

Abstract
Background
RNA interference (RNAi) is commonly applied in genome-scale gene functional screens. However, a one-on-one RNAi analysis that targets each gene is cost-ineffective and laborious. Previous studies have indicated that siRNAs can also affect RNAs that are near-perfectly complementary, and this phenomenon has been termed an off-target effect. This phenomenon implies that it is possible to silence several genes simultaneously with a carefully designed siRNA.
Results
We propose a strategy that is combined with a heuristic algorithm to design suitable siRNAs that can target multiple genes and a group testing method that would reduce the number of required RNAi experiments in a large-scale RNAi analysis. To verify the efficacy of our strategy, we used the Orchid expressed sequence tag data as a case study to screen the putative transcription factors that are involved in plant disease responses. According to our computation, 94 qualified siRNAs were sufficient to examine all of the predicated 229 transcription factors. In addition, among the 94 computer-designed siRNAs, an siRNA that targets both TF15 (a previously identified transcription factor that is involved in the plant disease-response pathway) and TF21 was introduced into orchids. The experimental results showed that this siRNA can simultaneously silence TF15 and TF21, and application of our strategy successfully confirmed that TF15 is involved in plant defense responses. Interestingly, our second-round analysis, which used an siRNA specific to TF21, indicated that TF21 is a previously unidentified transcription factor that is related to plant defense responses.
Conclusions
Our computational results showed that it is possible to screen all genes with fewer experiments than would be required for the traditional one-on-one RNAi screening. We also verified that our strategy is capable of identifying genes that are involved in a specific phenotype.
Keywords: RNA interference, RNAi screening, SiRNA design, Gene functional analysis, Group testing
Background
The study of plant genomes has increased dramatically over the last decade [1,2]. Many genome sequencing projects for important crops, such as maize [3,4], wheat [5] and rice [6,7], have recently been completed. The available plant genomic sequence data will continue to increase explosively because of the recently developed next-generation sequencing techniques [8-10]. Therefore, a major challenge to all biologists is how to efficiently translate the functions of the genetic code sequences. It is especially important to rapidly screen for genes that are related to certain important phenotypes, such as salt-tolerance, disease-resistance and flower color, because these genes will help to improve crops. For example, by transferring mannitol-1-phosphate dehydrogenase gene from Escherichia coli, researchers have created many genetically modified plants with increased salt tolerance, which is beneficial to farmers and consumers [11,12].
One of the most efficient tools in high-throughput gene functional screening is the application of RNA interference (RNAi) [13]. RNAi can be induced by introducing a small double-stranded (ds) RNA into a cell [14] and has been widely employed in genome-wide scale studies of model organisms, including animals and plants [15-20]. The primary goals of these applications are usually related to the screening of a single gene or multiple genes that convey(s) a specific phenotype. In these applications, each dsRNA is designed to target one specific gene and to facilitate the observation of the resulting phenotype change. This one-on-one approach works very well for model organisms, because whole genome RNAi libraries are available for these model organisms [21-23]. However, even when one-on-one RNAi experiments are feasible in model organisms, it is still laborious and costly because more than ten thousand coding and non-coding genes are typically found in an organism (e.g., approximately 20,000-25,000 genes were identified in the human genome [24] and approximately 25,500 genes were predicted in the Arabidopsis genome [25]). Thus, matches to the gene number in RNAi experiments should at least be performed. Although using a vector carrying multiple shRNAs/multi-miRNA hairpins can be applied to silence multiple genes simultaneously (“combinatorial RNAi”) [26,27] and can reduce the required experiments for high-throughput analysis, these approaches still need to design and synthesize as many siRNAs as the number of target gene sets.
RNAi libraries are not available for non-model organisms, and the cost and scale of high-throughput RNAi screening experiments are still difficult for most laboratories. In reality, RNAi screening can focus on screening specific groups of genes through strategies such as literature surveys or transcriptome analyses [28,29]. However, these approaches usually identify hundreds to thousands of candidate genes; the subsequent large-scale RNAi analysis would be still a challenge for most laboratories by using a one-on-one approach. In fact, siRNAs do not need to perfectly complement their targets. Genes that contain a sequence that is partially similar to the designed siRNA could also be down-regulated (off-target effect) [30]. The off-target effects have been widely discussed and studied in both animals and plants [13,31,32] , and they are widely considered to be a general problem for analyses; however, off-target effects also facilitate the silencing of multiple genes simultaneously with a carefully designed siRNA. Thus, utilizing the off-target effect to carefully design siRNA could provide opportunities for us to design a minimum of siRNA(s) to target a maximum of genes, which would reduce the labors and costs for high-throughput gene functional screening and would decrease the obstacles for most laboratories in facing high-throughput screening. This approach would be especially useful for applications on non-model organisms for which RNAi libraries are not available.
Here, we utilize the off-target effects to develop a hierarchical RNAi screening strategy to reduce the cost and labor of high-throughput RNAi analyses. Our strategy utilizes a heuristic algorithm to design siRNAs that have multiple target genes and uses group testing methods [33] to minimize the RNAi analysis for identifying genes that are involved in a specific phenotype. Our strategy can be applied to whole-genome RNAi analyses to reduce the number of initial experiments that are needed for phenotype screening, and it is especially suitable for a group of candidate genes that were first identified by an experimental approach. Moreover, to our knowledge, no design of an siRNA for multiple-gene targets has been described previously. We have verified the applicability of this method by analyzing transcription factors that are known to be involved in plant defense responses in orchids, and we also identified a new transcription factor that is involved in this process.
Methods
Orchid ESTs library
To verify the applicability of our approach, we used an established Orchid (Phalaenopsis equestris) expressed sequence tags (ESTs) library in our initial test. There are 233 previously predicted transcription factor (TF) sequences [34] and 8017 mRNA sequences that are available for download from the NCBI ESTs database. We used the BLAST tool [35] to remove the redundant sequences. First, we used each transcription factor sequence to perform a BLAST search against the transcription factor data to delete the redundant sequences. If the BLAST result revealed that two sequences had a >95% match ratio and a >85% coverage ratio, we removed the shorter sequence as a redundant sequence. The match ratio is defined as the ratio of the number of identical bases in the aligned sequences to the total length of the aligned sequences, and the coverage ratio is defined as the total length of the aligned sequences relative to the length of the longer sequence among the two transcription factor sequences. After the pre-processing, there were 229 non-redundant transcription factor (TF) sequences remaining. The sequences that we used are available at OrchidBase ( http://140.116.25.218/EST/) [36].
Because the mRNA data could overlap with TF data, we continued to remove the mRNA sequences that overlapped with the TF data. We performed a BLAST search against the mRNA data for each TF sequence. If the BLAST result revealed that one mRNA sequence had an >85% coverage ratio or 90% overlapping ratio with the TF sequence, then we removed the mRNA sequence as an overlapping sequence. The overlapping ratio was defined as the total length of the aligned sequences relative to the length of the shorter sequence among these two sequences.
Qualified siRNA model for our strategy
Our strategy was designed to screen all of the genes in the genome or to screen candidate genes that had been narrowed down using previous data or experimental analyses. Therefore, without loss of generality, we defined two disjoint sets of genes. We first defined the set of candidate-genes, C = {c1, c2,…, cn}, which contained the candidate genes of the RNAi analysis. Second, we defined the set of excluded-genes, E = {e1, e2,…, em}, which contained all of the genes except the candidate-genes. Because silencing these excluded-genes would have likely complicated further analysis, our RNAi design attempted to target the candidate-genes and avoid silencing the excluded-genes. Furthermore, to utilize the off-target effect to design the proper siRNAs for the analysis, we employed two user-defined parameters, dT and dN, to determine the specificity of the designed siRNAs. First, the user can employ dT to define the maximum number of sequence mismatches that will be tolerated between the designed siRNA and its target. Second, the user can adjust dN to define the minimum number of sequence mismatches that will be allowed between the designed siRNA and its non-target to prevent the designed siRNA from targeting unanticipated genes. Before we can provide a formal definition for a qualified siRNA and its target gene(s), we must first introduce the definition of a qualified sequence as follows:
Given a set of n candidate-genes, C = {c1, c2,…, cn}, a set of m excluded-genes, E = {e1, e2,…, em}, and three integers, dT, dN and L (L >dN>dT), a sequence r of length L is determined to be a qualified sequence if and only if there exists a subset of candidate-genes T, T ⊆ C, such that for each gene ti ∈ T, HD(r, ui) ≤dT for some length L substring ui of ti and for each gene gi ⊆ {E ∪ C−T}, HD(r, u′i) ≥dN for any length L substring u′i of gi. Here HD(,) represents the Hamming distance between two strings.
We defined a qualified siRNA r′ as the reverse complement sequence of a qualified sequence. Therefore, by definition, finding a qualified siRNA is equal to finding a qualified sequence. Moreover, because there are no more than dT mismatches between each gene ti ∈ T and the qualified siRNA r′, T would be the target gene set of the qualified siRNA r′. Conversely, {E ∪ C−T} is the non-target gene set of r′ because each gene gi ⊆ {E ∪ C−T} has at least dN mismatches with the qualified siRNA r′. Each qualified siRNA can silence some of the candidate-genes and avoids silencing the excluded-genes; therefore, they can be used to perform an RNAi experiment that will identify genes that are involved in a specific phenotype.
Enumerating subsequences and computing subsequence similarity
To obtain sequences for the qualified siRNAs, we first enumerated all of the subsequences of length L from the candidate-genes with a sliding scan (Figure 1a). The subsequences that contained an undetermined nucleotide “N” were discarded due to the bad sequencing quality of the sequence. Each subsequence from where the sequence was derived and its original position was recorded. Therefore, every subsequence could be differentiated by its original sequences and its position. We temporarily marked these subsequences as candidates for qualified sequences. Next, we computed the Hamming distances of the pairwise subsequences to measure their sequence similarity. If the Hamming distance between two subsequences was equal to or smaller than dT, these two subsequences were considered neighbors. However, if the Hamming distance between the two subsequences was larger than dT and smaller than dN, these two subsequences were considered far_neighbors.
Figure 1.

Schematic diagram of each step of our heuristic method. (a) The process of enumerating the subsequences from the candidate-genes. The green sequence represents a candidate-gene, and the blue sequences represent the subsequences that were derived from this candidate-gene by the sliding scan. (b) Diagram of the relationship between the subsequences. The blue lines C1, C2,…, C8 represent the candidate-genes, and S1, S2,…, S11 are the subsequences that were enumerated from these candidate-genes. A solid line between two subsequences indicates that the two subsequences are neighbors, and the dotted line indicates that the two subsequences are far_neighbors. Each subsequence is marked as a candidate for a qualified sequence. (c) Diagram of the relationship between the marked subsequences after the far_neighbor examination. The subsequences S5, S9 and S10 are unmarked because they all contained a far_neighbor. However, S2 is still marked because one of its neighbors was located in C4, and its far_neighborS5 was also located in C4. In this situation, we are not concerned about whether the siRNA that was designed based on S2 will recognize S5 because C4 is already the target gene of this siRNA. Therefore, S2 is still marked as a candidate for the qualified sequence. (d) Diagram of the relationship between the marked subsequences after the powerful subsequence examination. S1, S4 and S6 are unmarked because they are not powerful subsequences and are all dominated by S2. (e) Diagram of the relationship between the marked subsequences after the excluded-gene hit examination. Ei is one of the excluded-genes, and the dot-dashed line indicates that the Hamming distance between a marked subsequence and a substring that is located in an excluded-gene is less than dN; this scenario also indicates that this marked subsequence contains an excluded-gene hit. Because any subsequences that contain an excluded-gene hit will be unmarked, S11 is unmarked.
According to the sequence similarity, we obtained the neighborhood relationship of all of the similar subsequences (Figure 1b). Let N(r) denotes the set of all neighbors of the subsequence r. According to the off-target effect, because r is very similar to its neighbors, when the reverse complement sequence of r is used as an siRNA in an RNAi experiment, it is possible that this siRNA will also target the neighbors of r. Therefore, the neighborhood relationship of these subsequences allowed us to determine the potential target genes of the siRNAs that were designed based on these subsequences.
Far_neighbor and powerful subsequence examination
To prevent the targeting of unanticipated candidate-genes, each qualified sequence was required to contain a Hamming distance of at least dN with any substrings of each gene with the exception of its own target genes. However, because dT and dN are defined by users, a subsequence may have far_neighbors. If we designed an siRNA that was derived from a subsequence that contained far_neighbors, this designed siRNA would not be guaranteed to target these far_neighbors. Therefore, in this situation, our algorithm avoids selecting subsequences that contain far_neighbors to design the siRNA, and it would unmark the subsequence with far_neighbors unless the far_neighbors of a subsequence were derived from the genes from which its neighbors were derived (Figure 1c).
To reduce the computing time that was required to perform the next steps, we attempted to remove the less useful subsequences. To achieve this, we first looked for a “powerful subsequence”. Any subsequence P that satisfied equation (1), where ∪ is the union operator, was defined as a powerful subsequence, and the powerful subsequence dominated its neighbors. Because the powerful subsequence is expected to target all of its neighbors' target genes, the removal of these subsequences (their target genes can be covered by the powerful subsequence) will save computational resources without causing any side effects. (Additional file 1, Figure S1 shows an example of a powerful subsequence and the subsequences that would be unmarked.)
| (1) |
To identify the powerful subsequences, we examined each subsequence to determine whether it satisfied equation (1). After identifying the powerful subsequences, we continued to unmark the subsequences that were dominated and were not powerful subsequences (Figure 1d). These unmarked subsequences were ignored in our later analysis.
Excluded-gene hit examination
To prevent the targeting of excluded-genes, we continued to examine whether each marked subsequence displayed a Hamming distance at least a dN to every excluded-gene. For any subsequence t, if there existed a subsequence u of an excluded-gene such that HD(t, u) < dN, HD(t, u) represented the Hamming distance between t and u, then t is determined to contain an excluded-gene hit and this subsequence t would be abandoned. To perform this work, we enumerated all of the substrings of length L from the list of excluded-genes by utilizing a sliding scan. Next, we computed the Hamming distance between these substrings and the marked subsequences. We unmarked the subsequences that contained an excluded-gene hit (Figure 1e). The remaining marked subsequences were therefore determined to be qualified sequences, and the reverse complement sequences of these qualified sequences were qualified siRNAs.
Redundancy reduction
All of the qualified siRNAs are expected to silence the candidate-gene(s) without silencing the excluded-gene; however, several siRNAs could target the same genes. Because these qualified siRNAs are redundant to each other, only the effective siRNA will be selected. To properly evaluate the effective siRNA, we use existing siRNA designing rules to evaluate the effective siRNA. In plants, design rules for only artificial microRNAs, amiRNAs, (microRNAs are another class of small regulatory RNA [37]) have been proposed [38]. The proposed amiRNA targeting rules have been shown to share several features with the proposed siRNA designing rules in animal systems [39,40]. We used the siRNA designing rules proposed by Reynolds et al. [40] for selecting siRNA in our analysis. Three siRNA designing rules proposed by Reynolds [40], Hsieh [41] and Takasaki [42] are currently included in our program, allowing users to select their preferred design rules.
If more than one qualified siRNA passed the evaluation, then we continue to calculate their Hamming distance sum. The Hamming distance sum, HD-sum for short, is the sum of all of the Hamming distances between a qualified sequence and all of its neighbors. A smaller HD-sum indicates that a qualified sequence is more similar to its neighbors, and the qualified siRNA that is derived from it may have a higher gene silencing efficiency. Therefore, when two qualified sequences had the same target genes, we retained the qualified sequence that displayed the smaller HD-sum. If two qualified sequences both displayed the same HD-sum, we discarded the subsequences that displayed the maximum Hamming distance to one of its neighbors. After we eliminated the redundancies, we obtained the non-redundant qualified sequences.
Construction of a pB7GWIWG2-derived clone
The primer pairs PhaTF15-hpRNA F/PhaTF15-hpRNA R, PhaTF21-hpRNA F/PhaTF21-hpRNA R, PhaTF60 hpRNA F/PhaTF60 hpRNA R and PhaRNA hpRNA F/PhaRNA hpRNA R were used to anneal to double-stranded (dsDNA) (Table 1). Two microliters of each primer (15 μg/μl) was mixed with its complementary primer at 72°C for 10 min and the solution was transferred to 25°C for an additional 10 min. The dsDNA was then cloned into the pCR8®/GW/TOPO® Gateway entry vector (Invitrogen) by following the manufacturer's recommendations. The pCR8®/GW/TOPO dsDNA construct was sequenced to confirm the sequence of the cloned fragments. The LR Clonase II enzyme (Invitrogen) was used to transfer the cloned fragments into pB7GWIWG2 [43] to generate pB7GWIWG2-PhaTF15-hpRNA.
Table 1.
Primers used in this study
| Primer | Nucleotide sequence |
|---|---|
| TF106 F |
5′-CCAAAACGGCCGAGGACCCC-3′ |
| TF106 R |
5′-CGCCACACACCTGACGGTCC-3′ |
| TF157 F |
5′- TCATGAACTCGTCGCCTTCCGA-3′ |
| TF157 R |
5′-GCTCGCCCTTCATACGTGGCA-3′ |
| TF187 F |
5′-TGCAAGTTCCAGCAATGCTCCT-3′ |
| TF187 R |
5′-AGAACAACAGGGATGGTGTGCATC-3′ |
| PhaTF15-hpRNA F |
5′-CCTGCGATTTCGACTATAAGT-3′ |
| PhaTF15-hpRNA R |
5′-ACTTATAGTCGAAATCGCAGG-3′ |
| PhaTF21-hpRNA F |
5′-TTACGGAGACCATCTTAAAAC-3′ |
| PhaTF21-hpRNA R |
5′-GTTTTAAGATGGTCTCCGTAA-3′ |
| PhaTF60 hpRNA F |
5′-CAACTTATGCCACTATGCAAT-3′ |
| PhaTF60 hpRNA R |
5′-ATTGCATAGTGGCATAAGTTG-3′ |
| PhaRNA hpRNA F |
5′-TGGTTCCGGTGGTGGGGGCTA-3′ |
| PhaRNA hpRNA R |
5′-TAGCCCCCTCCTCCGGAACCA-3′ |
| CymMV CPF |
5′-GAAATAATCATGGGAGAGCC-3′ |
| CymMV CPR |
5′-AGTTTGGCGTTATT CAGTAGG-3′ |
| PR1 F |
5′-AGGACCCTGGCGTCTAAAG-3′ |
| PR1 R |
5′-TATTACAAATCAAACCGCTAAAG-3′ |
| NPR1 F |
5′-CGCATTTGTGTCGTAGTTTAT-3′ |
| NPR1 R |
5′-GGCCTTGCTCCTTTAGTTA-3′ |
| TF15 F |
5′-TGAATTAGCACAAGATCCGAT-3′ |
| TF15 R |
5′-AGCAATGGAAACAGACCACCAAT-3′ |
| TF21 F |
5′-AGGGACGGAGATGCCAAGAAGGC-3′ |
| TF21 R |
5′ -CAACCATCCAGCTAATCTATC-3′ |
| TF60 F |
5′-ATGATTTCAAAATTTTATCAGTT-3′ |
| TF60 R |
5′-CAATGGAATACTATTCTGACCA-3′ |
| Pha RNA F |
5′-AAGCCGTCAAGTACAAGGGC-3′ |
| Pha RNA R |
5′-GCTAATCAGTTGCAAGAATTA-3′ |
| Ubiqutin10 F |
5′-CCGGATCAGCAAAGGTTGA-3′ |
| Ubiqutin10 R | 5′-TCAGGCGGAGGACAAGATG-3′ |
Transient gene silencing assay
pCambia-CymMV-Gateway, pB7GWIWG2 and their derivatives were transformed into Agrobacterium tumefaciens LBA4404 by electroporation. The A. tumefaciens strains were cultured in 2 ml of YEB of medium containing 100 mg/l kanamycin and 100 μM acetosyringone overnight at 28°C . One milliliter of the bacterial culture was then transferred into 10 ml of YEB medium containing 100 mg/l kanamycin and 100 μM acetosyringone and was further incubated at 28°C until the OD reached approximately 1.0–1.2. The A. tumefaciens cultures were then centrifuged at 3000 ×g for 10 min; the cells were resuspended in 1 ml of infiltration medium (10 mM MES, 10 mM MgCl2, 100 mM acetosyringone) and incubated at room temperature for 3 h. Each Phalaenopsis plant was then infiltrated with 100 μl of the A. tumefaciens suspension. The plant RNA was extracted and analyzed 4 days after the agroinfiltration.
RNA isolation and semiquantitative RT-PCR analysis
To conduct the semiquantitative RT-PCR experiment, the total RNA was extracted from the orchid plant as previously described [44]. One microgram of RNA was treated with RNase-free DNase (Ambion, 1 unit) for 0.5 h to eliminate any genomic DNA contamination. Next, 0.5 μg of DNA-free RNA for each sample was used for the synthesis of the first strand of cDNA using Moloney murine leukemia virus (MMLV) reverse transcriptase following the manufacturer's instructions (Invitrogen). The PCR amplification was performed using gene-specific oligonucleotide primers according to a previously described method [45]. The PCR products were separated by electrophoresis and visualized with ethidium bromide staining. Ubiquitin 10 was used as an internal control. The primer pairs used in the semiquantitative RT-PCR analysis are listed in Table 1.
Results
A case study using the orchid ESTs library
To verify the applicability of our strategy, we used an established Orchid (Phalaenopsis equestris) expressed sequence tags (ESTs) library in our initial test. The ESTs library contained 229 predicted transcription factor sequences and 7721 mRNA sequences (see the subsection “OrchidEST library” in the Methods section).
To use our strategy to screen for transcription factors that are involved in plant defense responses, we defined the predicated transcription factors as candidate-genes and the remaining mRNAs as excluded-genes. A previously described transcription factor, TF15, was used as a positive control [46]. Before we applied our strategy to screen for transcription factors, we first needed to design qualified siRNAs to silence the candidate-genes (see the subsection “Qualified siRNAs for our strategy” in the Methods section). Obtaining qualified siRNAs that target the maximum number of genes was desirable to minimize the number of experiments that were performed for the RNAi screening. However, this problem is NP-hard [we used the distinguishing substring selection problem (DSSP) [47,48] to prove that finding a qualified siRNA that has the maximum number of target genes is NP-complete, and the proof is presented in Additional file 2. Moreover, to analyze every candidate-gene by RNAi screening, we needed to obtain sufficient numbers of qualified siRNAs to silence all of the candidate-genes. Therefore, we needed at least k qualified siRNAs such that ∪1≤i≤kTi = C, where Ti is the target gene set of siRNA r′i, and C is the set of candidate-genes. To obtain a sufficient number of qualified siRNAs and their corresponding target genes within a reasonable time, we provided a heuristic algorithm. This heuristic algorithm was incorporated into the workflow that was used to design the siRNAs that were needed in the first-round analysis of our strategy. The flowchart is depicted in Figure 2.
Figure 2.

Flowchart of the design of the qualified siRNAs that were used in the first-round analysis of our strategy. The first three stages demonstrate the flow of the heuristic algorithm that we proposed. The qualified sequences can be derived by this heuristic algorithm, and the complement sequences of these qualified sequences are the qualified siRNAs. Based on the availability of the qualified siRNAs, we can then select the siRNAs for the first-round RNAi analysis from the qualified siRNAs in stage 4.
Because previous experiments indicated that 21 nucleotides were sufficient to trigger RNAi in plants, we set the length (L) of the qualified siRNA to 21 [49]. Previous reports also indicated that siRNAs can target genes with four mismatches [13,50]. Therefore, we first tolerated the presence of four mismatches (dT) between an siRNA and its target gene. To avoid the target of unanticipated genes by the siRNA, each siRNA was designed to contain at least five mismatches (dN), with every gene excluding its own target genes. Therefore, in this case study, we selected the parameters dT = 4, dN = 5 and L = 21 for our heuristic algorithm to design the qualified siRNAs.
In the first stage of our heuristic algorithm, we enumerated 191,087 subsequences of length 21 from these 229 TF sequences and then computed the pairwise Hamming distances between these subsequences (see the subsection “Enumerating subsequences and computing subsequence similarity” in the Methods section). According to the similarity relationship of these subsequences, we obtained the information regarding the potential target genes of the siRNAs that were designed based on these subsequences. Next, to ensure that the designed siRNAs contained at least a dN distance to their non-target genes and to remove the less powerful subsequences, we performed a far_neighbor and powerful subsequence examination of these subsequences, and we unmarked the disqualified subsequences (see the subsection “Far_neighborand dominating subsequence examination” in the Methods section). After this examination, 188,889 subsequences were still marked. To prevent the designed siRNAs from targeting the excluded-genes, we continued to perform the excluded-gene hit examination of the remaining marked subsequences in the third stage (see the subsection “Excluded-gene hit examination” in the Methods section). In total, 106,188 subsequences passed this examination, and these subsequences were the qualified sequences.
In stage 4, the subsequences were selected from the qualified sequences, and they were used as templates to design the qualified siRNAs. However, some of the qualified siRNAs that were derived from these qualified sequences may have targeted the same genes, which would have resulted in redundancy; therefore, we incorporated some pre-proposed siRNA design rules [40-42] in the redundancy reduction stage to select the best sequences for this use (see the subsection “Redundancy reduction” in the Methods section). After the reduction was performed, 2,147 non-redundant qualified siRNAs remained. To screen every candidate-gene at least once, we needed at least k qualified siRNAs, r1 … rk, such that ∪1≤i≤kTi = S, where Ti is the target gene set of siRNA ri. Moreover, to minimize the cost, we also wanted to minimize the number of first-round RNAi experiments. The problem of satisfying ∪1≤i≤kTi = S with a minimum of k qualified siRNAs is exactly the set cover problem that has been proven to be an NP-hard problem [51]. Therefore, we adopted the greedy algorithm for the set cover problem to select the siRNAs [52,53]. Furthermore, to increase the examination frequency of each candidate-gene, we slightly modified this greedy algorithm (see Additional file 3 for more detail). A total of 94 siRNAs were selected from the 2,147 non-redundant qualified siRNAs by our modified greedy algorithm (the list of the selected siRNAs is reported in Additional file 4: Table S1). According to our computational result, these 94 selected siRNAs are sufficient to examine all 229 candidate-genes in the first-round of the RNAi experiments (Table 2). Any researchers who need our program can email to us or visit the website http://algorithm.cs.nthu.edu.tw/RNAi_screening.php.
Table 2.
The detailed results of our testing on the Phalaenopsis ESTs data with different parameters
| Parameters | dT = 3,dN = 5,L = 21 | dT = 4,dN = 5,L = 21 | dT = 4,dN = 6,L = 21 |
|---|---|---|---|
|
Number of enumerated subsequences |
191,087 |
191,087 |
191,087 |
|
Number of subsequences (afterFar_neighborand powerful subsequence examination) |
173,106 |
188,889 |
117,203 |
|
Number of qualified siRNAs |
100,890 |
106,188 |
6,443 |
|
Number of non-redundant qualified siRNAs |
487 |
2,147 |
352 |
|
Average number of target genes of a qualified siRNA |
1.0583 |
1.1163 |
1.0852 |
|
Number of qualified siRNAs (number of target gene > 1) |
5,565 |
10,863 |
491 |
|
Average number of target genes of qualified siRNAs (number of target gene > 1) |
2.0577 |
2.1370 |
2.1181 |
|
Number of selected siRNAs in first round analysis |
142 |
94 |
162 |
|
Number of candidate-genes examined in first round analysis |
229 |
229 |
227 |
| Average examination frequency of each candidate-gene | 1.153 | 1.170 | 1.110 |
From these result we can see that the number of qualified siRNA has important influence on the number of used siRNAs in first round analysis.
Hierarchical RNAi analysis strategy
Once we had selected the 94 siRNAs (See Additional file 4: Table S1 for more detail), we were able to perform our strategy, hierarchical RNAi analysis, which contained two rounds of RNAi analysis (Figure 3 shows the schematic diagram of the hierarchical examination process). In the first round of RNAi analysis, we observed phenotypes and correlated those phenotypes to at least one of the genes that were affected in the experiment. Thus, we can rapidly exclude the candidate-genes that did not affect the target phenotype from the suspect list, and we can also determine which candidate-genes could be related to the target phenotype. To identify the associated gene more precisely, a second round of analysis was needed. However, in an RNAi experiment, the selected siRNA and its reverse complement sequence (the selected siRNA sense strand) are employed to form a dsRNA that triggers RNAi. Previous studies have demonstrated that both of the dsRNA strands can be loaded into the RNA-induced silencing complex (RISC) and that both strands can facilitate RNAi [54-56]. Therefore, in addition to the target genes of the selected siRNA, we also needed to examine the possible target(s) of the complementary strand of the selected siRNA. The potential target genes contained a subsequence that had at most dT mismatches (in this case, dT = 4) with the selected siRNA sense strand. To precisely identify the associated gene from these affected genes, we designed dsRNAs that would specifically target all of the possible target genes in the second-round analysis (Figure 3). According to our statistical results from each sense strand of the qualified siRNA, the average number of potential targets of the sense strand was 0.6511; thus, only a limited number of genes could have been affected by the siRNA sense strands in each RNAi experiment in the first-round analysis.
Figure 3.

Schematic diagram of the hierarchical RNAi analysis for identifying genes that are involved in a specific phenotype. R = {r1, r2, …, rk} is the set of qualified siRNAs that were selected by our modified algorithm. The candidate-genes in each ellipse are the target genes of each available siRNA ri. In the first-round RNAi analysis, we can quickly determine which candidate-genes could be related to the target phenotype based on the RNAi experiment results. The green ellipse represents the target phenotype that was affected in the RNAi experiment with ri, whereas the blue ellipse indicates that the target phenotype was not affected. In the second-round RNAi analysis, we further examined the candidate-genes in the green ellipses to precisely identify the related genes. However, the complementary strand of the selected siRNA, r′i, can also be loaded into the RISC, and it could facilitate RNAi. A gene that contains a subsequence that contains no more than dT mismatches to r′i would be regarded as the possible target gene of r′i, which is defined as Xi. To precisely identify the associated gene from the genes that are affected by ri and r′i, we would silence those genes one-on-one to identify which genes were definitively associated with the target phenotype.
Experimental verification
To verify if siRNA can silence multiple genes with four mismatches, we first randomly selected an siRNA, siRNA-G41. The predicted targets of siRNA-G41 are TF106, TF157, and TF187. The number of mismatches between siRNA-G41 and its targets, TF106, TF157, and TF187, are 4, 0 and 2, respectively. The experimental results showed that siRNA-G41 successfully silenced PhaTF106, PhaTF157 and PhaTF187 (Figure 4), demonstrating that it is feasible to simultaneously silence multiple genes with a carefully designed siRNA and that siRNA can silence the target gene even with 4 mismatches to its target genes.
Figure 4.
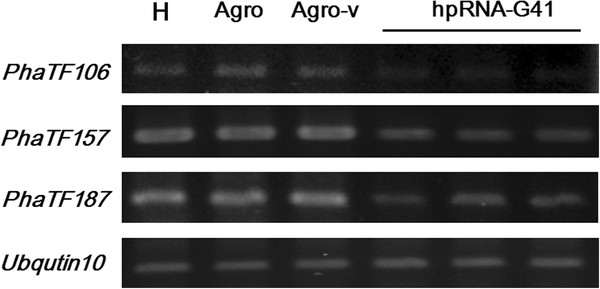
Using siRNA-G41 to simultaneously silence three target genes. The effects of silencing three putative transcription factors in plants that were transfected with a hairpin RNA-G41 are shown. The RNA levels of Pha106, Pha157 and PhaTF187 were analyzed in healthy (H) plants, in plants that were infiltrated with Agrobacterium (agro), in plants that were infiltrated with Agrobacterium that carried an empty pB7GWIW2 vector (agro-v) or in plants that were infiltrated with Agrobacterium that carried pB7GWIW2 to deliver hairpin RNAs designed from siRNA-G41 (hpRNA-G41). Phalaenopsis Ubiquitin 10 was used as an internal control.
Among the 94 siRNAs, we observed one siRNA, siRNA-G55, which targeted two transcription factors, TF15 and TF21. TF15 has been previously reported to be involved in the critical salicylic acid (SA)-related plant defense response [46]; the depletion of TF15 in SA-treated plants resulted in the decreased expression of orchid pathogenesis-related gene 1 (PhaPR1), which is a marker of the plant defense response, and the important central regulator PhaNPR1. To confirm that our strategy can help us to determine that TF15 is involved in plant defense responses, we selected siRNA-G55 to perform a first-round RNAi experiment that utilized Agrobacterium-mediated dsRNA delivery. The dsRNA that was derived from siRNA-G55 silenced both TF15 and TF21 but not the randomly selected transcription factor TF60.
The first-round experimental result showed that siRNA-G55 successfully silenced both PhaTF15 and PhaTF21, and the RNA levels of PhaPR1 and PhaNPR1 were affected (Figure 5). This scenario verified that we could observe a change in the target phenotype when a related gene is affected in the first-round analysis. According to our strategy, we then select two siRNAs from qualified siRNAs that are specific to PhaTF15 or PhaTF21 for the second-round analysis. Because the complementary strand of siRNA-G55 also targeted a Phalaenopsis gene (PhaRNA, accession number CB034920 [34]), we also designed an siRNA that was specific to this RNA sequence.
Figure 5.
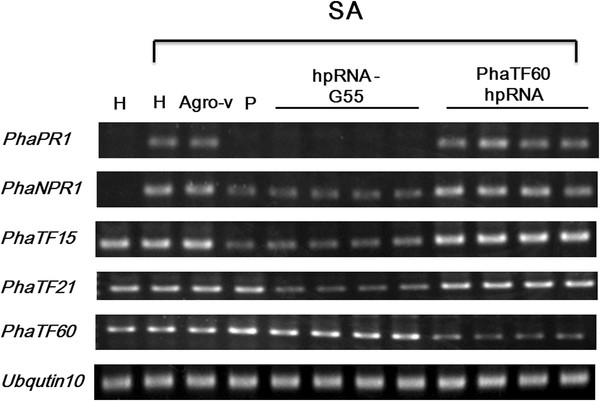
Using siRNA-G55 in first-round analysis. The effects of silencing the marker genes that are involved in the salicylic acid (SA)-related plant defense response pathway in plants that were transfected with a hairpin RNA-G55. The RNA levels of PhaPR1, PhaNPR1, PhaTF15, PhaTF21 and PhaTF60 were analyzed in healthy (H) plants, plants that were infiltrated with Agrobacterium that carried an empty pB7GWIW2 vector (agro-v), or Agrobacterium that carried pB7GWIW2 to deliver different hairpin RNAs that were designed from siRNA G55 (hpRNA-G55) or were specific to PhaTF60 (PhaTF60-hpRNA). The Agrobacterium strain that carried a partial PhaTF15 cDNA was used as a positive control (P), and PhaTF60 was used as a negative control. The plants that were pretreated with SA to induce the SA-related plant defense response are indicated. Phalaenopsis Ubiquitin 10 was used as an internal control.
The second-round experimental data indicated that both PhaPR1 and PhaNPR1 were silenced in the PhaTF15- and PhaTF21-silenced plants, but not in the PhaRNA-silenced plant (Figure 6). These data suggested that PhaTF15 and PhaTF21 are involved in the SA-related plant defense response pathway but that PhaRNA is not. This result showed that our hierarchical strategy is capable of identifying genes that are involved in a specific phenotype.
Figure 6.
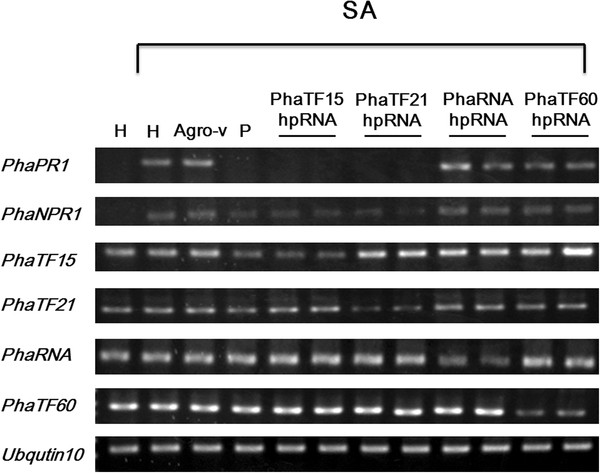
Identifying the associated gene more precisely in the second-round analysis. The knockdown effects of the marker genes involved in the salicylic acid (SA)-related plant defense response pathway in plants that were transfected with hairpin RNA and that are specific to PhaTF15 and PhaTF21. The RNA levels of PhaPR1, PhaNPR1, PhaTF15 and PhaTF60 were analyzed in healthy (H) plants, plants that were infiltrated with Agrobacterium that carried an empty pB7GWIW2 vector (agro-v), or Agrobacterium that carried pB7GWIW2 to deliver different hairpin RNAs that were specific to PhaTF15 (PhaTF15 hpRNA), PhaTF21 (PhaTF21 hpRNA), PhaRNA (PhaRNA), or PhaTF60 (PhaTF60 hpRNA). The Agrobacterium strain that carried the partial PhaTF15 cDNA was used as a positive control (P), and PhaTF60 was used as a negative control. The plants that were pretreated with salicylic acid (SA) to induce the SA-related plant defense response are indicated. Phalaenopsis Ubiquitin 10 was used as an internal control.
Discussion
Our strategy provides a method for discovering new phenotypes that is an alternative to the traditional one-on-one RNAi experiments. The analysis of the mutant collection of model organisms revealed that single-gene knockouts did not usually exhibit an observable phenotype [57-59]. Our strategy facilitates the more efficient screening of single genes that are involved in a phenotype and the simultaneous targeting of several genes; this simultaneous targeting provides an opportunity to observe additional phenotypes that can be observed only when multiple genes are silenced.
Although an siRNA can target its complementary genes, the number of mismatches that are allowed between the siRNA and its target gene(s) has not been strictly defined. According to our experimental results, at least 4 mismatches are allowed between the siRNA and its target gene(s) (Figure 4). Besides, previous reports have indicated that the targeting of the siRNA to the RNA could be affected by three, four or even five mismatches [13,50]. Therefore, using a threshold number of mismatches to distinguish the target and the non-target genes of an siRNA could generate a prediction error. To prevent the unexpected target genes of a certain siRNA, in our qualified siRNA model, two user-defined parameters, dT and dN, were employed to predict the target and non-target genes of the qualified siRNAs, respectively (the genes within a dT distance of the siRNA are its targets, whereas the genes that have at least a dN distance to the siRNA are its non-targets). Theoretically, a smaller dT / greater dN should benefit the accuracy of the prediction of target/non-target genes because the probability of being a target gene tends to increase as the number of mismatches decreases. Nevertheless, according to our experimental results for the Orchid EST data (Table 2), a smaller dT / greater dN decreased the number of non-redundant qualified siRNAs and limited the reduction of the RNAi experiments. When the pairings of dT and dN were designed differently, they affected the number of RNAi experiments that would be required to screen the entire genome; however, as long as the dT is set within a reasonable range (dT < 5), each candidate-gene will be examined by at least one qualified siRNA.
In addition, our strategy allows the user to design two disjoint sets of genes, which are the candidate-gene set and the excluded-gene set. After that researchers narrow down the genes that could be associated with a specific phenotype into a set of candidate-genes by utilizing existing knowledge or an experimental analysis; this design can help them to focus their resources on the study of these candidate-genes. However, if a user cannot exclude any genes in advance or wants to perform a whole-genome analysis, our heuristic algorithm and hierarchical strategy can still assist in the reduction of the number of RNAi experiments that are required to perform a whole-genome analysis. We found that additional RNAi experiments can be eliminated when the excluded-gene set is empty. For example, for the orchid ESTs data, when the candidate-genes included all of the mRNA sequences and the TF sequences and no genes were excluded, our heuristic algorithm identified 3,092,859 qualified siRNAs with the parameters L = 21, dT = 4 and dN = 5. Only 1,674 RNAi experiments would be required to analyze 7,796 candidate-genes in the first-round RNAi analysis because there were no excluded-gene hit restrictions on the qualified siRNAs; therefore, more qualified siRNAs that can target more genes could be obtained.
Previous studies have indicated that both strands of a dsRNA can be loaded into the RNA-induced silencing complex (RISC), and they can both facilitate RNAi [54-56]. Therefore, whenever a mutant phenotype is observed, the target genes of the qualified siRNA and the potential target genes of the complementary strand of the qualified siRNAs must be examined in the second-round RNAi analysis. This step is necessary because our qualified siRNA was initially designed to be complementary to the candidate-genes. According to our analysis, the average number of possible targets of the complementary strand of a qualified siRNA was 0.6511, which indicates that only a few additional genes required screening in the second-round analysis.
Schwab et al. [60] used artificial microRNAs (amiRNA) to investigate the specificity of miRNA and showed that amiRNA can target mRNAs with a few mismatches. Artificial microRNAs can knockdown single and multiple genes and thus are an effective tool for knocking down several related but not identical genes. Schwab et al. [60] developed a tool, Web MicroRNA Designer (WMD), to facilitate the design of amiRNAs for silencing of single and multiple genes. Our strategy and that by Schwab et al. [60] are similar in that they both design small RNA to target single or multiple genes. However, WMD allows users to design effective amiRNA to knockdown their interested genes, whereas our strategy aims to reduce the initial experiments needed in a genome-wide screening.
Our initial screening using hpRNA-G55 allowed us to knockdown PhaTF15 and PhaTF21 simultaneously (Figure 5), and a second round of analysis using siRNAs specific to PhaTF15 and PhaTF21 individually indicated that TF21, similar to the previously identified transcription factor TF15, is involved in plant disease resistance (Figure 6). Interestingly, the alignment of TF15 and TF21 revealed that these two TFs share a conserved region of approximately 180 nucleotides (see Additional file 1: Figure S2 for more detail). We also found that this conserved region contains a zinc finger AN1 domain, and the target region of siRNA-G55 is located in the region of this domain. To determine whether siRNA-G55 could silence all of the genes that contain the zinc finger AN1 domain, we queried the SWISS-PROT database for all of the genes of Arabidopsis thaliana that contain the zinc finger AN1 domain. Seventeen genes were reported to contain this domain, and only two of these seventeen genes may be affected by the siRNA-G55 (when set at dT = 4). It will be interesting to further determine their roles in the plant defense response.
Conclusions
In this study, we developed a strategy to minimize the efforts required to perform high-throughput RNAi screening to identify genes involved in a specific phenotype. Our strategy took advantage of the biological phenomenon of the off-target effect of RNAi and utilized the set cover algorithm and the group testing method. We also provided a heuristic algorithm to design the siRNAs that can target multiple genes for our strategy. In the case study using the Orchid ESTs library, our computational result showed that 94 qualified siRNAs are sufficient to screen all 229 candidate-genes in the first-round analysis, where 229 siRNAs are required in traditional one-on-one RNAi screening. Furthermore, we verified our strategy with a previously reported transcription factor (TF15) involved in plant defense responses. In the first-round analysis, we used siRNA-G55, which is one of the 94 selected siRNAs, to silence TF15 and TF21 simultaneously, and the experimental data showed that the RNA levels of PhaPR1 and PhaNPR1 were subsequently affected. Moreover, in the second-round analysis, we successfully confirmed that TF15 is involved in plant defense responses only with three additional RNAi experiments. Additionally, we also indicated that TF21 is a previously unidentified transcription factor that is involved in plant defense responses.
Abbreviations
RNAi: RNA interference; dsRNA: double-stranded RNA; RISC: RNA-induced silencing complex; TF: Transcription factor; ESTs: Expressed sequence tags; DSSP: Distinguishing substring selection problem; MMLV: Moloney murine leukemia virus; amiRNA: Artificial microRNA.
Competing interests
The authors declare that they have no competing interests
Authors’ contributions
CHC designed and implemented the algorithms for the analysis, performed the computational analysis, and drafted the paper. HHY and HIW conceived of the study, participated in the design and drafted the paper. HHY, HCL and CEC performed the wet experiments. HHC provided the transcription factor sequence data. HHY and CYT provided constructive opinions and refined the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Figures S1 and S2. A simple example of a powerful subsequence P, and the dotplot of TF15 and TF21 sequences.
Proof to prove that the problem of finding a qualified sequence r maximizing the size of T is NP-complete.
Methods the detail of our modified greedy algorithm for selecting qualified siRNAs to perform the first-round RNAi analysis.
Tables S1-3. The lists of the siRNAs that were selected for first-round RNAi analysis with different parameters.
Contributor Information
Chih-Hung Chang, Email: d948311@oz.nthu.edu.tw.
Hsiang-Iu Wang, Email: d926319@oz.nthu.edu.tw.
Hsiang-Chia Lu, Email: d93633005@ntu.edu.tw.
Cheng-En Chen, Email: f93633004@ntu.edu.tw.
Hong-Hwa Chen, Email: hhchen@mail.ncku.edu.tw.
Hsin-Hung Yeh, Email: hyeh@ntu.edu.tw.
Chuan Yi Tang, Email: cytang@pu.edu.tw.
Acknowledgements
This research was supported by grants from National Science Council, Taiwan [97-2221-E-126-012-MY3 and 97-2221-E-126-013-MY3] and partly supported by grants from National Science Council, Taiwan [98-2313-B-002-043-MY3 and 98-2321-B-002-019-MY3].
References
- Bennetzen JL, Kellogg EA, Lee M, Messing J. A plant genome initiative. Plant Cell. 1998;10:488–493. [Google Scholar]
- Dong Y, Burch-Smith TM, Liu Y, Mamillapalli P, Dinesh-Kumar SP. A ligation-independent cloning tobacco rattle virus vector for high-throughput virus-induced gene silencing identifies roles for NbMADS4-1 and -2 in floral development. Plant Physiol. 2007;145:1161–1170. doi: 10.1104/pp.107.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RK, Schnable PS, Ware D, Fulton RS, Stein JC, Wei FS, Pasternak S, Liang CZ, Zhang JW, Fulton L. et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Chandler VL, Brendel V. The maize genome sequencing project. Plant Physiol. 2002;130:1594–1597. doi: 10.1104/pp.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill BS, Appels R, Botha-Oberholster AM, Buell CR, Bennetzen JL, Chalhoub B, Chumley F, Dvorak J, Iwanaga M, Keller B. et al. A workshop report on wheat genome sequencing: international genome research on wheat consortium. Genetics. 2004;168:1087–1096. doi: 10.1534/genetics.104.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Burr B. International rice genome sequencing project: the effort to completely sequence the rice genome. Curr Opin Plant Biol. 2000;3:138–141. doi: 10.1016/S1369-5266(99)00047-3. [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Rounsley SD, Last RL. Shotguns and SNPs: how fast and cheap sequencing is revolutionizing plant biology. Plant J. 2010;61:922–927. doi: 10.1111/j.1365-313X.2009.04030.x. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Imelfort M, Edwards D. De novo sequencing of plant genomes using second-generation technologies. Brief Bioinform. 2009;10:609–618. doi: 10.1093/bib/bbp039. [DOI] [PubMed] [Google Scholar]
- Liu J, Huang S, Peng X, Liu W, Wang H. Studies on high salt tolerance of transgenic tobacco. Chin J Biotechnol. 1995;11:275–280. [PubMed] [Google Scholar]
- Abebe T, Guenzi AC, Martin B, Cushman JC. Tolerance of mannitol-accumulating transgenic wheat to water stress and salinity. Plant Physiol. 2003;131:1748–1755. doi: 10.1104/pp.102.003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schwab R, Weigel D. Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 2008;53:674–690. doi: 10.1111/j.1365-313X.2007.03328.x. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Keating CD, Kriek N, Daniels M, Ashcroft NR, Hopper NA, Siney EJ, Holden-Dye L, Burke JF. Whole-genome analysis of 60 G protein-coupled receptors in Caenorhabditis elegans by gene knockout with RNAi. Curr Biol. 2003;13:1715–1720. doi: 10.1016/j.cub.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Sugimoto A. High-throughput RNAi in Caenorhabditis elegans: genome-wide screens and functional genomics. Differentiation. 2004;72:81–91. doi: 10.1111/j.1432-0436.2004.07202004.x. [DOI] [PubMed] [Google Scholar]
- Agaisse H, Burrack LS, Philips JA, Rubin EJ, Perrimon N, Higgins DE. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science. 2005;309:1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- Furlong EE. A functional genomics approach to identify new regulators of Wnt signaling. Dev Cell. 2005;8:624–626. doi: 10.1016/j.devcel.2005.04.006. [DOI] [PubMed] [Google Scholar]
- McGinnis K, Chandler V, Cone K, Kaeppler H, Kaeppler S, Kerschen A, Pikaard C, Richards E, Sidorenko L, Smith T. et al. Transgene-induced RNA interference as a tool for plant functional genomics. Methods Enzymol. 2005;392:1–24. doi: 10.1016/S0076-6879(04)92001-0. [DOI] [PubMed] [Google Scholar]
- Hilson P, Allemeersch J, Altmann T, Aubourg S, Avon A, Beynon J, Bhalerao RP, Bitton F, Caboche M, Cannoot B. et al. Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Res. 2004;14:2176–2189. doi: 10.1101/gr.2544504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S. et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Yang JP, Fan W, Rogers C, Chatterton JE, Bliesath J, Liu G, Ke N, Wang CY, Rhoades K, Wong-Staal F, Li QX. A novel RNAi library based on partially randomized consensus sequences of nuclear receptors: identifying the receptors involved in amyloid beta degradation. Genomics. 2006;88:282–292. doi: 10.1016/j.ygeno.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Sun DQ, Melegari M, Sridhar S, Rogler CE, Zhu L. Multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. Biotechniques. 2006;41:59–63. doi: 10.2144/000112203. [DOI] [PubMed] [Google Scholar]
- Xia XG, Zhou HX, Xu ZS. Multiple shRNAs expressed by an inducible pol II promoter can knock down the expression of multiple target genes. Biotechniques. 2006;41:64–68. doi: 10.2144/000112198. [DOI] [PubMed] [Google Scholar]
- Ouyang B, Yang T, Li H, Zhang L, Zhang Y, Zhang J, Fei Z, Ye Z. Identification of early salt stress response genes in tomato root by suppression subtractive hybridization and microarray analysis. J Exp Bot. 2007;58:507–520. doi: 10.1093/jxb/erl258. [DOI] [PubMed] [Google Scholar]
- Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14:836–843. doi: 10.1261/rna.895308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Moffat J, Reiling JH, Sabatini DM. Off-target effects associated with long dsRNAs in Drosophila RNAi screens. Trends Pharmacol Sci. 2007;28:149–151. doi: 10.1016/j.tips.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Kulkarni MM, Booker M, Silver SJ, Friedman A, Hong P, Perrimon N, Mathey-Prevot B. Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods. 2006;3:833–838. doi: 10.1038/nmeth935. [DOI] [PubMed] [Google Scholar]
- Du D, Hwang F. Combinatorial group testing and its applications. 2. Singapore: World Scientific; 2000. [Google Scholar]
- Tsai WC, Hsiao YY, Lee SH, Tung CW, Wang DP, Wang HC, Chen WH, Chen HH. Expression analysis of the ESTs derived from the flower buds of Phalaenopsis equestris. Plant Sci. 2006;170:426–432. doi: 10.1016/j.plantsci.2005.08.029. [DOI] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Fu CH, Chen YW, Hsiao YY, Pan ZJ, Liu ZJ, Huang YM, Tsai WC, Chen HH. OrchidBase: a collection of sequences of the transcriptome derived from orchids. Plant Cell Physiol. 2011;52:238–243. doi: 10.1093/pcp/pcq201. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8:517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nat Struct Mol Biol. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Bo RH, Manola J, Vazquez F, Bare O, Khvorova A, Scaringe S, Sellers WR. A library of siRNA duplexes targeting the phosphoinositide 3-kinase pathway: determinants of gene silencing for use in cell-based screens. Nucleic Acids Res. 2004;32:893–901. doi: 10.1093/nar/gkh238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki S, Kotani S, Konagaya A. An effective method for selecting siRNA target sequences in mammalian cells. Cell Cycle. 2004;3:790–795. [PubMed] [Google Scholar]
- Karimi M, Inze D, Depicker A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002;7:193–195. doi: 10.1016/S1360-1385(02)02251-3. [DOI] [PubMed] [Google Scholar]
- Tian T, Klaassen VA, Soong J, Wisler G, Duffus JE, Falk BW. Generation of cDNAs specific to lettuce infectious yellows closterovirus and other whitefly-transmitted viruses by RT-PCR and degenerate oligonucleotide primers corresponding to the closterovirus gene encoding the heat shock protein 70 homolog. Phytopathology. 1996;86:1167–1173. doi: 10.1094/Phyto-86-1167. [DOI] [Google Scholar]
- Lu HC, Chen HH, Tsai WC, Chen WH, Su HJ, Chang DC, Yeh HH. Strategies for functional validation of genes involved in reproductive stages of orchids. Plant Physiol. 2007;143:558–569. doi: 10.1104/pp.106.092742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Hsieh MH, Chen CE, Chen HH, Wang HI, Yeh HH. A high-throughput virus-induced gene-silencing vector for screening transcription factors in virus-induced plant defense response in orchid. Mol Plant Microbe Interact. 2012;25:738–746. doi: 10.1094/MPMI-10-11-0266. [DOI] [PubMed] [Google Scholar]
- Deng X, Li G, Li Z, Ma B, Wang LS. Genetic design of drugs without side-effects. SIAM J Comput. 2003;32:1073–1090. doi: 10.1137/S0097539701397825. [DOI] [Google Scholar]
- Lanctot JK, Li M, Ma B, Wang SJ, Zhang LX. Distinguishing string selection problems. Inform and Comput. 2003;185:41–55. doi: 10.1016/S0890-5401(03)00057-9. [DOI] [Google Scholar]
- Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17:49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D. Control of leaf morphogenesis by microRNAs. Nature. 2003;425:257–263. doi: 10.1038/nature01958. [DOI] [PubMed] [Google Scholar]
- Garey MR, Johnson DS. Computers and intractability: a guide to the theory of NP-completeness. San Francisco: W. H. Freeman; 1979. [Google Scholar]
- Lovasz L. Ratio of Optimal Integral and Fractional Covers. Discrete Math. 1975;13:383–390. doi: 10.1016/0012-365X(75)90058-8. [DOI] [Google Scholar]
- Johnson DS. Approximation Algorithms for Combinatorial Problems. J Comput System Sci. 1974;9:256–278. doi: 10.1016/S0022-0000(74)80044-9. [DOI] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykanen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107:309–321. doi: 10.1016/S0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Hanada K, Kuromori T, Myouga F, Toyoda T, Li WH, Shinozaki K. Evolutionary persistence of functional compensation by duplicate genes in Arabidopsis. Genome Biol Evol. 2009;1:409–414. doi: 10.1093/gbe/evp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wagner A. Duplicate genes and robustness to transient gene knock-downs in Caenorhabditis elegans. Proc Biol Sci. 2004;271:89–96. doi: 10.1098/rspb.2003.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouche N, Bouchez D. Arabidopsis gene knockout: phenotypes wanted. Curr Opin Plant Biol. 2001;4:111–117. doi: 10.1016/S1369-5266(00)00145-X. [DOI] [PubMed] [Google Scholar]
- Schwab R, Ossowski S, Riester M, Warthmann N, Weigel D. Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell. 2006;18:1121–1133. doi: 10.1105/tpc.105.039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 and S2. A simple example of a powerful subsequence P, and the dotplot of TF15 and TF21 sequences.
Proof to prove that the problem of finding a qualified sequence r maximizing the size of T is NP-complete.
Methods the detail of our modified greedy algorithm for selecting qualified siRNAs to perform the first-round RNAi analysis.
Tables S1-3. The lists of the siRNAs that were selected for first-round RNAi analysis with different parameters.