

Abstract
Whole genome-wide scanning for susceptibility loci based on linkage disequilibrium (LD) has been proposed as a powerful strategy for mapping common complex diseases, especially in isolated populations. We recruited 389 individuals from 175 families in the US territory of American Samoa, and 96 unrelated individuals from American Samoa and the independent country of Samoa in order to examine background LD by using a 10 centimorgan (cM) map containing 381 autosomal and 18 X-linked microsatellite markers. We tested the relationship between LD and recombination fraction by fitting a regression model. We estimated a slope of -0.021 (SE 0.00354; p < 0.0001). Based on our results, LD in the Samoan population decays steadily as the recombination fraction between autosomal markers increases. The patterns of LD observed in the Samoan population are quite similar to those previously observed in Palau but markedly contrast with those observed in a non-isolated Caucasian sample, where there is essentially no marker-to-marker LD. Our analyses support the hypothesis of a recent bottleneck, which is consistent with the known demographic history of the Samoan population. Furthermore, population substructure tests support the hypothesis that self-identified Samoans represent one homogenous genetic population.
Keywords: association mapping, linkage disequilibrium, isolated population, Samoa
Introduction
Linkage disequilibrium (LD), the non-random association between loci, can aid in genetic mapping of complex diseases. It has been proposed that under high-density genetic maps, genome-wide LD scanning can be a powerful approach for searching for susceptibility genes determining complex diseases[1]. There have been several debates on the usefulness of isolated populations for mapping susceptibility genes determining complex diseases[2,3] and whether the extent of genome-wide LD is indeed larger in such populations compared with general populations[4-6]. Boehnke[7] notes that negative findings from a few populations, or simulation results,[8] should be elaborated with caution, and even if the extent of LD is similar across populations, the benefits of an isolated population living in a relatively homogeneous environment and the ease of study should not be ignored[9-13].
The Samoans of the Western Pacific represent one of the best examples of an isolated population. Archaeological and linguistic evidence[14] indicate a rapid eastward migration of populations into the western Pacific from Southern China, which took place about 4,000 to 5,000 years before the present (BP). By about 3,000 years BP, archaeological evidence indicates that Polynesian culture was established and flourished in Tonga and Samoa, well before further eastward expansion[15,16]. This Express Train model of Polynesian settlement[17] is supported by mtDNA data[18-20]. An alternative model, the Entangled Bank, proposed by Terrell,[21] asserts a neighbouring homeland for the Polynesians in Melanesia, in which the Polynesians evolved in a complex nexus of interactions among the already settled Pacific islanders. This is supported by nuclear DNA data[22]. Our previous work based on Y-chromosome SNP haplotypes, however, found no Melane-sian-specific haplotypes among the Polynesians, particularly the Samoans[23]. Samoans had only four haplotypes out of the 15 observed in the entire region of study comprising South-East Asia, Melanesia, Micronesia and Polynesia. In other work with microsatellite data, we also reported a significant reduction in genetic diversity among the Samoans compared with large cosmopolitan populations[24-26]. Further analysis of microsatellite data indicated a small effective population size and associated bottleneck events during Samoan history[27].
Reconstruction of the population history of the Samoan islands is difficult, and estimates of population size through time, including at the time of first European contact and for the subsequent 100 years, remain debatable[28,29]. Nonetheless, it is important to describe the archaeological evidence and population genetic interpretations of Samoan demographic history. The original settlers of Polynesia were thought to be small in number, however, the ideal nutritional and disease ecology allowed rapid and sustained population growth up until the time of first European contact[16]. Detailed and well-dated archaeological and ethnohistorical evidence indicates that prior to European contact, Samoan villages were located at all levels on the mountain slopes (including at the top) in both what is now Samoa and American Samoa[15,16]. The archaeological findings of widespread population, beyond the littoral area, are consistent with estimates of the maximum population density, or human carrying capacity, derived from ethnographic work on agricultural intensity and population on other Polynesian islands[16]. Based on the wealth of archaeological and ethnographic data, contemporary scholars assert that the pre-European contact population of the Samoan islands could not have comprised fewer than 100,000 people and could have been as large as 300,000 people[15,16].
The Samoan population suffered a significant depopulation after European contact, which has been attributed largely to the impact of introduced diseases[16]. The earliest reports of Samoan population size were made in the middle of the 19th century, many decades after European contact, which occurred first in 1722, and again in 1768 and 1787, with steady contact established only in 1836. From 1849 until 1900, the population size estimates for the Samoan islands vary between 30,000 to 40,000 individuals[28]. Throughout the 19th century, there were documented epidemics of infectious disease, chiefly influenza, and estimates of high mortality attributable to these waves of disease[29,30]. The impact of infectious diseases is assumed to have started soon after European contact in the later 18th century. By the early 20th century, the population had again increased. Approximate population sizes for the Samoan islands in the 20th century are provided by McArthur[29] for several time periods; these include: about 39,800 for the period 1900-1910; about 50,000 in 1930; and about 69,500 in 1940. Thus, it is quite clear that a massive depopulation occurred in the Samoas after European contact and that population growth was stagnant until the early 20th century, when it grew rapidly. Based on the 2000 census of American Samoa (performed with the aid of the US Census Bureau), the population of ethnic Polynesians in American Samoa is 55,704. Based on projections from the 2001 census of Samoa, the population of ethnic Samoans in 2003 was estimated at approximately 165,000 in independent Samoa.
The combination of genetic, archaeological and demographic evidence strongly suggests that the Samoan population was established by a relatively small number of founders, has been an isolated population and experienced a reduction in population size about 200-300 years ago. These population history events are very likely to have influenced the patterns of LD in the contemporary Samoan population.
In this study, we examined the distribution of genome-wide LD in Samoa as a function of recombination based on marker data from a 10 centimorgan (cM) genome-wide scan. We also tested for bottleneck effects and population substructure.
Materials and methods
Subjects
A sample of 390 individuals (201 males, 189 females) from 177 families was recruited in American Samoa using the Department of Health diabetes registry as part of a genome-wide study of type 2 diabetes susceptibility genes[31]. Subjects were asked about their Samoan ancestry in order to limit study participation to those who reported that all four grandparents were of Samoan ethnicity, without European or Asian ancestry. Study protocols were approved by the Institutional Review Board of the Miriam Hospital, Providence, RI, USA. Written informed consent was obtained from all participants.
We also sampled 96 unrelated individuals (50 males, 46 females), 40 recruited from American Samoa and 56 from Samoa. None of these individuals had diabetes and they all self-reported that all four grandparents were Samoan. These samples were derived from our previous longitudinal study of adiposity and cardiovascular disease risk factors in American Samoa and Samoa from 1990 to 1995[32,33].
To provide a measure of marker-to-marker LD in a 'typical' outbred population, 333 unrelated North American Caucasians of European ancestry (172 males, 161 females) were selected, regardless of their disease status, from 229 pedigrees, each of which contained at least one affected individual with confirmed ankylosing spondylitis. Specifically, the 'founders' or the parents of the pedigrees were selected. The samples were collected through the North American Spondylitis Consortium (NASC). The genotyping data analysed in the present study contain information neither on individual identification nor on disease status of each individual. Geno-typing protocols used in genotyping NASC individuals are identical to those used in this study.
Genotyping
Buffy coats were prepared from 10 ml of blood in the field following standard protocols and shipped on dry ice to the laboratory in Cincinnati. DNA was isolated from buffy coats using the Puregene DNA isolation kit (Gentra Systems Inc) quantitated and arrayed in 96-well microtitre plates.
The genome scan was conducted with the Applied Biosystems Inc (ABI) PRISM linkage mapping set version 2, consisting of 400 microsatellite markers, with fluorescently labelled primers, spaced at an average 10 cM distance between markers. We conducted multiplex polymerase chain reaction (PCR) amplification (three to five markers in each PCR reaction) in a 7.5 μl final reaction volume containing, ~20 ng of genomic DNA and ~0.8-1.0 μl of AmpliTaq GoldTM DNA polymerase (5 U/μl). Initial incubation occurred for 12 minutes at 95°C; the first amplification was carried out for approximately 10-15 cycles in the PE GeneAmpeTM 9600 thermal cycler using the following parameters: denaturation at 94°C for 15 seconds, annealing at 55°C for 15 seconds and extension at 72°C for 30 seconds. The second amplification was carried out for approximately 20-23 cycles using the following parameters: first denaturation at 89°C for 15 seconds, annealing at 55°C for 15 seconds and extension at 72°C for 30 seconds; then denaturation at 94° C for 15 seconds, annealing at 55°C for 15 seconds and extension at 72° C for 30 seconds; and then a final extension at 72°C for ten minutes and an overnight incubation at 4°C.
Amplified DNA products underwent gel electrophoresis on an ABI 377 DNA sequencer using internal standard Gene Scan-400 ROX (PE Applied Biosystems) for 2.5 hours at constant power (3000 V, 60 mA, 200 W) and at 51°C. For quality control, a negative control and two positive control samples of known genotype [Centre du Etude Polymor-phisme Humain (CEPH) sample 1347-02] were run on each gel. GeneScan 3.1 and Genotyper 2.5 (PE Applied Biosystems) software were used for sizing and genotyping, respectively.
Statistical method
Measure of LD. We computed a multi-allelic version of the D' statistic,[34] which we define here using the notation found in the GOLD program documentation[35]. Consider two markers A and B. Let ni• be the number of haplotypes carrying allele i at locus A, n•j be the number of haplotypes carrying allele j at locus B and nij be the number of hap-lotypes carrying allele i at locus A and allele j at locus B. If N is the total number of haplotypes, then the allele frequencies pi• and p•j and haplotypes frequencies pij can be estimated as:
The multi-allelic definition of D' is then:
Haplotype frequency estimation and LD testing
For both the Samoan and the NASC sample, we used the same sets of 381 autosomal microsatellite markers and 18 X-linked microsatellite markers to evaluate LD between all pairs of markers on the same chromosome; there were 3,531 autosomal marker pairs and 153 X-linked marker pairs. The 'ldmax' program, which is part of the GOLD package,[35] was used to estimate haplotype frequencies for each marker pair, using the expectation-maximisation (EM) algorithm,[36,37] and to calculate the multi-allelic version of the D' statistic[34] for the autosomal marker data and females' X-linked marker data (GOLD website). Since haplotypes of X-linked data for males can be established unequivocally, we computed the D' statistic for males' X-linked data using a function we wrote in R[38]. Haplotype frequencies were estimated ignoring familial relationships -- Broma[39] has shown that such estimates, while they may be slightly less precise, are unbiased[40,41].
When the sample size is small, Lewontin's D' measure can be biased upwards[42]. We corrected this bias by performing a permutation analysis. We permuted the alleles at the first marker of each pair, calculated and recorded the new D', repeated these two steps 1,000 times and took the average of the D' over 1,000 permutations as the permuted D'. We also recorded the corresponding empirical p-value for each marker pair. We then computed an adjusted D' by taking the difference between the observed D' and the permuted D'; the adjusted D', which we denote as , should be an unbiased measure of LD. Teare et al.[42] evaluated this permutation correction and found that it works well under the null hypothesis and is generally an over-correction under alternative hypotheses, which suggests that the levels of LD presented here may be underestimated.
To examine the distribution of LD across the entire autosomal genome, we regressed the from the autosomal markers against the inter-marker recombination fractions. We also investigated the distribution of LD on the X chromosome by using the measures obtained from male and female data.
To test for heterogeneity in LD across the autosomal genome, we performed analysis of covariance, in which the predictors in the analysis were the chromosome number and the inter-marker recombination fractions.
We used 5 Y-chromosomal short tandem repeat (STR) markers (DYS388, DYS390, DYS391, DYS394, DYS395) in 20 unrelated Samoan males to estimate the diversity of Y haplotypes, which we computed as 1 - ΣPi, where Pi is the frequency of the ith observed haplotype.
Population bottleneck
A population bottleneck followed by population expansion creates an imbalance between heterozygosity and allele size variance. Under such conditions, the variance will, for a time, be higher than expected. We measured this by computing the imbalance index (β) of Kimmel et al.[43] using, after data cleaning, 371 autosomal markers for the Samoan population and 380 autosomal markers for the NASC population.
Population substructure
Using 25 unlinked autosomal loci, we used a correlated allele frequency model to make inferences about the underlying population substructure. We chose the 25 unlinked loci from our microsatellite marker data; all of these loci were in Hardy- We inberg equilibrium (it was necessary to use unlinked loci because the statistical tests we employed assume that all loci are unlinked). We picked two loci from chromosomes 1-3 that are at least 200 cM apart, so that the two loci are essentially unlinked. For the other chromosomes, we picked one locus from each chromosome. For these analyses, we used the 'structure' program (structure website) to fit clustering models with K = 1, 2, and 3 clusters; the 'structure' program has been shown to produce accurate population assignments with modest numbers of loci[44].
Results
We used 381 autosomal microsatellite markers and 18 X-linked microsatellite markers to evaluate LD between all pairs of markers on the same chromosome. For each pair of markers, we computed a permutation-adjusted measure of LD, , which adjusts for biases in Lewontin's D' measure due to small sample sizes. For the autosomal marker pairs, we found that declines with increasing recombination fractions between marker pairs. When we averaged the measures within recombination fraction intervals, the average in the Samoans decreased as the recombination fraction increased (Table 1a). In general, 15.87 per cent of the measures are significantly different from zero. By contrast, in the NASC sample, the average is essentially zero and the percentage of significant values is close to the percentage expected by chance (Table 1a). Note that the per cent significant is a function of sample size and so interpretation of apparent differences can be confounded by sample size differences; however, the Samoan sample size and the NASC sample size are of the same order of magnitude.
Table 1a.
Mean and per cent significant (at the 0.05 level) as a function of the recombination fraction between all possible pairs of autosomal markers drawn from the same chromosome.
| Population | Statistic | Recombination fraction interval | ||||
|---|---|---|---|---|---|---|
| < 0.1 | 0.1- < 0.2 | 0.2- < 0.3 | 0.3- < 0.4 | > 0.4 | ||
| Samoa (N = 482) | Mean | 0.026 | 0.018 | 0.016 | 0.014 | 0.014 |
| % significant | 24.79 | 16.43 | 17.79 | 12.90 | 15.06 | |
| Palau* (N = 84) | Mean | 0.031 | 0.019 | 0.017 | 0.012 | 0.009 |
| % significant | 16.2 | 11.6 | 11.6 | 7.1 | 4.4 | |
| NASC (N = 333) | Mean | -0.001 | 0.001 | 0.000 | 0.000 | 0.000 |
| % significant | 4.48 | 3.97 | 3.87 | 4.44 | 4.63 | |
| Table 1b. Mean and per cent significant (at the 0.05 level) as a function of the recombination fraction between all possible pairs of X-linked markers. | ||||||
| Population | Statistic | Recombination fraction interval | ||||
| < 0.1 | 0.1- < 0.2 | 0.2- < 0.3 | 0.3- < 0.4 | > 0.4 | ||
| Samoan females (N = 201) | Mean | 0.129 | 0.062 | 0.080 | 0.059 | 0.082 |
| % significant | 55.6 | 33.3 | 56.3 | 48.2 | 48.2 | |
| Samoan males (N = 249) | Mean | 0.116 | 0.050 | 0.071 | 0.036 | 0.067 |
| % significant | 44.4 | 38.9 | 43.8 | 37.0 | 41.0 | |
| Palaun males* (N = 54) | Mean | 0.123 | 0.041 | 0.041 | 0.020 | 0.026 |
| % significant | 44.0 | 0 | 13.6 | 13.6 | 21.4 | |
| NASC females (N = 161) | Mean | -0.002 | 0.000 | -0.002 | 0.006 | -0.002 |
| % significant | 12.5 | 7.69 | 9.09 | 4.55 | 9.52 | |
| NASC males (N = 172) | Mean | -0.021 | -0.003 | -0.014 | -0.018 | 0.000 |
| % significant | 11.11 | 11.11 | 0 | 0 | 4.82 | |
* Palaun data from Devlin et al.[9]
We fitted a regression model to examine the relationship between and recombination fraction, and found that significantly decreases as the recombination fraction increases (Figure 1); we estimated a slope of -0.021 (SE 0.00354; p < 0.0001). If we remove the potentially influential outlier with a of 0.38, the results remain essentially the same, with a slope of - 0.019 (SE 0.00346; p < 0.0001).
Figure 1.
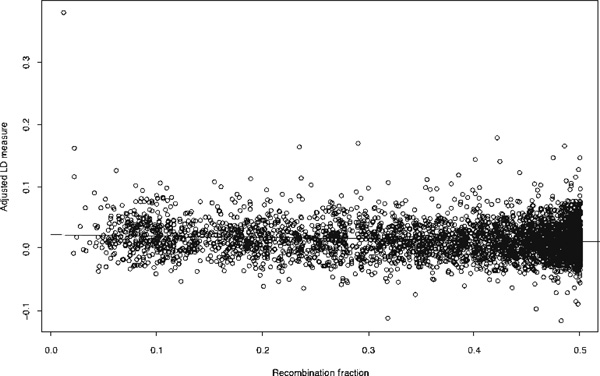
The adjusted linkage disequilibrium measure, ; of all autosomal marker pairs versus recombination fraction.
We evaluated whether there was heterogeneity in the values across the autosomal chromosomes by using analysis of covariance. The predictors in the analysis were the chromosome number (treated as a set of indicator functions) and inter-marker recombination fractions. We did not find evidence of heterogeneity across chromosomes (F 0.96; df 21; p = 0.509), whereas the recombination fraction was significant (F 38.10; df 1; p < 0.0001).
For the 18 X-linked microsatellite marker data, we did not observe any steady pattern in the Samoans between the averaged LD measure and recombination fractions, either in the male or female data (Table 1b). The average values and the per cent significant are much higher in the Samoans than in the NASC sample (Table 1b). We also fitted a regression model to examine the relationship between and recombination fraction in X-linked data. By regressing on recombination fractions, we obtained negative slopes in males and females, but the p-values of the slopes were not significant (data not shown).
We estimated the heterozygosity of the autosomal (X-linked) markers using data from the (female) unrelated individuals and the first (female) sib from each family. We then compared the Samoan heterozygote frequencies with the observed heterozygote frequencies in the CEPH and NASC families. We found the average heterozygosity (0.67) in Samoan data was 0.12 less than in the CEPH families (0.79) and 0.10 less than in the NASC families (0.77). Eighty-seven per cent of markers in the CEPH data and 83 per cent of markers in the NASC data had greater heterozygosity than the corresponding marker in the Samoan data; a sign test showed that both of these differences were highly significant (p < 0.0001 for both tests). A previous study in the Palauans found similar results[9].
The imbalance index (β), an index sensitive to bottleneck events,[43] was estimated for both the Samoan and NASC populations using our autosomal markers. In the presence of a bottleneck event, β is expected to exceed 1. The imbalance index in the Samoan population is 3.86, which is much larger than that in the NASC population (β = 1.31), indicating that the bottleneck event that occurred in the Samoans is a much more recent event. The β estimated in the NASC population is very similar to an earlier estimation of 1.33 in Europeans[43]. It should be noted that the presence of population substructure would also lead to an elevated β value, which is not the case for the Samoan population, given that it is genetically more homogeneous than the other populations.
Using 25 unlinked autosomal loci, we also tested for population substructure[44]. The number of alleles at these markers ranged from four to 17, with a median of ten, in the Samoan population; and from five to 18, with a median of 11, in the NASC population. Our population substructure analyses strongly support the hypothesis that the Samoans represent one genetic population, even though members of our sample are drawn from two different countries. Based on the 'structure' results, assuming a uniform prior on the number of clusters K, we obtain a posterior probability of 1.0 that K = 1. Note also that we have enhanced homogeneity by selecting individuals with four Samoan grandparents. The very low level of non-Polynesian alleles seen in our sample[24] indicates that our selection scheme reduced admixture to very low levels.
Discussion
The results we present here are relevant to both the potential use of LD to map disease genes in the Samoan population and to inferences about the evolutionary history of the Samoan population. From our results, we find that LD, as measured using multi-allelic markers, extends over substantial distances across the whole genome in Samoans. When we divide the LD values into groups according to recombination fractions, we observe that the average LD in most intervals is quite similar to that observed in Palau[9] (Table 1a). Compared with the auto-somal data, we find a higher level of LD in the X-linked data (as would be expected, based on its smaller effective population size), but when we regress LD on the recombination fractions, the slopes for the X-linked data are not significantly negative. Since we estimated LD for males and females separately, it is possible that the sample sizes were too small. The patterns of LD observed in the Samoans are quite different from those observed in the non-isolated Caucasian NASC sample (Tables 1a and 1b), with the NASC sample displaying essentially no marker-to-marker LD above that which would be expected by chance (except, perhaps, for markers close to each other on the X-chromosome).
We have also typed 43 SNPs from a fragment spanning 104 kb of DNA on chromosome 21 to study the relationship between LD and physical distance between SNP markers in Samoans and four outbred populations, namely, Benin from Nigeria, German, Japanese and Chinese, each representing the four major continental populations[45]. The results show that the Samoans have significantly elevated D' values compared with all four continental populations throughout the 100 kb region, without any sign of attenuation. It is very likely that this pattern of LD will extend much further than 100 kb in the Samoans. By contrast, in the other four populations, D' shows a declining slope when plotted against increasing physical distance.
Based on our Y-linked STR data, the Y haplotype diversity in Samoans is 0.855, which is lower than the observed diversity of 0.96 in the Palauan study; however, it is comparable to other isolated populations. From the five Y-linked STR data in two isolated Iberian samples (Basque and Catalan), the diversities of Y haplotypes are 0.85 and 0.89 in Basque and Catalan, respectively[46].
Our statistical tests on our genetic data support the scenario of a recent bottleneck, which is consistent with the known demographic history of the Samoan population. Furthermore, our population substructure results support the hypothesis that self-identified Samoans represent one homogenous genetic population. These results are consistent with our prior reports on the genetic structure of the Samoans[24].
Our finding of high levels of genome-wide LD among Samoans are consistent with the multiple lines of evidence from genetic, archaeological and demographic history research that indicate that the Samoan population was founded by a relatively small number of founders, remained isolated for about 3,000 years and experienced a reduction in population size about 200-300 years ago[15,16,23-27]. The findings of genome-wide LD in the Samoan population are similar to levels of LD observed in another isolated Pacific population, namely, Palau of Micronesia,[9] as well as an isolated population from the Central Valley of Costa Rica[10]. The present Samoan findings and these others[9] support the hypothesis that LD extends further in isolated populations than in continental populations[4-6].
The results presented here on patterns of LD using a relatively sparse set of markers indicate that a more thorough characterisation of LD patterns in the Samoans is likely to be of interest. In particular, it would be of interest to compare the Samoan population with other isolated populations that have grown rapidly. While we have begun to generate higher density data in limited regions, as described above for chromosome 21,[45] the results presented here provide important baseline information about the levels of background LD, but provide little information about the extent of 'useful' LD, as required for genome-wide association scans[47]. While the precise definition of what levels of LD are useful for association mapping remains debatable, in outbred northern European populations LD is thought to be useful only over a relatively short range of 10-30 kb[47]. The dramatic difference between our results for the Samoan population and those from the NASC sample favour the hypothesis that the level of useful LD will be higher in the Samoan population.
Electronic database information
URLs for data presented herein are as follows:
GOLD, Abecasis and Cookson, http://www.sph.umich.edu/csg/abecasis/GOLD/index.html, University of Michigan (accessed 20th April, 2004).
structure, Pritchard, J.K., Stephens, M. and Donnelly, P., http://pritch.bsd.uchicago.edu/software.html, University of Chicago, [cited 2004 April 20]
Acknowledgements
This research was supported by NIH grants AG09375, HL52611, DK55406 and DK59642. We thank the members of the Department of Health, Government of Samoa and the American Samoan Department of Health for their assistance in data collection; local political officials for their cooperation; and the participants for their patience. We would like to thank Dr John Reveille, the Principal Investigator of the North American Spondylitis Consortium (NASC) project for his generous support in sharing the genotype data. The genotyping of the NASC samples was supported by NIH grant RO1-AR46208. We thank Dr Ning Wang of the Center for Genome Information, University of Cincinnati, for her help in computing the imbalance indices. We thank the two anonymous reviewers whose comments helped us to markedly improve this manuscript.
References
- Risch N, Merikangas K. 'The future of genetic studies of complex human diseases'. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Kruglyak L. 'Genetic isolates: Separate but equal?'. Proc Natl Acad Sci USA. 1999;96:1170–1172. doi: 10.1073/pnas.96.4.1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen L, Palotie A, Lange K. 'Use of population isolates for mapping complex traits'. Nat Rev Genet. 2000;1:182–190. doi: 10.1038/35042049. [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardina I, Saccone NL. et al. 'Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28'. Nat Genet. 2000;25:324–328. doi: 10.1038/77100. [DOI] [PubMed] [Google Scholar]
- Eaves IA, Merriman TR, Barber RA. et al. 'The genetically isolated populations of Finland and Sardinia may not be a panacea for linkage disequilibrium mapping of common disease genes'. Nat Genet. 2000;25:320–323. doi: 10.1038/77091. [DOI] [PubMed] [Google Scholar]
- Lonjou C, Collins A, Morton NE. 'Allelic association between marker loci'. Proc Natl Acad Sci USA. 1999;96:1621–1626. doi: 10.1073/pnas.96.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehnke M. 'A look at linkage disequilibrium'. Nat Genet. 2000;25:246–247. doi: 10.1038/76980. [DOI] [PubMed] [Google Scholar]
- Kruglyak L. 'Prospects for whole-genome linkage disequilibrium mapping of common disease genes'. Nat Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K, Otto C. et al. 'Genome-wide distribution of linkage disequilibrium in the population of Palau and its implications for gene flow in Remote Oceania'. Hum Genet. 2001;108:521–528. doi: 10.1007/s004390100511. [DOI] [PubMed] [Google Scholar]
- Service SK, Ophoff RA, Freimer NB. 'The genome-wide distribution of background linkage disequilibrium in a population isolate'. Hum Mol Genet. 2001;10:545–551. doi: 10.1093/hmg/10.5.545. [DOI] [PubMed] [Google Scholar]
- Shifman S, Darvasi A. 'The value of isolated populations'. Nat Genet. 2001;28:309–310. doi: 10.1038/91060. [DOI] [PubMed] [Google Scholar]
- Mohlke KL, Lange EM, Valle TT. et al. 'Linkage disequilibrium between microsatellite markers extends beyond 1 cM on chromosome 20 in Finns'. Genome Res. 2001;11:1221–1226. doi: 10.1101/gr.173201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D, Wijsman EM, Roos JL. et al. 'Extended inter-marker linkage disequilibrium in the Afrikaners'. Genome Res. 2002;12:956–961. doi: 10.1101/gr.136202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellwood PS. Man's Conquest of the Pacific: The Prehistory of Southeast Asia and Oceania. Oxford University Press, New York, NY; 1979. [Google Scholar]
- Green RC, Davidson JM, Bernice Pauahi Bishop Museum. Archaeology in Western Samo. Auckland Institute and Museum, Auckland, NZ; 1969. [Google Scholar]
- Kirch PV. On the Road of the Winds: An Archaeological History of the Pacific Islands before European Contact. University of California Press, Berkeley, CA; 2000. [Google Scholar]
- Diamond JM. 'Express train to Polynesia'. Natur. 1988;336:307–308. doi: 10.1038/336307a0. [DOI] [Google Scholar]
- Sykes B, Leiboff A, Low-Beer J. et al. 'The origins of the Polynesians: An interpretation from mitochondrial lineage analysis'. Am J Hum Genet. 1995;57:1463–1475. [PMC free article] [PubMed] [Google Scholar]
- Redd AJ, Takezaki N, Sherry ST. et al. 'Evolutionary history of the COII/tRNALys intergenic 9 base pair deletion in human mito-chondrial DNAs from the Pacific'. Mol Biol Evol. 1995;12:604–615. doi: 10.1093/oxfordjournals.molbev.a040240. [DOI] [PubMed] [Google Scholar]
- Melton T, Clifford S, Martinson J. et al. 'Genetic evidence for the proto-Austronesian homeland in Asia: mtDNA and nuclear DNA variation in Taiwanese aboriginal tribes'. Am J Hum Genet. 1998;63:1807–1823. doi: 10.1086/302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell J. 'History as a family tree, history as an entangled bank: constructing images and interpretations of prehistory in the South Pacific'. Antiquity. 1988;62:642–657. [Google Scholar]
- Martinson JJ. In: Molecular Biology and Human Diversity. Boyce, A.J. and Mascie-Taylor, C.G.N, editor. Cambridge University Press, Cambridge, UK; 1996. 'Molecular perspectives on the colonization of the Pacific'; pp. 171–195. [Google Scholar]
- Su B, Jin L, Underhill P. et al. 'Polynesian origins: Insights from the Y chromosome'. Proc Natl Acad Sci USA. 2000;97:8225–8228. doi: 10.1073/pnas.97.15.8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka R, McGarvey ST, Ferrell RE. et al. 'Genetic characterization of American and Western Samoans'. Hum Biol. 1994;66:805–822. [PubMed] [Google Scholar]
- Deka R, Jin L, Shriver MD. et al. 'Population genetics of dinucleotide (dC-dA)n.(dG-dT)n polymorphisms in world populations'. Am J Hum Genet. 1995;56:461–474. [PMC free article] [PubMed] [Google Scholar]
- Deka R, Shriver MD, Yu LM. et al. 'Genetic variation at twentythree microsatellite loci in sixteen human populations'. J Genet. 1999;78:99–121. doi: 10.1007/BF02924561. [DOI] [Google Scholar]
- Shriver MD, Jin L, Ferrell RE. et al. 'Microsatellite data support an early population expansion in Africa'. Genome Res. 1997;7:586–591. doi: 10.1101/gr.7.6.586. [DOI] [PubMed] [Google Scholar]
- McArthur N. Island Populations of the Pacific. Australian National University Press, Canberra, Australia; 1967. [Google Scholar]
- McArthur NA. The Populations of the Pacific. Part III American Samoa and Part IV We stern Samoa and the Tokelau Islands, Australian National University, Department of Demography, Canberra, Australia; 1956. [Google Scholar]
- Gilson RP. Samoa 1830 to 1900: The Politics of a Multi-cultural Community. Oxford University Press, New York, NY; 1970. [Google Scholar]
- Tsai H-J, Sun G, Weeks DE. et al. 'Type 2 diabetes and three calpain-10 gene polymorphisms in Samoans: No evidence of association'. Am J Hum Genet. 2001;69:1236–1244. doi: 10.1086/324646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarvey ST, Levinson PD, Bausserman L. et al. 'Population-change in adult obesity and blood-lipids in American-Samoa from 1976-1978 to 1990'. Am J Hum Biol. 1993;5:17–30. doi: 10.1002/ajhb.1310050106. [DOI] [PubMed] [Google Scholar]
- Galanis DJ, McGarvey ST, Quested C. et al. 'Dietary intake of modernizing Samoans: Implications for risk of cardiovascular disease'. J Am Diet Assoc. 1999;99:184–190. doi: 10.1016/S0002-8223(99)00044-9. [DOI] [PubMed] [Google Scholar]
- Lewontin RC. 'The interaction of selection and linkage. I. General considerations; heterotic models'. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cookson WO. 'GOLD -- Graphical overview of linkage disequilibrium'. Bioinformatics. 2000;16:182–183. doi: 10.1093/bioinformatics/16.2.182. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M. 'Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population'. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M. 'An E-M algorithm and testing strategy for multiple-locus haplotypes'. Am J Hum Genet. 1995;56:799–810. [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. 'R: A language for data analysis and graphics'. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- Broman KW. 'Estimation of allele frequencies with data on sib-ships'. Genet Epidemiol. 2001;20:307–315. doi: 10.1002/gepi.2. [DOI] [PubMed] [Google Scholar]
- Chakraborty R. 'Number of independent genes examined in family surveys and its effect on gene frequency estimation'. Am J Hum Genet. 1978;30:550–552. [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R. 'Inclusion of data on relatives for estimation of allele frequencies'. Am J Hum Genet. 1991;49:242–244. [PMC free article] [PubMed] [Google Scholar]
- Teare MD, Dunning AM, Durocher F. et al. 'Sampling distribution of summary linkage disequilibrium measures'. Ann Hum Genet. 2002;66:223–233. doi: 10.1046/j.1469-1809.2002.00108.x. [DOI] [PubMed] [Google Scholar]
- Kimmel M, Chakraborty R, King JP. et al. 'Signatures of population expansion in microsatellite repeat data'. Genetics. 1998;148:1921–1930. doi: 10.1093/genetics/148.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 'Inference of population structure using multilocus genotype data'. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka R, McGarvey ST, Weeks DE. et al. 'Genetic variation in an isolated population, the Samoans of Polynesia: Implications for mapping complex traits'. Am J Hum Genet. 2003;73(Suppl):187. [Google Scholar]
- Perez-Lezaun A, Calafell F, Seielstad M. et al. 'Population genetics of Y-chromosome short tandem repeats in humans'. J Mol Evol. 1997;45:265–270. doi: 10.1007/PL00006229. [DOI] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. 'Patterns of linkage disequilibrium in the human genome'. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]