

Abstract
Events that occur in rheumatoid arthritis synovial tissues are responsible for the signs and symptoms of joint inflammation and for the eventual destruction of articular and periarticular structures that lead to joint dysfunction and disability. The three most abundant cell populations in RA synovium are synovial macrophages (type A synoviocytes), synovial fibroblasts (type B synoviocytes) and infiltrating T lymphocytes. Other important cell populations include B lymphocytes, dendritic cells, plasma cells, mast cells and osteoclasts. Our current understanding of rheumatoid arthritis is moving beyond previous concepts that view this disease as the consequence of a specific and focused humoral or cellular autoimmune response to a single autoantigen. Rather, a new view of rheumatoid arthritis is emerging, which seeks to understand this disease as the product of pathologic cell–cell interactions occurring within a unique and defined environment, the synovium. T lymphocytes in rheumatoid arthritis synovium interact closely with dendritic cells, the most potent antigen-presenting cell population in the immune system. T cells also interact with monocytes and macrophages and cytokine-activated T cells may be, especially, suited to trigger production of the important cytokine TNFα by synovial macrophages. Recent evidence also suggests a potent bidirectional interaction between synovial T cells and synovial fibroblasts, which can lead to activation of both cell types. An important role for synovial B lymphocytes has been emphasized recently, both by experimental data and by results of clinical interventions. B cells in synovium can interact with fibroblasts as well as with other cells of the immune system and their potential role as antigen-presenting cells in the joint is as yet underexplored. Rheumatoid arthritis synovium may be one of the most striking examples of pathologic, organ-specific interactions between immune system cells and resident tissue cell populations. This view of rheumatoid arthritis also leads to the prediction that novel approaches to treatment will more logically target the intercellular communication systems that maintain such interactions, rather than attempt to ablate a single cell population.
Keywords: T cells, B cells, Fibroblasts, Dendritic cells, Monocytes
1. Introduction
The hallmark lesion of rheumatoid arthritis (RA) is synovial inflammation—synovitis—that leads to erosion and destruction of cartilage, bone and periarticular structures. While the etiology of RA remains unknown and controversy persists concerning the role of humoral and cellular autoimmunity in the pathogenesis of RA, substantial insight has been achieved into the processes and molecular mediators that characterize the synovial biology of RA. RA synovium contains a variety of cell types many of which are listed in Table 1. No single cell population is capable of causing RA, although aggressive fibroblasts from RA pannus possess autonomous tissue invasive properties, even after extraction from established synovial lesions. It has become increasingly clear that interactions among the important cell populations in the RA synovium not only define many aspects of the synovial biology of this disease, but also offer targets for therapeutic interventions.
Table 1.
Cellular components of the rheumatoid synovium
Abundant cell populations
|
Other cell populations
|
Interactions between cell populations in RA synovium can be thought of as falling into two classes: first, interactions mediated by secreted molecules, such as cytokines and second, cognate cell–cell interactions that require direct contact between two different types of cells and that alter the activation or differentiation state of one or both of the cell types. Some of these interactions that are relevant to the pathogenesis of RA are listed in Table 2. This review will focus on interactions between lymphocytes in RA synovium and the other key cell populations, such as dendritic cells, monocyte-macrophage cells and synovial fibroblasts and will emphasize recent findings.
Table 2.
Cell–cell interactions in rheumatoid arthritis synovium
|
2. Interactions between synovial dendritic and T cells
Dendritic cells are, especially, powerful initiators of immune responses, even when present in small numbers. In RA, dendritic cells are actually abundant both in synovial tissue and in synovial fluid. Attention, therefore, has been directed at possible roles of dendritic cells in initiation and perpetuation of rheumatoid synovitis (recently reviewed in Ref. [1]). Cells with dendritic morphology were initially recognized in RA synovial tissue over 2 decades ago. Key observations were made in 1982 by Klareskog et al. who proposed that RA synovitis was a delayed-type hypersensitivity reaction generated by the interaction of synovial dendritic and T lymphocytes [2]. These dendritic cells, which were noted to be functionally similar to Langerhan cells of the skin, were partially purified and were shown to be powerful immune stimulators.
Subsequently, such cells were also found in synovial fluid, comprising as many as 5% or more of RA synovial fluid mononuclear cells. Synovial fluid dendritic cells were capable of attracting a cluster of T lymphocytes and activating antigen-specific T cell responses. In synovial tissue, dendritic cells were found within both large and small lymphoid aggregates adjacent to vascular structures. These cells express a variety of co-stimulatory ligands known to be important in interactions with T lymphocytes.
Dendritic cells have been proposed to be critical for the development of the architecture of inflamed RA synovium, which can vary from an appearance similar to a lymph node to a diffuse lymphocytic infiltrate. The entry of dendritic cells and their positioning within synovial tissue may be dependent on specific chemokines, such as CXCL12. This chemokine binds to the receptor CXCR4 and both are highly expressed in perivascular and sublining regions of RA synovium.
Secretion of cytokines by dendritic cells can skew the nature of the T cell immune response. Th1-inducing cytokines include IL-12, IL-23 and IL-27. IL-23, discovered recently, may be of particular importance because of its ability to induce expression of IL-17, a T cell cytokine capable of activating synovial fibroblasts to express pro-inflammatory and tissue-destructive mediators. Dendritic cells are also one of the sources of IL-1, IL-6 and TNFα, important pro-inflammatory cytokines that are targets of biologic therapeutic agents in RA.
A critical question is the nature of the autoantigens or foreign antigens to which the immune response is directed and focused in RA. A wide variety of such antigens are known to be capable of preferentially stimulating T cells, especially, synovial compartment T cells in RA. The association of RA with the HLA-DR4(0401) Class II MHC allele has raised the possibility that specific MHC molecules present arthritogenic antigens and initiate or perpetuate RA. However, a variety of other mechanisms to explain the association of RA with MHC alleles have been proposed and none has yet been proven. Whichever antigens turn out to be most important, it is likely that dendritic cells are the most critical antigen-presenting cell population involved in initiating these responses. An, especially, intriguing issue is the location of such responses: does RA begin in the joints or begin systemically with immune responses triggered by events in other tissues?
3. Monocyte activation by T cells in RA
Infiltrating T cells and macrophages reside in close proximity in the inflamed RA synovium. This intimate association provides many opportunities for interactions between the cells [3,4]. Evidence supporting T cell participation in TNFα production comes from experiments in which depletion of CD3+ cells from RA synovial cell cultures resulted in decreased TNFα production, whereas depletion of CD3+ cells from OA cultures did not [5]. These observations suggest that T cells have a direct impact on TNFα induction in RA joints. To further investigate the role of T cells in TNFα production, T cells activated by a cytokine cocktail (Tck) [6] were used, as a model for RA T cells, to stimulate monocytes. Tck were able to induce TNFα from monocytes via a cell–cell contact-dependent mechanism that mimicked RA T cells, but differed from T cells activated through their TCR and CD28 [5]. Another study indicates that Tck can also induce production of the anti-inflammatory cytokine IL-10 by M-CSF-treated monocytes (i.e. macrophages) [7]. These studies imply that RA synovial T cells are similar to bystander-activated Tck in their phenotype and their effects on monocyte/macrophage cytokine production.
The receptor activator of nuclear factor κB ligand (RANKL) has been detected in RA synovial tissue. RANK/ RANKL interactions are necessary for the differentiation of osteoclasts from monocytic precursor cells. RANKL was found to localize specifically to CD3+ CD4+ cells and not other mononuclear cells, in synovial histological sections [8]. In the same study, T cells activated with PHA upregulated RANKL and effectively induced monocytes to differentiate into osteoclasts [8]. Although this system induces RANKL on T cells using the lectin PHA, it provides evidence for a potentially important pathogenic mechanism in synovium.
4. T cell activation by monocytes
T cell homeostasis is thought to be aberrant in RA. Using in vitro co-cultures of autologous RA or normal T cells with monocytes, it was found that the mechanism of homeostatic proliferation differed between the two T cell types [9]. While both RA and normal T cells exhibited proliferation when co-cultured with autologous monocytes, blockade of MHC II with antibody inhibited proliferation in normal T cell controls. In contrast, RA T cells showed diminished proliferation when TNFα was blocked. The required TNFα was determined to be membrane bound, as separation of RA T cells and autologous monocytes by transmembrane inserts prevented proliferation. It is also interesting to note that psoriatic arthritis T cells show homeostatic proliferation similar to normal controls (i.e. dependence on MHC II) and distinct from RA T cells [9]. It would seem that T cells can induce TNFα from monocytes and that this TNFα can then send a proliferative signal to the T cells.
Although no unique autoantigen or autoreactive T cell clone has been shown to cause RA, the association of RA with a restricted set of MHC II alleles in the Caucasian population is strong [10]. The autoantigens human cartilage glycoprotein 39 (HCgp39) and human collagen-type II (HcII) are expressed within RA synovium [11]. Interesting results have been reported using mice transgenic for RA-associated MHC II, HLA-DR4 (DRB1*0401). T cells from this mouse recognize antigens presented by human MHC II. T cell clones specific for peptides of HCgp39 or HcII from this mouse were fused to generate T cell hybridomas, which recognize antigen presented by specific human MHC II. When human DRB1*0401 monocytes were loaded with peptides derived from HCgp39 or HcII, the corresponding T cell hybridoma was able to recognize the cognate peptide–MHC II complex, but not irrelevant peptide–MHC II complexes [11]. This is strong evidence that moncytes can process and load a functional peptide–MHC II complex containing autoantigen.
5. Synovial fibroblast activation by T cells
Early experiments on co-cultures of T cells and fibroblast-like synoviocytes (FLS) demonstrated that interaction between these cells types could activate FLS. Phorbol myristate acetate (PMA)-activated T cells triggered IL-1β transcription in FLS and release of IL-1 into culture supernatants, dependent on interactions between leukocyte functional antigen-1 (LFA–1/CD11aCD18) and intercellular adhesion molecule-1 (ICAM-1/CD54) [12]. LFA–1/ICAM-1 interactions are important in T cell adhesion and immunologic synapse formation between T cells and conventional antigen-presenting cells (APC). It was noted that resting T cells did not adhere within a time course of 30 min, while PMA-treated T cells had firm LFA–1/ICAM-1-mediated adhesion. Potential limitations of this study involve its use of SV-40-transformed FLS lines and PMA treatment of T cells. The transformed FLS constitutively expressed ICAM-1 at a high level that was uninfluenced by IL-1β, suggesting a preactivated state. PMA activation of T cells might not be physiologically relevant. Nonetheless, this study demonstrates the potential for antigen-independent interaction of T cells with FLS, leading to inflammatory mediator production.
Work in our laboratory has also focused on the interaction between T cells and FLS. Using resting T cells as a stimulus, we have documented activation of FLS and release of pro-inflammatory mediators [13]. We found that autologous or allogeneic resting T cells have similar activating potential on FLS. This effector function of resting T cells is not restricted to a particular T cell population. Various subsets of T cells, CD4+, CD8+, CD45RO+ and CD45RA+ all had comparable ability to induce synovial fibroblast activation. Activated FLS showed induction or augmentation of mRNA for stromelysin, IL-6 and IL-8, gene products important in joint inflammation and joint destruction. Furthermore, increased production of IL-6 and IL-8 was quantitated both by ELISA and by intracellular cytokine staining and flow cytometry. Another striking observation was that the T cell specific cytokine IL-17 synergized with T cells to activate FLS. These co-culture systems spanned up to 24 h and no T cell activation was noted, with no upregulation of CD154/CD40L or CD69. Thus, using resting T cells as a stimulus on untransformed FLS lines, we documented further evidence of T cell–FLS antigen-independent interactions resulting in induction of an inflammatory profile of FLS (Fig. 1).
Fig. 1.
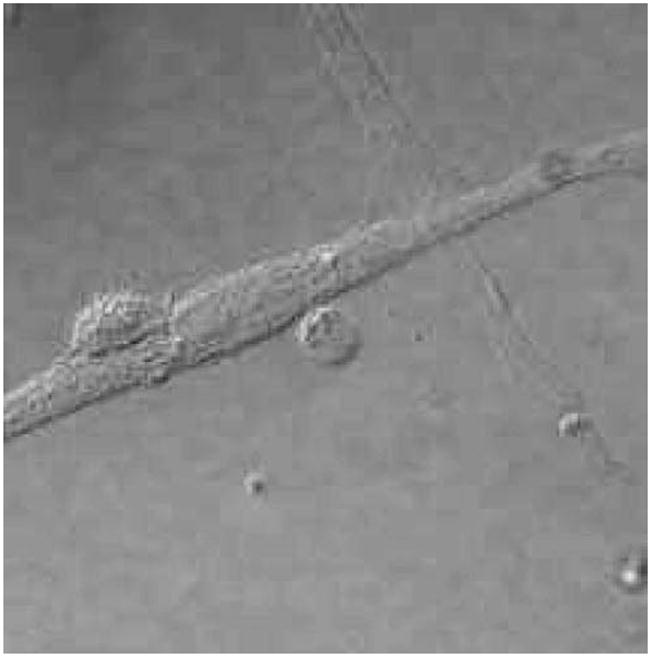
T cell–FLS interaction. Co-culture of RA FLS and purified peripheral blood T lymphocytes (smaller rounded cells) demonstrates intimate cell–cell contact.
6. Cross-talk between T cells and FLS
Two studies have recently provided further support for a role of T cell activation of FLS in antigen-independent systems. One study used unstimulated purified T cells [14] and the other used collagen-type II (CII) responsive T cells [15]. Both studies cite the importance of IL-15 expression by FLS and its upregulation after stimulation by T cells [14,15]. The first study utilized unstimulated T cells purified from peripheral blood or RA synovial fluid. After 96 h of co-culture with these T cells, induction of ICAM-1, IL-8, IL-6 and IL-15 was noted on the FLS [14]. This induction was dependent on cell–cell contact, as evident when transwell inserts separating T cells from FLS blocked activation. Similarly blockade of CD69, CD11a, IL-17, TNFα and IFNγ also inhibited activation of FLS. Another interesting observation was that T cells showed signs of activation by induction of CD69, CD25, IL-17, TNFα and IFNγ after co-culture with FLS [14]. This is contrary to our findings described above [13] and the discrepancy is explained by the duration of co-culture (96 h versus 24 h). T cell responses were blunted by blocking antibodies towards ICAM-1 and IL-15. When CII-activated T cells were used as the stimulus, FLS displayed production of TNFα, IL-15 and IL18 [15]. These CII-activated T cells were generated by culture of T cells with bovine CII and autologous irradiated APC for extended periods. An intriguing observation was that the increased length of stimulation by CII before co-culture with FLS resulted in increased IL-17 and IFNγ production by T cells.
7. FLS as APC
Initial experiments using human dermal fibroblasts showed that fibroblasts were poor generators of allogeneic responses [16]. This defect was not due to inadequate expression of MHC II, but was due to lack of an accessory molecule that could be provided by conventional APC. However, it was noted that dermal fibroblasts could stimulate previously activated allogeneic T cells [16]. Expanding on this work, the capacity of dermal fibroblasts to function in antigen presentation was evaluated. Dermal fibroblasts were able to process antigen, but did not function well as APC without accessory cell help [17]. In both of these studies, INFγ was used to induce MHC II and antigens relevant to RA were not evaluated. These studies do document fibroblast expression of functional MHC II.
FLS of RA synovium express high levels of MHC II ex vivo [18], indicating the potential for antigen presentation by FLS in RA. Early studies suggest that FLS can process antigen similarly to professional APC [19]. In those experiments, FLS were able to take up and present various antigens and present them to T cell clones via an MHC II-restricted mechanism [19]. This gives support to potential antigen-specific interaction between T cells and FLS (as APC). However, antigens relevant to RA were not assayed nor were observed responses robust.
We have also observed MHC II-dependent signaling between FLS and T cells. Superantigens activate FLS to secrete inflammatory mediators and potentially participate in RA pathology. Thus, we assessed the ability of IFNγ-treated FLS to present superantigens to T cells [20]. FLS can indeed present superantigens, inducing resting T cells to proliferate. T cell proliferation to superantigens was dependent on MHC II, CD2, LFA-1 and the cytokine IL-2. This study provides a mechanism for FLS to activate naïve T cells, but does not demonstrate an “antigen-specific” response.
There is also evidence that FLS might not activate T cells, but instead induce anergy [21]. These experiments assessed the APC and allostimulatory functions of FLS. Similar to previous studies, FLS were able to load antigen onto MHC II. However, allogeneic responses depended upon the addition of accessory cells expressing CD80 and blockade of CD80 abolished the response. When FLS without accessory cells were cultured with T cells, they adopted a phenotype resembling anergy: upregulation of CD25, reduced proliferation and reconstitution of proliferation by exogenous IL-2 [21]. Interestingly, CD69 on T cells was also upregulated after T cell culture with FLS. This study implies that FLS cause anergy due to a lack of co-stimulatory molecules, but that bystander cells expressing co-stimulatory molecules could overcome this. The potential for accessory costimulation exists abundantly within RA synovium due to the close proximity of FLS with B cells, macrophages and dendritic cells.
8. The role of B cells in RA synovium
Several lines of evidence highlight the important roles played by B lymphocytes in the pathogenesis of RA. Recent clinical trials, in which B cells have been depleted by antibody treatment, have shown efficacy of this approach as treatment for RA [22,23]. In many patients, the improvements following B cell depletion, include decreased joint swelling and tenderness and reoccurrence of disease symptoms correlate to regeneration of B cells [23,24]. Some B cells produce autoreactive antibodies to citrullinated proteins or anti-immunoglobulin rheumatoid factors that lead to formation of immune complexes and complement deposition in the joints (reviewed elsewhere in this issue). B cells are also sources of cytokines that contribute to cellular activation, germinal center formation and inflammation in rheumatoid synovium. Additionally, B cells may play an important role in the pathogenesis of RA through cell–cell interaction with T cells, dendritic cells, synovial nurse-like cells and fibroblasts.
In secondary lymphoid organs, such as lymph nodes, spleen and tonsils, lymphocytes organize into complex structures called germinal centers (GC). At the center of the GCs are follicular dendritic cells surrounded by an area rich in B cells. Surrounding the B cell-rich region is an area containing mostly T cells and dendritic cells (DC) with a mantle zone between them in which B and T cells interact. The functional importance of GC is to provide an environment in which rare, antigen-specific cells can encounter APC bearing relevant antigens, thus, leading to signaling and activation through cell surface interactions and cytokine networks. Immunoglobulin genes in activated B cells undergo somatic hypermutation leading to increased antibody affinity for target antigens and B cells differentiate within GC into plasma cells specialized to secrete antibodies. In addition, the GC provides an environment in which B cells may take up antigens and become more potent APCs by upregulating MHC and co-stimulatory molecule expression.
Approximately, 20% of patients with RA develop GC-like structures within the synovial tissue [25,26]. The mechanisms underlying the neogenesis of GC in RA synovium are consistent with formation of GC in lymph nodes and spleen. Endothelial cells, synovial fibroblasts and follicular DC express CXCL13 (BLC), a chemokine involved in attracting B cells into the GC [26,27]. The B cells, in turn, express lymphotoxins (LT) α and β on their cell surface, cytokines involved in the activation of follicular dendritic cells, regulation of T cell attracting chemokines and the structural organization of GC [28,29]. A unique feature of GC formation in rheumatoid synovium appears to be the dependence on CD40L+ /CD8+ T cells, these cells may activate B cells, macrophages and DC through ligation of CD40 on the cell surface [30].
8.1. T cell–B cell interaction in RA synonium
B–T cell interactions lead to mutual activation, maturation and proliferation. Analysis of a series of RA patients revealed three distinct patterns of B–T cell interaction in rheumatoid synovium [25]. The majority of patients had a diffuse distribution of T cells in the synovium. In these patients, very low levels of B cells were found in the synovium despite the presence of interdigitating DC. Other patients had organized GC as outlined above, while some patients had small aggregates of T and B cells in the synovium that lacked follicular DC, and therefore, did not form GC. These patterns of lymphoid organization were not random since patients with multiple lesions in distinct joints, had the same type of T–B cell interaction. In order to study the dependency of T cell activation on synovial lymphoid architecture, Weyand et al. identified three T cell clones from a patient with GC-like synovial structures [31]. These clones were distributed in separate follicles in distinct synovial tissue biopsies, which suggested that they were antigen-specific. Adoptive transfer of these T cell clones to SCID mice engrafted with human synovial tissue revealed that the T cells could orchestrate intra-synovial, pro-inflammatory cytokine production in MHC Class II-matched but not mismatched synovial tissue. Evidence that T–B cell interaction was required for pro-inflammatory cytokine production was obtained by adoptive transfer to SCID mice that were engrafted with synovial tissue from patients with the diffuse pattern of T cell localization. To confirm the role of B cells in T cell activation in this experimental model, mice engrafted with GC-like synovial tissue were depleted of human B cells with anti-human CD20 antibodies prior to adoptive transfer. Pro-inflammatory cytokine production was reduced by B cell depletion in an antibody dose-dependent manner. However, the lack of infiltrating T cells into the engrafted synovial tissue that was depleted of B cells does not allow for determination of whether the essential role of B cells has to do with a T cell homing defect or with direct cell–cell interaction between T and B cells.
Antigen presentation by human RA synovial B cells has not been as well characterized as the APC role played by synovial dendritic cells. However, it is likely that B cells specific to autoantigens or B cells producing rheumatoid factors are able to bind, internalize and process antigens for presentation to autoreactive T cells in the ectopic germinal centers of rheumatoid synovium [32,33].
8.2. Other synoviocyte interactions with B cells
The terminal differentiation of B cells into antibody-secreting plasma cells is an antigen-driven and cytokine-dependent process. Experiments involving microdissection of RA synovium have demonstrated that B cells clonally expand, develop a memory phenotype and differentiate into plasma cells within the ectopic germinal centers formed in some RA patients synovial tissues [34,35]. B cell maturation and differentiation may be driven by non-lymphoid synoviocytes since antibody secretion and increased surface expression of the plasma cell markers CD38 and CD44 were observed in co-cultures of tonsillar B cells with adherent synovial cells from normal individuals and RA patients [36].
Candidate synoviocytes that may drive the development of B cells include follicular dendritic cells, synovial nurse-like cells and synovial fibroblasts. Follicular dendritic cells (FDC) are found in synovium of RA patients with ectopic germinal centers and are known to function in B cell maturation in lymph nodes [26,37]. Lymph node FDC express CD106 (VCAM-1) which is a survival and differentiation factor for GC B cells [38,39]. In addition, complement decay-accelerating factor (DAF) and complement receptor 2 (CR2) are important molecules expressed by lymph node FDC that prevent complement deposition and promote B cell survival [40,41]. FDC also trap antigens at their cell surfaces for long periods of time allowing them to act as an antigenic sink for B cells [40–42]. To date, only CR2 expression has been reported on the FDC found in RA synovium [37].
Nurse-like cells (NLC) are a subset of stromal cells, distinct from FDC or fibroblasts, that can be cultured out of the bone marrow and synovial tissue of RA patients [43,44]. RA–NLC constitutively express IL-6, IL-8 and granulocyte colony-stimulating factor (GCSF) and upregulate expression of IL-1β and TNFα following co-culture with B cells. These cytokines are known to be important mediators of inflammation in RA synovium. Reciprocally, RA–NLC stimulate B cell proliferation, immunoglobulin production, migration under the NLC monolayer and survival. Cell–cell contact mediated by CD106 was required for RA–NLC-enhanced migration and survival of B cells, but a non-CD106/CD49d (VLA-4) cell adhesion mechanism accounts for B cell-induced expression of pro-inflammatory cytokines IL-6 and IL-8 by RA–NLC [44]. Colocalization of RA–NLC and synovial B cells in the joint may occur through the chemokine SDF-1/CXCR4 receptor axis since SDF-1 was produced by a cultured RA–NLC line [44].
Similar effects on B cell migration characteristics, including dependency on SDF-1 and CD106, were demonstrated in synoviocytes that resembled fibroblasts [45]. Fibroblast-like synoviocytes express CD106 within the joints of RA patients and upregulate expression of the B cell survival factors: CD106, DAF and CR2 upon in vitro stimulation with TNF-α and IFN-γ [46]. RA-FLS express the receptor for LT-β, which is important in formation of lymph nodes and ectopic synovial germinal centers and FLS can be stimulated through LT-α1β2, a molecule expressed by synovial B cells, to express cell adhesion molecules, pro-inflammatory cytokines, chemokines and matrix metalloproteinases [47].
9. Conclusion
The cell–cell interactions that occur in rheumatoid arthritis synovium are multiple, complex and fundamentally important to the pathogenesis and outcome of this disease. In addition to the cell populations considered in detail in this review, osteoclasts, chondrocytes, mast cells, plasma cells and other cell types are important in the events that occur within RA pannus and in adjacent cartilage and bone. The synovial fluid contains large numbers of neutrophils, which are critical to inflammatory events that occur outside synovial tissue but within synovial fluid. RA synovial tissue can be viewed as a prototypical lesion in which cells of the immune system, ectopically located, interact with a variety of resident tissue cells. It can also be viewed as a lesion in which the distinct roles of the innate and adaptive components of the immune response become blurred. For example, T lymphocytes, central cells in the adaptive immune response, can function in an antigen-independent, innate manner to activate synovial fibroblasts. On the other hand, the synovial fibroblast, a resident tissue cell not normally considered to be part of the immune system, can activate T cell responses to superantigens and perhaps even peptide antigens. Further elucidation of the critical cell–cell interactions in RA synovium should provide additional therapeutic targets for new biologic agents and will provide a rational basis for safe and effective combinations of biologic interventions.
References
- 1.Sarkar S, Fox DA. Dendritic cells in rheumatoid arthritis. Frontiers Biosci. 2005;10:656–665. doi: 10.2741/1560. [DOI] [PubMed] [Google Scholar]
- 2.Klareskog L, Forsum U, Scheynius A, Kabelitz D, Wigzell H. Evidence in support of a self-perpetuating HLA-DR-dependent delayed-type cell reaction in rheumatoid arthritis. Proc Natl Acad Sci USA. 1982;79:3632–3636. doi: 10.1073/pnas.79.11.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burger D, Dayer JM. The role of human T lymphocyte–monocyte contact in inflammation and tissue destruction. Arthritis Res. 2002;4(Suppl 3):169–176. doi: 10.1186/ar558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McInnes IB, Leung BP, Liew FY. Cell–cell interactions in synovitis: interactions between T lymphocytes and synovial cells. Arthritis Res. 2000;2(5):374–378. doi: 10.1186/ar115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BMJ, Feldmann M. Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 2002;46(1):64–70. doi: 10.1002/1529-0131(200201)46:1<31::AID-ART10029>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Unutmaz D, Pileri P, Abrignani S. Antigen-independent activation of naïve memory resting T cells by a cytokine combination. J Exp Med. 1994;180(3):1159–1164. doi: 10.1084/jem.180.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foey A, Green P, Foxwell B, Feldmann M, Brennan F. Cytokine-stimulated T cells induce macrophage IL-10 production dependent on phosphatidylinositol 3-kinase and p70S6K: implications for rheumatoid arthritis. Arthritis Res. 2002;4(1):64–70. doi: 10.1186/ar385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E, Takahashi K, Furuya T, Ishiyama S, Kim K-J, Saito S, Nishikawa T, Takahashi N, Togari A, Tomatsu T, Suda T, Kamatani N. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2001;44(5):1003–1012. doi: 10.1002/1529-0131(200105)44:5<1003::AID-ANR179>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 9.Wagner U, Pierer M, Wahle M, Moritz F, Kaltenhäuser S, Häntzschel H. Ex vivo homeostatic proliferation of CD4+ T cells in rheumatoid arthritis is dysregulated and driven by membrane-anchored TNFα. J Immunol. 2004;173(4):2825–2833. doi: 10.4049/jimmunol.173.4.2825. [DOI] [PubMed] [Google Scholar]
- 10.Gregersen P, Silver J, Winchester R. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 11.Tsark EC, Wang W, Teng Y-C, Arkfeld D, Dodge GR, Kovats S. Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J Immunol. 2002;169(11):6625–6633. doi: 10.4049/jimmunol.169.11.6625. [DOI] [PubMed] [Google Scholar]
- 12.Nakatsuka K, Tanaka Y, Shubscher, Abe M, Wake A, Saito K, Morimoto I, Eto S. Rheumatoid synovial fibroblasts are stimulated by the cellular adhesion to T cells through lymphocyte function associated antigen-1/intercellular adhesion molecule-1. J Rheumatol. 1997;24(3):458–464. [PubMed] [Google Scholar]
- 13.Yamamura Y, Gupta R, Morita Y, He X, Pai R, Endres J, Freiberg A, Chung K, Fox DA. Effector function of resting T cells: activation of synovial fibroblasts. J Immunol. 2001;166(4):2270–2275. doi: 10.4049/jimmunol.166.4.2270. [DOI] [PubMed] [Google Scholar]
- 14.Miranda-Carus ME, Balsa A, Benito-Miguel M, Pérex de Ayala C, Martíin-Mola E. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate. J Immunol. 2004;173(2):1463–1476. doi: 10.4049/jimmunol.173.2.1463. [DOI] [PubMed] [Google Scholar]
- 15.Cho ML, Yoon C-H, Hwang S-Y, Park M-K, Min S-Y, Lee S-H, Park S-H, Kim H-Y. Effector function of type II collagen-stimulated T cells from rheumatoid arthritis patients: cross-talk between T cells and synovial fibroblasts. Arthritis Rheum. 2004;50(3):776–784. doi: 10.1002/art.20106. [DOI] [PubMed] [Google Scholar]
- 16.Geppert TD, Lipsky PE. Antigen presentation by interferon-gamma-treated endothelial cells and fibroblasts: differential ability to function as antigen-presenting cells despite comparable Ia expression. J Immunol. 1985;135(6):3750–3762. [PubMed] [Google Scholar]
- 17.Geppert TD, Lipsky PE. Dissection of defective antigen presentation by interferon-gamma-treated fibroblasts. J Immunol. 1987;138(2):385–392. [PubMed] [Google Scholar]
- 18.Zimmermann T, Kunisch E, Pfeiffer R, Hirth A, Stahl H-D, Sack U, Laube A, Liesaus E, Roth A, Palombo-Kinne E, Emmrich F, Kinne RW. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture: primary culture cells markedly differ from fourth-passage cells. Arthritis Res. 2001;3(1):72–76. doi: 10.1186/ar142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boots AM, Wimmers-Bertens AJ, Rijnders AW. Antigen-presenting capacity of rheumatoid synovial fibroblasts. Immunology. 1994;82(2):268–274. [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai C, Diaz LA, Jr, Singer NG, Li LL, Kirsch AH, Mitra R, Nickoloff BJ, Crofford LJ, Fox DA. Responsiveness of human T lymphocytes to bacterial superantigens presented by cultured rheumatoid arthritis synoviocytes. Arthritis Rheum. 1996;39(1):125–136. doi: 10.1002/art.1780390117. [DOI] [PubMed] [Google Scholar]
- 21.Corrigall VM, Solau-Gervais E, Panayi GS. Lack of CD80 expression by fibroblast-like synoviocytes leading to anergy in T lymphocytes. Arthritis Rheum. 2000;43(7):1606–1615. doi: 10.1002/1529-0131(200007)43:7<1606::AID-ANR26>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Leandro MJ, Edwards JC, Cambridge G. Clinical outcome in 22 patients with rheumatoid arthritis treated with B lymphocyte depletion. Ann Rheum Dis. 2002;61(10):883–888. doi: 10.1136/ard.61.10.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 24.Moore J, Ma D, Will R, Cannell P, Handel M, Milliken S. A phase II study of rituximab in rheumatoid arthritis patients with recurrent disease following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004;34(3):241–247. doi: 10.1038/sj.bmt.1704570. [DOI] [PubMed] [Google Scholar]
- 25.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167(2):1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 26.Weyand CM, Goronzy JJ. Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci. 2003;987:140–149. doi: 10.1111/j.1749-6632.2003.tb06042.x. [DOI] [PubMed] [Google Scholar]
- 27.Shi K, Hayashida K, Kaneko M, Hashimoto J, Tomita T, Lipsky PE, Yoshikawa H, Ochi T. Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001;1661(1):650–655. doi: 10.4049/jimmunol.166.1.650. [DOI] [PubMed] [Google Scholar]
- 28.Fu YX, Huang G, Wang Y, Chaplin DD. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha-dependent fashion. J Exp Med. 1998;187(7):1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin’s role in inflammation and development. Immunol Res. 1999;19(2–3):119–125. doi: 10.1007/BF02786481. [DOI] [PubMed] [Google Scholar]
- 30.Wagner UG, Kurtin PJ, Wahner A, Brackertz M, Berry DJ, Goronzy JJ, Weyand CM. The role of CD8+ CD40L+ T cells in the formation of germinal centers in rheumatoid synovitis. J Immunol. 1998;161(11):6390–6397. [PubMed] [Google Scholar]
- 31.Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167(8):4710–4718. doi: 10.4049/jimmunol.167.8.4710. [DOI] [PubMed] [Google Scholar]
- 32.Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen–antibody complexes by rheumatoid factor B cells. J Exp Med. 1991;173(2):487–489. doi: 10.1084/jem.173.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patil NS, Hall FC, Drover S, Spurrell DR, Bos E, Cope AP, Sonderstrup G, Mellins ED. Autoantigenic HCgp39 epitopes are presented by the HLA-DM-dependent presentation pathway in human B cells. J Immunol. 2001;166(1):33–41. doi: 10.4049/jimmunol.166.1.33. [DOI] [PubMed] [Google Scholar]
- 34.Schröder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 1996;93(1):221–225. doi: 10.1073/pnas.93.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HJ, Krenn V, Steinhauser G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. 1999;162(5):3053–3062. [PubMed] [Google Scholar]
- 36.Dechanet J, Merville P, Durand I, Banchereau J, Miossec P. The ability of synoviocytes to support terminal differentiation of activated B cells may explain plasma cell accumulation in rheumatoid synovium. J Clin Invest. 1995;95(2):456–463. doi: 10.1172/JCI117685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imai Y, Yamakawa M, Masuda A, Sato T, Kasajima T. Function of the follicular dendritic cell in the germinal center of lymphoid follicles. Histol Histopathol. 1986;1(4):341–353. [PubMed] [Google Scholar]
- 38.Freedman AS, Munro JM, Rice GE, Bevilacqua MP, Morimoto C, McIntyre BW, Rhynhart K, Pober JS, Nadler LM. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science. 1990;249(4972):1030–1033. doi: 10.1126/science.1697696. [DOI] [PubMed] [Google Scholar]
- 39.Huang MJ, Osborn L, Svahn J, Schiffer SB, Eliseo L, Zhou LJ, Rhynhart K, Benjamin CD, Freedman AS. Expression of vascular cell adhesion molecule-1 by follicular dendritic cells. Leuk Lymphoma. 1995;18(3–4):259–264. doi: 10.3109/10428199509059616. [DOI] [PubMed] [Google Scholar]
- 40.Lambert N, Lescoulie PL, Yassine-Diab B, Enault G, Mazieres B, De Preval C, Cantagrel A. Substance P enhances cytokine-induced vascular cell adhesion molecule-1 (VCAM-1) expression on cultured rheumatoid fibroblast-like synoviocytes. Clin Exp Immunol. 1998;113(2):269–275. doi: 10.1046/j.1365-2249.1998.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prodinger WM. Complement receptor type two (CR2, CR21): a target for influencing the humoral immune response and antigen-trapping. Immunol Res. 1999;20(3):187–194. doi: 10.1007/BF02790402. [DOI] [PubMed] [Google Scholar]
- 42.Tew JG, Wu J, Qin D, Helm S, Burton GF, Szakal AK. Follicular dendritic cells and presentation of antigen and co-stimulatory signals to B cells. Immunol Rev. 1997;156:39–52. doi: 10.1111/j.1600-065x.1997.tb00957.x. [DOI] [PubMed] [Google Scholar]
- 43.Shimaoka Y, Attrep JF, Hirano T, Ishihara K, Suzuki R, Toyosaki T, Ochi T, Lipsky PE. Nurse-like cells from bone marrow and synovium of patients with rheumatoid arthritis promote survival and enhance function of human B cells. J Clin Invest. 1998;102(3):606–618. doi: 10.1172/JCI3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi E, Tomita T, Toyosaki-Maeda T, Kaneko M, Takano H, Hashimoto H, Sugamoto K, Suzuki R, Ochi T. Establishment and characterization of nurse cell-like stromal cell lines from synovial tissues of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42(2):221–228. doi: 10.1002/1529-0131(199902)42:2<221::AID-ANR3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 45.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1 and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107(3):305–315. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards JC, Leigh RD, Cambridge G. Expression of molecules involved in B lymphocyte survival and differentiation by synovial fibroblasts. Clin Exp Immunol. 1997;108(3):407–414. doi: 10.1046/j.1365-2249.1997.4061306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braun A, Takemura S, Vallejo AN, Goronzy JJ, Weyand CM. Lymphotoxin beta-mediated stimulation of synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2004;50(7):2140–2150. doi: 10.1002/art.20356. [DOI] [PubMed] [Google Scholar]