


Abstract
Transcription initiation complexes formed by bacterial RNA polymerases (RNAPs) exhibit dramatic species-specific differences in stability, leading to different strategies of transcription regulation. The molecular basis for this diversity is unclear. Promoter complexes formed by RNAP from Thermus aquaticus (Taq) are considerably less stable than Escherichia coli RNAP promoter complexes, particularly at temperatures below 37°C. Here, we used a fluorometric RNAP molecular beacon assay to discern partial RNAP-promoter interactions. We quantitatively compared the strength of E. coli and Taq RNAPs partial interactions with the −10, −35 and UP promoter elements; the TG motif of the extended −10 element; the discriminator and the downstream duplex promoter segments. We found that compared with Taq RNAP, E. coli RNAP has much higher affinity only to the UP element and the downstream promoter duplex. This result indicates that the difference in stability between E. coli and Taq promoter complexes is mainly determined by the differential strength of core RNAP–DNA contacts. We suggest that the relative weakness of Taq RNAP interactions with DNA downstream of the transcription start point is the major reason of low stability and temperature sensitivity of promoter complexes formed by this enzyme.
INTRODUCTION
Transcription is the first step of gene expression and a target of extensive regulation. The bacterial DNA-dependent RNA polymerase (RNAP) is the principal enzyme of transcription. The bacterial RNAP initiates transcription in the form of the holoenzyme (subunit composition αIαIIββ′ωσ). The dissociable specificity subunit σ is required for promoter recognition and melting (1,2). Bacterial genomes encode multiple σ factors, each targeting RNAP core to a particular group of promoters with common sequence (3). One σ, called primary, is usually present at the highest amounts and enables RNAP to recognize the majority of promoters required for expression of genes under normal physiological conditions.
Despite the functional and sequence similarities between the primary σ subunits from various bacteria, promoter complexes formed by corresponding holoenzymes exhibit dramatic species-specific differences in their stability and efficiency of transition from initiation to elongation stages of transcription. These differences have been clearly revealed in the context of model enzymes of bacterial transcription—RNAPs from Escherichia coli, Bacillus subtilis and two closely related thermophilic bacteria, Thermus aquaticus (Taq) and Thermus thermophilus (Tth). These holoenzymes recognize the same conserved promoter elements centered around −10 and −35 positions (consensus sequences are TATAAT and TTGACA, respectively). B. subtilis RNAP forms unstable open complexes (RPo) at the majority of promoters. These complexes are in equilibrium with closed complexes and, in turn, with free RNAP (4). To provide the energy required to stimulate RPo formation, thermophilic bacteria depend on the high temperatures of their environment. Holoenzymes from Taq (Taq EσA), Tth and other related thermophilic bacteria form moderately stable open complexes at ∼60°C. These complexes become very unstable at temperatures below 37°C (5–11). In contrast, open complexes formed by E. coli RNAP holoenzyme (Ec Eσ70) at many promoters are stable even at physiologically suboptimal temperatures (20–25°C), and, in many cases, the RPo formation is essentially irreversible (2). However, at some promoters, Ec Eσ70 does form intrinsically unstable open complexes, with E. coli rrnB P1 promoter being the best-studied model. Stability of the rrnB P1 open complex is a subject of genetic regulation and is determined by concentration of the initiating NTP and by ppGpp and DksA (12). Efficient transcription from promoters with unstable open complexes is often observed and could be caused by facilitated promoter escape, which should increase the rate of transition from transcription initiation to transcription elongation. Indeed, B. subtilis and Taq RNAPs are less prone to abortive RNA synthesis than the E. coli enzyme during transition to elongation from the same promoters (4,9,11). The pronounced differences in RNAPs behavior during initiation and transition to elongation steps may result in different strategies of control of the transcription process (4,13).
Species-specific variations of strength of RNAP interactions with certain promoter segments can account for overall differences in promoter complex properties, including their stability and temperature sensitivity. In principle, promoter contacts with RNAP core, σ or both can be responsible for these differences. Schroeder and deHaseth (10) found that oligonucleotides and upstream fork junction promoter fragments containing the consensus −10 and −35 element sequences bound to Taq RNAP similarly or even stronger than to E. coli RNAP, as judged by sensitivity of the RNAP–DNA probe complexes to heparin. This result suggests that the strengths of Taq and E. coli RNAPs interactions with the basal promoter elements may not correlate with efficiencies of the RPo formation by these enzymes. In contrast to the E. coli RNAP, no sequence-specific interaction of Thermus RNAP α subunits with promoter UP element was found (14). However, as the upstream promoter interactions are necessary for the initiation complex formation only at a fraction of Ec Eσ70 promoters, the difference in strength of these interactions can not solely explain the generally higher stability of Ec Eσ70 promoter complexes. Several studies suggested that many differences between bacterial transcription complexes can be due to the changes in contacts made between the downstream DNA duplex and the core RNAP β and β′ subunits (4,15–17). Miropolskaya et al.(11) recently showed that the structures of the N-terminal regions 1.1 and 1.2 of the E. coli σ70 and Taq σA subunits in part determine higher stability of E. coli RNAP promoter complexes. It seems plausible that the effect of σ region 1.1 on promoter complex stability is also related with modulating strength of RNAP interactions with the downstream promoter duplex (11). However, direct data on comparative strengths of downstream contacts for RNAPs from different bacteria are currently unavailable. Thus, although the species-specific peculiarities of RPo formation in bacteria are well established, the molecular basis for this diversity is unclear.
A recently reported E. coli RNAP beacon assay (18) was previously used to measure specific affinity of Ec Eσ70 to various model promoter fragments and quantitatively characterize partial E. coli RNAP promoter interactions (18–21). Here, we developed a similar beacon assay for Taq EσA. This approach allowed us to comprehensively compare the strengths of partial RNAP-promoter interactions that are known to be essential for RPo formation by Ec Eσ70 and Taq EσA. The data form a basis for quantitative rationalization of a number of previously described biochemical observations. In particular, we found that in the context of RNAP complexes with model promoter fragments, Taq EσA interacts with the downstream promoter duplex much weaker than does Ec Eσ70. This result provides direct quantitative evidence that the strength of downstream RNAP-promoter contacts varies considerably in different bacteria and can therefore be an important factor determining species-specific stability and, by extension, modes of regulation of promoter complexes.
MATERIALS AND METHODS
Proteins
Expression plasmids pET28TaqσA and pET28TthσA encoding T. aquaticus and T. thermophilus σA subunits, respectively, were used for site-specific mutagenesis and subsequent purification of polyhistidine-tagged mutant protein derivatives with single Cys residues. Taq core RNAP, wild-type and Cys σA derivatives were purified as described in (8). The recombinant system used for the preparation of Taq RNAP yields highly active enzyme that is indistinguishable from the ‘native’ enzyme purified from Taq cell cultures in biochemical assays (8,22). Tth RNAP and Cys σA derivatives were purified as described in (23). Fluorescent labels were incorporated into single-Cys σ70 and σA derivatives using Cys-specific chemical modification [procedures as described in (24), efficiencies of labeling were >70%]. RNAP holoenzymes containing the labeled σ derivatives were prepared as in (24).
DNA probes
DNA oligonucleotides were synthesized by Integrated DNA Technologies. Fork junction and double-stranded DNA probes were prepared as in (18).
Fluorometric assays
Fluorescence measurements were performed using a QuantaMaster QM4 spectrofluorometer (PTI) in transcription buffer [40 mM Tris–HCl (pH 8.0), 100 mM NaCl, 5% glycerol, 1 mM DTT and 10 mM MgCl2] containing 0.02% Tween 20 at 25 or 45°C. Final assay mixtures (800 μl) contained 1 nM labeled RNAP holoenzyme and DNA probes at various concentrations. The Tetramethylrhodamine-5-maleimide (TMR) fluorescence intensities were recorded with an excitation wavelength of 550 nm and an emission wavelength of 578 nm.
To obtain equilibrium dissociation constants (Kd), the experimental dependence of the fluorescent signal amplitude (F) on DNA probe concentration was fit to Equation 1, unless otherwise noted.
| (1) |
where X=(F-F0)/(Fmax-Fo), Fo is the initial value of the amplitude, and Fmax is the limiting value of the amplitude at [DNA]=∞. The data were analysed using SigmaPlot software (SPSS, Inc.).
To prevent dissociation of relatively short duplexes in downstream fork junctions, in particular at 45°C, 100 nM excesses of template strand fragments of these probes were added to the assayed samples. Control experiments verified that the template strand oligos generated negligibly low signals and did not interfere with binding of the downstream fork junctions.
An equilibrium competition binding assay was used to measure affinity of tight E. coli RNAP complexes with fork junction probes 8, 12 and 13 (Figure 2 and Supplementary Figure S2). A double-stranded [−58/−14] fragment of N25cons producing negligible signal on binding to (211Cys-TMR) σ70 holo RNAP was used as a competitor, as described in (18). Time-dependent fluorescence changes were monitored after manual-mixing of RNAP beacon (800 µl) and a DNA probe (<20 µl) in a cuvette; the mixing dead-time was 15 s.
Figure 2.

Structures of DNA probes used. The probe names used in the text are in red. In fork junction probe names, numbers in left and right parentheses correspond to borders of upper and bottom strands of fork junctions with respect to the transcription start located at +1 (underlined). The −10 and −35 promoter element sequences are highlighted in larger size font. Asterisks above the [−40/+20] probe indicate positions where the fragments used in experiment shown in Figure 5 were truncated.
In vitro abortive initiation assay
Abortive transcription reactions were performed in a final volume of 10 μl and contained 200 nM Ec Eσ70 (Epicentre) or Taq EσA, and 50 nM N25cons promoter DNA in transcription buffer [30 mM Tris–HCl (pH 7.9), 40 mM KCl, 10 mM MgCl2, 2 mM β-mercaptoethanol]. Reactions were mixed and incubated for 10 min at optimal temperatures 37°C (for E. coli RNAP) or at 55°C (for Taq RNAP) followed by 5 min incubation with 50 µg/ml heparin or 0.5 µM of fork junction DNA competitor (where indicated). Next, the reactions were incubated for a further 10 min at optimal 37°C or 55°C with added transcription hot mix [200 μM CpA RNA dinucleotide primer, 20 μM cold UTP, and (α-32P)UTP (3000 Ci/mmol)] and then terminated with an equal volume of urea-formamide loading buffer. Alternatively, the reaction mixtures after incubation at the optimal temperatures were placed at suboptimal temperature 25°C, supplemented with heparin (where indicated) and incubated for a further 10 min with transcription hot mix at the same temperature. The reaction products were resolved on a 20% (w/v) polyacrylamide denaturing gel and visualized using a PhosphorImager. Abortive initiation experiments were repeated two or three times, with standard deviation ≤20%.
RESULTS
Development of a protein beacon assay to study the interactions of thermophilic RNAPs with promoters
A recently developed RNAP molecular beacon assay allows one to monitor the Ec Eσ70 interactions with promoters and a wide variety of model DNA substrates mimicking DNA structures in the open promoter complex (18). The assay relies on the detection of fluorescence signal from RNAP holoenzyme containing the σ70 subunit with fluorescent label site-specifically incorporated in proximity to region 2.3, the part of σ that recognizes the −10 promoter element. The base-line fluorescence of labeled RNAP holoenzyme is low owing to quenching by σ70 region 2 Trp and Tyr residues via photoinduced electron transfer mechanism. Efficient quenching usually occurs at length scales below 1 nm. On RNAP interaction with promoter DNA or promoter fragments, the aromatic amino acids lose contact with the fluorescent probe, decreasing the quenching efficiency leading to increased fluorescence.
In this work, we developed new RNAP beacons based on RNAPs from two bacteria of the Thermus genus, T. aquaticus and T. thermophilus. Taq EσA beacons were developed similarly to the previously described Ec Eσ70 beacons (18). Single-cysteine mutants of the Taq RNAP σA subunit that allow site-specific introduction of fluorescent labels were prepared. The sites where unique cysteine residues were introduced (aminoacid positions 243 and 245) were chosen, based on available structures of Taq σA domain 2 (21,25). Fluorescent labels attached to cysteines at these positions should be close to Trp residues of σA region 2.3. Therefore, these Trp residues may quench the fluorescence of site-specifically attached fluorophores. The positions of Cys243 and Cys245 on a structural model of the σA–DNA complex (21) are shown in Figure 1. As can be seen, the bases of the −10 element, in particular the -11A base, are located just between the modified SH groups and the Trp256/Trp257 side chains, suggesting that specific binding to promoter should unquench the signal from the fluorescent label.
Figure 1.
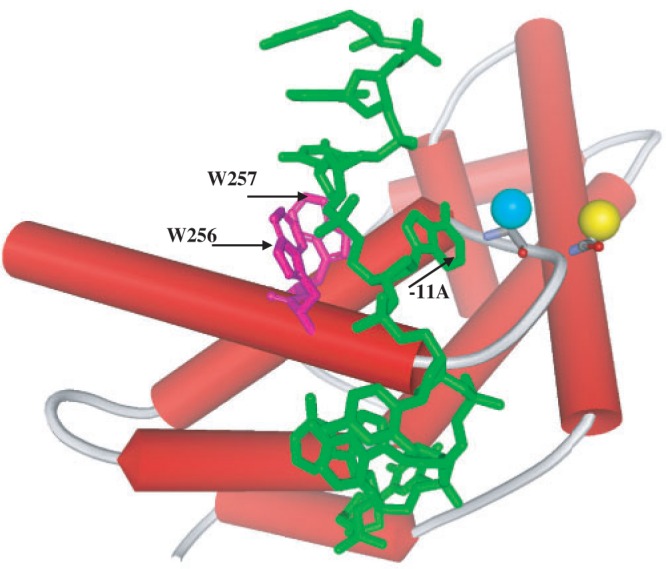
Structural model of σA domain 2 bound to single-stranded DNA (in green) showing the positions of SH groups in single-cystein σA derivatives 245Cys (yellow sphere) and 243Cys (blue sphere). The structure is from (21).
To determine whether σA-based beacons function as expected, single-cysteine σA mutants were modified with TMR (the efficiency of labeling was at least 80%) and used to reconstitute Taq RNAP holoenzyme. Both mutant holoenzymes were functional (at least 70% of the wild-type Taq EσA activity in abortive initiation assay on the galP1 promoter, data not shown). When EσA holoenzymes containing fluorescent labels at σA positions 243 and 245 were combined with DNA fragments containing the T5 N25 promoter (Figure 2), the fluorescence signal increased ∼1.7 and 2.3-fold, respectively. A similar result was obtained with other promoters (N25cons and galP1). Data presented below show that increased fluorescence was owing to specific RNAP interaction with the −10 promoter element, allowing us to conclude that Taq σA-based beacons function as expected.
We also tested a number of similarly purified and labeled Tth σA derivatives. The Tth RNAP holoenzyme containing σA with TMR attached at 230 position demonstrated readily measurable ‘beacon effect’ similar to those observed with the Taq RNAP beacons.
In what follows, we use the Taq EσA beacon based on (245Cys-TMR)σA and a previously described Ec Eσ70 beacon (18) based on (211Cys-TMR)σ70 to quantitatively compare RNAP affinities to various promoter fragments. Dissociation constants (Kd) measured by the E. coli RNAP beacon assay were slightly higher than Kd values measured using other methods, presumably because in the beacon assay, some binding free energy must be used to disrupt the van der Waals contacts between the fluorophore and the quenching aromatic amino acids (18). The strengths of the fluorophore-quencher contacts may be not equal in the Taq EσA and Ec Eσ70 beacons, though the use of the same fluorescent label in the both beacons should help diminish the difference. Be that as it may, this effect could somewhat affect comparison of the Taq EσA and Ec Eσ70 beacon interactions with the −10 element per se and immediately adjacent bases, but it should not influence the RNAP affinities to promoter parts that are remote from the −10 element.
DNA probes
Certain promoter fragments are known to interact with RNAP, and resulting complexes mimic RNAP interactions with corresponding promoter segments in complexes with full-sized promoters. These model substrates include DNA oligos containing sequences corresponding to the non-template strand of the −10 promoter element (26), upstream fork junctions (27–29), downstream fork junctions (19) and short double-stranded promoter fragments (19,21). Studies of RNAP interactions with such fragments can be used to dissect partial RNAP-promoter interactions. The RNAP beacon assay is well-suited for this purpose owing to its high sensitivity and low intensity of non-specific background signal.
We compared the strength of interactions of Taq EσA and Ec Eσ70 with model promoter fragments schematically shown in Figure 2. Together, these DNA probes encompass the entire promoter length and include all essential promoter elements. The DNA probes are based on T5 N25 and N25cons promoter sequences (Figure 2). N25cons is a ‘full consensus’ derivative of T5 N25 with consensus −35 element, an optimized UP element, and the extended −10 element (20). For E. coli RNAP, the strength of interactions with many of the probes shown in Figure 2 was measured previously using the beacon assay (18–20).
Taq EσA is highly active on N25cons in the abortive transcription initiation assay at 55°C. The activity is reduced ∼10-fold when an open complex formed at 55°C is shifted to 25°C and then supplemented with abortive transcription reaction substrates (Figure 3). To evaluate Taq EσA-N25cons complex stability, we measured activity of preformed complex after a 5-min incubation with 50 -µg/ml heparin, which is known to compete with DNA for the binding to RNAP. As shown in Figure 3, Taq EσA-N25cons complex was very unstable at 25°C: heparin treatment resulted in a 14-fold drop in activity. Heparin sensitivity at 55°C was less pronounced (1.7-fold drop in activity, Figure 3). In contrast, the activity of preformed Ec Eσ70-N25cons complex decreased only slightly (by ∼15%) after incubation with heparin at either 25°C or 37°C (Figure 3). In principle, Taq EσA and Ec Eσ70 may bind heparin with different strength, which could modulate the relationship between promoter stabilities and heparin sensitivities. Therefore, we compared stabilities of Taq EσA and Ec Eσ70 complexes with T5 N25 and N25cons promoter DNA fragments at 55°C and 37°C, respectively, using a fork junction fragment of N25cons (shown in Supplementary Figure S1A), rather than heparin, as a competitor. The fork junction competitor formed very tight and inactive complexes with both Taq and Ec enzymes (data not shown). In agreement with the result obtained with heparin, we found that activities of the Taq EσA promoter complexes decreased considerably faster in the presence of the fork junction competitor than activities of Ec Eσ70 promoter complexes (Supplementary Figure S1B).
Figure 3.

Stabilities of transcription initiation complexes of T. aquaticus and E. coli RNAPs. The plot shows relative efficiencies of abortive transcription initiation reactions by E. coli and T. aquaticus RNAP holoenzymes at N25cons promoter DNA fragment. All samples were initially incubated for 10 min at optimal (37°C for E. coli and 55°C for T. aquaticus) temperatures. Next, four reactions (1–4) were carried out as following: at the optimal temperatures, either immediately after the initial incubation (1) or after 5 min additional incubation with 50 µg/ml heparin (2). After the initial incubation, samples 3 and 4 were further incubated at 25°C for 15 min, followed by the reactions either without (3) or after the heparin treatment (4).
Thus, promoter complexes formed by Taq and E. coli RNAPs on N25cons and T5N25 demonstrate differences in stability that are consistent with results obtained previously on other promoters (6–11).
The affinities of DNA probes to Taq EσA and Ec Eσ70 beacons were characterized by determining dissociation constants values and are listed in Tables 1 and 2. Most of the measurements were carried out at 25°C; some experiments with Taq EσA were also performed at 45°C. Representative experimental data for Taq EσA are shown in Figure 4.
Table 1.
Dissociation constants for the binding of promoter fragments to Taq EσA and Ec Eσ70 at 25°C
| Promoter fragments | DNA probe | Kdtaq, nM | Kdec, nM | Kdtaq/Kdec |
|---|---|---|---|---|
| Oligos | 1. −12/+2 | 100 | 160 | 0.63 |
| 3. −12/+2; ggga | 15 | 110 | 0.14 | |
| 4. −12/+2; ccct | 2200 | 690 | 3.2 | |
| 5. −12/−6 | 54000 | 80000 | 0.68 | |
| Upstream fork junctions | 6. [−38/−11][−38/−12] | 13.7 | 5 | 2.8 |
| 7. [−38/−11][−38/−12]TG | 1.2 | 0.43 | 2.8 | |
| 8. [−38/−8][−38/−12] | 0.8 | 0.14 | 5.7 | |
| 9. [−38/−7][−38/−12] | <0.2 | <0.2 | ||
| 10. [−38/−7][−38/−12] −35mut | 0.57 | 2.1 | 0.27 | |
| 11. [−26/−3][−26/−12] | 0.65 | 0.46 | 1.4 | |
| 12. [−59/−11][−59/−12]UPec | 4.1 | 0.008 | 510 | |
| 13. [−59/−11][−59/−12]UPth | 3.4 | 0.35 | 9.7 | |
| Downstream fork junctions | 14. [−12/+20][+3/+20] | 1.6 | <0.2 | >8 |
| 15. [−12/+20][+3/+20]ggga | 0.2 | <0.2 | ||
| 16. [−11/+14][+2/+14] | 42 | 0.25 | 170 |
The Kd values presented are averages obtained from two to three individual experiments, the error is ±15%.
Table 2.
Comparison of dissociation constants for the binding of selected promoter fragments to Taq EσA at 25°C and 45°C
| DNA probe | Kdtaq, nM |
|
|---|---|---|
| 25°C | 45°C | |
| 3. −12/+2; ggga | 15 | 180 |
| 7. [−38/−11][−38/−12]TG | 1.2 | 2.6 |
| 14. [−12/+20][+3/+20] | 1.6 | 30 |
| 21. [−40/+2] | 7.1 | 3.2 |
The Kd values presented are averages obtained from two to three individual experiments, the error is ±15%.
Figure 4.
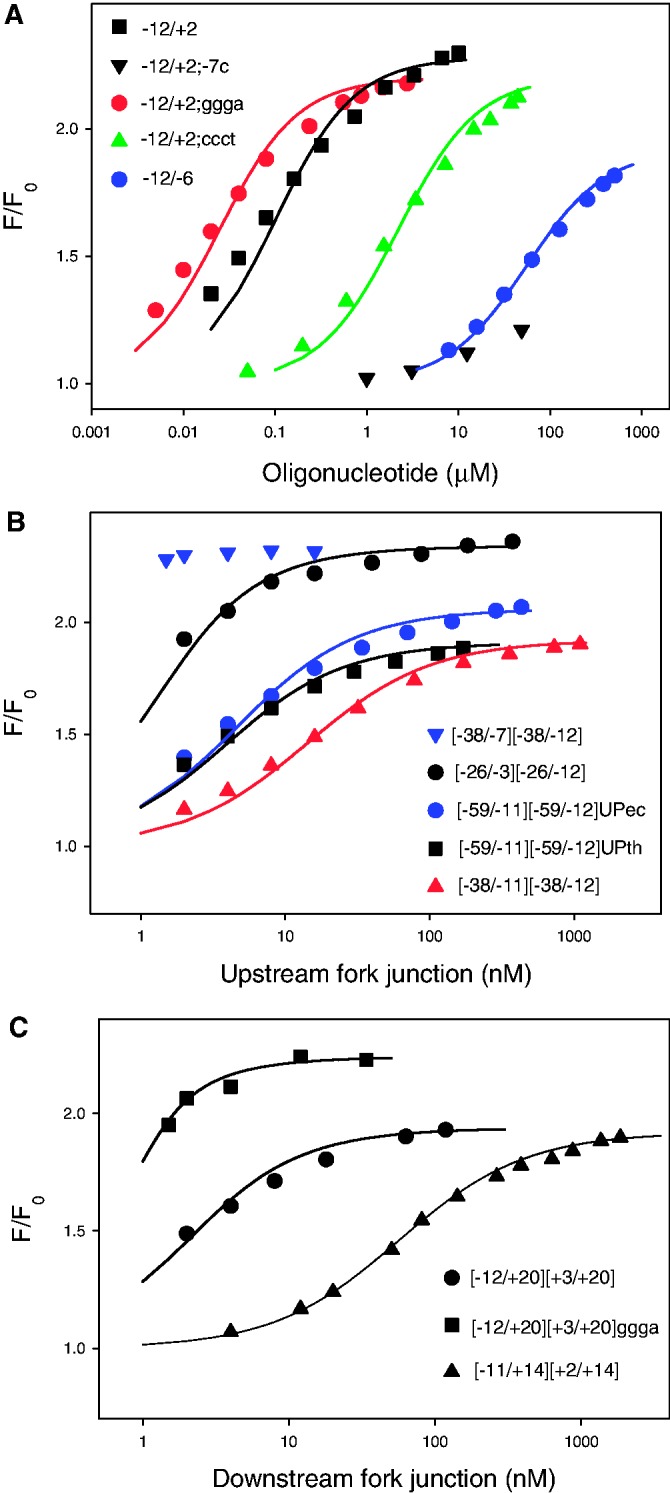
Binding of promoter fragments to T. aquaticus RNAP beacon. Titration of Taq EσA beacon with oligonucleotides (A) or upstream (B) and downstream (C) fork junction probes is shown. Solid lines correspond to a non-linear regression fit of the data. The experimental variation of F/F0 among replicate measurements usually did not exceed 15% of the average value.
Interactions of Taq EσA and Ec Eσ70 with oligos containing the −10 promoter element sequence
As a starting point, we compared Taq EσA and Ec Eσ70 affinities with an oligonucleotide corresponding to non-transcribed strand positions −12/+2 of the T5 N25 promoter (probe 1). A mutant oligo 2 with a T for C substitution at a highly conserved position −7 was used as control. The −12/+2 oligo bound Taq EσA RNAP about 2-fold better than Ec Eσ70 (Kd = 100 and 160 nM, respectively). The same ratio of affinities was found for the shorter −12/−6 probe 5. The control, −12/+2;−7C oligo, bound Taq EσA poorly (Figure 4A), testifying that Taq σA-based beacon is specific. The GGGA motif located immediately downstream of the −10 promoter element contributes to the strength of promoters recognized by Taq σA RNAP and is likely recognized by region 1.2 of σ (30,31). We measured the effect of substitution of the −12/+2 oligo positions −6/−3 (AGAT) for the optimal sequence GGGA (probe 3) and unfavorable sequence CCCT (probe 4) on RNAP binding. The substitutions caused considerable, ∼100-fold, change in affinity to Taq EσA, whereas only ∼10-fold change was observed with Ec Eσ70 (Figure 4A, Table 1). These data correlate well with reported effects caused by similar sequence changes on efficiencies of transcription initiation by these enzymes, suggesting that the downstream promoter motif is more important for Taq RNAP than for E. coli RNAP (32).
Interactions of Taq EσA and Ec Eσ70 with upstream fork junctions
To compare Taq EσA and Ec Eσ70 interactions with the part of promoter located upstream of the −10 element, we measured the affinities of several upstream fork junctions (probes 6–13). The measurements could not be carried out using probes with identical single-stranded fragments because Kd values for probes with the shortest and longest double-stranded segments differed by >105-fold, making quantitaive analysis technically impossible in some cases. To circumvent this problem, probe affinities were adjusted by changing the length of the single-stranded segment. Probe 11, bearing the shortest duplex segment with upstream end at position −26, bound Taq EσA and Ec Eσ70 with similar affinities (Kd = 0.65 and 0.46 nM, respectively). Other upstream forks contained longer double-stranded segments. One series of probes extended to position −38 and allowed to evaluate the contribution of the −35 promoter element to RNAP-promoter interaction. Ec Eσ70 bound probes 6–8 (contain consensus −35 element sequence) 3- to 6-fold stronger than Taq EσA. Conversely, we found that probe 10 bearing a mutated −35 element had ∼4-fold higher affinity to Taq EσA than to Ec Eσ70. The result suggests that Taq EσA may have higher affinity to non-consensus −35 element bases than Ec Eσ70.
The structures of probes 6 and 7 are identical, except for the TG extended −10 promoter motif present in probe 7 (positions −14/−15). The respective Kd values of Taq EσA and Ec Eσ70 binding to probe 6 are 12-fold higher than to probe 7 (Table 1). Thus, the contribution of TG to the binding of both enzymes is equal.
On the whole, the data obtained with probes extending up to position −38 suggest that although the modes of Taq EσA and Ec Eσ70 interactions with the −35 element may be somewhat different, the overall difference of interaction with the −10 and −35 promoter elements between EσA and Eσ70 is not very large and is therefore unlikely to account for observed large differences in stabilities of promoter complexes formed by these enzymes.
The [−59/−11][−59/−12]UPec and [−59/−11][−59/−12]UPth (probes 12 and 13) were designed to reveal additional RNAP interactions upstream of the −35 element. The sequences of these probes downstream of position −39 coincide with the [−38/−11][−38/−12] fork junction (probe 6), whereas the −39 to −59 bases represent either an optimized E. coli UP element sequence (probe 12) or the upstream sequence of 16 S rRNA promoter from T. thermophilus (33) (probe 13). As can be seen from Table 1, the affinities of probes 12 and 13 to Taq EσA were similar and only ∼4-fold higher than that of the shorter parent probe 6. In sharp contrast, Ec Eσ70 bound probes 12 and 13, respectively, 620 - and 14-fold stronger, than probe 6. Thus, in agreement with previous report (14), we observe only weak non-specific interactions between upstream promoter segment and Taq EσA. In contrast, and as expected, Ec Eσ70 bound to the optimal UP element sequence much stronger than to upstream DNA present in probe 13. Higher stability of Ec RNAP complexes compared with Taq RNAP complexes is observed on promoters without UP elements (6–11) and, therefore, the upstream interactions cannot be the sole cause of higher stability of E. coli RNAP complexes.
Interactions of Taq EσA and Ec Eσ70 with downstream fork junctions
Downstream fork junctions (probes 14–16) were previously used to characterize E. coli RNAP promoter interactions (19). Positions of the junction points in these probes (+2 or +3) are optimal for the binding to RNAP (19) and correspond to the position of the downstream boundary of transcription bubble in RPo. Therefore, RNAP interactions with the duplex segment of these downstream fork junctions likely mimic the corresponding downstream RNAP-promoter interactions in RPo. A Kd value of 0.25 nM was found for Ec Eσ70 binding to [−11/+14][+2/+14] downstream fork junction (probe 16) with a relatively short duplex segment, whereas the binding to a longer [−12/+20][+3/+20] downstream fork junction (probe 14) was so strong that only a lower estimate of Kd (<0.2 nM) could be obtained (19). Taq RNAP affinity to probe 14 was clearly below that of Ec Eσ70 (Kd = 1.6 nM, Figure 4C, Table 1). The affinity of probe 16 to Taq EσA was 170-fold lower than to Ec Eσ70 (Figure 4C and Table 1). The single-stranded parts of the downstream fork junctions are similar to the oligo probes, which, as shown above, bind to the T. aquaticus and E. coli RNAPs with similar affinities. Therefore, the considerably weaker binding of the downstream fork junctions to Taq RNAP than to the Ec enzyme is likely due to weaker interaction with duplex segments located downstream of position +1.
Probes 14 and 15 differ in their sequence at positions −6/−3 (AGAT and GGGA, respectively). Comparison of Kd values for Taq EσA binding with these probes (Kd = 1.6 and 0.2 nM, Figure 4C and Table 1) demonstrates that Taq EσA recognizes the GGGA motif in the context of downstream fork junctions.
Temperature dependence of Taq EσA binding to promoter fragments
The temperature dependence of Taq EσA and Ec Eσ70 binding to promoter DNA, upstream fork junctions and oligo templates was studied by Schroeder and deHaseth (10) using a heparin resistance assay. They found that both RNAPs bound promoter fragments better at lower temperatures, whereas the binding of DNA containing complete promoters improved when the temperature was increased (10). The downstream RNAP-promoter contacts are mainly made by RNAP domains called the lobe and the clamp, which can move relative to the central part of the enzyme (34,35). This conformational flexibility may be required for RP0 formation. We considered a possibility that the low affinity of Taq EσA to downstream fork junctions at 25°C could be improved at higher temperatures, for example, as a consequence of a conformational transition in Taq EσA that stimulates downstream DNA binding. Accordingly, we measured Kd for Taq EσA binding to four representative probes 1, 7, 14 and 21 (an oligo, an upstream and a downstream fork junction and a double-stranded promoter fragment without downstream DNA, correspondingly) at 45°C, a condition when the Taq enzyme is active, and at 25°C, when efficiency of open complex formation by EσA is low (Table 2). We found that only the binding of the [−40/+2] probe was stronger at 45°C than at 25°C. The affinity of other probes dropped at higher temperature. The decreases in the oligo and downstream fork junction affinities were close (12- and 19-fold, respectively), suggesting that the temperature dependence of fork junction binding is mainly determined by its single-stranded part. The −12/+2 segment of the [−40/+2] template corresponds to the position of the transcription bubble in promoter DNA. The improvement of [−40/+2] probe affinity at higher temperature suggests that its binding to RNAP is coupled with melting of this segment, mimicking the binding of full promoter DNA. Overall, we conclude that there is no increase of intrinsically weak binding of downstream DNA to Taq RNAP at elevated temperatures.
Effect of DNA downstream of the −10 element on the binding of double-stranded promoter fragments to Taq and Ec RNAPs
Previously, the strength of Ec Eσ70 beacon interaction with double-stranded DNA fragments whose downstream ends extended from −3 to +20 (probes 17–22) was measured (19). The key finding was the demonstration that a certain threshold length of downstream DNA was needed for efficient binding. We measured Taq EσA binding to probes 17–22 to compare it with Ec Eσ70 binding. The parent probe was a [−40/+20] fragment of the T5 N25 promoter. The binding of this fragment was specific, as a control fragment bearing a C to A substitution at the critically important −11 position [−40/+20; −11 C] generated much lower signal than the wild-type probe with both enzymes (Figure 5). The binding of [−40/+20]–based probes truncated at base pairs −3, +2, +10 and +15 was assessed by measuring Taq EσA beacon signal intensities generated by 1 nM RNAP beacon in the presence of 4 nM of each probe. The reaction temperature was 45°C. The resulting fluorescence intensities normalized to maximal signal amplitude (a signal generated by the [−40/−3] probe at a saturating concentration of 40 nM) are shown in Figure 5, along with similar data from (19) obtained with the E. coli RNAP beacon at 25°C. Both Taq and E. coli RNAPs generated the highest and lowest signals on the binding to [−40/−3] and [−40/+10] probes, respectively, whereas complexes with [−40/+2] generated intermediate signal amplitudes. Signals caused by the E. coli beacon binding to probes extended to positions +15 and +20 were close to the signal obtained with the [−40/+2] probe. In contrast, signals resulting from Taq EσA beacon interactions with [−40/+15] and [−40/+20] were considerably lower than that observed with [40/+2]. This result suggests that Taq EσA interactions with downstream segments of [−40/+15] and [−40/+20] are weaker than the corresponding interactions with Ec Eσ70, in agreement with the data obtained with downstream fork junctions.
Figure 5.
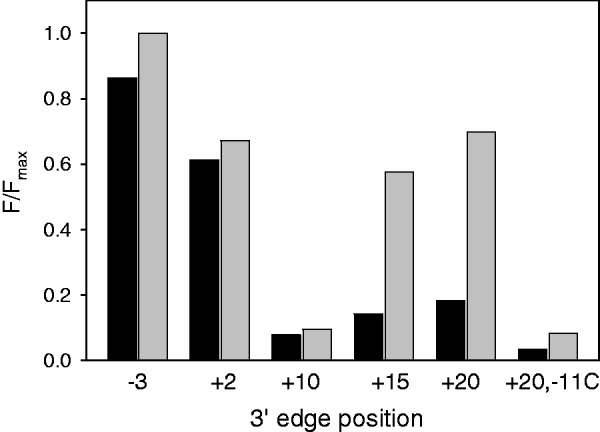
Measuring RNAP interaction with double-stranded promoter fragments using RNAP beacon assay. Normalized beacon signal amplitudes were measured in samples containing 1 nM Taq EσA (dark bars) or Ec Eσ70 (light bars) beacons and probes truncated at the indicated positions; the probe concentrations and temperatures were 4 nM, 45°C and 2 nM, 25°C in the experiments with Taq EσA and Ec Eσ70, respectively. The experimental variation of F/Fmax among replicate measurements did not exceed 20% of the average value.
Interaction of E. coli and Taq core RNAP enzymes with DNA
E. coli RNAP core forms tight, slowly dissociating non-promoter complexes with DNA in vitro (36). Formation of such long-lived complexes of RNAP core with DNA can affect transcription from promoters and likely should be somehow prevented in vivo (37). Although the structure of these non-specific complexes is unknown, the weakness of Taq core RNAP-mediated contacts with the downstream and upstream promoter segments in the context of Taq EσA suggests that Taq core RNAP may not form stable complexes with DNA as observed for E. coli core RNAP. A filter-binding assay for Taq and E. coli core RNAPs interactions with N25cons promoter DNA fragment supports this suggestion (Supplementary Figure S3).
We further evaluated the inhibition effect exerted by the RNAP core interactions with DNA on the formation of Taq and E. coli RNAP holoenzymes using the beacon assay. The −10 element-containing oligos bind to Ec Eσ70 and Taq EσA >100-fold stronger than to isolated σ subunits (18). Therefore, the kinetics of Taq and E. coli core RNAP binding to corresponding labeled σ subunits was measured by detecting the beacon signal increase caused by the addition of RNAP core to a sample containing a σ subunit and oligo probe −12/+2. Maximal signal intensities were reached in about 1 min [Supplementary Figure S4A and B, curves ‘(σA + oligo) + Taq core’ and ‘(σ70 + oligo) + Ec core’]. The kinetics of fluorescence intensity increase was apparently determined by the kinetics of holo RNAP formation, as oligo binding to preformed holo RNAP beacons occurred considerably faster, as shown in Supplementary Figure S4B [curve ‘(σA + Taq core) + oligo’]. Next, we performed similar experiments with a promoterless DNA fragment shown in Supplementary Figure S4C. The non-promoter DNA itself generated only negligible signals on interaction with preformed Taq EσA and Ec Eσ70 beacons under conditions used. In these experiments, the RNAP core enzymes were pre-incubated with the non-promoter DNA for 5 min. This pre-incubation exerted only a small effect on the rate of Taq EσA formation [Supplementary Figure S4A, compare curves ‘(σA + oligo) + Taq core’ and ‘(σA + oligo) + (Taq core + DNA)’] but considerably delayed the formation of Ec Eσ70 (Supplementary Figure S4B). Overall, the data show that Taq core RNAP-DNA complexes are much less stable than those formed by Ec core RNAP.
DISCUSSION
Quantification of RNAP binding to model promoter fragments and comparisons of the data with known properties of promoter complexes allow one to analyse the fine details of the RPo formation. Here, we systematically applied this approach to Taq and E. coli RNAP-promoter interactions using the RNAP beacon assay. The main goal was to identify RNAP-promoter contacts, which can be responsible for the relatively low stability of promoter complexes formed by Taq EσA. Consistent with a previous report (10), our data indicate that Taq RNAP is not compromised in terms of its intrinsic affinity to the single-stranded non-template segment of transcription bubble bearing the −10 element. However, in agreement with the known biochemical results (32), we found that the dependence of RNAP affinity on the sequence of discriminator is different in Taq and E. coli RNAPs: the ratio of affinities of oligos 3 and 4 bearing, respectively, the GGGA and CCCT discriminator sequences is 150 for Taq EσA and only 7 for to Ec Eσ70.
Our assay did not reveal noticeable differences in the strength of Taq EσA and Ec Eσ70 binding to the TG motif of the extended −10 element, which is widespread among Taq and Tth promoters, or to an upstream fork junction bearing a short duplex segment (probe 11). The data obtained with longer fork junctions containing the −35 element (probes 5–7) indicate that the affinity of Taq EσA to the consensus −35 element is moderately lower than that of Ec Eσ70. However, affinity of probe 10 with mutated −35 element to Taq EσA is higher than to Ec Eσ70. This result is consistent with the observation that non-specific interactions of Taq EσA region 4 with DNA suffice for activity of a T7 A1 promoter derivative (38).
The RNAP α subunit plays an important role in bacterial transcription regulation. In E. coli, interactions of two α subunits with various protein regulators and direct contacts of the α C-terminal domains (αCTD) with the upstream promoter DNA modulate the strength of RNAP-promoter interactions (39). In agreement with these data, our assay revealed that the consensus E. coli UP element stimulated Ec Eσ70 binding to probe 12 by >600-fold. An upstream segment of 16S rRNA Tth promoter bearing no significant sequence similarity to the UP element stimulated the Ec Eσ70 binding 14-fold, in the context of probe 13. However, in the context of the same probes, the UP element or the 16S rRNA Tth promoter segment stimulated the binding of Taq EσA by only ∼3.5-fold. Such weakness of upstream Taq EσA–promoter contacts suggests that αCTD-promoter interactions are unlikely to play an essential role in transcription regulation process in Thermus. Our results are consistent with the proposal that certain residues important for formation of the DNA-binding surface are not present in Taq αCTD, despite the overall similarity between the Ec αCTD and Taq αCTD structures (14).
Numerous studies indicate that downstream RNAP-promoter contacts, formed mostly by the β′ subunit, stabilize transcription initiation and elongation complexes formed by bacterial RNAPs and may decrease the energy barrier for promoter melting (15–17,40–45). A regulator of bacterial stringent response DksA decreases stability of RPo formed by Ec Eσ70 at the rrnB P1 promoter by disrupting downstream RNAP-promoter contacts (46). In the context of the transcription elongation complex, the downstream contacts participate in response of RNAP to pause and termination signals (4). An important new finding of this study is that RNAP contacts with promoter segment located downstream of the transcription start point are much weaker in Taq EσA complexes than in complexes formed by Ec Eσ70. In the context of downstream fork junctions, the difference between the strength of E. coli and Taq RNAPs interactions with downstream DNA duplex is ∼100-fold. We propose that similar difference of downstream RNAP-promoter interactions also exists in native promoter complexes formed by these RNAPs. Thus, the weakness of downstream interactions in Taq EσA promoter complexes could be one of the major reasons of their low stability at moderate temperatures. At elevated temperatures, the stability of Taq EσA transcription initiation complexes naturally increases owing to facilitation of promoter melting and the ability of the enzyme to withstand high temperature. The E. coli β′ subunit harbors an extensive β′In6 lineage-specific domain insertion (also called β′SI3 and β′GNCD) that is missing in Taq (47). Deletion of the β′In6 in E. coli results in destabilization of open complexes (15). Structural modeling suggests that this domain plays a role in stabilizing RNAP interactions with downstream DNA (48). Thus, the β′In6 domain likely contributes to the high affinity of Ec Eσ70 to downstream promoter DNA duplex. As noted earlier, a possibility of existence of species-specific differences in the strength of downstream RNAP-promoter contacts was already predicted basing on comparison of kinetic characteristics of E. coli and B. subtilis transcription complexes (4). Our work provides direct confirmation for this hypothesis.
On the whole, our data indicate that the difference in stability between Ec Eσ70 and Taq EσA promoter complexes is in large part determined by difference in strength of contacts mediated by the RNAP core. In this regard, it is not surprising that the non-specific interaction of Taq core RNAP with DNA was found to be much weaker than in the case of Ec RNAP core. We speculate that this property of Taq RNAP may help prevent formation of long-lived Taq core RNAP-DNA complexes in vivo.
Future studies will allow more systematic elucidation of relationships between partial RNAP-promoter interactions and functional properties of complexes formed by RNAPs from various bacteria.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–4.
FUNDING
National Institutes of Health [R01 GM64530 and R01 GM59295 to K.S.]; Molecular and Cell Biology Program grant from the Russian Academy of Sciences Presidium (to K.S.); “Scientific and scientific-pedagogical personnel of innovative Russia 2009–2013” [8475 to V.M.]. Funding for open access charge: “Scientific and scientific-pedagogical personnel of innovative Russia 2009–2013” [8475].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 2003;13:31–39. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 2.Saecker RM, Record MT, Jr, Dehaseth PL. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011;412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross CA, Chan C, Dombroski A, Gruber T, Sharp M, Tupy J, Young B. The functional and regulatory roles of sigma factors in transcription. Cold Spring Harbor Symp. Quant. Biol. 1998;63:141–155. doi: 10.1101/sqb.1998.63.141. [DOI] [PubMed] [Google Scholar]
- 4.Artsimovitch I, Svetlov V, Anthony L, Burgess RR, Landick R. RNA polymerases from Bacillus subtilis and Escherichia coli differ in recognition of regulatory signals in vitro. J. Bacteriol. 2000;182:6027–6035. doi: 10.1128/jb.182.21.6027-6035.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meier T, Schickor P, Wedel A, Cellai L, Heumann H. In vitro transcription close to the melting point of DNA: analysis of Thermotoga maritima RNA polymerase-promoter complexes at 75 degrees C using chemical probes. Nucleic Acids Res. 1995;23:988–994. doi: 10.1093/nar/23.6.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue Y, Hogan BP, Erie DA. Purification and initial characterization of RNA polymerase from Thermus thermophilus. Biochemistry. 2000;39:14356–14362. doi: 10.1021/bi0012538. [DOI] [PubMed] [Google Scholar]
- 7.Minakhin L, Nechaev S, Campbell EA, Severinov K. Recombinant Thermus aquaticus RNA polymerase, a new tool for structure-based analysis of transcription. J. Bacteriol. 2001;183:71–76. doi: 10.1128/JB.183.1.71-76.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuznedelov K, Minakhin L, Severinov K. Preparation and characterization of recombinant Thermus aquaticus RNA polymerase. Methods Enzymol. 2003;370:94–108. doi: 10.1016/S0076-6879(03)70009-3. [DOI] [PubMed] [Google Scholar]
- 9.Kulbachinskiy A, Bass I, Bogdanova E, Goldfarb A, Nikiforov V. Cold sensitivity of thermophilic and mesophilic RNA polymerases. J. Bacteriol. 2004;186:7818–7820. doi: 10.1128/JB.186.22.7818-7820.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schroeder LA, deHaseth PL. Mechanistic differences in promoter DNA melting by Thermus aquaticus and Escherichia coli RNA polymerases. J. Biol. Chem. 2005;280:7422–17429. doi: 10.1074/jbc.M501281200. [DOI] [PubMed] [Google Scholar]
- 11.Miropolskaya N, Ignatov A, Bass I, Zhilina E, Pupov D, Kulbachinskiy A. Distinct functions of regions 1.1 and 1.2 of RNA polymerase σ subunits from Escherichia coli and Thermus aquaticus in transcription initiation. J. Biol. Chem. 2012;287:23779–23789. doi: 10.1074/jbc.M112.363242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annu. Rev. Genet. 2004;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa S, Oshima T, Kurokawa T, Kusuya Y, Ogasawara N. RNA polymerase trafficking in Bacillus subtilis cells. J. Bacteriol. 2010;192:5778–5787. doi: 10.1128/JB.00489-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wada T, Yamazaki T, Kyogoku Y. The structure and the characteristic DNA binding property of the C-terminal domain of the RNA polymerase alpha subunit from Thermus thermophilus. J. Biol. Chem. 2000;275:16057–16063. doi: 10.1074/jbc.275.21.16057. [DOI] [PubMed] [Google Scholar]
- 15.Artsimovitch I, Svetlov V, Murakami KS, Landick R. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- 16.Bartlett MS, Gaal T, Ross W, Gourse RL. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J. Mol. Biol. 1998;279:331–345. doi: 10.1006/jmbi.1998.1779. [DOI] [PubMed] [Google Scholar]
- 17.Ederth J, Artsimovitch I, Isaksson LA, Landick R. The downstream DNA jaw of bacterial RNA polymerase facilitates both transcriptional initiation and pausing. J. Biol.Chem. 2002;277:37456–37463. doi: 10.1074/jbc.M207038200. [DOI] [PubMed] [Google Scholar]
- 18.Mekler V, Pavlova O, Severinov K. The interaction of Escherichia coli RNA polymerase σ70 subunit with promoter elements in the context of free σ70, RNA polymerase holoenzyme, and the β′–σ70 complex. J. Biol. Chem. 2011;286:270–279. doi: 10.1074/jbc.M110.174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mekler V, Minakhin L, Severinov K. A critical role of downstream RNA polymerase-promoter interactions in the formation of initiation complex. J. Biol. Chem. 2011;286:22600–22608. doi: 10.1074/jbc.M111.247080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mekler V, Minakhin L, Sheppard C, Wigneshweraraj S, Severinov K. Molecular mechanism of transcription inhibition by phage T7 gp2 protein. J. Mol. Biol. 2011;413:1016–1027. doi: 10.1016/j.jmb.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feklistov A, Darst SA. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase σ subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuznedelov K, Lamour V, Patikoglou G, Chlenov M, Darst SA, Severinov K. Recombinant Thermus aquaticus RNA polymerase for structural studies. J. Mol. Biol. 2006;359:110–121. doi: 10.1016/j.jmb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Sevostyanova A, Djordjevic M, Kuznedelov K, Naryshkina T, Gelfand MS, Severinov K, Minakhin L. Temporal regulation of viral transcription during development of Thermus thermophilus bacteriophage phiYS40. J. Mol. Biol. 2007;366:420–435. doi: 10.1016/j.jmb.2006.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis AN, Niu W, Ebright YW, Levy R, Ebright RH. Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase promoter open complex. Cell. 2002;108:599–614. doi: 10.1016/s0092-8674(02)00667-0. [DOI] [PubMed] [Google Scholar]
- 25.Campbell EA, Muzzin O, Chlenov, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, Darst SA. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell. 2002;9:527–539. doi: 10.1016/s1097-2765(02)00470-7. [DOI] [PubMed] [Google Scholar]
- 26.Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- 27.Guo J, Gralla JD. Promoter opening via a DNA fork junction binding activity. Proc. Natl Acad. Sci. USA. 1998;95:11655–11660. doi: 10.1073/pnas.95.20.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heyduk E, Heyduk T. Conformation of fork junction DNA in a complex with Escherichia coli RNA polymerase. Biochemistry. 2002;41:2876–2883. doi: 10.1021/bi012133i. [DOI] [PubMed] [Google Scholar]
- 29.Tsujikawa L, Tsodikov OV, deHaseth PL. Interaction of RNA polymerase with forked DNA: evidence for two kinetically significant intermediates on the pathway to the final complex. Proc. Natl Acad. Sci. USA. 2002;99:3493–3498. doi: 10.1073/pnas.062487299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haugen SP, Berkmen MB, Ross W, Gaal T, Ward C, Gourse RL. rRNA promoter regulation by nonoptimal binding of sigma region 1.2: an additional recognition element for RNA polymerase. Cell. 2006;16:1069–1082. doi: 10.1016/j.cell.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 31.Feklistov V, Barinova N, Sevostyanova A, Heyduk E, Bass I, Vvedenskaya I, Kuznedelov K, Merkiene E, Stavrovskaya E, Klimasauskas S, et al. A basal promoter element recognized by free RNA polymerase sigma subunit determines promoter recognition by RNA polymerase holoenzyme. Mol. Cell. 2006; 23:97–107. doi: 10.1016/j.molcel.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Barinova N, Kuznedelov K, Severinov K, Kulbachinskiy A. Structural modules of RNA polymerase required for transcription from promoters containing downstream basal promoter element GGGA. J. Biol. Chem. 2008;283:22482–22489. doi: 10.1074/jbc.M802445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartmann RK, Erdmann VA. Thermus thermophilus 16S rRNA is transcribed from an isolated transcription unit. J. Bacteriol. 1989;171:2933–2941. doi: 10.1128/jb.171.6.2933-2941.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darst SA, Opalka N, Chacon P, Polyakov A, Richter C, Zhang G, Wriggers W. Conformational flexibility of bacterial RNA polymerase. Proc. Natl Acad. Sci. USA. 2002;99:4296–4301. doi: 10.1073/pnas.052054099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, et al. Opening and closing of the bacterial RNA polymerase clamp. Science. 2012;337:591–595. doi: 10.1126/science.1218716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.deHaseth PL, Lohman TM, Burgess RR, Record MT., Jr Nonspecific interactions of Escherichia coli RNA polymerase with native and denatured DNA: differences in the binding behavior of core and holoenzyme. Biochemistry. 1978;17:1612–1622. doi: 10.1021/bi00602a006. [DOI] [PubMed] [Google Scholar]
- 37.Shaw G, Gan J, Zhou YN, Zhi H, Subburaman P, Zhang R, Joachimiak A, Jin DJ, Ji X. Structure of RapA, a Swi2/Snf2protein that recycles RNApolymerase during transcription. Structure. 2008;16:1417–1427. doi: 10.1016/j.str.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuzenkova Y, Tadigotla VR, Severinov K, Zenkin N. A new basal promoter element recognized by RNA polymerase core enzyme. EMBO J. 2011;30:3766–3775. doi: 10.1038/emboj.2011.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DJ, Minchin SD, Busby SJ. Activating Transcription in Bacteria. Annu. Rev. Microbiol. 2012;66:125–152. doi: 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 40.Nudler E, Avetissova E, Markovtsov V, Goldfarb A. Transcription processivity: protein-DNA interactions holding together the elongation complex. Science. 1996;273:211–217. doi: 10.1126/science.273.5272.211. [DOI] [PubMed] [Google Scholar]
- 41.Naryshkin N, Revyakin A, Kim Y, Mekler V, Ebright RH. Structural organization of the RNA polymerase-promoter open complex. Cell. 2000;101:601–611. doi: 10.1016/s0092-8674(00)80872-7. [DOI] [PubMed] [Google Scholar]
- 42.Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov V, Goldfarb A, Darst SA. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- 43.Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 44.Saecker RM, Tsodikov OV, McQuade KL, Schlax PE, Jr, Capp MW, Record MT., Jr Kinetic studies and structural models of the association of E. coli sigma(70) RNA polymerase with the lambdaP(R) promoter: large scale conformational changes in forming the kinetically significant intermediates. J. Mol. Biol. 2002;319:649–671. doi: 10.1016/S0022-2836(02)00293-0. [DOI] [PubMed] [Google Scholar]
- 45.Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007;448:157–162. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- 46.Rutherford ST, Villers CL, Lee JH, Ross W, Gourse RL. Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev. 2009;23:236–248. doi: 10.1101/gad.1745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J. Mol. Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chlenov M, Masuda S, Murakami KS, Nikiforov V, Darst SA, Mustaev A. Structure and function of lineage-specific sequence insertions in the bacterial RNA polymerase beta' subunit. J. Mol. Biol. 2005;353:138–154. doi: 10.1016/j.jmb.2005.07.073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.