and tBHQ treated astrocytes, significant increases were observed in endogenous genes regulated by the Nrf2-ARE pathway, including glutamate cysteine-ligase, catalytic subunit (GCLC), glutamate cysteine-ligase, modifier subunit (GCLM), and NAD(P)H:quinine oxidoreductase 1 (NQO1) (Table 1). In all cases, mRNA changes were restored to control levels by 7 days post administration in tBHQ-treated astrocytes, but were sustained in the siKeap1-treated astrocytes. There was an associated increase in ARE-driven hPAP activity following siKeap1 treatment at 2, 3, and 4 days (Fig. 1A). Administration of 4 different siRNA sequences specific for Keap1, but not Lipofectamine™ 2000 alone or siNT, resulted in increased hPAP activity 4 days post transfection (Fig. 1B). The extent of increased activity was similar to that observed with the chemical inducer tBHQ, but slightly delayed since a significant increase in hPAP mRNA and activity was present by 24 hr with tBHQ and 48 hr with siKeap1 (Table 1 and Fig. 1A).
Table 1. Astrocytes treated with siKeap1 or tBHQ upregulate Nrf2-ARE controlled genes.
Confluent astrocytes were collected at 1,2, 4, and 7 days after a 24 hour administration of siKeap1 or tBHQ. Data are represented as fold change from controls, using siNT as the control for siKeap1 and 0.05% EtOH vehicle for tBHQ. * p<0.05 relative to control. n=3, error SEM.
| Gene | siKeap1 1 day |
2 day | 4 day | 7 day | 1 day tBHQ |
2 day | 4 day | 7 day |
|---|---|---|---|---|---|---|---|---|
| Keap1 | 0.27±0.13* | 0.13±0.03* | 0.38±0.13* | 0.22±0.07* | 2.95±0.62* | 0.84±0.01 | 1.32±0.57 | 0.89±0.16 |
| Nrf2 | 0.88±0.38 | 1.50±0.62 | 0.81±0.13 | 0.66±0.11 | 1.21±0.05 | 1.73±0.08* | 0.80±0.16 | 0.93±0.07 |
| GCLM | 5.92±2.30* | 13.8 ±7.39 | nd | 6.09±1.02* | 17.9±2.11* | 0.92±0.21 | nd | 1.20±0.13 |
| GCLC | 2.00±0.84* | 3.52±0.31* | 1.80±0.44 | 2.88±0.40* | 5.97±0.88* | 0.82±0.10 | 0.92±0.55 | 0.84±0.14 |
| NQO1 | 2.77±0.37* | 5.34±0.74* | 13.9±4.18 | 4.00±1.24* | 22.0±4.22* | 1.62±0.05 | 1.13±0.27 | 0.73±0.21 |
| hPAP | 17.5±11.3 | 109±21.0* | 280±158 | 695±183* | 84.8±15.0* | 36.1±10.7* | 9.65±1.68* | 0.99±.01 |
Figure 1. Keap1 knockdown upregulates of ARE-hPAP activity in vitro in a time-dependent and siKeap1 dependent manner.
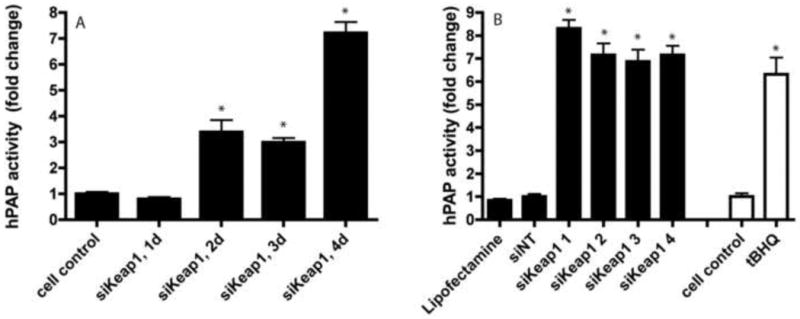
A) Confluent astrocytes were treated with siKeap1 for 24 hours followed by media change and ARE-hPAP activity levels were assayed at timepoints from 1-4 days and compared to untreated cell control. B) Four different siRNA sequences all unique to Keap1 (siKeap1 1-4) were administered for 24 hours and tested for hPAP reporter activity in confluent astrocyte culture 4 days post administration. hPAP activity of tBHQ after 24 hours administration was used as a positive control. n=6; error bars SEM; * p<0.05, siKeap1 vs. siNT (black bars) and tBHQ vs vehicle (white bars).
2.2. Keap1 knockdown confers protection through an Nrf2-dependent mechanism
Four days after treatment with siKeap1, astrocytes were challenged with a 24 hour administration of tert-butyl hydroperoxide (tBOOH). Keap1 knockdown conferred protection against tBOOH in Nrf2 WT astrocytes (Fig. 2A) but not Nrf2 KO astrocytes (Fig. 2B). To determine whether siKeap1 astrocytes could protect neurons from oxidative stress-induced toxicity, siKeap1 astrocytes were plated onto a previously prepared neuronally enriched culture at a ratio of 10,000 astrocytes to 100,000 neurons (Fig. 2C). Astrocytes were treated with siKeap1 for 24 hours, incubated in fresh medium for another 24 hours, and then co-plated and co-incubated with the neuronal cultures for an additional 48 hours prior to tBOOH challenge (24 hours). Nrf2 WT siKeap1 astrocytes conferred protection to neurons when compared to Nrf2 WT siNT astrocytes. In contrast, Nrf2 KO siKeap1 astrocytes conferred little to no protection to neurons when compared to Nrf2 KO siNT astrocytes. As expected, the addition of any astrocytes increased the resistance of the neurons to toxic insult. However, the conferred neuroprotection was greater for Nrf2 WT compared to Nrf2 KO astrocytes in the presence or absence of siKeap1 (Fig. 2C).
Figure 2. Keap1 knockdown protects primary astrocytes and neuronally enriched cultures incubated with siKeap1 treated astrocytes against tert-butyl hydrogen peroxide in an Nrf2-dependent manner.

Confluent astrocytes were treated with siKeap1 or siNT for 24 hours four days prior to a 24 hour tBOOH challenge in A) wild-type (WT) or B) Nrf2 knockout (KO) primary astrocyte cultures; n=4, error bars SEM, * p<0.05 siKeap1 vs siNT treated cells. C) Confluent primary astrocytes were treated with siNT or siKeap1 for 24 hours, incubated in fresh medium for 24 hours and then lifted, dissociated, and coplated onto neuronally enriched cultures at a plating density of 10,000 astrocytes per 100,000 neurons for 48 hours, followed by a 24 hour tBOOH challenge. Astrocytes were either WT or KO, and plated onto WT neurons. n=3, error bars are SEM. * p< 0.05, no astrocyte addition vs siNT treated astrocytes on neuronally enriched cultures. # p <0.05 WT siNT vs WT siKeap1 astrocytes on neuronally enriched cultures; † p <0.05 KO siNT vs KO siKeap1 on neuronally enriched cultures.
2.3. Nrf2-ARE activation after Keap1 knockdown is persistent in in vitro astrocyte culture
In an initial experiment using four different siKeap1 sequences, hPAP activity was sustained for 3 weeks after one 24 hour transfection (Fig. 3A). A full time course revealed that siKeap1 astrocytes maintained activation of hPAP (Fig 3B) and NQO1 (Fig. 3C) activity up to six weeks post transfection with a maximum activity at 21 (25-fold) and 14 days (3-fold), respectively. In contrast, tBHQ treated astrocytes displayed transient activation of both hPAP (up to 7 days) and NQO1 (up to 14 days) activity post transfection before returning to baseline.
Figure 3. Nrf2-ARE regulated proteins are increased for 6 weeks after a single treatment of siKeap1.
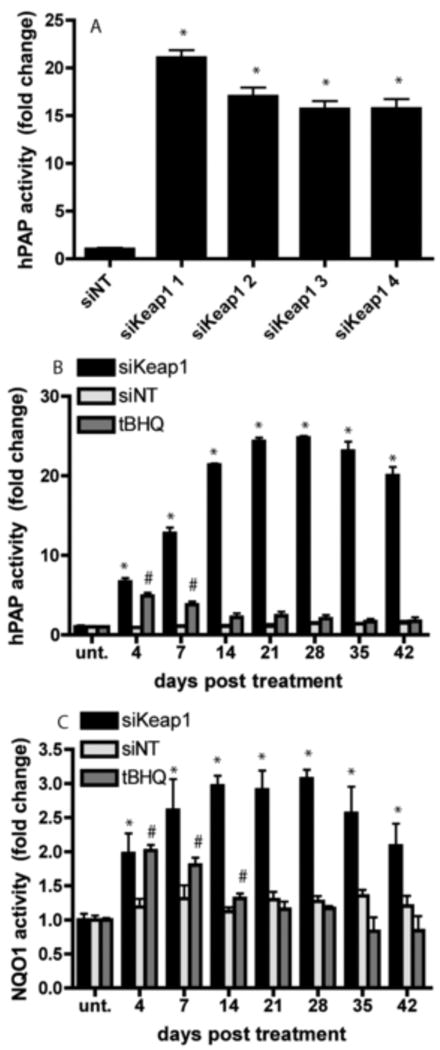
A) Four different siRNA sequences all unique to Keap1 (siKeap1 1-4) and siNT were administered for 24 hours and tested for hPAP reporter activity in confluent astrocyte culture 3 weeks post treatment. * p<0.05, siKeap1 vs siNT. B) hPAP reporter activity and C) NQO1 activity was assayed from 4-42 days after a single 24 hour treatment with siKeap1 (black bars), siNT (light grey bars) or tBHQ (dark grey bars) in confluent astrocytes. * p<0.05, siKeap1 vs siNT; #p<0.05, tBHQ vs siNT.
To determine if siKeap1 astrocytes maintained resistance to oxidative stress induced cell death, siKeap1 astrocytes were exposed to tBOOH after 1, 2 or 3 weeks post transfection as compared to siNT astrocytes (Fig. 4A). A single exposure of siKeap1 conferred continued resistance to tBOOH, indicating that activation of the Nrf2-ARE pathway was maintained at extended time points. The sensitivity of siNT astrocytes to tBOOH at 7 days (Fig. 4A) was not significantly different for those at 14 or 21 days (data not shown).
Figure 4. Transplantation of siRNA treated astrocytes onto neuronally enriched culture protects against tBHP mediated toxicity for three weeks after siRNA administration.
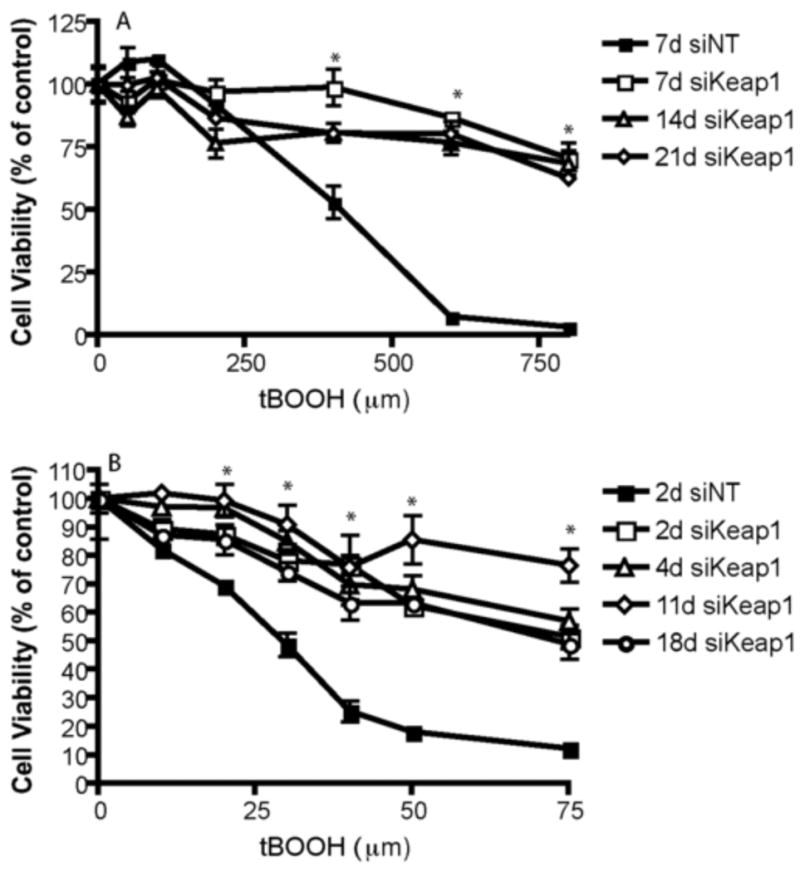
A) Cell viability was assayed in confluent primary astrocytes treated with siKeap1 or siNT for 24 hours and challenged with tBOOH 1,2, or 3 weeks post siRNA treatment. n=3, error bars SEM. * p<0.05, all siKeap1 vs. siNT. B) Confluent astrocytes were treated with siRNA for 24 hours, followed by medium change and subsequent lifting, dissociation, and coplating with neurons at a density of 10,000 astrocytes onto 100,000 cells in a neuronally enriched culture per well of a 96 well plate at the timepoints indicated; at all timepoints astrocytes were incubated with neurons for 48 hours followed by tBOOH challenge and assay of cell viability. n=3, error bars SEM. * p<0.05 all siKeap1 timepoints are significantly different than nontargeting siRNA treated.
To further evaluate if siKeap1 astrocytes could protect neurons from oxidative stress induced toxicity in vitro long after administration with siKeap1, siKeap1 astrocytes were incubated for 24 hours with siRNA, then maintained for a total of 2-18 days post transfection, lifted and co-plated with neuronally enriched cultures for 48 hours prior to challenge with tBOOH for 24 hours (Fig. 4B). The siKeap1 astrocytes conferred protection to neurons at all timepoints as compared to siNT astrocytes. The sensitivity of neurons to tBOOH when co-plated with siNT astrocytes at 2 days (Fig. 4A) was not significantly different from those at 4, 11 or 18 days (data not shown).
2.4. Keap1 siRNA in vivo after direct brain injection
To determine if siKeap1 could activate the Nrf2-ARE pathway in vivo, the striatum of ARE-hPAP mice was stereotactically injected with either siNT (contralateral control) or siKeap1 (ipsilateral). There was a dramatic increase in hPAP histochemical staining in the siKeap1 injected striatum relative to the siNT striatum one week post injection (Fig. 5A). This was quantified by measuring hPAP activity in the striatum nine days after injection (Fig. 5B). SiKeap1 was administered uncomplexed with any transfection reagents. However, unlike the in vitro data, the increase in the Nrf2-ARE pathway in striatum returned to baseline levels by 3 weeks post-injection (data not shown).
Figure 5. hPAP activity is increased after siKeap1 injection in the striatum of ARE:hPAP animals.
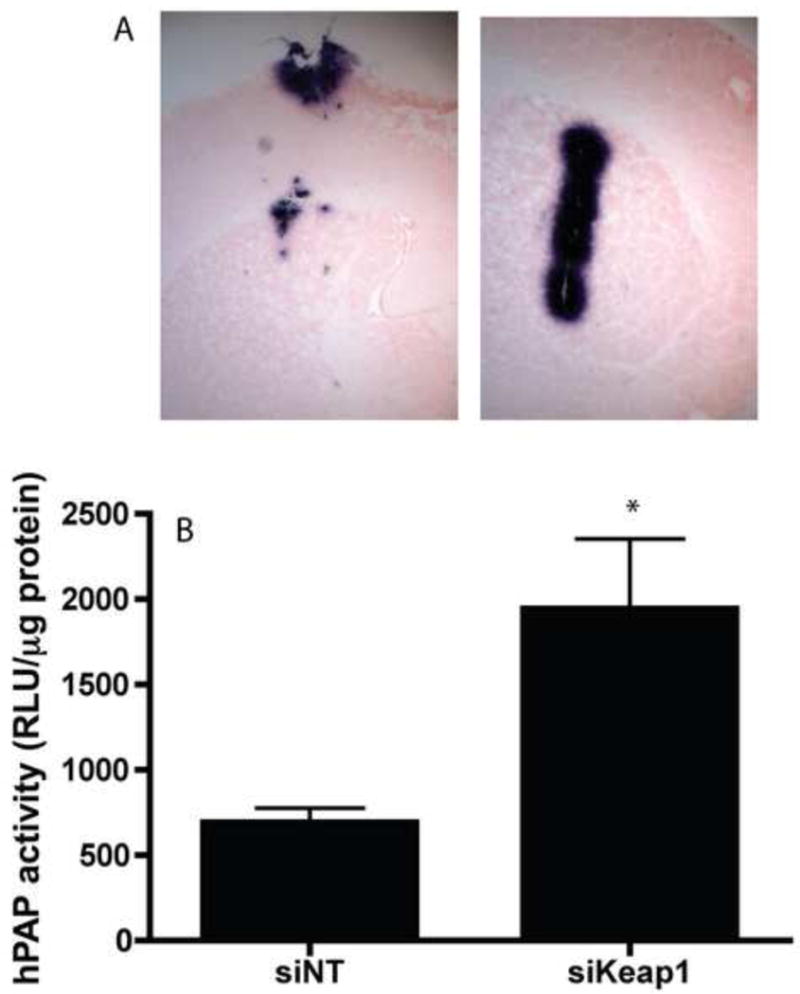
A) ARE-hPAP reporter mice were injected with one μL 20 μM siNT (left panel) or 20 μM siKeap1 (right panel) in the striatum and harvested 1 week postinjection and sectioned and stained for hPAP reporter activity via BCIP/NBT staining. Images are taken at 5×. B) ARE-hPAP reporter mice treated as described above and sacrificed 9 days post injection, the striatum was dissected out and snap frozen, and hPAP activity was quantitated. n=3, SEM, *p<0.05.
To determine whether siKeap1 pre-administration was sufficient to protect against a toxicity challenge in vivo, siKeap1 was injected into the striatum of mice four days prior to a subchronic MPTP model of Parkinson's disease. The loss of protein levels of tyrosine hydroxylase (TH), a marker for dopaminergic neurons and projections, were significantly reduced in siKeap1 MPTP mice in the striatum relative to siNT MPTP mice as determined by immunohistochemistry (Fig. 6A) and western blot (Fig. 6C). Astrocytic gliosis was also reduced in siKeap1 MPTP treated mice (Fig. 6B) in the striatum relative to siNT MPTP treated mice. This was not, however, reflected in a reduced loss of striatal dopamine (Fig. 6D) or dopamine metabolite (DOPAC and HVA; data not shown) levels.
Figure 6. siKeap1 is mildly protective in the striatum after subchronic MPTP challenge in vivo.
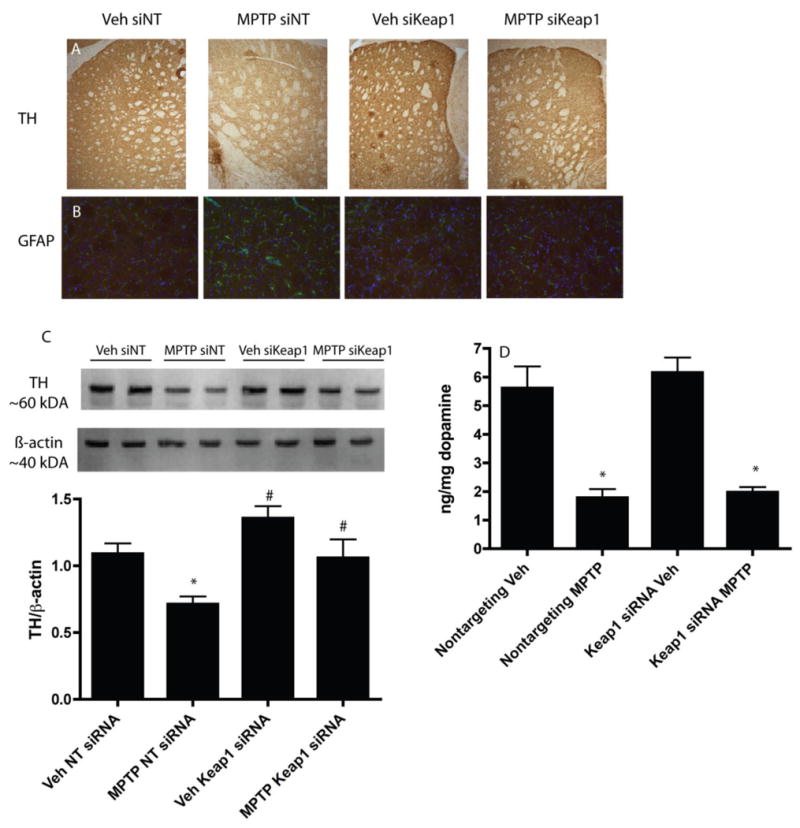
A) Mice were injected intrastriatally with 1 μL of 20 μM siKeap1 or siNT 4 days before the initiation of the subchronic MPTP protocol, and collected 7 days after MPTP injections and stained for tyrosine hydroxylase (TH) in the striatum. Images are taken at 20×. B) The striatum was also stained for GFAP (G) and DAPI (B), images taken at 40×. C) TH levels in the striatum were quantified via western blot. * p<0.05, MPTP vs vehicle. # p<0.05 siKeap1 vehicle vs siNT vehicle; # p<0.05, siKeap1 MPTP vs siNT MPTP. n=6 for quantification, with a representative n=2 shown on western blot. D) Dopamine levels were quantified via HPLC after completion of the MPTP protocol. Dopamine content shown as a ratio of ng dopamine per mg tissue wet weight. n=7-8, *p<0.05, vehicle vs MPTP treated.
3. Discussion
SiRNA knockdown of Keap1 resulted in upregulation of the Nrf2-ARE pathway in the brain and brain-derived primary cultures. In primary astrocytes, siKeap1 knockdown confers protection in vitro in an Nrf2-dependent manner, suggesting that activation of endogenous intracellular levels of Nrf2 can provide a therapeutic benefit. In addition, siKeap1 astrocytes also protect neurons from oxidative stress-induced cell death, adding support to the central hypothesis that astrocytic Nrf2 activation increases the ability of the astrocyte to support neurons.
Interestingly, the siKeap1-dependent activation of the Nrf2-ARE pathway persisted in vitro much longer than expected after a single treatment of siRNA. Specifically, activation of the Nrf2-ARE pathway was observed out to at least six weeks post transfection in confluent astrocytes. Since the primary astrocytes used in this study were not treated with siRNA until they were confluent, the lack of cell division could relate to the persistence of the siRNA in the cells. Previous reports have suggested that persistent effects of siRNA can be observed in non-dividing but not dividing cells (Bartlett and Davis, 2006, Omi et al., 2004, Song et al., 2003). No persistent activation of the Nrf2-ARE pathway was observed in tBHQ treated cells, implying that extended activation is a function of the siKeap1 rather than an inherent feature of the Nrf2-ARE pathway.
We observed modest protection in the striatum of mice treated with siKeap1 prior to challenge with the subchronic MPTP model. Despite the less than robust protection, it is encouraging to see some protection in this model after activation of the endogenous Nrf2-ARE pathway, rather than through overexpression of Nrf2 (Chen et al., 2009). The limited protection observed may be due to the constraints on diffusion of siRNA in the brain. As observed in Fig. 6A, the distribution of the ARE activation was restricted to the area around the needle track. Because siRNA is a negatively charged oligonucleotide, it does not diffuse extensively through the extracellular space in the brain (Thorne and Nicholson, 2009), which is populated by the negatively charged glycoproteins studded on the cell surfaces. Since most of the striatum was dissected out for analysis after MPTP treatment, it is possible that the unprotected areas were masking the more localized protection in the area of Nrf2-ARE activation. Indeed, the TH and GFAP staining that was done on sections near the injection site (Fig. 6A and 6B) would argue that there is greater localized protection than that reflected by the biochemical measures (Fig. 6C and 6D). Other research has found similar diffusion limitations when siRNA is injected locally with an osmotic minipump to the brain parenchyma (Agrawal et al., 2009) or to the brain ventricles (Senn et al., 2005). However, sustained distribution using an osmotic minipump in the ventricles has been reported to have more diffuse effects, albeit with modest knockdown of about 30% (Thakker et al., 2004, Thakker et al., 2005). Other work has attempted to deliver siRNA intravascularly across the blood brain barrier. Although there have been successful reports of siRNA knockdown after brain delivery across the blood brain barrier (Kumar et al., 2007), the technology is still highly experimental and not commercialized or widely available.
Given our in vitro finding that the siKeap1-induced Nrf2-ARE activation is persistent in non-dividing brain derived cells, it was interesting that the Nrf2-ARE pathway was not persistently activated in the striatum after siKeap1 administration. This in vitro versus in vivo difference could be due to many factors in vivo that are not major issues in vitro. Some possible explanations could be a modest inflammatory response to the needle stick leading to astrogliosis and microglial activation, as well as potential cell division and wound healing around the needle stick of the injection site. If siRNA could be safely administered to un-damaged and non-dividing brain tissue using some of the delivery methods being developed, it may persist longer than in areas of the body where cell division occurs more rapidly. This would significantly reduce the dosing schedule and improve the treatment of neurodegenerative diseases where therapeutics may need to be administered regularly for long periods of time.
The results show that modulation of Keap1 levels can have direct effects on the Nrf2-ARE pathway, both in the brain and in brain-derived primary cultures, and that modulation of Keap1 can result in Nrf2-ARE dependent protection. This indicates that therapeutic modulation of the endogenous levels of Nrf2 instead of genetically induced overexpression can be therapeutic if drugs or siRNA treatments can be developed that cross the blood brain barrier and activate the Nrf2-ARE pathway in the brain. Indeed, researchers have reported some protection after systemic administration of Nrf2-activating small molecules (Jazwa et al., 2011, Yang et al., 2009) in the MPTP model of Parkinson's disease. A benefit of using siRNA, rather than small molecule activators that could have numerous off-target effects, is target specificity. Herein, we have shown the validity of such an approach by targeting Keap1 and its potential for extended modulation of the Nrf2-ARE pathway in post-mitotic differentiated astrocytes in vitro. Targeting Keap1 in vivo using siRNA also appears to have significant therapeutic potential, but remains dependent on the development of improved siRNA delivery methods.
*Highlights.
In this study we determine the effect of Keap1 siRNA on the Nrf2-ARE pathway.
Keap1 siRNA activated the Nrf2-ARE pathway and protected against oxidative stress.
In vivo, Keap1 siRNA had modest protection in the MPTP model of Parkinson's disease.
We conclude that Keap1 siRNA is a potential therapeutic for Parkinson's disease.
Acknowledgments
We thank Sara Amirahmadi, Jennifer Kutzke, and Jonathan Resch for generation of the animals used in these experiments. This work was supported by a grant from the National Institute of Environmental Health Sciences ES10042. TPW was supported by award number T32GM008349 from the National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal A, Min DH, Singh N, Zhu H, Birjiniuk A, von Maltzahn G, et al. Functional delivery of siRNA in mice using dendriworms. ACS Nano. 2009;3:2495–504. doi: 10.1021/nn900201e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev. 2003;67:657–85. doi: 10.1128/MMBR.67.4.657-685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006;34:322–33. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao TT, Ma L, Kandpal G, Warren L, Hess JF, Seabrook GR. Increased nuclear factor-erythroid 2 p45-related factor 2 activity protects SH-SY5Y cells against oxidative damage. J Neurochem. 2005;95:406–17. doi: 10.1111/j.1471-4159.2005.03377.x. [DOI] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–8. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: Critical role for the astrocyte. Proc Natl Acad Sci U S A. 2009;106:2933–8. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devling TW, Lindsay CD, McLellan LI, McMahon M, Hayes JD. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc Natl Acad Sci U S A. 2005;102:7280–5A. doi: 10.1073/pnas.0501475102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Johnson DA, Johnson JA. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur J Neurosci. 2010;31:967–77. doi: 10.1111/j.1460-9568.2010.07138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–75. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–91. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernandez-Ruiz J, Cuadrado A. Pharmacological Targeting of the Transcription Factor Nrf2 at the Basal Ganglia Provides Disease Modifying Therapy for Experimental Parkinsonism. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2010.3731. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–46. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–41. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol. 2006 doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, Resch JM, Johnson DA, Johnson JA. Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp Neurol. 2007;207:107–17. doi: 10.1016/j.expneurol.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Hee Kim M, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–57. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Omi K, Tokunaga K, Hohjoh H. Long-lasting RNAi activity in mammalian neurons. FEBS Lett. 2004;558:89–95. doi: 10.1016/S0014-5793(04)00017-1. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–9. [PubMed] [Google Scholar]
- Satoh T, Harada N, Hosoya T, Tohyama K, Yamamoto M, Itoh K. Keap1/Nrf2 system regulates neuronal survival as revealed through study of keap1 gene-knockout mice. Biochem Biophys Res Commun. 2009;380:298–302. doi: 10.1016/j.bbrc.2009.01.063. [DOI] [PubMed] [Google Scholar]
- Schafer M, Dutsch S, auf dem Keller U, Navid F, Schwarz A, Johnson DA, et al. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010;24:1045–58. doi: 10.1101/gad.568810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn C, Hangartner C, Moes S, Guerini D, Hofbauer KG. Central administration of small interfering RNAs in rats: a comparison with antisense oligonucleotides. Eur J Pharmacol. 2005;522:30–7. doi: 10.1016/j.ejphar.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohe R, et al. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35:639–50. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Lee SK, Dykxhoorn DM, Novina C, Zhang D, Crawford K, et al. Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J Virol. 2003;77:7174–81. doi: 10.1128/JVI.77.13.7174-7181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa S, Fujii M, Hou DX. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med. 2007;42:1690–703. doi: 10.1016/j.freeradbiomed.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Thakker DR, Natt F, Husken D, Maier R, Muller M, van der Putten H, et al. Neurochemical and behavioral consequences of widespread gene knockdown in the adult mouse brain by using nonviral RNA interference. Proc Natl Acad Sci U S A. 2004;101:17270–5. doi: 10.1073/pnas.0406214101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, Natt F, Husken D, van der Putten H, Maier R, Hoyer D, et al. siRNA-mediated knockdown of the serotonin transporter in the adult mouse brain. Mol Psychiatry. 2005;10:782–9. 14. doi: 10.1038/sj.mp.4001687. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C. In vivo optical imaging of oligonucleotide diffusion in brain extracellular space: Dramatically restricted transport compared to polyethylene glycol 39th Annual meeting abstracts. Society for Neuroscience. 2009;643:15. [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–81. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–45. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS One. 2009;4:e5757. doi: 10.1371/journal.pone.0005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–9. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]