

Abstract
Exchange protein directly activated by cAMP (EPAC) and cAMP-dependent protein kinase (PKA) are two intracellular receptors that mediate the effects of the prototypic second messenger cAMP. Identifying pharmacological probes for selectively modulating EPAC activity represents a significant unmet need within the research field. Herein, we report the identification and characterization of 3-(5-tert-butyl-isoxazol-3-yl)-2-[(3-chloro-phenyl)-hydrazono]-3-oxo-propionitrile (ESI-09), a novel noncyclic nucleotide EPAC antagonist that is capable of specifically blocking intracellular EPAC-mediated Rap1 activation and Akt phosphorylation, as well as EPAC-mediated insulin secretion in pancreatic β cells. Using this novel EPAC-specific inhibitor, we have probed the functional roles of overexpression of EPAC1 in pancreatic cancer cells. Our studies show that EPAC1 plays an important role in pancreatic cancer cell migration and invasion, and thus represents a potential target for developing novel therapeutic strategies for pancreatic cancer.
Introduction
The major physiologic effects of cAMP in mammalian cells are transduced by two ubiquitously expressed intracellular cAMP receptor families: the classic protein kinase A/cAMP-dependent protein kinase (PKA/cAPK) and the more recently discovered exchange protein directly activated by cAMP/cAMP-regulated guanine nucleotide exchange factor (EPAC/cAMP-GEF) (Cheng et al., 2008). Similar to PKA, EPAC contains an evolutionarily conserved cAMP-binding domain (CBD) that acts as a molecular switch for sensing intracellular levels of the second messenger cAMP, and activates the downstream signaling molecules small GTPases Rap1 and Rap2 (de Rooij et al., 1998; Kawasaki et al., 1998). In addition, EPAC proteins exert their functions through interactions with other cellular partners at specific cellular loci. For example, EPAC1 is known to associate with mitotic spindle, plasma membrane, and nuclear membrane by interacting with tubulin (Qiao et al., 2002; Mei and Cheng, 2005), ezrin-radixin-moesin (ERM) proteins (Gloerich et al., 2010; Ross et al., 2011), and nucleoporin RanBP2 (Liu et al., 2010; Gloerich et al., 2011), respectively. On the other hand, EPAC2 can interact with Rim (Rab3 interacting molecule) and Rim2 (Ozaki et al., 2000; Kashima et al., 2001), as well as a structurally related calcium sensor Piccolo (Fujimoto et al., 2002). In pancreatic β cells, interactions among EPAC2, Rim2, and Piccolo are critical for cAMP-mediated insulin secretion (Ozaki et al., 2000; Kashima et al., 2001; Fujimoto et al., 2002).
More than a decade of extensive studies have now firmly established that many cAMP-related cellular processes previously thought to be controlled by PKA alone are also mediated by EPAC (Gloerich and Bos, 2010). For example, EPAC proteins have been implicated in regulating exocytosis and secretion (Ozaki et al., 2000; Maillet et al., 2003; Seino and Shibasaki, 2005; Li et al., 2007), cell adhesion (Rangarajan et al., 2003; Enserink et al., 2004), endothelial barrier junctions (Cullere et al., 2005; Kooistra et al., 2005), leptin signaling (Fukuda et al., 2011), and cardiac functions (Metrich et al., 2010). Interestingly, although PKA and EPAC are activated by the same second messenger cAMP, they can act antagonistically in controlling various cellular functions such as Akt/PKB phosphorylation (Mei et al., 2002; Brennesvik et al., 2005) and hedgehog signaling (Ji et al., 2007). On the other hand, depending upon the specific cellular context, EPAC and PKA can exert synergistic effects on downstream signaling such as stimulation of neurotensin secretion (Li et al., 2007) and attenuation of cAMP signaling through phosphodiesterases (Dodge-Kafka et al., 2005). Despite these significant advances in our knowledge, the lack of EPAC-specific antagonists has greatly limited our ability to discern the physiologic functions of this family of proteins and understand their role in numerous diseases. For example, EPAC1 is overexpressed in human pancreatic ductal adenocarcinoma (PDA) specimens (Lorenz et al., 2008), but the implications of this overexpression are unclear. To address the lack of EPAC inhibitors, we have developed a robust high-throughput screening (HTS) assay for identifying new chemical probes specifically targeting EPAC proteins (Tsalkova et al., 2012a). In this study, we describe the identification and characterization of 3-(5-tert-butyl-isoxazol-3-yl)-2-[(3-chloro-phenyl)-hydrazono]- 3-oxo-propionitrile (ESI-09), a novel EPAC-specific inhibitor, which enabled us to demonstrate that EPAC1 plays an important role in pancreatic cancer cell migration and invasion.
Materials and Methods
Reagents.
8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate, acetoxymethyl ester (007-AM), initially provided by and subsequently purchased from BioLog Life Science Institute (Bremen, Germany), is the highly membrane-permeant and potent acetoxymethyl ester prodrug (Vliem et al., 2008; Chepurny et al., 2009) of 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (007), the first selective EPAC agonist that is more than 100-fold less potent for PKA (Enserink et al., 2002; Christensen et al., 2003). After cell penetration of the uncharged prodrug, the active parental compound 007 is released by endogenous esterases and trapped inside the cell due to the negatively charged cyclic phosphate moiety. 8-[[2-[(7-Nitro-4-benzofurazanyl)amino]ethyl]thio] adenosine-3′,5′-cyclic monophosphate (8-NBD-cAMP) was also obtained from BioLog Life Science Institute. 2′-/3′-O-(N′-methylanthraniloyl)guanosine-5′-O-diphosphate (Mant-GDP) was purchased from Molecular Probes, Invitrogen (Carlsbad, CA). All other reagents were purchased through Sigma-Aldrich (St. Louis, MO).
Antibodies.
Antibodies specific against Akt (#9272), phospho-Akt (T308) (#4056), phospho-Akt (S473) (#9271), EPAC1 (#4155), and EPAC2 (#4156) were purchased from Cell Signaling Technology (Danvers, MA), while anti-Rap1 antibody (SC-65) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Protein Expression and Purification.
Recombinant EPAC1, EPAC2, and C-terminal truncated Rap1B(1-167) were purified as described previously (Mei and Cheng, 2005; Li et al., 2011). PKA RIα, RIIβ, and catalytic subunits were recombinantly expressed in Escherichia coli and purified to homogeneity as reported (Cheng et al., 2001). Type I and II PKA holoenzymes were reconstituted from individually purified recombinant PKA R and C subunits (Yu et al., 2004). All proteins used in this study were at least 95% pure, as judged by SDS-PAGE.
HTS.
Fluorescence intensity of 8-NBD-cAMP in complex with EPAC2 has been used as a primary signal in HTS (Tsalkova et al., 2012a). A primary screen of the Maybridge HitFinder collection of compounds (Maybridge; Thermo Fisher Scientific, Trevillett, UK) was performed in black 384-well low-volume microplates (Corning Costar, Cambridge, MA). In brief, a protein solution containing 65 nM EPAC2 and 75 nM 8-NBD-cAMP was dispensed into 384-well plates (35 µl/well) using the Biomek FX Laboratory Automation Workstation equipped with a 96-multichannel pipetting head (Beckman Coulter, Brea, CA). Test compounds (1 µl/well) were added from 10 mM stock solutions in DMSO. DMSO or cAMP at 10 mM was used as a negative or positive control, respectively. Fluorescence intensity signal was recorded at room temperature before and after tested compounds were added using the SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA) with excitation/emission wavelengths set at 470/540 nm.
In Vitro GEF Activity Assay of EPAC Proteins.
In vitro EPAC activity was measured using purified Rap1B(1-167) loaded with Mant-GDP as previously described (Tsalkova et al., 2009).
In Vitro PKA Activity Assay.
PKA kinase activities of the type I and II PKA holoenzymes were measured spectrophotometrically with a coupled enzyme assay using purified and reconstituted recombinant PKA holoenzymes (Cook et al., 1982).
Cell Culture.
AsPC-1 and PANC-1 cells with suppressed EPAC1 expression were generated using MISSION TRC (Sigma-Aldrich, St. Louis, MO) lentiviral-based short hairpin RNA according to the manufacturer’s protocol. AsPC-1 and PANC-1 cells were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS). INS-1 cells (passage numbers 70–90) were maintained in RPMI 1640 medium containing 10 mM Hepes, 10% FBS, 2.0 mM l-glutamine, 1.0 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. All cells were kept in a humidified incubator (95% air, 5% CO2) at 37°C.
Phosphorylation of Akt.
Cells were starved of serum for 24 hours before treatment with ESI-09 for 5 minutes. Cells were then treated with 10 µM 007-AM for 15 minutes, and cellular proteins were extracted in SDS lysis buffer (2% SDS, 10% glycerol, 60 mM Tris, pH 6.8). Nontreated cells and cells treated with 10 µM 007-AM only were used as negative and positive controls, respectively. Approximately 10 µg of total protein extract was separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Akt phosphorylation was probed with anti-phospho-Akt T308 and S473 antibodies (1:1000). Phosphorylation levels were determined with densitometry and expressed as a percentage of basal Akt phosphorylation (negative control). At least three independent experiments were performed for each Western blot.
Insulin Secretion Assay.
INS-1 cells were plated in 96-well plates precoated with polylysine at a density of 1 × 105 cells/well. After overnight incubation, the medium was replaced with Krebs-Ringer bicarbonate (KRB) containing 2.9 mM glucose. After an additional 2-hour incubation, the cells were treated with ESI-09 or DMSO vehicle as a control in fresh KRB containing 11.8 mM glucose for 10 minutes, followed by a 30-minute stimulation with 10 µM 007-AM. The supernatant was collected, and insulin was quantified using an Ultra Sensitive Rat Insulin ELISA kit from Crystal Chem Inc. (Downers Grove, IL).
Transwell Migration/Invasion Assay.
The top chambers of 8-micron inserts (Costar Inc.) were coated with BD Matrigel Basement Membrane Matrix (50 µg/ml) (BD Biosciences, San Jose, CA). Cells (2 × 105) pretreated with ESI-09 for 24 hours were added to the top chamber of the inserts in serum-free RPMI 1640 medium containing 0.25% bovine serum albumin. The bottom chamber was filled with 600.0 μl of RPMI containing 10% FBS and ESI-09. The cells were then incubated at 37°C in 5% CO2 for 20 hours. Cells were removed from the top chamber, and migrated cells were fixed in methanol and stained with crystal violet. The numbers of migrated cells were counted in four different fields. Three independent experiments were performed for each ESI-09 concentration.
Wound Healing Assay.
Cells were grown to 95%–100% confluence before a scratch wound was made. The medium was changed to RPMI 10% FBS containing 10 μM ESI-09 or vehicle. The cells were then incubated at 37°C in 5% CO2. The wound was imaged at 0 and 22 hours after changing the medium. Healing rate was determined by calculating the percentage of wound closure according to the following equation: % wound closure = (initial wound width – wound width 22 hours post-treatment with ESI-09 or vehicle)/initial wound width × 100.
To make the results comparable across all assays, the widths of the initial wounds were all normalized to a 1-mm distance.
Cell Adhesion Assay.
Cells were serum-starved for 24 hours in RPMI 1640, detached using 0.25% trypsin-EDTA, and washed twice with serum-free RPMI 1640 before treatment with ESI-09 for 15 minutes. The cells were then treated with 10 µM 007-AM and ∼3 × 104 cells in 100 µl RPMI 1640 were immediately added to each well of a non-tissue-culture-treated EIA/RIA 96-well plate (Corning Life Sciences (Lowell, MA). Nontreated cells and cells treated with 10 µM 007-AM only were used for comparison. The plates were previously coated with collagen I for 2 hours (10 µg/ml in phosphate-buffered saline [PBS]) and blocked with 5% bovine serum albumin in PBS for 1 hour at 37°C. The plates were incubated at 37°C for 1 hour, and unattached cells were removed by washing twice with PBS. The cells were fixed with 3.7% paraformaldehyde in PBS for 10 minutes and stained with trypan blue. The attached cells were counted, and the percentage of adherent cells in each treatment group was calculated using the following equation: % adhesion = number of adherent cells in treatment group/number of adherent cells in nontreatment group × 100.
The adhesion assays were repeated three times.
Results
Identification and Biochemical Characterization of ESI-09.
We developed a sensitive and robust HTS assay (Tsalkova et al., 2012a) based on the principle that fluorescence intensity of a cAMP analog, 8-NBD-cAMP, is environmentally sensitive. Binding of 8-NBD-cAMP to purified full-length EPAC2 led to a >100-fold increase in fluorescence signal. This increase was competitively reversed by the binding of cAMP (Supplemental Fig. 1). Through HTS of the Maybridge HitFinder diversity library with 14,400 drug-like chemicals, we identified 3-[5-(tert-butyl)isoxazol-3-yl]-2-[2-(3-chlorophenyl)hydrazono]-3-oxopropanenitrile, referred to as ESI-09 (Fig. 1), which was capable of reducing both EPAC1 and EPAC2 GEF activity to basal levels at 25 µM concentration in the presence of an equal concentration of cAMP (Tsalkova et al., 2012b). After the initial identification of the hit, individually vialed and highly purified ESI-09 was purchased from Maybridge and its chemical structure and purity were validated by 1H and 13C NMR as well as mass spectrometry analysis. We then performed dose-dependent titrations to determine the relative binding affinity of ESI-09. When various amounts of cAMP or ESI-09 were added to the reaction mixture, with fixed concentrations of EPAC2 and 8-NBD-cAMP, a concentration-dependent decrease in 8-NBD-cAMP fluorescence was observed (Fig. 2A). While cAMP competed with 8-NBD-cAMP binding with an apparent IC50 of 39 ± 2.0 µM, ESI-09 showed an increased potency with an apparent IC50 of 10 ± 1.2 µM. To determine whether this apparent higher-affinity binding of EPAC2 antagonists can be translated to a high potency in suppressing the GEF activity of EPAC2, we determined the inhibition curves of EPAC-catalyzed Rap1-GDP exchange activity for ESI-09 in the presence of 25 µM cAMP. As shown in Fig. 2B, ESI-09 inhibited cAMP-mediated EPAC2 GEF activity with an apparent IC50 of 1.4 ± 0.1 µM. Similarly, ESI-09 was also effective in suppressing cAMP-mediated EPAC1 GEF activity with an apparent IC50 of 3.2 ± 0.4 µM. To determine the specificity of ESI-09, we performed counterscreening assays that measured type I and II PKA holoenzyme activities. An ESI-09 concentration of 25 µM did not significantly alter cAMP-induced type Iα and IIβ PKA holoenzyme activation, whereas H89, a selective PKA inhibitor, blocked type I and II PKA activities completely (Supplemental Fig. 2). When tested at 100 µM concentration, ESI-09 inhibited type II PKA activity up to 20% in the presence of 100 µM cAMP. These data suggest that ESI-09 is at least 100 times more selective for EPAC proteins than PKA.
Fig. 1.

Chemical structure of ESI-09.
Fig. 2.

Relative potency of ESI-09. (A) dose-dependent competition of ESI-09 (open circles) and cAMP (closed squares) with 8-NBD-cAMP in binding to EPAC2. (B) dose-dependent inhibition of EPAC1 (closed circles) or EPAC2 (open circles) GEF activity by ESI-09 in the presence of 25 µM cAMP.
Molecular Docking of ESI-09 into the CBDs of EPAC1 and EPAC2.
To gain insights into the structural basis for the binding of ESI-09 to the EPAC proteins, the intermolecular interactions of ESI-09 with EPAC1 and EPAC2 were analyzed and compared by molecular docking (Trott and Olson, 2010) using the cAMP-bound EPAC2 crystal structure (3CF6) (Rehmann et al., 2008) and a three-dimensional homology model of EPAC1 (Arnold et al., 2006). AutoDock Vina docking results revealed that ESI-09 could be docked to the CBD of EPAC1 and EPAC2, with better energetic scores than those for cAMP. ESI-09 could fit well into the functional cAMP-binding pocket of EPAC1, establishing favorable hydrophobic and hydrogen bonding interactions with the protein’s active-site residues. The tert-butyl-isoxazolyl moiety of ESI-09 could extend to a deep hydrophobic pocket around Phe268 and Met312, with its 3-chloro-phenyl fragment close to the hydrophobic pocket around Ala281 and Leu314. Moreover, ESI-09 could form five putative hydrogen bonds with the residues Gly269, Gln270, Leu271, Ala280, and Ala281 (Fig. 3, A and B). With EPAC2, the tert-butyl-isoxazolyl moiety could interact with a hydrophobic pocket formed by Phe367, Leu406, Ala407, and Ala415, while residues Val386, Val394, Leu397, Ala416, and Leu448 could form hydrophobic interactions with the 3-chloro-phenyl fragment of ESI-09. Meanwhile, the tert-butyl-isoxazolyl moiety could form a hydrogen bond with the residue Gly404 of EPAC2 (Fig. 3, C and D). These docking observations are in full agreement with the experimental EPAC inhibition results as shown in Fig. 2, and provide useful information to assist our ongoing efforts to design and synthesize more potent and specific inhibitors based on ESI-09 as a chemical lead.
Fig. 3.

Predicted binding mode and molecular docking of ESI-09 into the CBD of EPAC1 protein (homology modeling) and EPAC2 protein (Protein Data Bank code 3CF6). (A) binding mode of ESI-09 with EPAC1 protein (homology modeling); (B) surface of the CBD of EPAC1 protein; (C) binding mode of ESI-09 with EPAC2 protein; (D) surface of the CBD of EPAC2 protein. Important residues are drawn in sticks. Hydrogen bonds are shown as dashed green lines. ESI-09 is shown in pink, and cAMP is shown in pale yellow.
Cellular Characterization of ESI-09.
To test whether our newly identified EPAC antagonist ESI-09 is capable of modulating EPAC activation in living cells, we monitored its ability to suppress Akt phosphorylation, as EPAC proteins are also known to activate Akt signaling, whereas PKA inhibits it (Mei et al., 2002). To determine whether ESI-09 is capable of blocking EPAC-mediated Akt activation, the phosphorylation status of T308 and S473 of Akt in the pancreatic cancer cell line AsPC-1, which overexpresses EPAC1, was followed using anti-phospho-Akt antibodies. As shown in Fig. 4, ESI-09 inhibited 007-AM-stimulated Akt phosphorylation at T308 and S473 in a dose-dependent manner. Similar results were observed in the rat pancreatic β-cell line INS-1 (Supplemental Fig. 3). On the other hand, ESI-09 failed to suppress epidermal growth factor (EGF)-induced phosphorylation of Akt in AsPC1 cells (Fig. 4).
Fig. 4.
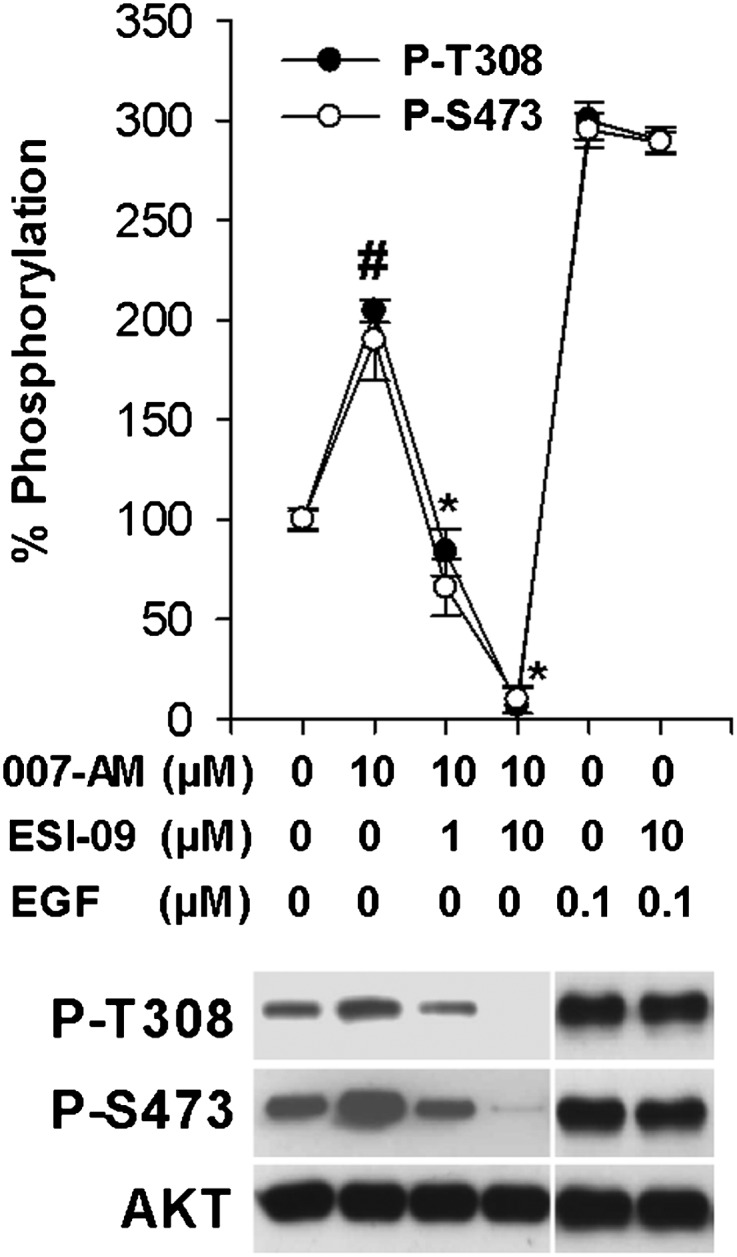
ESI-09 inhibits EPAC- but not EGF-mediated Akt phosphorylation in AsPC-1 pancreatic cancer cells. Serum-starved AsPC-1 cells were stimulated with vehicle, 10 µM 007-AM, or 0.1 µM EGF after pretreatment with the indicated concentrations of ESI-09. Cell lysates were subjected to Western blot analyses using anti-phospho-Akt T308 and S473 antibodies (representative blot shown). Data points represent mean ± S.D. (n = 3). #Significantly higher than vehicle group (P < 0.02). *Significantly lower that 007-AM-stimulated group (P < 0.02).
After demonstrating that ESI-09 was capable of inhibiting EPAC-mediated Akt phosphorylation in cell culture, we proceeded to determine whether ESI-09 was capable of suppressing EPAC-mediated physiologic functions. EPAC proteins, particularly EPAC2, have been implicated in regulating cAMP-mediated exocytosis in a variety of secretory cells (Seino and Shibasaki, 2005). In pancreatic endocrine β cells, EPAC mediates the glucose-induced insulin secretion potentiated by glucagon-like peptide 1 and gastric inhibitory polypeptide, which bind to receptors on the surface of β cells and promote intracellular cAMP production (Leech et al., 2000; Kashima et al., 2001; Kang et al., 2003; Shibasaki et al., 2007). When INS-1 was stimulated by 007-AM, a steady increase in insulin secretion was observed. This EPAC-mediated increase in the level of insulin secretion was inhibited in a dose-dependent manner by preincubation of the cells with ESI-09 (Fig. 5). The impact of ESI-09 on EPAC-mediated Akt phosphorylation and insulin secretion is consistent with the biochemical Rap1 nucleotide exchange data shown in Fig. 2B and demonstrates that this small molecule is a pan-EPAC inhibitor that is capable of suppressing EPAC cellular functions.
Fig. 5.

ESI-09 inhibits EPAC2-mediated insulin secretion in INS-1 cells. INS-1 cells were stimulated with vehicle or 10 µM 007-AM after pretreatment with the indicated concentrations of ESI-09. Bars represent mean ± S.D. (n = 3). #Significantly higher than vehicle group (P < 0.05). *Significantly lower that 007-AM-stimulated group (P < 0.05).
ESI-09 inhibits Pancreatic Cancer Migration.
Increased levels of EPAC1 expression have been observed in human PDA compared with normal pancreas or surrounding tissue (Lorenz et al., 2008). However, the functional role of EPAC1 elevation in this neoplasm remains unclear. We sought to employ ESI-09 to determine the role of EPAC1 signaling in pancreatic cancer. Treatment of pancreatic cancer cells with ESI-09 did not significantly affect cell proliferation and viability (Supplemental Fig. 4). On the other hand, when the pancreatic cancer cell lines AsPC-1 and PANC-1 were pretreated with ESI-09, a significant decrease in cell migration was observed using both Transwell migration/invasion and wound healing assays (Fig. 6, A and B). To determine whether the observed impact on cell migration is EPAC1-specific, we examined the effect of suppressing EPAC1 expression on AsPC-1 and PANC-1 migration using RNA interference. Short hairpin construct shEPAC1 clone C28 led to a near complete knockdown of EPAC1 expression and significantly inhibited migration of both cell lines, whereas a partial reduction of EPAC1 expression by shEPAC1 clone C32 decreased AsPC-1 and PANC-1 cell migration slightly, but the changes were not significant (Fig. 6C). These results, combined with the fact that pancreatic cancer cells do not express detectable levels of EPAC2 (Supplemental Fig. 5), suggest that EPAC1 might play an important role in pancreatic cancer cell migration and invasion. To further determine how ESI-09 inhibits PDA cell migration and invasion, we performed a cell adhesion assay on a collagen I matrix. As shown in Fig. 7, 007-AM led to an increase in cell adhesion for both AcPC-1 and PANC-1 cells, whereas pretreatment with ESI-09 decreased 007-AM-induced cell adhesion dose-dependently.
Fig. 6.

EPAC1 inhibition decreases pancreatic cancer cell migration and invasion. AsPC-1 and PANC-1 cells were pretreated with the indicated concentrations of ESI-09 for 24 hours before migration and invasion were measured by Transwell (A) and wound healing assays (B). EPAC1 expression was suppressed by shEPAC1-C28 and shEPAC1-C32 and migration and invasion were measured by Transwell assays (C). Bars represent mean ± S.D. (n = 3). *Significantly lower than vehicle group (P < 0.05). #Significantly lower than parental cells group (P < 0.02).
Fig. 7.

ESI-09 inhibits EPAC1-mediated adhesion of PDA cells on collagen I. AsPC-1 and PANC-1 cells were stimulated with vehicle or 10 µM 007-AM after treatment with the indicated concentrations of ESI-09 for 5 minutes. Bars represent mean ± S.D. (n = 3). #Significantly higher than vehicle group (P < 0.03). *Significantly lower than 007-AM-stimulated group (P < 0.02).
Discussion
Taken together, our results show that we have successfully identified ESI-09, a noncyclic nucleotide small molecule that specifically inhibits EPAC1 and EPAC2. Consistent with its EPAC inhibition activity observed in a biochemical assay, ESI-09 also inhibited several well known EPAC-mediated cellular functions, including Akt phosphorylation and insulin secretion. To the best of our knowledge, this is the first report of discovery and characterization of a small-molecule inhibitor of the EPAC family of proteins since their initial discovery in 1998. McPhee et al. (2005) reported the discovery of compounds capable of competing with 3H-cAMP for binding with the isolated CBDs of EPAC1 and EPAC2. However, it has not been shown that these compounds are capable of inhibiting EPAC functions biochemically or in cells. The discovery of an EPAC-specific antagonist may have immense implications for cAMP biology, as it will provide a means to pharmacologically dissect the roles of EPAC proteins in various cellular and diseases processes.
PDA is one of the most lethal human diseases, largely due to the fact that pancreatic cancer is resistant to treatments that are usually effective for other types of cancer. A better understanding of the molecular mechanism of PDA development and metastasis and novel, effective therapeutics are desperately needed. Recently, it has been shown that EPAC1 is markedly elevated in human PDA cells compared with normal pancreas or surrounding tissue (Lorenz et al., 2008). Although EPAC1 has been implicated in promoting cellular proliferation in prostate cancer (Misra and Pizzo, 2009, 2012) and migration and metastasis in melanoma (Baljinnyam et al., 2009, 2010, 2011), the role of EPAC1 in pancreatic cancer has never been investigated. Using our novel EPAC inhibitor ESI-09, we have demonstrated a functional role of EPAC1 overexpression in pancreatic cancer cell migration and invasion. These findings are consistent with similar results based on RNA interference silencing techniques, suggesting that EPAC1 may represent a potential target for developing novel therapeutic strategies for PDA.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. George Holz for providing the INS-1 cells and advice on insulin secretion analysis. The authors also thank Dr. Alfred Wittinghofer (Abteilung Strukturelle Biologie Max-Planck-Institut für molekulare Physiologie) and Dr. Holger Rehmann and Professor Johannes Bos (UMC Utrecht Universiteitsweg) for providing E. coli CK600K cells and pGEX-4T-EPAC2 constructs.
Abbreviations
- 007
8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate
- 007-AM
007 acetoxymethyl ester
- CBD
cAMP-binding domain
- DMSO
dimethyl sulfoxide
- EPAC
exchange protein directly activated by cAMP
- FBS
fetal bovine serum
- GEF
guanine nucleotide exchange factor
- HTS
high-throughput screening
- Mant-GDP
2′-/3′-O-(N′-methylanthraniloyl)guanosine-5′-O-diphosphate
- 8-NBD-cAMP
8-[[2-[(7-nitro-4-benzofurazanyl)amino]ethyl]thio]adenosine-3′,5′-cyclic monophosphate
- PBS
phosphate-buffered saline
- PDA
pancreatic ductal adenocarcinoma
- PKA
protein kinase A/cAMP-dependent protein kinase
- Rap
Ras-proximate
Author Contributions
Participated in research design: Almahariq, Tsalkova, Mei, Chen, Zhou, Sastry, Cheng.
Conducted experiments: Tsalkova, Mei, Almahariq, Chen, Cheng.
Contributed new reagents or analytic tools: Schwede.
Performed data analysis: Tsalkova, Mei, Almahariq, Chen, Zhou, Cheng.
Wrote or contributed to the writing of the manuscript: Tsalkova, Mei, Almahariq, Chen, Zhou, Cheng.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medicine [Grant R01GM066170], a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia funded by the National Institutes of Health National Institute of General Medicine [Grant T32GM089657] and, in part, by the John S. Dunn Foundation through the Gulf Coast Consortium for Chemical Genomics.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Arnold K, Bordoli L, Kopp J, Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201 [DOI] [PubMed] [Google Scholar]
- Baljinnyam E, De Lorenzo MS, Xie LH, Iwatsubo M, Chen S, Goydos JS, Nowycky MC, Iwatsubo K. (2010) Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res 70:5607–5617 [DOI] [PubMed] [Google Scholar]
- Baljinnyam E, Iwatsubo K, Kurotani R, Wang X, Ulucan C, Iwatsubo M, Lagunoff D, Ishikawa Y. (2009) Epac increases melanoma cell migration by a heparan sulfate-related mechanism. Am J Physiol Cell Physiol 297:C802–C813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baljinnyam E, Umemura M, De Lorenzo MS, Iwatsubo M, Chen S, Goydos JS, Iwatsubo K. (2011) Epac1 promotes melanoma metastasis via modification of heparan sulfate. Pigment Cell Melanoma Res 24:680–687 [DOI] [PubMed] [Google Scholar]
- Brennesvik EO, Ktori C, Ruzzin J, Jebens E, Shepherd PR, Jensen J. (2005) Adrenaline potentiates insulin-stimulated PKB activation via cAMP and Epac: implications for cross talk between insulin and adrenaline. Cell Signal 17:1551–1559 [DOI] [PubMed] [Google Scholar]
- Cheng X, Ji Z, Tsalkova T, Mei F. (2008) Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 40:651–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Phelps C, Taylor SS. (2001) Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms Ialpha and IIbeta to the catalytic subunit. J Biol Chem 276:4102–4108 [DOI] [PubMed] [Google Scholar]
- Chepurny OG, Leech CA, Kelley GG, Dzhura I, Dzhura E, Li X, Rindler MJ, Schwede F, Genieser HG, Holz GG. (2009) Enhanced Rap1 activation and insulin secretagogue properties of an acetoxymethyl ester of an Epac-selective cyclic AMP analog in rat INS-1 cells: studies with 8-pCPT-2′-O-Me-cAMP-AM. J Biol Chem 284:10728–10736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, et al. (2003) cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 278:35394–35402 [DOI] [PubMed] [Google Scholar]
- Cook PF, Neville ME, Jr, Vrana KE, Hartl FT, Roskoski R., Jr (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21:5794–5799 [DOI] [PubMed] [Google Scholar]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. (2005) Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105:1950–1955 [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–477 [DOI] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437:574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4:901–906 [DOI] [PubMed] [Google Scholar]
- Enserink JM, Price LS, Methi T, Mahic M, Sonnenberg A, Bos JL, Taskén K. (2004) The cAMP-Epac-Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the alpha3beta1 integrin but not the alpha6beta4 integrin. J Biol Chem 279:44889–44896 [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Shibasaki T, Yokoi N, Kashima Y, Matsumoto M, Sasaki T, Tajima N, Iwanaga T, Seino S. (2002) Piccolo, a Ca2+ sensor in pancreatic beta-cells. Involvement of cAMP-GEFII⋅Rim2⋅Piccolo complex in cAMP-dependent exocytosis. J Biol Chem 277:50497–50502 [DOI] [PubMed] [Google Scholar]
- Fukuda M, Williams KW, Gautron L, Elmquist JK. (2011) Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab 13:331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. (2010) Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 50:355–375 [DOI] [PubMed] [Google Scholar]
- Gloerich M, Ponsioen B, Vliem MJ, Zhang Z, Zhao J, Kooistra MR, Price LS, Ritsma L, Zwartkruis FJ, Rehmann H, et al. (2010) Spatial regulation of cyclic AMP-Epac1 signaling in cell adhesion by ERM proteins. Mol Cell Biol 30:5421–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Vliem MJ, Prummel E, Meijer LA, Rensen MG, Rehmann H, Bos JL. (2011) The nucleoporin RanBP2 tethers the cAMP effector Epac1 and inhibits its catalytic activity. J Cell Biol 193:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Mei FC, Johnson BH, Thompson EB, Cheng X. (2007) Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J Biol Chem 282:37370–37377 [DOI] [PubMed] [Google Scholar]
- Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. (2003) Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem 278:8279–8285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. (2001) Critical role of cAMP-GEFII⋅Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276:46046–46053 [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282:2275–2279 [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Corada M, Dejana E, Bos JL. (2005) Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579:4966–4972 [DOI] [PubMed] [Google Scholar]
- Leech CA, Holz GG, Chepurny O, Habener JF. (2000) Expression of cAMP-regulated guanine nucleotide exchange factors in pancreatic beta-cells. Biochem Biophys Res Commun 278:44–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, O’Connor KL, Cheng X, Mei FC, Uchida T, Townsend CM, Jr, Evers BM. (2007) Cyclic adenosine 5′-monophosphate-stimulated neurotensin secretion is mediated through Rap1 downstream of both Epac and protein kinase A signaling pathways. Mol Endocrinol 21:159–171 [DOI] [PubMed] [Google Scholar]
- Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D, Woods VL, Jr, Cheng X. (2011) Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J Biol Chem 286:17889–17897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Takahashi M, Li Y, Dillon TJ, Kaech S, Stork PJ. (2010) The interaction of Epac1 and Ran promotes Rap1 activation at the nuclear envelope. Mol Cell Biol 30:3956–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Aleksic T, Wagner M, Adler G, Weber CK. (2008) The cAMP/Epac1/Rap1 pathway in pancreatic carcinoma. Pancreas 37:102–103 [DOI] [PubMed] [Google Scholar]
- Maillet M, Robert SJ, Cacquevel M, Gastineau M, Vivien D, Bertoglio J, Zugaza JL, Fischmeister R, Lezoualc’h F. (2003) Crosstalk between Rap1 and Rac regulates secretion of sAPPalpha. Nat Cell Biol 5:633–639 [DOI] [PubMed] [Google Scholar]
- McPhee I, Gibson LC, Kewney J, Darroch C, Stevens PA, Spinks D, Cooreman A, MacKenzie SJ. (2005) Cyclic nucleotide signalling: a molecular approach to drug discovery for Alzheimer’s disease. Biochem Soc Trans 33:1330–1332 [DOI] [PubMed] [Google Scholar]
- Mei FC, Cheng XD. (2005) Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol Biosyst 1:325–331 [DOI] [PubMed] [Google Scholar]
- Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. (2002) Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem 277:11497–11504 [DOI] [PubMed] [Google Scholar]
- Métrich M, Berthouze M, Morel E, Crozatier B, Gomez AM, Lezoualc’h F. (2010) Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflugers Arch 459:535–546 [DOI] [PubMed] [Google Scholar]
- Misra UK, Pizzo SV. (2009) Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J Cell Biochem 108:998–1011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Misra UK, Pizzo SV. (2012) Upregulation of mTORC2 activation by the selective agonist of EPAC, 8-CPT-2Me-cAMP, in prostate cancer cells: assembly of a multiprotein signaling complex. J Cell Biochem 113:1488–1500. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, et al. (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2:805–811 [DOI] [PubMed] [Google Scholar]
- Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. (2002) Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem 277:26581–26586 [DOI] [PubMed] [Google Scholar]
- Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL. (2003) Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J Cell Biol 160:487–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. (2008) Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 455:124–127 [DOI] [PubMed] [Google Scholar]
- Ross SH, Post A, Raaijmakers JH, Verlaan I, Gloerich M, Bos JL. (2011) Ezrin is required for efficient Rap1-induced cell spreading. J Cell Sci 124:1808–1818 [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85:1303–1342 [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, et al. (2007) Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 104:19333–19338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Blumenthal DK, Mei FC, White MA, Cheng X. (2009) Mechanism of Epac activation: structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J Biol Chem 284:23644–23651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Cheng X. (2012a) A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS ONE 7:e30441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. (2012b) Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA DOI: 10.1073/pnas.1210209109 109:18613– 18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. (2008) 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. ChemBioChem 9:2052–2054 [DOI] [PubMed] [Google Scholar]
- Yu S, Mei FC, Lee JC, Cheng X. (2004) Probing cAMP-dependent protein kinase holoenzyme complexes I alpha and II beta by FT-IR and chemical protein footprinting. Biochemistry 43:1908–1920 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.